

Abstract
Background
The effect of lixisenatide—a prandial once‐daily glucagon‐like peptide‐1 receptor agonist—on glycaemic control in patients with inadequately controlled type 2 diabetes mellitus (T2DM), stratified by baseline β‐cell function, was assessed.
Methods
The 24‐week GetGoal‐M, ‐P and ‐S trials evaluated the efficacy and safety of lixisenatide in combination with oral antidiabetic agents. This post hoc analysis used data from patients receiving lixisenatide in these trials, divided into matched cohorts by propensity scoring, and stratified according to baseline homeostasis model assessment of β‐cell function (HOMA‐β) index levels, high HOMA‐β: > median HOMA‐β (28.49%); low HOMA‐β: ≤ median.
Results
The matched “low” and “high” HOMA‐β index cohorts (N = 546 patients) had comparable baseline parameters. Mean change from baseline in glycated haemoglobin (HbA1c) was −0.85% and −0.94% for low and high HOMA‐β cohorts, respectively (P = .2607). Reductions from baseline in fasting plasma glucose (FPG; −0.77 vs −1.04 mmol/L; P = .1496) and postprandial plasma glucose (PPG; −5.82 vs −5.61 mmol/L; P = .7511) were similar in the low versus high HOMA‐β index cohorts. Reduction in body weight was significantly greater in the low versus high HOMA‐β index cohort (–2.06 vs –1.13 kg, respectively; P = .0006).
Conclusions
In patients with T2DM, lixisenatide was associated with reduction in HbA1c and improvements in both FPG and PPG, regardless of β‐cell function, indicating that lixisenatide is effective in reducing hyperglycaemia, even in patients with more advanced stages of T2DM and poor residual β‐cell function.
Keywords: HOMA‐β index, lixisenatide, type 2 diabetes mellitus, β‐cell function
1. INTRODUCTION
Type 2 diabetes mellitus (T2DM) is a progressive disease characterized by both disrupted glucose and lipid metabolism, and by hyperglycaemia, which leads to vascular complications and a variety of clinical comorbidities, including myocardial infarction, stroke, blindness, and kidney failure.1, 2, 3, 4 Treatment of T2DM is aimed at reducing hyperglycaemia, usually assessed by the level of glycated haemoglobin (HbA1c) and requires intensification of therapy and additional interventions over time. Both fasting plasma glucose (FPG) and postprandial plasma glucose (PPG) affect overall HbA1c levels,5 with PPG having a greater impact than FPG in suboptimally controlled patients with T2DM with comparatively low levels of HbA1c.6 As the disease progresses, β‐cell mass is reduced7 and β‐cell function deteriorates, mirroring the observed increase in FPG levels.8 Thus, the use of oral antidiabetic drugs (OADs) and adjunctive therapies, such as injectable Glucagon‐like peptide‐1 (GLP‐1) receptor agonists (RAs) or dipeptidyl peptidase‐4 (DPP‐4) inhibitors, may become insufficient to maintain normoglycaemia and an insulin regimen must be initiated. Guidelines from the American Diabetes Association/European Association for the Study of Diabetes recommend that insulin therapy begins with once‐daily injection of a long‐acting basal insulin, such as insulin glargine, to regulate FPG levels.9, 10, 11, 12
GLP‐1 RAs mimic the activity of GLP‐1 while being resistant to degradation by DPP‐4, leading to prolonged activity compared with endogenous GLP‐1. Prandial (or short‐acting) and long‐acting GLP‐1 RAs have been defined based on their pharmacokinetic, pharmacodynamic, and mechanistic differences. While prandial GLP‐1 RAs exert glycaemic benefits through their effects on both slowing of gastric emptying and the incretin pathway, it is the former that has a profound impact on PPG.13 Long‐acting GLP‐1 RAs exert their glycaemic effects primarily through stimulation of insulin secretion and reduction in glucagon levels and, as such, have a dominant effect on FPG.13 The stimulation of insulin release from pancreatic β cells and reduction of glucagon levels brought about by GLP‐1 RAs are both glucose dependent.14 Currently, it remains unclear whether the degree of loss of β‐cell function affects the efficacy of these agents. The present analysis was designed to investigate the influence of residual β cell function on the efficacy of a prandial GLP‐1 RA and, to our knowledge, is the first of its kind on an agent of this class.
