

Abstract
The synthetic transformation of polypeptides with molecular accuracy holds great promise for providing functional and structural diversity beyond the proteome. Consequently, the last decade has seen an exponential growth of site‐directed chemistry to install additional features into peptides and proteins even inside living cells. The disulfide rebridging strategy has emerged as a powerful tool for site‐selective modifications since most proteins contain disulfide bonds. In this Review, we present the chemical design, advantages and limitations of the disulfide rebridging reagents, while summarizing their relevance for synthetic customization of functional protein bioconjugates, as well as the resultant impact and advancement for biomedical applications.
Keywords: bioactive hybrids, disulfide rebridging, drug delivery, multimodal proteins, site-selective protein modifications
Introduction
Nature imparts diversity in peptides and proteins by chemical modifications to confer additional functional and structural properties after their biosynthesis.1, 2 Inspired by Nature's machinery, there have been tremendous efforts to develop synthetic methodologies that allow the structure and function of existing proteins to be reengineered, thereby providing new avenues for the innovation of biocatalysts,3 biomaterials4 and therapeutic research.5
Given the stringent demands for stability, biocompatibility and biosafety in modern applications, the formation of a homogeneous, well‐defined bioconjugate is crucial. To achieve this, chemical modifications need to be introduced at a single, distinct site (regioselectivity/site‐selectivity) in high yields under mild conditions, preferably with retained tertiary structure and function. However, the precise modification of proteins is challenging since a single functionality needs to be addressed in the presence of abundant unprotected and reactive groups. Although non‐canonical amino acids containing halogen,6 ethynyl,7 or azido8 groups can be engineered at distinct positions in the polypeptide sequence9 for subsequent bioorthogonal reactions, their applications are often restricted by low yields and tedious synthesis of aminoacylated tRNA.10 Thus, the development of chemical tools for the modification of endogenous amino acids that are present in a broad spectrum of proteins and exhibit high degree of chemoselectivity and regioselectivity is highly desirable.
The relatively rare amino acid cysteine is most often exploited due to the selective reactivity of its thiol group towards maleimide moieties under pH control.11 Nevertheless, very few native proteins contain unpaired and accessible cysteine residues12, 13 and recombinant techniques are often required, which could be inconvenient or impossible.14, 15 Moreover, the stability of thioethers formed from the maleimide–cysteine reaction in plasma can hamper its application.16 N‐terminal modifications have been reported as an efficient alternative chemical strategy.17, 18 Tyrosine and tryptophan modifications can be applied to a broader spectrum of proteins but these residues are normally less accessible as they are often buried in the hydrophobic interior of proteins.19, 20, 21
In this context, disuflides have emerged as eminent candidates for site‐selective modification since most proteins contain disulfide bonds that control their stability and biological activity.22 Disulfides are either buried within the protein's folding region or exposed on its solvent accessible surface.23 The solvent accessible disulfides mainly impart stability to the protein, while the disulfides located within the protein's hydrophobic pocket contribute primarily to the biological activity.12, 24 Most therapeutically relevant proteins have at least one disulfide bond close to the surface,25 representing an opportunity for site‐selective modifications. Since its first inception by Brocchini,26, 27 disulfide functionalization has gained wide applications for site‐selective protein modifications to derive various peptide/protein hybrids that expand the repertoire in Nature. The disulfide rebridging strategy has been highlighted recently.28, 29 However, a timely review of the implementation of the function in native biopolymers together with head‐to‐head comparison between the different classes of disulfide rebridging reagents has not appeared yet. The last review of this topic was in 2008 where the implication for protein PEGylation using bis‐sulfones was mainly discussed.25 Thus, this Review article discusses the latest developments of their key features and the progress of disulfide rebridging agents to impart function into proteins or cyclic peptides and to guide the reader in the selection of the appropriate reagents and strategies for disulfide modifications. Reagents used for disulfide stapling to increase stability but without addition of new functional features will be excluded. Highlights of emerging applications of the resultant hybrids and a perspective of the future of this field will also be provided.
I. Disulfide Rebridging Agents: Syntheses, Features and Bioconjugation Products
The syntheses, features and resultant products of the disulfide reagents (Figure 1, I–VII) will be discussed to give an overview of the design of the appropriate reagents. Where possible, the yields and conversion of comparable bioconjugates will be stated. Comments on the stability of some of the bioconjugates will also be given. A Table summarizing some of the key features and properties (Table 1) is provided to guide the reader in the selection of the appropriate reagents for their intended applications using this contemporary modification strategy.
Figure 1.
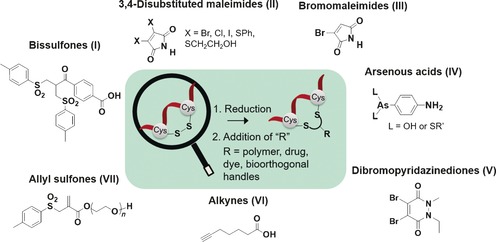
Overview of disulfide rebridging reagents discussed in the text. Bissulfones (I); disubstituted maleimides (II); bromomaleimides (III); arsenous acids (IV); dibromopyridazinediones (V); alkynes (VI); allyl sulfones (VII).
Table 1.
Summary of the key features of the different disulfide rebridging agents and the stability of the resultant conjugates.
| Reagent | Requires activation | Multifunctional | Conjugation yield [%][a] | Selectivity over unpaired Cys | Water soluble | Substrate | Stability of products |
|---|---|---|---|---|---|---|---|
| I | Y[b] | N | 20–40[d] | N | N | peptides, Fab, proteins, antibodies | cleaved by GSH, stable in serum |
| II | N | N | >80[e] | N | N | peptides, Fab, proteins, antibodies | cleaved in GSH, serum and mammalian cells |
| III | N | N | 70[e] | N | N | peptides, Fab | – |
| IV | N | N | quantitative[e] | Y | N | peptides | – |
| V | N | Y | quantitative[e] | N | N | peptides, Fab, antibodies | stable in GSH, serum and buffers |
| VI | Y[c] | N | 3–22[d] | N | N | peptides | – |
| VII | N | Y | 49[d] | N | Y | peptides, proteins | stable in buffers and serum |
[a] Based on conjugation yields to peptides. [b] Pre‐incubation for elimination of protecting group. [c] Requires photoactivation. [d] Isolated yields. [e] Determined from chromatography or spectroscopy.
Criteria for disulfide rebridging
Bissulfones (I) and dibromomaleimides (DBMs, II; Figure 1) produced by the groups of Brocchini, Baker and Caddick represent the earliest developed and most established disulfide rebridging reagents.23, 26, 30 From the findings of these earliest reagents, some of the key steps and criteria in disulfide modification can be summarized as follows: 1) the “activation” of the disulfide by reduction to release two active cysteines for reaction, 2) followed by a fast addition reaction of the reagent “R” (Figure 1) with one reduced thiol group to minimize unfolding, aggregation or disulfide scrambling, and most critically, 3) the second rebridging step of the other proximal cysteine residue to retain the 3D structure and function of the protein. In order to preserve the tertiary structure, potential irreversible denaturation and aggregation of the proteins need to be minimized, and disulfide scrambling reactions should be avoided as well in the case that more than one disulfide is reduced.26, 27, 31 Site‐selectivity is finally achieved by: 1) examination of solvent accessible disulfides from the protein structure, and 2) stoichiometric control of the disulfide rebridging reagent used.23, 26, 30, 32 In the case where there are two disulfides that are equally accessible, a mixture of different isomers could be obtained for the mono‐functionalized hybrid.26
Disulfide reduction
Reduction of the solvent accessible disulfide bridge is usually the first step in the bioconjugation sequence. Here, the selection of the optimal reductant needs to be considered carefully. For example, the mild reducing agent tris(2‐carboxyethyl)phosphine (TCEP) hydrochloride is often used and in some cases, it can even be applied in situ together with the disulfide rebridging reagent without further purification.32, 33, 34, 35 The stronger reductants dithiothreitol (DTT) or 2‐mercaptoethanol usually require neutral pH as they have a narrow effective pH (≥7)36 as compared to TCEP (1.5<pH<8.5), as well as poorer stability in the presence of metal ions.37 Moreover, unreacted DTT must be removed through an additional purification step25 as it reacts more readily with sulfhydryl‐reactive agents.
Bissulfones
The group of Brocchini pioneered the modification of disulfides by developing bis‐alkylation reagents to achieve the PEGylation of a protein's native disulfide bond with high selectivity and efficiency.27 The bis‐sulfone reagent (I) can be prepared from p‐acetylbenzoic acid over a three‐steps synthesis with an overall yield of 28 % (Scheme 1 A).26
Scheme 1.
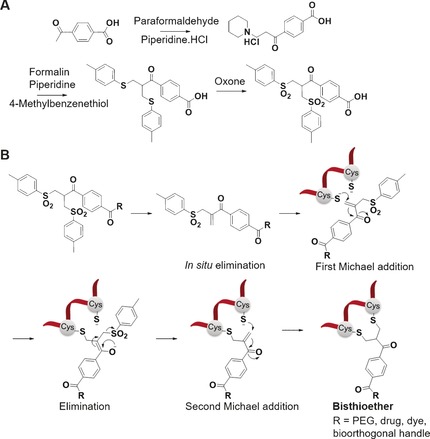
A) Synthesis of bis‐sulfones from p‐acetylbenzoic acid. B) Mechanism of bis‐sulfone reagents in the bis‐alkylation of disulfides to form a three‐carbon bridge between two cysteines of proteins and cyclic peptides.
The bis‐sulfone reagents reported thus allowed the “reannealing” of accessible disulfide sites of peptides or proteins through sequential addition–elimination reactions without the loss of their biological activity or structural integrity.25, 26, 34
Generally, the reactive monosulfone is obtained from the respective bis‐sulfone (Scheme 1 B) after elimination of p‐toluene sulfinic acid, which is formed as by‐product.26, 32 This activation reaction is usually conducted in situ at neutral or slightly basic pH values. The monosulfone comprises an electron‐withdrawing group (e.g., a carbonyl group), an α,β‐unsaturated double bond and a β’‐sulfonyl group. Disulfide rebridging is then initiated by the mild reduction of a solvent accessible disulfide to release its two cysteine thiols while preserving the protein's tertiary structure. In the next step, one thiol group reacts with the corresponding monosulfone to eliminate the second p‐toluene sulfinic acid group, yielding the second Michael system. Thereafter, the second thiol group in close vicinity undergoes a subsequent Michael addition with the concomitant formation of a three‐carbon bridge to yield the bisthioether (Scheme 1 B). The mechanism of disulfide rebridging by bis‐sulfones has been further elucidated by NMR spectroscopy and it has been shown that diastereomers are formed.31 In the absence of thiol groups, side reactions with water can occur at basic pH (>8) due to the reactivity of the alkene group in the monosulfone,38 and thus such high pH should be avoided. Bioconjugation is normally carried out in situ starting with the bis‐sulfones at slightly basic pH values. For peptides and proteins, which are less stable in alkaline solutions, bioconjugation can still be achieved at acidic pH but this requires isolation of the monosulfones.