Lixisenatide (Lyxumia®, Sanofi, Paris, France) is a once‐daily GLP‐1 RA indicated for the treatment of patients with T2DM and was first approved in the European Union in 2013. In the GetGoal programme of randomized phase III clinical trials, lixisenatide compared with placebo significantly improved HbA1c levels and significantly reduced PPG in patients with T2DM.15, 16, 17, 18, 19, 20 In other studies, lixisenatide brought about significantly greater improvements in PPG than liraglutide, a long‐acting GLP‐1 RA, but showed more limited effects on FPG.21, 22 Nevertheless, significant improvements in FPG were seen in the GetGoal trials in which lixisenatide was used as monotherapy or added to existing OAD treatment.15, 16, 23, 24 Pharmacological studies have demonstrated that, although lixisenatide acts through multiple mechanisms, the marked reduction in PPG excursions associated with lixisenatide is largely caused by slowing of gastric emptying.25 Furthermore, in vivo preclinical studies have shown that the extent of inhibition of gastric emptying with lixisenatide is so great that the reduction in PPG concentrations is associated with a reduction rather than an increase in plasma insulin.26 This indicates that lixisenatide acts through a pathway independent of islet function, resulting in a marked alleviation of the prandial β‐cell secretory burden.27 The weight of evidence in the trials described above suggests that lixisenatide should be efficacious even in patients with markedly reduced residual β‐cell function.
The homeostasis model assessment of β‐cell function (HOMA‐β)28 is a widely used clinical and epidemiological tool for the assessment of β‐cell function using FPG and insulin (or C‐peptide) concentrations, with a higher HOMA‐β index value representing better β‐cell function. The HOMA‐β has been validated against several other more sophisticated methods for the assessment of β‐cell function, including the hyperglycaemic clamp, the acute insulin response (following an intravenous glucose tolerance test), and the continuous infusion glucose model assessment.28, 29, 30, 31, 32
To investigate the hypothesis that residual β‐cell function is not a major determinant of the efficacy of lixisenatide, this post hoc analysis evaluated the efficacy (and safety) of lixisenatide according to patients' baseline β‐cell function, as determined by HOMA‐β index, using pooled data from 3 studies in the GetGoal clinical trial programme.
2. SUBJECTS AND METHODS
2.1. Study design
This was a descriptive, post hoc analysis of individual patient data extracted from the intent‐to‐treat populations of 3 randomized, double‐blind, placebo‐controlled, multicentre, 24‐week GetGoal trials that evaluated the efficacy and safety of adding lixisenatide to OAD treatment in patients with T2DM. GetGoal‐M (NCT00712673), GetGoal‐P (NCT00763815), and GetGoal‐S (NCT00713830)15, 20, 23 assessed lixisenatide as add‐on therapy to OADs in patients inadequately controlled with metformin, pioglitazone (with or without metformin), or sulphonylureas (with or without concomitant metformin), respectively. These 3 trials were chosen for inclusion in this analysis because they were the studies in the GetGoal programme in which plasma insulin levels were assessed. The present analysis included all patients in the intent‐to‐treat population who had been randomized to the lixisenatide treatment arms of these 3 studies with baseline and endpoint visit HbA1c measurements, and baseline and endpoint values for HOMA‐β index. Patients were stratified into 2 cohorts according to their HOMA‐β index value at baseline relative to the median value (28.49%). Those in the “low” HOMA‐β index cohort had an index value ≤ median HOMA‐β index of all eligible patients, while those in the “high” HOMA‐β index group had an index value > median HOMA‐β index. HOMA‐β index values were calculated as 20 × fasting plasma insulin [μU/mL])/ FPG [mmol/L] –3.5. For the 3 studies included in this analysis, the trial protocols complied with the recommendations of the Declaration of Helsinki and were approved by independent ethics committees and institutional review boards. The protocols also complied with the laws and regulations, as well as any applicable guidelines, of the countries where the studies were conducted.
2.2. Endpoints assessed
Key efficacy endpoints included the mean change from baseline to endpoint in HbA1c, PPG, glucose excursion, FPG, body weight, body mass index (BMI), and HOMA‐β index, and the proportion of patients achieving a treatment target of HbA1c < 7%, achieving a FPG treatment target of <6.11 mmol/L, and switching HOMA‐β index group between baseline and endpoint.
To assess the change in PPG and glucose excursion in GetGoal‐M and ‐S,15, 20 a standardized breakfast meal challenge test consisting of a 600‐kcal liquid meal (400 mL of Ensure Plus®, Abbott Nutrition, Columbus, OH, USA; composed of 53.8% carbohydrate, 16.7% protein, and 29.5% fat) was performed 30 minutes after drug administration at baseline and at Week 24. Glucose excursion was calculated as 2‐hour PPG minus plasma glucose levels 30 minutes prior to the meal test before lixisenatide administration.
Safety endpoints included symptomatic hypoglycaemia, defined as symptoms of hypoglycaemia with an accompanying plasma glucose level of <3.33 mmol/L or prompt recovery with oral carbohydrate or glucagon administration, and severe hypoglycaemia, defined as an event requiring assistance of another person due to acute neurological impairment directly resulting from the hypoglycaemic event, with a plasma glucose level of <2.00 mmol/L or prompt recovery following carbohydrate or glucagon administration.
Composite endpoints were endpoint HbA1c levels of <7% with no symptomatic hypoglycaemia; endpoint HbA1c levels of <7% with no severe hypoglycaemia; endpoint HbA1c levels of <7% with no weight gain (defined as a change in weight from baseline to endpoint of ≤0 kg); endpoint HbA1c levels of <7%, no weight gain and no symptomatic hypoglycaemia; and endpoint HbA1c levels of <7%, no weight gain and no severe hypoglycaemia.
2.2.1. Statistical analysis
A multivariable logistic regression model was used to assess the main variables affecting the probability of patients having high versus low HOMA‐β index scores. The high versus low HOMA‐β index status was the dependent variable, and age, sex, baseline BMI, duration of diabetes, baseline HbA1c, baseline FPG, and sulphonylurea usage status were the independent variables. The propensity scores, evaluated as the probabilities from the logistic regression model, were then matched between patients with high versus low HOMA‐β index scores, resulting in a population matched for the independent variables. Thereafter, study endpoints were assessed with these matched study cohorts. Additionally, the same analyses of study endpoints were also performed on the original, unmatched cohort.
In the unmatched cohort, the multivariable logistic regression analysis was also used to determine independent predictors of baseline HOMA‐β scores. Furthermore, a Pearson correlation analysis was performed on the unmatched population to assess HOMA‐β index at baseline versus clinical and demographic features.
Baseline demographics, clinical characteristics, and efficacy and safety outcomes were evaluated according to HOMA‐β index cohort. Outcomes for HOMA‐β index cohorts were compared with one another, with P values calculated using a chi‐square test for categorical variables or analysis of variance for continuous variables. Paired t tests were used to compare baseline and endpoint continuous measurements among patients within each cohort, with a P value of .05 used to determine the level of statistical significance.
3. RESULTS
3.1. Baseline characteristics
Of the 980 patients with T2DM treated with lixisenatide in the 3 GetGoal clinical trials who met the inclusion criteria, a total of 546 patients were included in the propensity score‐matched population, 273 in each cohort (please refer to the Statistical analysis section for details on how the propensity score matching was done). Baseline characteristics were comparable across the high versus low HOMA‐β index cohorts, with the exception of PPG and glucose excursion, which were not matched in the analysis and were thus higher in the low versus high HOMA‐β index cohort (Table 1). Baseline characteristics of the unmatched cohort are shown in Table S1.
Table 1.