Post‐functionalization of the bis‐sulfone can be achieved by amide coupling or ester formation to introduce a broad range of functionalities. To overcome the low solubility of the bis‐sulfone reagent, we have further incorporated a triethyleneoxide linker, which is particularly attractive for protein chemistry. Using this approach, we have also derived a library of biohybrids of the cyclic peptide hormone somatostatin (SST)33, 35 with a single disulfide bond. The conjugation to SST typically leads to isolated yields in the range of 20–40 % after purification by high performance liquid chromatography (HPLC).33, 35 It has been shown that the rebridging reaction does not proceed with monosulfones bearing sterically demanding functionalities such as drug molecules or dendrons.33 Such groups should be attached directly to the respective protein or peptide and not at the level of the sulfone reagent.
An extensive investigation to PEGylate interferon (IFN), a representative immunotherapeutic protein with accessible disulfide bonds, demonstrated that the mono‐functionalized IFN is the major product (57 %) when stoichiometric amount of bis‐sulfone reagent is applied.26 A doubly modified IFN forms the minor product (9 %) with recovery of 32 % unreacted IFN and 2 % of aggregated IFN. A doubly bridged IFN can be obtained as the major product when an excess of bis‐sulfone reagent is applied.26 A 78 % conversion of an antibody into antibody–drug conjugates (ADC) was achieved with the humanized antibody trastuzumab.39 The drug‐to‐antibody ratio (DAR) ratio can be manipulated through variation of the concentration of reductant and the stoichiometries of reactants.39 In addition, the site‐specific disulfide rebridging by applying the bis‐sulfones has been also demonstrated by the successful modification of antibody fragments39, 40 and therapeutic proteins while retaining their activities.27, 32
Next generation substituted maleimides
Several groups including Baker, Caddick, Chudasama, Jackson and Haddleton have elaborated on the inherent reactivity of maleimides toward thiols to design next‐generation maleimides for disulfide rebridging (Figure 1, II, III; Scheme 2 A).30, 41, 42, 43, 44, 45 Baker and Caddick proposed that the fast reaction kinetics of the maleimides compared to the bis‐sulfone reagents will facilitate reaction in tandem with the reducing agent resulting in less protein loss due to aggregation or disulfide scrambling.30, 41 The earliest examples reported are disubstituted maleimides (DSM), which carry substituents in the 3‐ and 4‐position with good leaving groups that allow reaction with two nucleophilic thiol groups such as the two thiols of a reduced cystine to rebridge the disulfides through a rigid two‐carbon spacer to afford the bisthiomaleimide bioconjugates (Scheme 2 B).30, 41, 42, 43, 44, 45
Scheme 2.
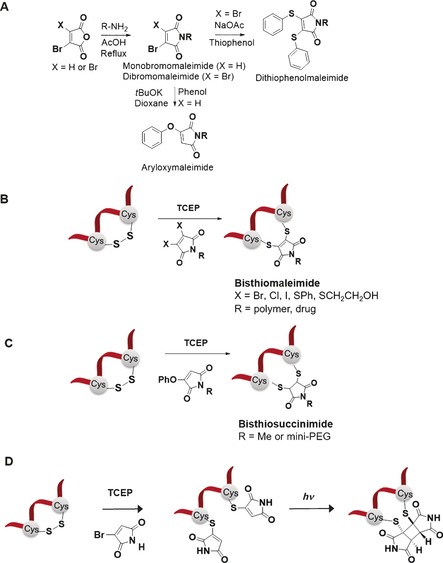
A) Syntheses of DSMs and aryloxymaleimide from bromomaleic anhydride. B) Conjugation of DSMs to peptides or proteins to form two‐carbon bridged bisthiomaleimides. C) Bioconjugation with aryloxymaleimides to form two‐carbon bridged bisthiosuccinimides. D) Photochemical rebridging of disulfide bonds with bromomaleimides.
Additionally, DSMs offer a broad pH range (6<pH<8) for bioconjugation in comparison to bis‐sulfones that require slightly basic pH for the elimination reaction. 3,4‐DBMs are commercially available and post‐functionalization can be achieved by the Mitsunobu reaction but in relatively low yields41 or dehydrative cyclization from dibromomaleic anhydride.46 However, these reagents can undergo side reactions with the reducing reagent, TCEP, and thus the rebridging reaction cannot be carried out in situ and large excess of the reductant should be avoided in the bioconjugation.41
Subsequently, a selection of 3,4‐DSMs was screened and dithiophenolmaleimides (DTM) were proven to be most efficient for in situ disulfide bridging as they are less susceptible to side reactions.41 DTMs can be prepared from dibromomaleic anhydride over two steps synthesis in moderate yield (Scheme 2 A).46 Benzeneselenol can be applied together with dithiophenolmaleimide to achieve reduction of the disulfide. The selection of the reductant/co‐reductant ratios has been particularly useful for example, in controlling the specific number of drugs attached to antibodies.46
The dibromomaleimide was applied successfully for selectively decorating a 32‐amino acid hormone peptide, salmon calcitonin (sCT), which consists of a single disulfide with synthetic polymers.47 Nonetheless, the direct incorporation of dibromomaleimides into polymers synthesized by atom transfer radical polymerization (ATRP) was impossible, presumably due to the radical addition to maleimides. Therefore, the post‐polymerization modification approach was employed to introduce the dibromomaleimide group onto the synthetic polymer for disulfide modification.47 Later, Haddleton and co‐workers developed a radical‐compatible dithiophenol maleimide functional ATRP initiator, which is less susceptible to nucleophilic attack.42 This reagent does not require protection of the maleimide end group for polymerisation under ATRP reaction conditions42, 44 and can offer more possibilities for the attachment of polymers to peptides and proteins to modulate their properties. In addition, the dithioalkyl maleimide molecules after bioconjugation show fluorescence and the group of O'Reilly has recently capitalized on this intrinsic property of DTM reagents for site‐specific fluorescent labelling.48
As in the case of the bis‐sulfones, the DSMs have also been successfully applied to the disulfide of cyclic peptides,44 fragment antigen‐binding (Fab) protein,49 a region on an antibody that binds to antigens, and antibodies.46 The bioconjugation reactions using DSMs often demonstrated high (>80 %) to quantitative conversion to afford the bisthiomaleimide conjugate (Scheme 2 B).41, 44, 46
Aryloxymaleimides have also been reported since phenol could serve as suitable leaving group in the disulfide rebridging reactions (Scheme 2 C). They can be prepared from the addition of the corresponding phenol to N‐methyl bromomaleimide in moderate to high (40–70 %) yield.50 Unlike DBMs, the addition‐elimination is slower and the conversion to the product can vary from 30 % to quantitative, presumably through increasing the reaction rate by incorporation of electron‐withdrawing groups in the aryloxy ring. Aryloxymaleimides do not react with TCEP, can be easily PEGylated to increase water solubility and do not form bis‐addition product, yielding only the bisthiosuccinimide.50
A recent development of next generation maleimides that have immense potential in the disulfide rebridging arena is the concept of phototriggered rebridging (Scheme 2 D). This can be achieved by functionalizing reduced dithiols with bromomaleimides to form thiomaleimides51 and thereafter controlling the disulfide rebridging by photochemistry by [2+2] cycloadditions of photoactive tags on both cysteine residues (Scheme 2 D).52 Such strategy could prove valuable as a photoswitchable tool to manipulate protein structure and function. Moreover, photorebridging offers improved solubility in aqueous media of the substrates after the first bioconjugation. Here, commercially available 2‐bromomaleimide was used to rebridge the cyclic peptide hormone analogue octreotide and the monoclonal antibody fragment, Herceptin‐Fab which contains one solvent accessible disulfide bond cross‐linking the heavy and light chains.52 Contrary to the other rebridging reagents, this method requires large excess of the bromomaleimide to be used to form the monothiol. Thereafter, photoactivation of bioactive peptides by manipulating disulfide bonds with spatial–temporal control was achieved to generate a cyclobutane product which contains a rigid two‐carbon bridge between the two thiols. In this instance, a mixture of the different diastereo‐ and regioisomers could be formed. The rebridged product was found to be the major species with the use of LCMS, with enamides as minor side products as a result of decarboxylative release of the thiomaleimide. The favourable properties of this photorebridging strategy are near‐quantitative rebridging efficiency, biocompatibility, fast reaction times and the formation of thiol‐stable products. Nevertheless, there are still challenges that have to be overcome. For example, the possibility to functionalize the nitrogen in the structure still needs to be investigated to determine the effect of the substituents on rebridging. Therefore, the introduction of additional functional features to the modified peptide/protein has to be explored to further broaden its scope of application.
The extensive work carried out with bis‐sulfones and next‐generation maleimides clearly underlines the viability of the disulfide rebridging strategy to achieve site‐selective modifications that are beyond what the genome, proteome and post‐translational modifications found in organisms can provide. In addition, the specific number of functional groups introduced per protein can be varied in proteins with more than one accessible disulfide bond through stoichiometric control of the reagent or choice of reductant.40, 46 Nevertheless, these rebridging reagents also exhibit certain limitations. For instance, both the bis‐sulfones and DSMs often have poor water solubility,26, 30, 32 do not exhibit any selectivity between thiols and disulfides under reductive conditions and site‐selective modification of disulfides in the presence of an unpaired cysteine residue could thus prove to be challenging. Consequently, alternative strategies are still being developed to further advance this protein modification strategy and a few new‐generation disulfide reagents are discussed in the next section to show the development in this aspect.
Optimized disulfide rebridging reagents
The molecular scaffold of disulfide rebridging reagents has been further modified (Figure 2 A, IV–VII) and optimized features such as improved hydrolytic stability, selectivity of disulfide over unpaired thiols, water solubility and the possibility to incorporate multiple functional groups in a single step have been introduced successfully.
Figure 2.
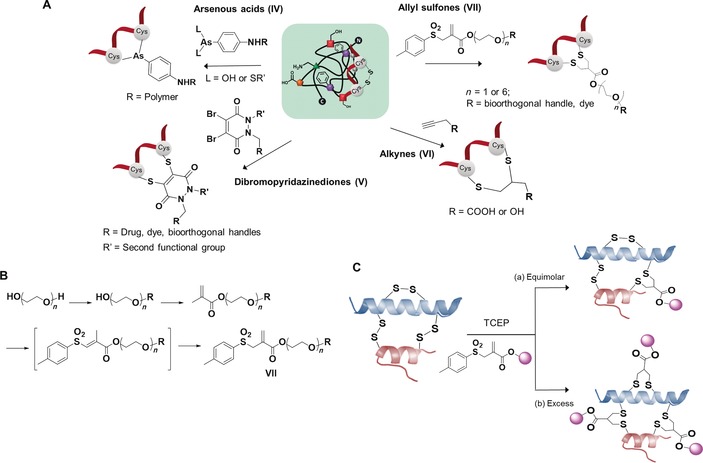
A) Optimized disulfide rebridging reagents and resultant conjugates. Arsenous acids (IV); dibromopyridazinediones (V); alkynes (VI); allyl sulfones (VII). B) Synthesis of allyl sulfone. C) Modification of peptides with: a) equimolar reagents leading to mono‐functionalized peptide, and b) excess reagents forming multiply functionalized peptide.
Recently, Wilson and Davis have reported trivalent arsenous acid (AsIII) derivatives as a new class of disulfide rebridging reagent (Figure 2 A, IV).53 The trivalent arsenous acid can be derived from reduction of commercially available p‐arsanilic acid in 38 % yield. Here, the authors exploited the entropy‐driven affinity of trivalent organic arsenicals towards dithiols in close proximity to achieve modification at the disulfide bridge in a specific manner. Since the formation of the monothiol adduct is entropically unfavourable, the AsIII reagent can even be used in large stoichiometric excess in contrast to most disulfide rebridging reagents to rebridge the disulfide by a single arsenic. In this way, the disulfide of the salmon calcitonin (sCT) was rebridged efficiently (t<2 min) in situ or through sequential reduction–conjugation.53 The application of the reactive intermediate to sCT showed quantitative conversion to the desired bioconjugate in less than an hour. The release of sCT from its conjugate was achieved in the presence of an excess of strong chelating agents, such as ethanedithiol. The organic arsenical exhibited limited reactivity (20 %) to native BSA (bovine serum albumin), a protein with an unpaired cysteine compared to N‐ethyl maleimide and dibromomaleimide which reacted with ≈90 % of the accessible thiol.53 The arsenic reagents offer significant advantage over the previously reported bis‐alkylation reagents in terms of their specificity towards disulfide bonds as compared to free cysteine residues and the authors have shown that the arsenic (AsV) and dithiaarsane (AsIII) polymers exhibit low levels of toxicity compared to free arsenic salts.53 Still, it remains to be seen if this class of reagents can be employed for in vivo applications at therapeutically relevant concentrations.
A drawback of the AsIII reagent might be its sensitivity to oxidation to form AsV species, especially during extensive purification by dialysis in water.53 This could be avoided by introducing an additional step of reducing AsV to AsIII by using an excess of glutathione followed by purification to isolate the reactive intermediate.53
Dibromopyridazinediones (Figure 2 A, V) were reported by Caddick and Chudasama for targeting disulfide bridges in peptides, Fab and antibodies.54, 55, 56, 57 The synthesis of dibromopyridazinediones can be achieved over three steps by the initial condensation of maleic anhydride with disubstituted hydrazine followed by sequential dibromination/elimination steps with a moderate overall yield (≈60 %).57 Complete conversion to bisthiopyridazinedione was achieved with a rigid two‐carbon bridge when reacted with SST for 2 h.57
To overcome the limitation of the reduction and rebridging in distinct steps which requires excess of reducing and bridging agents for in situ reaction, a dithioaryl(TCEP)pyridazinedione has been reported, which can address both the reduction and rebridging steps at the same time.58 In this way, disulfide scrambling can be avoided in the case of functional disulfide rebridging in a multidisulfide system, due to the minimization of the residency time of the cysteines liberated from disulfide reduction. The reagents are prepared over a four‐step synthesis with an overall yield of about 10 %. The all‐in‐one reagents were applied to peptides (octreotide and SST) and Fab and complete conversion was observed from liquid chromatography–mass spectrometry analysis.58 Reduced proteins were also not observed during the rebridging process.
A photochemical thiol‐yne coupling approach was recently reported.59 Commercially available bifunctional alkyne (Figure 2 A, VI) can be used in the presence of a water‐soluble radical initiator, lithium phenyl‐2,4‐6‐trimethylphosphinate and irradiated with UV‐light (365 nm) to achieve disulfide rebridging to form a two‐carbon bridged bioconjugate. Nevertheless, the conjugation yield to terlipressin, a cyclic peptide with a single disulfide, was found to be extremely low (3 %) with formation of regio‐ and stereoisomers which cannot be separated. Variation of terminal alkynes and peptides only improve the yield marginally (10–22 %). The approach can also be applied using cyclooctyne as a model for internal alkyne but similarly suffers low yield and a mixture of isomers are obtained which cannot be separated by chromatography.59 Further improvement in conjugation yields and purification needs to be achieved in order to stimulate broader applications.
The majority of the disulfide rebridging reagents reported so far are limited by poor solubility in aqueous media, so that bioconjugation of proteins usually is accomplished in the presence of organic–aqueous co‐solvents of varying ratios. The presence of organic solvents during bioconjugation can lead to denaturation of the protein structure and therefore, there is a great interest in the development of entirely water‐soluble reagents.33 To overcome this limitation, several groups have incorporated short polyethylene glycol (mini‐PEG) to the existing scaffolds to improve their water solubility.33, 50, 55 Nevertheless, this “mini‐PEGylation” approach has limited effects on bis‐sulfones due to the large number of hydrophobic aromatic groups in their scaffolds. Recently, we have introduced allyl sulfones as efficient disulfide rebridging reagents, providing improved reactivity, high water solubility and site specificity for precise protein modifications.60 Water‐soluble allyl sulfones (Figure 2 A, VII) were designed based on the essential features of disulfide rebridging reagents. Compared to the reported and widely used bis‐sulfones, the allyl sulfones possess two fewer hydrophobic benzene groups and therefore provide low n‐octanol–water partition coefficients (log Po/w), indicating higher water solubility, and the synthetic route was further simplified (Figure 2 B) to obtain the functionalized allyl sulfones in moderate to high yields (≈35–70 %). Additionally, more effective disulfide rebridging was achieved since the commonly used bis‐sulfones require in situ activation to form the reactive monosulfones and this equilibrium step usually does not proceed quantitatively. The broad applicability of this approach was demonstrated for the site‐directed modification of SST with one disulfide bridge and bovine insulin with three disulfide bridges, with yields of 49 and 28 %, respectively. Monofunctionalization of lysozyme, an enzyme containing four disulfide bridges, can also be achieved in 19 %.60 In this case, 30 % of unreacted native protein could even be recovered and could be recycled for subsequent reaction. High site‐selectivity and predictability allowed the functionalization of only the most solvent accessible disulfide of insulin and lysozyme while retaining tertiary structure and functional activity, which is of great significance for the construction of precise peptide and protein conjugates. The modification of the less accessible disulfides could be achieved when excess reducing agent and allyl sulfone VII (Figure 2 C) were added, suggesting future possibilities for multiple functionalization reactions by capitalizing on their differential accessibility.
Although limitations such as selectivity over unpaired cysteine or water solubility have been addressed partially by individual reagents, there is still no single reagent that has been reported to circumvent these problems simultaneously and a careful choice of the respective reagent for the intended applications is required.
Synthetic toolbox for functionalization
With the advent and multitude of disulfide rebridging reagents that are now available, it is imperative to scrutinize how this toolbox can be applied as a mean for posttranslational modifications of peptides and proteins to introduce novel synthetic entities.
The modification of various peptides to increase their functionalities has been demonstrated using the disulfide rebridging strategy. We have expanded extensively the functions of peptide/proteins by adopting the disulfide toolbox based on bis‐sulfones and allyl sulfones to derive a library of modified SST conjugates.33, 34, 35, 60, 61 Specifically, SST bioconjugates comprising reactive handles for bioorthogonal reactions (e.g., alkyne, azido, thiol, phenyl boronic acid and tetrazole),33, 61 affinity ligands (e.g., biotin and phenyl boronic acid)33 and imaging probes35, 60 have been derived through this versatile conjugation strategy. The resultant bioconjugates exhibit unique reactivities and properties from the chemical fusion of each constituent.34, 35 A fluorescent tag was also incorporated site‐selectively into lysozyme, an enzyme which hydrolyses the β(l–4) glycosidic bonds, and the mono‐modified enzyme retains 89 % activity.60 On the other hand, DSMs and dibromopyridazinediones have been employed extensively in the modification of peptides and antibodies to introduce bioorthogonal handles,55 macroinitiators,62 imaging probes,43 spin labels63 and drugs.46, 49
Multimodal disulfide rebridging reagents
Besides, mono‐functionalization, the development of multimodal reagents for increasing the functionality of proteins has received emerging interest over the past years. Despite the repertoire of reagents to functionalize proteins, site‐selective addition of multiple functional groups onto a single protein still remains a significant challenge.14, 64 Common strategies to introduce multiple functional groups still focus solely on the labelling of the N terminus or costly engineering of reactive unnatural amino acids into the protein sequence. To this end, several groups have previously demonstrated the opportunity to prepare bioconjugation reagents that can introduce heterofunctional groups into proteins in a single tag.54, 60, 65
The presence of two nitrogen atoms in the chemical structure of dibromopyridazinedione allows an asymmetric scaffold with two distinct functional groups to be instilled (Figure 3 A).54 Using the pyridazinedione motif, orthogonal “clickable” handles that undergo cycloaddition reactions were incorporated onto each nitrogen atom to generate a versatile platform that can be adopted to attach dual modalities.54 In this fashion, a multifunctional, active antibody comprising of a fluorescent dye and an anticancer agent has been prepared with a single reagent.54 A dual optical and nuclear imaging using the DTM scaffold has also been reported from a one‐pot, three‐component, copper(II)‐mediated reaction of azides, alkynes, and radiolabelled iodide (Figure 3 B).65
Figure 3.