Patient demographics and baseline characteristics of the matched population
| Characteristic | Low HOMA‐β Index (n = 273) | High HOMA‐β Index (n = 273) | P Value |
|---|---|---|---|
| HOMA‐β score | 18.5 (6.0) | 54.3 (71.2) | <.0001 |
| Age, years | 55.7 (9.6) | 55.8 (9.7) | .8453 |
| Sex, male/female, % | 52.8/47.3 | 54.2/45.8 | .7314 |
| Race, % | .3087 | ||
| Asian | 15.4 | 12.8 | |
| Black/African American | 3.3 | 3.3 | |
| White | 77.7 | 82.4 | |
| Other | 3.7 | 1.5 | |
| BMI, kg/m2 | 31.8 (6.1) | 32.4 (5.7) | .2102 |
| Known diabetes duration, years | 6.9 (4.4) | 7.4 (5.3) | .2460 |
| Duration of OAD therapy, years | 4.0 (3.5) | 4.6 (4.5) | .0899 |
| Metformin at baseline, % | 88.6 | 91.6 | .2514 |
| Sulphonylurea at baseline, % | 18.3 | 22.7 | .2034 |
| HbA1c, % | 8.1 (0.8) | 8.1 (0.9) | .8308 |
| PPG, mmol/L | 16.9 (3.9) | 15.3 (3.7) | .0012 |
| Glucose excursion, mmol/L | 7.0 (3.4) | 6.1 (3.1) | .0352 |
| FPG, mmol/L | 9.2 (1.7) | 9.3 (2.0) | .3910 |
Abbreviations: BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; HOMA‐β, homeostasis model assessment of residual β‐cell function; OAD, oral antidiabetic drug; PPG, postprandial plasma glucose; SD, standard deviation.
Data are mean (SD) unless stated otherwise.
A multivariate regression analysis of the unmatched data identified both BMI and duration of diabetes as independent predictors of HOMA‐β index category at baseline (Table 2). FPG and HbA1c at baseline were also identified as predictors of HOMA‐β values, although this was to be expected as FPG is involved in the calculation of HOMA‐β and contributes to overall HbA1c levels. Interestingly, while the use of a sulphonylurea was not found to be a predictor of baseline HOMA‐β index cohort, the P value showed a trend towards significance.
Table 2.
Predictors of high versus low baseline HOMA‐β index among study patients treated with lixisenatide in the unmatched population
| Parameters | Odds Ratio | Lower Limit | Upper Limit | P Value* |
|---|---|---|---|---|
| Age, years | 0.993 | 0.977 | 1.010 | .4274 |
| Sex, female vs male | 1.250 | 0.932 | 1.676 | .1364 |
| Baseline BMI, kg/m2 | 1.147 | 1.114 | 1.182 | <.0001 |
| Duration of diabetes, years | 0.956 | 0.929 | 0.985 | .0031 |
| Baseline HbA1c, % | 0.799 | 0.655 | 0.975 | .0270 |
| Baseline FPG, mmol/L | 0.983 | 0.978 | 0.988 | <.0001 |
| Other vs white | 0.434 | 0.176 | 1.068 | .1284 |
| Black or African American vs white | 0.925 | 0.420 | 2.035 | .5025 |
| Asian vs white | 0.772 | 0.465 | 1.282 | .8828 |
| Baseline sulphonylurea usage: Yes vs no | 0.672 | 0.452 | 1.000 | .0502 |
Abbreviations: BMI, body mass index; FPG, fasting plasma glucose; HbA1c, glycated haemoglobin; HOMA‐β, homeostasis model assessment of residual β‐cell function.
Bolded values reached statistical significance (P < .05).
P value derived from maximum likelihood estimates.
3.2. Efficacy endpoints
Reductions from baseline in HbA1c were −0.94% and −0.85% for the high and low HOMA‐β index cohorts, respectively (Figure 1). The difference in change from baseline in HbA1c between the cohorts was not statistically significant (P = .2607). A similar proportion of patients in each cohort achieved an endpoint HbA1c < 7% (45.79% and 43.59% of patients in the high versus low HOMA‐β index cohorts, respectively; P = .6055).
Figure 1.