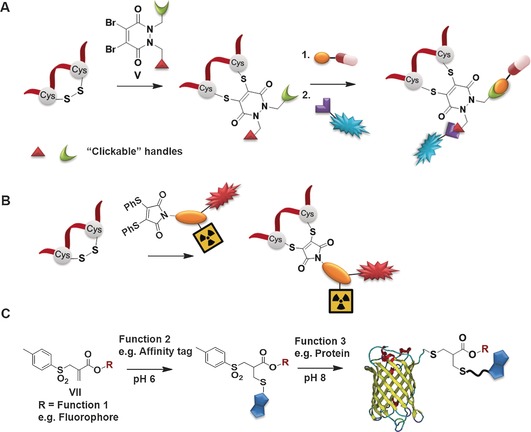
A) Dual functionalization using bifunctional dibromopyridazinedione.54 B) Dithiophenolmaleimide as a monomolecular multimodal imaging agent for peptide/protein labelling.65 C) Allyl sulfones as trifunctional linker through control of pH.60
Using the allyl sulfone reagent, we have further demonstrated that through control of the pH of the reaction media, two‐thiol‐containing molecules can be incorporated to the modified protein in a step‐wise fashion.60 More specifically, an affinity tag as well as a chromophore have been introduced facilitating separation and detection of the modified protein, exemplified by the functionalization of the green fluorescent protein (GFP; Figure 3 C). This multicomponent reaction addresses a significant challenge for example, for purification and tracking of chemically modified proteins since more than one tag or marker could be imparted. Such multimodal rebridging reagents hold immense potential for the preparation of precise multifunctional hybrids that are ahead of what biosynthesis and post‐translation processes in organisms can achieve.
Stability of bioconjugates
The stability of the bioconjugates after disulfide rebridging is also an important consideration for their intended applications in body liquids or cellular environments and has been investigated in some cases.
Bisthioether conjugates prepared from bis‐sulfones and allyl sulfones have been evaluated applying different methods such as Förster energy transfer or hyphenated liquid chromatography techniques.35, 39, 60 Serum stability was assessed in face of possible deconjugation of the payload and subsequent cross conjugation of the drug to other thiol‐containing species such as the blood plasma protein serum albumin.39, 40 A bisthioether antibody adduct was incubated in either 50 % rat or undiluted human serum, showing that the conjugate was extremely stable to serum exposure for up to 96 h at 37 °C.39 Furthermore, the antibody–drug conjugates formed by using the bis‐alkylating linker were stable in the presence of high concentrations of albumin over 5 days and the bisthioether is less susceptible to DTT (10 mm) compared to the original disulfide.39 A bisthioether SST adduct formed from the reaction with allyl sulfones was determined to be hydrolytically stable up to 48 h in various pH and 10 % fetal calf serum.60 Interestingly, millimolar concentrations of glutathione (GSH) present inside cells were found to cleave bisthioether conjugates.35 It is worth noting that in the blood serum, GSH is present in much lower concentrations compared to the cytoplasm of human cells (1–10 mm)66 or human cancer cells, providing an opportunity for GSH‐induced cleavage inside cancer cells.
The stability of bisthiomaleimide conjugates in mammalian cells was assessed, showing cleavage with first‐order kinetics both in vitro and in cells.67 The bisthiolmaleimide conjugates can be displaced by GSH (1 mm)44, 67 but the reversibility can be tuned68 through hydrolytic cleavage to form thiomaleamic acid, which also improves the serum stability of the conjugate significantly.43 Caddick and Smith have shown that the hydrolysis of bisthiomaleimide to thiomaleamic acid can lead to a pH‐responsive linker.69 On the other hand, bisthiosuccinimides formed from aryloxymaleimides, do not react even when large excess of GSH was applied for up to 48 h at room temperature but this is dependent on the biomolecular structure and is difficult to predict without further studies.50
The very high stability of the bisthiopyridazinediones is exemplified by recent studies by the groups of Chudasama and Caddick.54, 55 Here, they reported that the conjugate exhibits excellent stability in blood plasma mimicking conditions in the presence of high concentrations of human serum albumin and glutathione for 7 days, stability after 8 months of storage at 4 °C in phosphate‐buffered saline and stability in both low and high pH at 37 °C over an extended period.54
Clearly, the stability and degradability of the bioconjugates are influenced by the selection and application of the respective disulfide reagents and this could be capitalized upon to tailor for different biomedical applications in the microenvironment of diseased cells.
II. Preparation of Functional Biohybrids and Application Highlights
The disulfide‐rebridging strategy can be adopted to prepare different classes of structurally defined bioconjugates and these conjugates can be of immense therapeutic relevance. Bioconjugates consisting both biogenic and non‐biogenic molecules are often denoted biohybrids. Biohybrids hold great value for medical applications due to the synergy from the combination of synthetic functionalities and biopolymers found in Nature, as exemplified by protein–polymer or peptide–drug conjugates.70, 71, 72
In this context, the disulfide rebridging reagents could be powerful chemical ligation tools for the preparation of bioactive hybrid macromolecules with precise architectures. They exhibit a great degree of control over the molecular structures, functions, and activities of the resultant bioconjugates. In face of rigorous demands in modern biomedicine for customized formulation, this bioconjugation strategy provides access to diverse structures with fully tunable functions.
In the following, we will highlight the three major classes of biohybrid macromolecules (Figure 4) that have been achieved using this versatile platform, as well as emphasizing some key applications in emerging areas such as chemistry inside living cells or therapeutic applications.
Figure 4.
Precise biohybrids prepared by disulfide modification. A) Peptide biohybrids, B) protein biohybrids, and C) antibody biohybrids.
Peptide biohybrids
The conjugation of synthetic polymers such as poly(ethylene glycol) (PEG) enhances the in vivo stability of peptide therapeutics. For instance, SST, which plays a key role in regulating the endocrine system, has a short half‐life in vivo and cannot be used clinically.73 The disulfide rebridging strategy has been employed to introduce PEG to SST using DSMs and the SST hybrid retains agonist activity comparable to the native peptide.41 Functionalization of peptides with α‐functional polymer prepared from living radical polymerization can also be achieved to modulate their pharmacological properties and biodistribution.42, 44, 74 Oxytocin, a cyclic hormone nonapeptide used in treatment of postpartum haemorrhaging, was conjugated to poly(methoxy poly(ethylene glycol) acrylate) and the bioconjugate is resistant to thermal degradation even after 28 days in comparison to unmodified oxytocin.44 On the other hand, reactive, orthogonal handles have been introduced into peptides, which allow further chemical modifications to modulate the self‐assembly behaviour and form bioactive supramolecular peptide architectures. The preparation of cyclic peptide amphiphiles carrying hydrophobic components such as pyrene and lipids is challenging but has attracted considerable attention owing to various applications of self‐assembling peptide amphiphiles in drug delivery and regenerative medicine.75, 76, 77 Amphiphiles have been achieved through the post‐functionalization of a thiol‐modified SST with maleimide molecules carrying the hydrophobic group.61 These SST amphiphiles are valuable for the supramolecular formation of multivalent bioactive architectures. Additionally, well‐defined, homogenous peptide–drug conjugates can be formulated through post‐functionalization of peptide with reactive bioorthogonal handles34, 35 or to prepare imaging probes in living cells through photochemistry33 in a specific fashion and the applications are discussed in the following sections.
Peptide biohybrids for targeted drug delivery and controlled release
SST has been extensively investigated for targeted drug delivery78, 79 as it acts on multiple cell targets through a family of G protein‐coupled receptors (SSTRs) that are overexpressed on various tumour cells and tissues.80, 81 The SST conjugates act as potent SSTR2 agonists with EC50 value in the low nanomolar range (13 nm),35 and are therefore highly attractive as new generation biotherapeutics that impart novel bioactivities such as cell‐type selectivity. In combination with a photosensitizer (PS), we studied its role on photodynamic applications (PDT).34 PDT has been developed for the effective chemotherapeutic treatment of various cancers (e.g., lung, oesophagus, skin tumours).82, 83 In PDT, light activation within the tumour tissue produces radicals or singlet oxygen (1O2) to initiate apoptosis and/or necrosis and eventually lead to cell death.84 A SST‐PS conjugate comprising of a polypyridyl–ruthenium PS was prepared by the Huisgen 1,3‐dipolar cycloaddition (“click” reaction) of the ethynyl‐PS with an azido‐functionalized SST (Figure 5 A).34 The SST‐PS conjugate has the advantage of dual selectivity, namely preferential tumour uptake of the PS and selective co‐localization of light irradiation within tumours, thus improving therapeutic efficiency while avoiding collateral damage to healthy tissues. Its potent phototoxicity was found in the low micromolar range at very short drug‐to‐light intervals and low dosage of visible light and exhibits low cytotoxicity without light activation (Figure 5 B).
Figure 5.
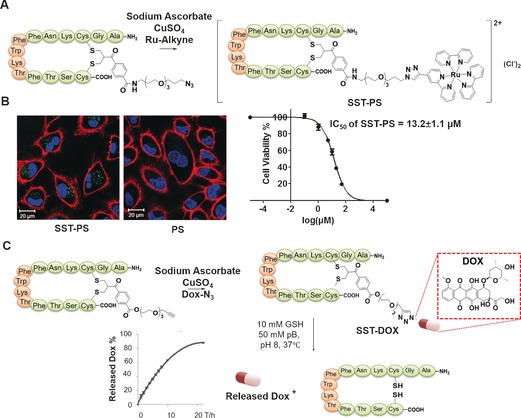
A) Synthesis of SST‐PS conjugate through CuI‐catalysed cycloaddition. B) Confocal microscopy images of A549 cells incubated with SST‐PS (left) and PS (right).34 C) Synthesis of SST‐DOX conjugate and GSH‐mediated cleavage of bisthioether conjugates and quantification of DOX release.35
Besides targeted drug delivery, on‐demand drug release to the diseased site represent a promising endeavour for enhancing the therapeutic efficiency and concurrently minimizing systemic toxicity.85, 86, 87 In this context, the redox sensitivity of the bisthioethers formed from disulfide rebridging agents can be exploited for programming redox responsiveness in the cellular environment. Cytosolic glutathione (GSH) in hypoxic tumour cells has been found at least four times higher compared with normal cells, and elevated GSH concentration of 10–25 mm have been reported for tumour cells such as bladder cancer cells.88 The significant difference of GSH levels thus offers potential for the controlled, tumour selective drug release. Somatostatin–doxorubicin (SST–DOX) conjugate can be synthesized from the “click” reaction of ethynyl‐SST with azido‐DOX (Figure 5 C). The SST–DOX conjugate achieved drug release and improved cellular toxicity at high GSH concentrations (10 mm, Figure 5‐C).35 The thioether bond of the bis‐sulfide conjugate is chemically more stable89 in the cell plasma of healthy cells with high structural integrity under physiological conditions since no disulfide scrambling occurs in contrast to disulfides which are conventionally used for redox‐responsiveness. Thus, the bis‐sulfide conjugation represents a powerful strategy to access structurally defined bioconjugates that combines tumour targeting and controlled intracellular release of therapeutic drug molecules in response to elevated GSH levels. In addition, the bis‐sulfone scaffold offers further functional versatility to include acid‐sensitive linkage, for example, ester bonds to introduce dual stimuli‐responsiveness to the system.
Exploring peptide bioconjugation inside live cells
The ability of peptides for cell‐type specific uptake and intracellular release could also be exploited to direct chemical reactions inside cellular compartments. Accomplishing bioorthogonal reactions inside living cells offer great opportunity to unravel the roles of individual biomolecules inside a complex array and inhomogenous environment.90 In addition, the reengineering of cellular behaviour by using the chemical biology toolbox discussed herein is gaining burgeoning interests with the great potential to ultimately control cellular behaviour or cell–cell interactions.91, 92
Controlling chemical reactions in a living environment requires the formation of the reactive functionalities with high spatial precision and light stimulus is ideal for such purposes. SST is well suited for exploring reactions in living cells since it is taken up and released into the cytoplasm of various cells expressing somatostatin receptors. Here, the disulfide rebridging strategy offers the unique opportunity to control reactions inside a living system spatially and temporally by light irradiation through incorporation of a photoactive moiety.93 A photoactive SST‐tetrazole was prepared which allowed bioconjugation inside living cells by photoinduced, bioorthogonal nitrile imine mediated tetrazole‐ene cycloaddition (NITEC) reaction with a functionalized maleimide.33 The NITEC involves a concerted reaction mechanism (Figure 6 A), where nitrogen is rapidly released from the diaryltetrazole upon photoirradiation and a reactive nitrile imine intermediate is generated in situ. Then, the nitrile imine readily undergoes a slower rate‐determining 1,3‐dipolar cycloaddition reaction with an alkene dipolarophile in a concerted manner to afford a stable pyrazoline‐based covalent linkage. The reaction of tetrazoles with maleimides is usually completed within 5 min at ambient temperature and represents one of the avant garde tools for rapid click conjugation.94 The formation of a photoluminescent product allows the study of the reaction kinetics.95, 96, 97 The NITEC reaction between a maleimide‐PEG polymer and SST‐tetrazole was successfully applied inside living cells (Figure 6 B and C).33 This could be of immense interest for pre‐targeting in which the tetrazole‐functionalized cell‐targeting peptide or antibody is first allowed to accumulate in desired areas, and the reaction with the counterpart compound could be sequentially triggered by light irradiation to form therapeutic species or a specific imaging probe with high spatial and temporal precision inside a living system.
Figure 6.
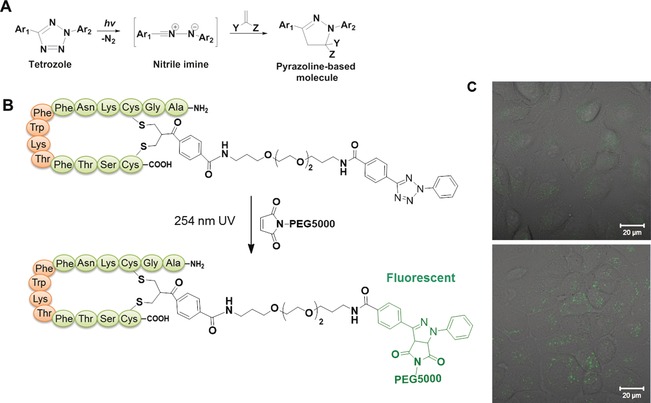
A) The mechanism of NITEC reaction. B) The NITEC reaction between SST‐tetrazole and maleimide‐PEG5000. C) Confocal images of A549 cells to monitor the intracellular NITEC reaction with (bottom) and without (top) light irradiation.33
Protein biohybrids
Proteins play a significant role in biological functions and are one of the most versatile building blocks in biology due to their diverse structural and functional properties.98 Due to their unique bioactivities and high biocompatibility, the therapeutic usage of proteins is emerging.99, 100 However, protein therapeutics also have some inherent drawbacks such as poor stability, sometimes high immunogenicity and antigenicity or low blood circulation times.101, 102 PEGylation of proteins represents a common strategy where the polymers are adopted as high‐molecular‐weight substituents to enhance pharmacokinetic parameters and promote accumulation of the proteins in tumour tissue.102, 103 Statistical PEGylation in solution could affect the activity of proteins adversely due to blocking or conformational change of the active site and site‐specific PEGylation is hence more desirable.102 In this context, the disulfide rebridging strategy is a valuable tool for the site‐selective modification of native proteins to form a homogenous protein–polymer hybrid. Several groups have prepared protein–polymer hybrids with a defined number of polymers through application of disulfide rebriging reagents that target the disulfide bonds.27, 32 The suitability of this approach is exemplified by the mono‐PEGylation of interferon (INF) using bis‐sulfones.27, 32 The antiviral activity of the three‐carbon disulfide single‐bridged PEG‐IFN was 8 % of IFN and, is comparable to the reported activity of the mono‐PEGylated IFN (≈7 % of IFN) in clinical use.32
The functional and structural complexity of proteins can also be expanded in a modular fashion through the cross‐conjugation of two biomacromolecules. Although various conjugation strategies have been reported, the orthogonal and spatially controlled conjugation of two sterically demanding biomacromolecules could be challenging to achieve. Consequently, there is an increasing need to expand the tools available to address a broader spectrum of biomacromolecules.
We have shown that conjugation of sterically bulky and rigid groups to proteins using bis‐sulfones can be challenging and an alternative strategy is required.33 By manipulating the difference in the reaction kinetics of the bis‐sulfone functionality and maleimide moiety, an orthogonal thiol‐reactive cross‐conjugation reagent was designed, allowing the pH‐controlled, step‐wise and site‐specific conjugation of two thiol containing biomacromolecules (Figure 7).38 At pH 6, the first Michael addition reaction between maleimide and thiol is accomplished rapidly without affecting the bis‐sulfone functionality (Figure 7). By slightly increasing the pH value to 8, in situ elimination of p‐toluene sulfinic acid occurs with the formation of monosulfone that allows another Michael addition reaction with the second thiol‐containing macromolecule under mild conditions (Figure 7). The versatility of this reagent has been demonstrated by cross conjugation of two different sterically demanding proteins (serum albumins) or between a cell‐penetrating peptide, TAT (transactivator of transcription) and an enhanced yellow fluorescent protein (EYFP; Figure 7) resulting in the formation of protein biohybrids with defined molecular structures and additive features from their individual constituents.38 For example, the TAT–EYFP hybrid prepared in this fashion is taken up into cancer cells while the native EYFP does not translocate across cell membranes. This attractive strategy is not limited to protein hybrids but can also be used to prepare a covalent oligonucleotide (ON)–peptide conjugate. Based on the established bis‐sulfone chemistry, the bottom‐up chemical approach can be extended for the modification of proteins by targeting disulfides and expand the repertoire of available protein biohybrids. This platform thereby is attractive for the construction of defined protein–peptide, protein–protein or protein–DNA biohybrids under physiological conditions in a straightforward fashion which could be of high significance for tailored biotherapeutics.
Figure 7.
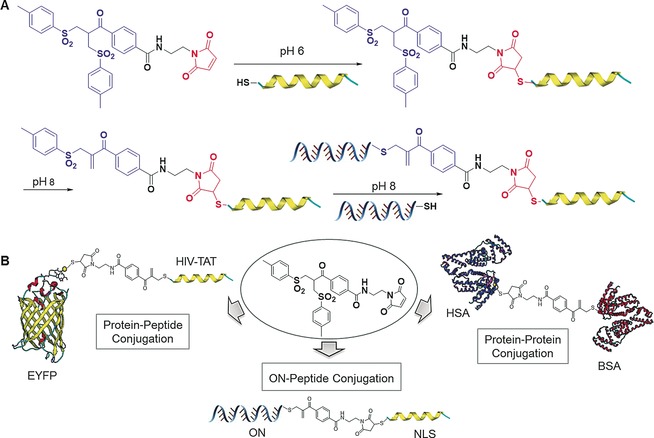
Illustration of the step‐wise conjugation of two different thiol‐containing biomolecules by thiol cross‐conjugation reagent through the pH switch, and its application for preparing heterobioconjugates.38
Antibody biohybrids
Antibodies (Abs) are one of the most important class of biopharmaceutics and have found applications in both immunoassays and immunotherapy.104, 105 They are considered emerging anticancer therapeutics due to their long serum half‐lives and their high binding specificity and affinity to surface markers on target cells. For example, antibody–drug conjugates (ADCs, Figure 8 A) represent powerful treatment options through a combination of the targeting ability of antibodies towards specific cell types together with the ability of potent cytotoxic drugs to induce apoptosis.106 ADCs improve the antibody's overall efficacy and also may address the challenge of drug resistance.40, 107 Although there are already two US Food and Drug Administration‐approved ADCs already available on the market and over 30 ADCs43 in clinical trials, addressing antibody modification while maintaining structural integrity and product homogeneity for precise formulation remains challenging.108, 109 Conventionally, this has been achieved by conjugation strategies that have poor site‐selectivity,110 resulting in heterogeneous mixtures with different drug loadings. The bis‐sulfones, DSMs and dibromopyridazinediones are suitable candidates for the preparation of well‐defined ADCs through modification of the accessible disulfides in the hinge region,43, 46, 54, 55, 63 without the need to engineer unnatural amino acids or cysteine mutants. Homogenous ADCs comprising of Herceptin (HER), a monoclonal IgG1 that targets the internalizing HER2 receptor for the treatment of HER2 positive breast cancer have been prepared successfully.43, 46, 55 Anticancer drugs such as monomethyl auristatin E (MMAE),39, 43 doxorubicin,46, 54 and photosensitizers49, 55 have been conjugated to these antibodies or antibody fragments with a highly precise antibody to drug ratios not greater than four, which is crucial for retaining their antigen binding and stability.111, 112
Figure 8.