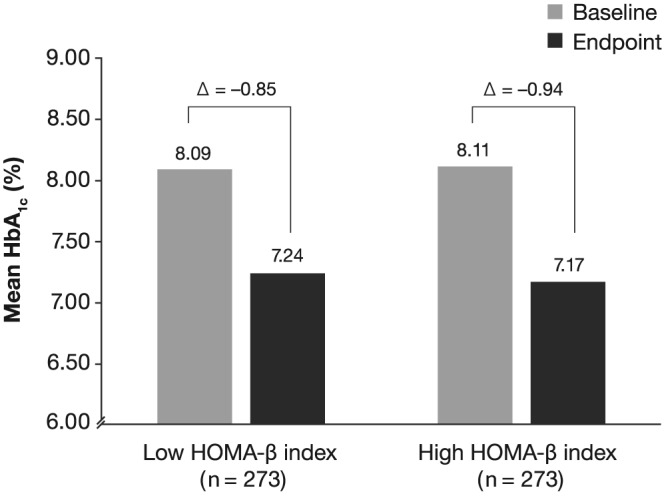
Mean change in HbA1c with lixisenatide by HOMA‐β index in the matched cohort. HbA1c, glycated haemoglobin; HOMA‐β, homeostasis model assessment of residual β‐cell function
Similar mean reductions from baseline were observed in the high and low HOMA‐β index cohorts in both PPG (P = .7511; Figure 2A) and glucose excursion (P = .9592; Figure 2B), despite a difference in baseline PPG, which was not accounted for in the propensity score‐matching analysis.
Figure 2.
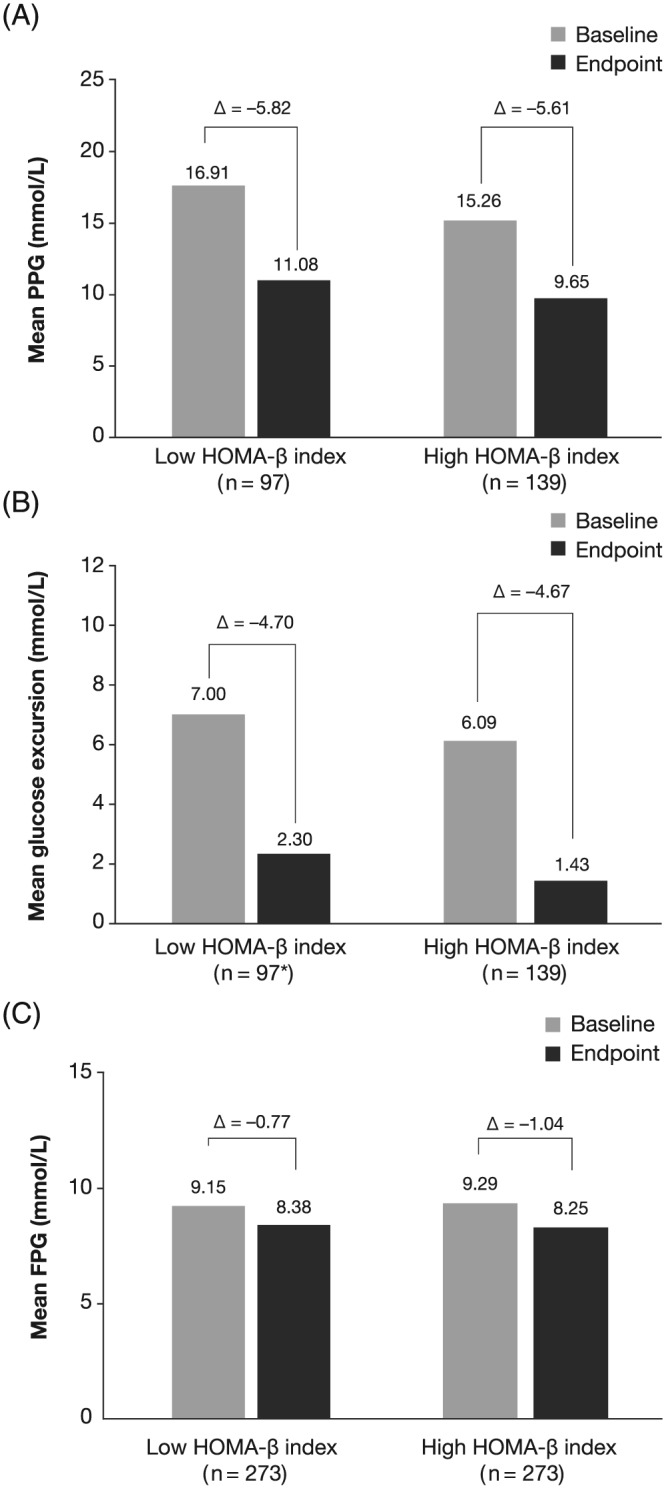
Mean change in, A, PPG; B, glucose excursion; and C, FPG with lixisenatide by HOMA‐β index in the matched cohort. FPG, fasting plasma glucose; HOMA‐β, homeostasis model assessment of residual β‐cell function; PPG, postprandial plasma glucose. *n = 96 for baseline and change from baseline measurements
Reductions from baseline in FPG of –1.04 and –0.77 mmol/L were observed for the high and low HOMA‐β index cohorts, respectively (Figure 2C). The change from baseline in FPG did not differ significantly between cohorts (P = .1496). A greater proportion of patients in the high HOMA‐β index cohort achieved the FPG treatment target of <6.11 mmol/L: 15.75% versus 9.89% in the high and low HOMA‐β index cohorts, respectively (P = .0405).