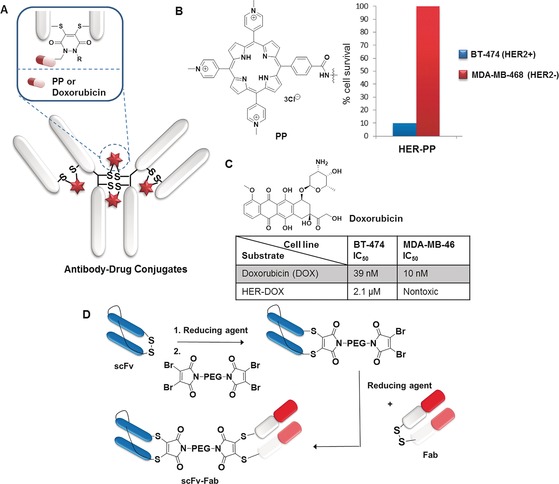
A) Overview of antibody–drug conjugates (ADCs) design. In vitro evaluation of: B) HER–PP in broad‐spectrum light shows internalization and phototoxicity in HER2+ cell lines,55 and C) HER–DOX and free drug (DOX) show cytotoxicity in HER2+ compared to HER2− cell lines.54 D) Construction of the bispecific antibody conjugate by disulfide rebridging.113
Using dibromopyridazinediones, the antibody HER has been modified with a cyclooctyne handle which is used in subsequent post‐functionalization to introduce payloads such as the anticancer drug doxorubicin (DOX)54 and the photosensitizer, porphyrin (PP;55 Figure 8 A). The HER–PP hybrid exhibited high selectivity towards HER2 positive cells (BT‐474) over HER2 negative cells (MDA‐MB‐468; Figure 8 B) and phototoxicity at nanomolar concentration (625 nm) to eradicate HER2+ cells with 90 % kill, while HER2− cells were left unaffected.55 The HER–DOX conjugate was subjected to biological evaluation against breast cancer cell lines and exhibited remarkable selectivity towards HER2 positive cells (IC50=2.1 μm) over HER2 negative cells (nontoxic) with enhanced therapeutic efficacy compared to the free drug (Figure 8 C).54 Homogenous ADC with a clinically validated auristatin payload prepared by the disulfide rebridging strategy was evaluated for its antigen binding affinity, efficacy in xenograft tumour models and pharmacokinetics and toxicity in rats.45 The exemplary investigation by Jackson and co‐workers showed that the homogenous ADC demonstrate improved pharmacokinetics, superior efficacy, and reduced toxicity in vivo compared to analogous conventional heterogeneous ADCs.45 In vivo efficacy of HER–Fab–MMAE was accessed with BT‐474 xenografts in mice.39 The antibody fragments selected as whole‐molecule IgGs may be limited in their tumour penetration due to their large size. The HER–Fab–MMAE conjugate not only retained binding and demonstrated antigen‐selective in vitro cytotoxicity but also showed a marked delay in tumour growth in vivo, and the ADC is well‐tolerated with no dose‐related effects on the body weight of the mice tested.39
In cases where high dye/drug loads are required for sensitive antigen detection by signal amplification or efficient drug delivery, it is not possible to increase the DAR beyond four with direct disulfide rebridging of the payload to the antibody which will result in decrease of antigen binding affinity.112 To further improve on the design, Gao et al. have recently used DSMs to install a macroinitiator into interchain disulfide in antibodies for the in situ atom‐transfer radical‐polymerization (ATRP) of dye or drug‐labelled polymer conjugates to introduce high payload and greater possibility to fine‐tune dye‐to‐antibody ratios in a homogenous polymer–antibody hybrid.62 The antibody hybrid reported in this proof‐of‐concept work shows enhanced antigen detection and is far superior to a conventional antibody–dye conjugate in antigen detection by signal amplification.62
On the other hand, the preparation of “bispecifics” that comprise two antibody fragments that can simultaneously bind two different antigens has been introduced to improve the therapeutic efficacy.113 In this case, Fab and disulfide‐stabilized single‐chain variable antibody fragments (scFv), which are commonly applied in a range of bispecific topologies, are chosen as the two components. Using a bis‐dibromomaleimide cross‐linker and strict control of the stoichiometry, the stepwise, efficient rebridging of disulfide bonds in Fab and scFv was achieved to give a homogenous antibody–antibody hybrid in high yield (Figure 8 D) while maintaining their activity, as determined from ELISA and cell binding assay.113 This powerful method requires only a simple two step synthesis to prepare the reagent and can potentially be important for the facile generation of bispecifics from a range of antibody fragments for clinical applications and be complementary to recombinant techniques.
The chemical platforms presented herein could pave the way to provide structurally defined ADC conjugates possessing both improved in vitro and in vivo efficacy and cell‐type selectivity and holds great promise for the development of a new generation of high efficacy biotherapeutics.
Conclusions
In summary, disulfide bonds in proteins represent attractive targets for the synthetic transformations of peptides/proteins tailored with molecular accuracy. In this respect, disulfide rebridging offers a robust and versatile strategy to impart a variety of reactive functionalities and to form well‐defined polypeptide conjugates beyond what Nature offers. Modified peptides and proteins could be used for the exploration of bioorthogonal reactions in live cells, the construction of stimulus‐responsive peptide–drug for targeted on‐demand drug delivery, as well as antibody–drug conjugates for targeted cancer therapy.
The new‐generation disulfide reagents provide additional complexity with the possibility to form precise protein biohybrids or introduce multifunctional groups in a single chemical tag. In addition, the differential reactivity based on the accessibility of the disulfides in a single protein can be exploited and holds great promise for achieving dual labelling of a single protein, which still remains a significant challenge despite the availability of a large number of site‐selective reagents. Nonetheless, dual functionalization or the construction of higher‐order protein conjugates necessitates more complex requirements and criteria, and the application of disulfide rebridging strategy alone may be insufficient. Nature has often exploited the features of each individual post‐translation modification process in sequential steps to control the functions of single proteins. Recombinant technologies can therefore be exploited to introduce unpaired cysteines and disulfide bonds to exploit the rebridging strategy, although efficiency, activity and scalability can be limited due to disulfide scrambling. In addition, one should not be limited to a single strategy and should capitalize on the combination of the advantages of the different strategies in the expanding site‐selective modification toolbox to prepare more complex macromolecular structures such as heterofunctional proteins and higher‐order protein conjugates.
Biographical Information
Tanja Weil received her Ph.D. from Mainz University in 2002. She was an Associate Professor at the National University of Singapore (2008–2010) and in 2010 became the Director at the Institute of Organic Chemistry III at Ulm University. In 2016, she was appointed as Director at the Max Planck Institute for Polymer Research in Mainz, Germany. She is the recipient of an ERC Synergy grant. Her research centres on designing precision macromolecules and hybrid materials for biomedical and materials science applications.

Biographical Information
Seah Ling Kuan obtained her Ph.D. (2009) from the National University of Singapore and received the Alexander von Humboldt fellowship (2011–2012). She was a group leader (2013–2016) at the Institute of Organic Chemistry III (Ulm University) and is now a project leader at the Max Planck Institute for Polymer Research in Mainz. Her research is focused on designing bioinspired architectures for medical applications.

Biographical Information
Tao Wang obtained her M.Sc. (2011) from the National University of Singapore and received her Ph.D. (2014) from Ulm University in the area of site‐selective protein chemistry under Professor Weil. In 2016 she became an Associate Professor at the Southwest Jiaotong University in China.

Acknowledgements
T.W. is grateful to the financial support from the Innovative Training Network (ITN) under the Horizon2020 program, the ERC Synergy Grant 319130‐BioQ, the EU Project “Hyperdiamond” supported under Horizon 2020 program and the German Ministry of Research and Education (BMBF) for supporting the project Selekomm within the Biotechnologie2020+ initiative. The authors thank Sean Harvey for proofreading the manuscript.
S. L. Kuan, T. Wang, T. Weil, Chem. Eur. J. 2016, 22, 17112.
References
- 1. Wang Y.-C., Peterson S. E., Loring J. F., Cell Res. 2013, 24, 1–18. [Google Scholar]
- 2. Beltrao P., Bork P., Krogan N. J., Van Noort V., Mol. Syst. Biol. 2013, 9, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Duschmalé J., Wennemers H., Chem. Eur. J. 2012, 18, 1111–1120. [DOI] [PubMed] [Google Scholar]
- 4. Holmes T. C., Trends Biotechnol. 2002, 20, 16–21. [DOI] [PubMed] [Google Scholar]
- 5. Schumacher D., Hackenberger C. P. R., Curr. Opin. Chem. Biol. 2014, 22, 62–69. [DOI] [PubMed] [Google Scholar]
- 6. Jackson J. C., Hammill J. T., Mehl R. A., J. Am. Chem. Soc. 2007, 129, 1160–1166. [DOI] [PubMed] [Google Scholar]
- 7. Miyake-Stoner S. J., Miller A. M., Hammill J. T., Peeler J. C., Hess K. R., Mehl R. A., Brewer S. H., Biochemistry 2009, 48, 5953–5962. [DOI] [PubMed] [Google Scholar]
- 8. Zhu S., Riou M., Yao C. A., Carvalho S., Rodriguez P. C., Bensaude O., Paoletti P., Ye S., Proc. Natl. Acad. Sci. USA 2014, 111, 6081–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Link A. J., Mock M. L., Tirrell D. A., Curr. Opin. Biotechnol. 2003, 14, 603–609. [DOI] [PubMed] [Google Scholar]
- 10. Sletten E. M., Bertozzi C. R., Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang H., Gao H., Guo N., Niu G., Ma Y., Kiesewetter D. O., Chen X., Theranostics 2012, 2, 607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Thornton J. M., J. Mol. Biol. 1981, 151, 261–287. [DOI] [PubMed] [Google Scholar]
- 13. Petersen M. T. N., Jonson P. H., Protein Eng. 1999, 12, 535–548. [DOI] [PubMed] [Google Scholar]
- 14. Stephanopoulos N., Francis M. B., Nat. Chem. Biol. 2011, 7, 876–884. [DOI] [PubMed] [Google Scholar]
- 15. Fahey E. M., Chaudhuri J. B., Binding P., Separ. Sci. Technol. 2000, 35, 1743–1760. [Google Scholar]
- 16. Shen B.-Q., Xu K., Liu L., Raab H., Bhakta S., Kenrick M., Parsons-Reponte K. L., Tien J., Yu S.-F., Mai E., Li D., Tibbitts J., Baudys J., Saad O. M., Scales S. J., McDonald P. J., Hass P. E., Eigenbrot C., Nguyen T., Solis W. A., Fuji R. N., Flagella K. M., Patel D., Spencer S. D., Khawli L. A., Ebens A., Wong W. L., Vandlen R., Kaur S., Sliwkowski M. X., Scheller R. H., Polakis P., Junutula J. R., Nat. Biotech. 2012, 30, 184–189. [DOI] [PubMed] [Google Scholar]
- 17. Obermeyer A. C., Jarman J. B., Francis M. B., J. Am. Chem. Soc. 2014, 136, 9572–9579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. MacDonald J. I., Munch H. K., Moore T., Francis M. B., Nat. Chem. Biol. 2015, 11, 326–331. [DOI] [PubMed] [Google Scholar]
- 19. Bauer D. M., Ahmed I., Vigovskaya A., Fruk L., Bioconjugate Chem. 2013, 24, 1094–1101. [DOI] [PubMed] [Google Scholar]
- 20. Ban H., Gavrilyuk J., Barbas C. F., J. Am. Chem. Soc. 2010, 132, 1523–1525. [DOI] [PubMed] [Google Scholar]
- 21. Antos J. M., Francis M. B., J. Am. Chem. Soc. 2004, 126, 10256–10257. [DOI] [PubMed] [Google Scholar]
- 22. Hogg P. J., Trends Biochem. Sci. 2003, 28, 210–214. [DOI] [PubMed] [Google Scholar]
- 23. Zloh M., Shaunak S., Balan S., Brocchini S., Nat. Protoc. 2007, 2, 1070–1083. [DOI] [PubMed] [Google Scholar]
- 24. Saunders A. J., Young G. B., Pielak G. J., Protein Sci. 1993, 2, 1183–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brocchini S., Godwin A., Balan S., Choi J.-W., Zloh M., Shaunak S., Adv. Drug Delivery Rev. 2008, 60, 3–12. [DOI] [PubMed] [Google Scholar]
- 26. Brocchini S., Balan S., Godwin A., Choi J.-W., Zloh M., Shaunak S., Nat. Protoc. 2006, 1, 2241–2252. [DOI] [PubMed] [Google Scholar]
- 27. Shaunak S., Godwin A., Choi J.-W., Balan S., Pedone E., Vijayarangam D., Heidelberger S., Teo I., Zloh M., Brocchini S., Nat. Chem. Biol. 2006, 2, 312–313. [DOI] [PubMed] [Google Scholar]
- 28. Boutureira O., Bernardes G. J. L., Chem. Rev. 2015, 115, 2174–2195. [DOI] [PubMed] [Google Scholar]
- 29. Gunnoo S. B., Madder A., ChemBioChem 2016, 17, 529–553. [DOI] [PubMed] [Google Scholar]
- 30. Smith M. E. B., Schumacher F. F., Ryan C. P., Tedaldi L. M., Papaioannou D., Waksman G., Caddick S., Baker J. R., J. Am. Chem. Soc. 2010, 132, 1960–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pfisterer A., Eisele K., Chen X., Wagner M., Müllen K., Weil T., Chem. Eur. J. 2011, 17, 9697–9707. [DOI] [PubMed] [Google Scholar]
- 32. Balan S., Choi J. W., Godwin A., Teo I., Laborde C. M., Heidelberger S., Zloh M., Shaunak S., Brocchini S., Bioconjugate Chem. 2007, 18, 61–76. [DOI] [PubMed] [Google Scholar]
- 33. Wang T., Wu Y., Kuan S. L., Dumele O., Lamla M., Ng D. Y. W., Arzt M., Thomas J., Christopher B.-K., Weil T., Chem. Eur. J. 2015, 21, 228–238. [DOI] [PubMed] [Google Scholar]
- 34. Wang T., Zabarska N., Wu Y., Lamla M., Fischer S., Monczak K., Ng D. Y. W., Rau S., Weil T., Chem. Commun. 2015, 51, 12552–12555. [DOI] [PubMed] [Google Scholar]
- 35. Wang T., Ng D. Y. W., Wu Y., Thomas J., TamTran T., Weil T., Chem. Commun. 2014, 50, 1116–1118. [DOI] [PubMed] [Google Scholar]
- 36. Getz E. B., Xiao M., Chakrabarty T., Cooke R., Selvin P. R., Anal. Biochem. 1999, 273, 73–80. [DOI] [PubMed] [Google Scholar]
- 37. Han J. C., Han G. Y., Anal. Biochem. 1994, 220, 5–10. [DOI] [PubMed] [Google Scholar]
- 38. Wang T., Pfisterer A., Kuan S. L., Wu Y., Dumele O., Lamla M., Muellen K., Weil T., Mullen K., Weil T., Chem. Sci. 2013, 4, 1889–1894. [Google Scholar]
- 39. Badescu G., Bryant P., Bird M., Henseleit K., Swierkosz J., Parekh V., Tommasi R., Pawlisz E., Jurlewicz K., Farys M., Camper N., Sheng X.-B., Fisher M., Grygorash R., Kyle A., Abhilash A., Frigerio M., Edwards J., Godwin A., Bioconjugate Chem. 2014, 25, 1124–1136. [DOI] [PubMed] [Google Scholar]
- 40. Bryant P., Pabst M., Badescu G., Bird M., McDowell W., Jamieson E., Swierkosz J., Jurlewicz K., Tommasi R., Henseleit K., Sheng X.-B., Camper N., Manin A., Kozakowska K., Peciak K., Laurine E., Grygorash R., Kyle A., Morris D., Parekh V., Abhilash A., Choi J.-W., Edwards J., Frigerio M., Baker M. P., Godwin A., Mol. Pharm. 2015, 12, 1872–1879. [DOI] [PubMed] [Google Scholar]
- 41. Schumacher F. F., Nobles M., Ryan C. P., Smith M. E. B., Tinker A., Caddick S., Baker J. R., Bioconjugate Chem. 2011, 22, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jones M. W., Strickland R. A., Schumacher F. F., Caddick S., Baker J. R., Gibson M. I., Haddleton D. M., Chem. Commun. 2012, 48, 4064–4066. [DOI] [PubMed] [Google Scholar]
- 43. Nunes J. P. M., Morais M., Vassileva V., Robinson E., Rajkumar V. S., Smith M. E. B., Pedley R. B., Caddick S., Baker J. R., Chudasama V., Chem. Commun. 2015, 51, 10624–10627. [DOI] [PubMed] [Google Scholar]
- 44. Collins J., Tanaka J., Wilson P., Kempe K., Davis T. P., Mcintosh M. P., Whittaker M. R., Haddleton D. M., Bioconjugate Chem. 2015, 26, 633–638. [DOI] [PubMed] [Google Scholar]
- 45. Behrens C. R., Ha E. H., Chinn L. L., Bowers S., Probst G., Fitch-Bruhns M., Monteon J., Valdiosera A., Bermudez A., Liao-Chan S., Wong T., Melnick J., Theunissen J.-W., Flory M. R., Houser D., Venstrom K., Levashova Z., Sauer P., Migone T.-S., van der Horst E. H., Halcomb R. L., Jackson D. Y., Mol. Pharm. 2015, 12, 3986–3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schumacher F. F., Nunes J. P. M., Maruani A., Chudasama V., Smith M. E. B., Chester K. A., Baker J. R., Caddick S., Org. Biomol. Chem. 2014, 12, 7261–7269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jones M. W., Strickland R. A., Schumacher F. F., Caddick S., Baker J. R., Gibson M. I., Haddleton D. M., J. Am. Chem. Soc. 2011, 133, 18865. [DOI] [PubMed] [Google Scholar]
- 48. Robin M. P., Wilson P., Mabire A. B., Kiviaho J. K., Raymond J. E., Haddleton D. M., O'Reilly R. K., J. Am. Chem. Soc. 2013, 135, 2875–2878. [DOI] [PubMed] [Google Scholar]
- 49. Bryden F., Maruani A., Savoie H., Chudasama V., Smith M. E. B., Caddick S., Boyle R. W., Bioconjugate Chem. 2014, 25, 611–617. [DOI] [PubMed] [Google Scholar]
- 50. Marculescu C., Kossen H., Morgan R. E., Mayer P., Fletcher S., Tolner B., Chester K., Jones L. H., Baker J. R., Chem. Commun. 2014, 50, 7139–7142. [DOI] [PubMed] [Google Scholar]
- 51. Smith M. E. B., Caspersen M. B., Robinson E., Morais M., Maruani A., Nunes J. P. M., Nicholls K., Saxton M. J., Caddick S., Baker J. R., Chudasama V., Org. Biomol. Chem. 2015, 13, 7946–7949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Richards D. A., Fletcher S. A., Nobles M., Kossen H., Tedaldi L., Chudasama V., Tinker A., Baker J. R., Org. Biomol. Chem. 2016, 14, 455–459. [DOI] [PubMed] [Google Scholar]
- 53. Wilson P., Anastasaki A., Owen M. R., Kempe K., Haddleton D. M., Mann S. K., Johnston A. P. R., Quinn J. F., Whittaker M. R., Hogg P. J., Davis T. P., J. Am. Chem. Soc. 2015, 137, 4215–4222. [DOI] [PubMed] [Google Scholar]
- 54. Maruani A., Smith M. E. B., Miranda E., Chester K. A., Chudasama V., Caddick S., Nat. Commun. 2015, 6, 6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maruani A., Savoie H., Bryden F., Caddick S., Boyle R., Chudasama V., Chem. Commun. 2015, 51, 15304–15307. [DOI] [PubMed] [Google Scholar]