Although both mean weight and BMI were comparable across the 2 cohorts at baseline, the change was smaller in the high HOMA‐β index cohort than that in the low HOMA‐β index cohort (weight: –1.13 vs –2.06 kg, respectively; P = .0006; BMI: –0.41 vs –0.77 kg/m2, respectively; P = .0004).
HOMA‐β index scores increased in both cohorts over the study period. HOMA‐β index scores increased above the cut‐off value in 34.07% (93/273) of patients in the low HOMA‐β index cohort, and decreased below the cut‐off in 12.45% (34/273) of patients in the high HOMA‐β index cohort.
Efficacy endpoints for the unmatched cohorts are shown in Table S2.
3.3. Pearson correlation analysis
The Pearson correlation analysis of the unmatched cohort showed that HOMA‐β index at baseline correlated strongly with change in HOMA‐β index over the treatment period (Table S3). HOMA‐β index at baseline also showed a strong negative correlation with age, and a moderate positive correlation with HOMA‐β index at study end.
3.4. Safety endpoints
An equal proportion of patients in the high versus low HOMA‐β index cohorts experienced symptomatic hypoglycaemia (5.49% vs 5.13%, respectively; P = .8487). There were no events of severe hypoglycaemia reported in either study cohort.
3.5. Composite endpoints
There were no statistically significant differences between the study cohorts in achieving the composite endpoints of HbA1c < 7% with no symptomatic hypoglycaemia, HbA1c < 7% with no weight gain and HbA1c < 7% with no weight gain and no symptomatic hypoglycaemia (Table 3). Composite endpoints for the unmatched population are shown in Table S4.
Table 3.
Composite endpoints of study cohorts treated with lixisenatide in the matched population
| Composite endpoint | Low HOMA‐β Index (n = 273) | High HOMA‐β Index (n = 273) | P Value | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| HbA1c < 7% and no symptomatic hypoglycaemia | 112 | 41.03 | 116 | 42.49 | .7285 |
| HbA1c < 7% and no weight gain | 99 | 36.26 | 89 | 32.60 | .3678 |
| HbA1c < 7% no weight gain and no symptomatic hypoglycaemia | 92 | 33.70 | 82 | 30.04 | .3584 |
Abbreviations: HbA1c, glycated haemoglobin; HOMA‐β, homeostasis model assessment of residual β‐cell function.