- 56. Lee M. T. W., Maruani A., Chudasama V., J. Chem. Res. 2016, 40, 1–9. [Google Scholar]
- 57. Chudasama V., Smith M. E. B., Schumacher F. F., Papaioannou D., Waksman G., Baker J. R., Caddick S., Chem. Commun. 2011, 47, 8781–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee M. T. W., Maruani A., Baker J. R., Caddick S., Chudasama V., Chem. Sci. 2016, 7, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Griebenow N., Dilmaç A. M., Greven S., Bräse S., Bioconjugate Chem. 2016, 27, 911–917. [DOI] [PubMed] [Google Scholar]
- 60. Wang T., Riegger A., Lamla M., Wiese S., Oeckl P., Otto M., Wu Y., Fischer S., Barth H., Kuan S. L., Weil T., Chem. Sci. 2016, 7, 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuan S. L., Wang T., Raabe M., Liu W., Lamla M., Weil T., ChemPlusChem 2015, 80, 1347–1353. [DOI] [PubMed] [Google Scholar]
- 62. Zhang L., Zhao W., Liu X., Wang G., Wang Y., Li D., Xie L., Gao Y., Deng H., Gao W., Biomaterials 2015, 64, 2–9. [DOI] [PubMed] [Google Scholar]
- 63. Schumacher F. F., Sanchania V. A., Tolner B., Wright Z. V. F., Ryan C. P., Smith M. E. B., Ward J. M., Caddick S., Kay C. W. M., Aeppli G., Chester K. A., Baker J. R., Sci. Rep. 2013, 3, 1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Grünewald J., Jones D. H., Brock A., Chiu H.-P., Bursulaya B., Ng K., Vo T., Patterson P., Uno T., Hunt J., Spraggon G., Geierstanger B. H., ChemBioChem 2014, 15, 1787–1791. [DOI] [PubMed] [Google Scholar]
- 65. Fletcher S. A., Sin P. K. B., Nobles M., Årstad E., Tinker A., Baker J. R., Org. Biomol. Chem. 2015, 13, 9559–9563. [DOI] [PubMed] [Google Scholar]
- 66. Smith C. V., Jones D. P., Guenthner T. M., Lash L. H., Lauterburg B. H., Toxicol. Appl. Pharmacol. 1996, 140, 1–12. [DOI] [PubMed] [Google Scholar]
- 67. Moody P., Smith M. E., Ryan C. P., Chudasama V., Baker J. R., Molloy J., Caddick S., ChemBioChem 2012, 13, 39–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ryan C. P., Smith M. E. B., Schumacher F. F., Grohmann D., Papaioannou D., Waksman G., Werner F., Baker J. R., Caddick S., Chem. Commun. 2011, 47, 5452–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Castañeda L., Maruani A., Schumacher F. F., Miranda E., Chudasama V., Chester K. A., Baker J. R., Smith M. E. B., Caddick S., Chem. Commun. 2013, 49, 8187–8189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Deng C., Wu J., Cheng R., Meng F., Klok H.-A., Zhong Z., Prog. Polym. Sci. 2014, 39, 330–364. [Google Scholar]
- 71. Wurm F., Steinbach T., Klok H.-A., Chem. Commun. 2013, 49, 7815–7817. [DOI] [PubMed] [Google Scholar]
- 72. Fuhrmann G., Grotzky A., Lukić R., Matoori S., Luciani P., Yu H., Zhang B., Walde P., Schlüter A. D., Gauthier M. A., Leroux J. C., Nat. Chem. 2013, 5, 582–589. [DOI] [PubMed] [Google Scholar]
- 73. Harris A. G., Gut 1994, 35, S1–S4.8307427 [Google Scholar]
- 74. Vugmeyster Y., Entrican C. A., Joyce A. P., Lawrence-Henderson R. F., Leary B. A., Mahoney C. S., Patel H. K., Raso S. W., Olland S. H., Hegen M., Xu X., Bioconjugate Chem. 2012, 23, 1452–1462. [DOI] [PubMed] [Google Scholar]
- 75. Brea R. J., Reiriz C., Granja J. R., Chem. Soc. Rev. 2010, 39, 1448–1456. [DOI] [PubMed] [Google Scholar]
- 76. Hudalla G., Sun T., Gasiorowski J. Z., Han H., Tian Y. F., Chong A. S., Collier J. H., Nat. Mater. 2014, 13, 829–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Webber M. J., Kessler J. A., Stupp S. I., J. Intern. Med. 2010, 267, 71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Grötzinger C., Wiedenmann B., Ann. N. Y. Acad. Sci. 2004, 1014, 258–264. [DOI] [PubMed] [Google Scholar]
- 79. Rolleman E. J., Kooij P. P. M., De Herder W. W., Valkema R., Krenning E. P., De Jong M., Eur. J. Nucl. Med. Mol. Imaging 2007, 34, 1854–1860. [DOI] [PubMed] [Google Scholar]
- 80. Cescato R., Schulz S., Waser B., Eltschinger V., Rivier J. E., Wester H.-J., Culler M., Ginj M., Liu Q., Schonbrunn A., Reubi J. C., J. Nucl. Med. 2006, 47, 502–511. [PubMed] [Google Scholar]
- 81.F. Barbieri, A. Bajetto, A. Pattarozzi, M. Gatti, R. Würth, S. Thellung, A. Corsaro, V. Villa, M. Nizzari, T. Florio, Int. J. Pept 2013; DOI: 10.1155/2013/926295. [DOI] [PMC free article] [PubMed]
- 82. Dolmans D. E. J. G. J., Fukumura D., Jain R. K., Nat. Rev. Cancer 2003, 3, 380–387. [DOI] [PubMed] [Google Scholar]
- 83. Vrouenraets M. B., Visser G. W., Snow G. B., van Dongen G. A., Anticancer Res. 2003, 23, 505–522. [PubMed] [Google Scholar]
- 84. Frei A., Rubbiani R., Tubafard S., Blacque O., Anstaett P., Felgenträger A., Maisch T., Spiccia L., Gasser G., J. Med. Chem. 2014, 57, 7280–7292. [DOI] [PubMed] [Google Scholar]
- 85. Blum A. P., Kammeyer J. K., Rush A. M., Callmann C. E., Hahn M. E., Gianneschi N. C., J. Am. Chem. Soc. 2015, 137, 2140–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wong S., Shim M. S., Kwon Y. J., J. Mater. Chem. B 2014, 2, 595–615. [DOI] [PubMed] [Google Scholar]
- 87. Jhaveri A., Deshpande P., Torchilin V., J. Controlled Release 2014, 190, 352–370. [DOI] [PubMed] [Google Scholar]
- 88. Pendyala L., Velagapudi S., Toth K., Zdanowicz J., Glaves D., Slocum H., Perez R., Huben R., Creaven P. J., Raghavan D., Clin. Cancer Res. 1997, 3, 793–798. [PubMed] [Google Scholar]
- 89. Knerr P. J., Tzekou A., Ricklin D., Qu H., Chen H., van der Donk W. A., Lambris J. D., ACS Chem. Biol. 2011, 6, 753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Baskin J. M., Bertozzi C. R., QSAR Comb. Sci. 2007, 26, 1211–1219. [Google Scholar]
- 91. Pawlak J. B., Gential G. P. P., Ruckwardt T. J., Bremmers J. S., Meeuwenoord N. J., Ossendorp F. A., Overkleeft H. S., Filippov D. V., van Kasteren S. I., Angew. Chem. Int. Ed. 2015, 54, 5628–5631; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 5720–5723. [Google Scholar]
- 92. Elahipanah S., Radmanesh P., Luo W., O'Brien P. J., Rogozhnikov D., Yousaf M. N., Bioconjugate Chem. 2016, 27, 1082–1089. [DOI] [PubMed] [Google Scholar]
- 93. Ramil C. P., Lin Q., Curr. Opin. Chem. Biol. 2014, 21, 89–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wang Y., Song W., Hu W. J., Lin Q., Angew. Chem. Int. Ed. 2009, 48, 5330–5333; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5434–5437. [Google Scholar]
- 95. Dürr C. J., Lederhose P., Hlalele L., Abt D., Kaiser A., Brandau S., Barner-Kowollik C., Macromolecules 2013, 46, 5915–5923. [Google Scholar]
- 96. Mueller J. O., Guimard N. K., Oehlenschlaeger K. K., Schmidt F. G., Barner-Kowollik C., Polym. Chem. 2014, 5, 1447–1456. [Google Scholar]
- 97. Dietrich M., Delaittre G., Blinco J. P., Inglis A. J., Bruns M., Barner-Kowollik C., Adv. Funct. Mater. 2012, 22, 304–312. [Google Scholar]
- 98. Nygaard J., Munch H. K., Thulstrup P. W., Christensen N. J., Hoeg-Jensen T., Jensen K. J., Arleth L., Langmuir 2012, 28, 12159–12170. [DOI] [PubMed] [Google Scholar]
- 99. Ng D. Y. W., Wu Y., Kuan S. L., Weil T., Acc. Chem. Res. 2014, 47, 3471–3480. [DOI] [PubMed] [Google Scholar]
- 100. Wu Y., Ng D. Y. W., Kuan S. L., Weil T., Biomater. Sci. 2015, 3, 214–230. [DOI] [PubMed] [Google Scholar]
- 101. Gauthier M. A., Klok H.-A., Chem. Commun. 2008, 2591–2611. [DOI] [PubMed] [Google Scholar]
- 102. Pisal D. S., Kosloski M. P., Balu-Iyer S. V., J. Pharm. Sci. 2010, 99, 2557–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Frokjaer S., Otzen D. E., Nat. Rev. Drug Discovery 2005, 4, 298–306. [DOI] [PubMed] [Google Scholar]
- 104. Darwish I. A., Int. J. Biomed. Sci. 2006, 2, 217–235. [PMC free article] [PubMed] [Google Scholar]
- 105. Andrady C., Sharma S. K., Chester K. A., Immunotherapy 2011, 3, 193–211. [DOI] [PubMed] [Google Scholar]
- 106. Senter P. D., Curr. Opin. Chem. Biol. 2009, 13, 235–244. [DOI] [PubMed] [Google Scholar]
- 107. Bouchard H., Viskov C., Garcia-Echeverria C., Bioorg. Med. Chem. Lett. 2014, 24, 5357–5363. [DOI] [PubMed] [Google Scholar]
- 108. Chudasama V., Maruani A., Caddick S., Nat. Chem. 2016, 8, 114–119. [DOI] [PubMed] [Google Scholar]
- 109. Akkapeddi P., Azizi S.-A., Freedy A. M., Cal P. M. S. D., Gois P. M. P., Bernardes G. J. L., Chem. Sci. 2016, 7, 2954–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Wang L., Amphlett G., Blättler W. A., Lambert J. M., Zhang W., Protein Sci. 2005, 14, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Junutula J. R., Raab H., Clark S., Bhakta S., Leipold D. D., Weir S., Chen Y., Simpson M., Tsai S. P., Dennis M. S., Lu Y., Meng Y. G., Ng C., Yang J., Lee C. C., Duenas E., Gorrell J., Katta V., Kim A., McDorman K., Flagella K., Venook R., Ross S., Spencer S. D., Wong W. L., Lowman H. B., Vandlen R., Sliwkowski M. X., Scheller R. H., Polaki P., Mallet W., Nat. Biotechnol. 2008, 26, 925–932. [DOI] [PubMed] [Google Scholar]
- 112. Hamblett K. J., Senter P. D., Chace D. F., Sun M. M. C., Lenox J., Cerveny C. G., Kissler K. M., Bernhardt S. X., Kopcha A. K., Zabinski R. F., Meyer D. L., Francisco J. A., Am. Assoc. Cancer Res. 2004, 10, 7063–7070. [DOI] [PubMed] [Google Scholar]
- 113. Hull E. A., Livanos M., Miranda E., Smith M. E. B., Chester K. A., Baker J. R., Bioconjugate Chem. 2014, 25, 1395–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]