4. DISCUSSION
This study of patients with T2DM inadequately controlled by OADs suggests that lixisenatide can lower HbA1c levels regardless of residual β‐cell function, as assessed by HOMA‐β index, ie, that treatment with lixisenatide is effective at reducing hyperglycaemia, even in the more advanced stages of T2DM when β‐cell function is markedly diminished. Low β‐cell function at baseline (as assessed by HOMA‐β index) was associated with older age, a lower BMI, longer known duration of diabetes, and longer duration of OAD treatment. These data are supported by the typical subphenotypes and natural history of T2DM. For instance, obese patients are characterized by more severe insulin resistance33 and, as a result, their critical β‐cell function threshold to develop diabetes is higher than that of nonobese patients.34 Furthermore, β‐cell mass has been shown to gradually decline with age in both diabetic and nondiabetic individuals, thereby accounting, at least in part, for the well‐established role of age as a risk factor of T2DM.7 Additionally, the same study showed that β‐cell mass also declines with diabetes duration.7 Other studies have shown that the functional impairment of β‐cells often significantly exceeds the deficit in β‐cell mass, as the secretory burden for the remaining β‐cells is increased by 100% if their overall mass is reduced by just 50%.35, 36, 37
Recent studies indicate that propensity score matching is an effective tool for comparing treatment regimens in those with diabetes and can decrease treatment selection bias between groups.38, 39 The use of the propensity score‐matching technique was a major strength of this analysis because there were several differences in baseline characteristics between patients in the high and low HOMA‐β index cohorts that may have influenced the effect of lixisenatide treatment. By using propensity score matching, we were able to reduce the bias caused by these potential confounding factors. Thus, baseline HbA1c, FPG, BMI, duration of diabetes, and sulphonylurea use were accounted for in the matched population and were similar between cohorts. As a result, reductions from baseline for both HbA1c and FPG were comparable between high versus low HOMA‐β index cohorts, and there was no difference between them in the proportion of patients achieving HbA1c < 7%. However, despite comparable baseline and endpoint FPG, higher proportions of patients in the high versus low HOMA‐β index‐matched cohorts achieved FPG treatment targets. Furthermore, in the matched population, baseline PPG and glucose excursions were greater for the low versus high HOMA‐β index cohort. Nevertheless, comparable reductions from baseline in both of these parameters were seen between cohorts.
On the whole, these data strongly suggest that lixisenatide confers glycaemic control in HbA1c targets and reductions in hyperglycaemia, particularly PPG, irrespective of β‐cell function. However, the FPG results are compatible with some impact of β‐cell function in modulating the action of lixisenatide on fasting glycaemia, with other mechanisms, ie, glucagon inhibition, also potentially involved. Furthermore, as HOMA‐β is essentially a static index of β‐cell function in the fasting state, it is reasonable that within the cohort with better fasting β‐cell function, a greater number of patients achieved a FPG lower than 6.1 mmol/L. However, lixisenatide is expected to act predominantly through control of PPG, mediated primarily through its role in delaying gastric emptying (which is not necessarily affected by β‐cell function) and in suppressing glucagon secretion,25 and to have a lesser effect on FPG. Indeed, these expectations are supported by the marked reductions in PPG observed in this pooled analysis, which are in line with previous reports15, 16, 17, 18, 19, 20, 24 and with the classification of lixisenatide as a prandial GLP‐1 RA. Fasting plasma glucose also decreased, but to a lesser extent than PPG, as expected and consistent with previous studies.15, 16, 23, 24 Finally, although not assessed in the present study, it is expected that lixisenatide will have reduced the total amount of insulin secreted in the fed state25 and, as a result, the “workload” of β cells. This may have potential long‐term benefits with respect to the preservation of β‐cell function. Hence, we speculate that part of the observed reduction in hyperglycaemia in this analysis may be the result of improved overall β‐cell function brought about by lixisenatide.
While findings from the unmatched population suggested that changes in weight and BMI were not different between the study cohorts, the propensity score‐matched analysis found that patients in the low HOMA‐β index cohort experienced significantly greater changes in both parameters, despite them being comparable at baseline. Interestingly, although these changes in weight and BMI in the matched analysis were statistically significant, the absolute difference between the study cohorts was modest and, hence, of questionable clinical relevance.
There were no significant differences between cohorts of the composite endpoints. This indicates that lixisenatide can improve glycaemic control irrespective of β‐cell function, with neither of the cohorts being more at risk of hypoglycaemia or weight gain, and further strengthens the rationale for the use of lixisenatide, even in patients with poor residual β‐cell function.
Mean HOMA‐β levels in both cohorts increased over the course of the 24‐week study, with 34.07% of patients in the low HOMA‐β index cohort increasing their score above the original cutoff for inclusion into the high HOMA‐β index cohort. This finding strongly suggests that HOMA‐β function is improved with lixisenatide treatment. The relative roles played by the direct effects of lixisenatide on β cells or the relief of glucose toxicity in improving β‐cell function cannot be determined with our experimental design. However, previous research27 has shown that the delay of gastric emptying brought about by exogenous GLP‐1 limits the rate and extent of PPG presented to β cells, leading to “β‐cell rest” and the partial recovery of β cells and the endogenous insulin response. This effect is also evident when incretin therapies are combined with basal insulin.40 In this scenario, the presence of exogenous insulin supplements endogenous insulin production, promoting β‐cell rest. Improvements in HOMA‐β index scores have also been seen with other GLP‐1 RAs,41, 42 DPP‐4 inhibitors,43 and some meglitinides.44 In the UK Prospective Diabetes Study, an increase in β‐cell function was seen at 1 year in patients receiving sulphonylureas, although this increase was not maintained over subsequent years.45 Baseline HOMA‐β levels in the present study were comparable with those seen in UK Prospective Diabetes Study, where 6 years after diagnosis and initiation of treatment the mean HOMA‐β index score were 28%.45 Consistency of treatment effect regardless of β‐cell function has also been observed previously for DPP‐4 inhibitors.46 A recent study has identified both C‐peptide and islet autoantibodies as potential biomarkers that could allow patients with a good predicted response to GLP‐1 RA therapy to be selected for treatment based on their β‐cell function.47
A limitation of this study is that there was no placebo comparator arm, as patient data were extracted from the single lixisenatide arms of 3 clinical trials. Furthermore, these trials were conducted in several countries, at various times, and with different background oral therapies. Although a slight increase in β‐cell function was observed between baseline and endpoint in this analysis, the 6‐month duration of the studies included was insufficient to determine whether lixisenatide is clearly associated with a beneficial effect on β‐cell function. Longer studies would be needed to explore this further. Additionally, as lixisenatide exerts its effects predominantly on PPG, it would be of interest to investigate the correlation between changes in HbA1c and PPG/FPG according to β‐cell function and in patients being treated with lixisenatide in general. The HOMA‐β index is only an approximate measure of β‐cell function but is nevertheless clinically useful.48 A further consideration is that the analyses presented here were conducted using the original HOMA1 model, even though this has been improved and a newer HOMA2 model is available,49 because it is simpler and facilitates comparison with existing literature.
While different oral agents (metformin, pioglitazone, or sulphonylureas) were used in the 3 studies included in this analysis, the real‐life, double‐blind, randomized multicentre A Diabetes Outcomes Progression Trial has also successfully used HOMA, albeit HOMA2, to compare the effects of thiazolidinediones, metformin, and sulphonylureas on long‐term glycaemic control and β‐cell function.50
Our findings indicate that treatment with once‐daily lixisenatide is effective in reducing hyperglycaemia, is well tolerated and associated with weight loss, even when β‐cell function is low, and highlights the importance of the non–β‐cell‐mediated actions of lixisenatide in improving glycaemic control.
CONFLICTS OF INTEREST
RCB has served on an advisory panel for Amgen USA, Inc, Eli Lilly and Company, Merck Sharp & Dohme Limited and Sanofi, and has participated in a speaker bureau for AstraZeneca Pharmaceuticals LP, Bristol‐Myers Squibb Company, Eli Lilly and Company, and Sanofi. LB has served as a consultant for AstraZeneca, GlaxoSmithKline, Intarcia Therapeutics, Inc, Janssen Pharmaceuticals, Inc, Merck & Co, Inc, Novo Nordisk, Inc, and Sanofi; has participated in a speaker bureau for AstraZeneca, Janssen Pharmaceuticals, Inc, Merck & Co, Novo Nordisk Inc, and Sanofi; and grant/research support to Dr Blonde and/or his institution has been received from AstraZeneca, Janssen Pharmaceuticals, Inc, Lexicon Pharmaceuticals, Inc, Merck & Co, Novo Nordisk, Inc, and Sanofi. MA has no disclosures. RB is an employee of Sanofi. PG has served as a consultant and acted as an author for AstraZeneca Pharmaceuticals LP, Boehringer Ingelheim Pharmaceuticals, Inc, Bristol‐Myers Squibb Company, Eli Lilly and Company, GlaxoSmithKline, Janssen Pharmaceuticals, Merck Sharp & Dohme Limited, Novartis Pharmaceuticals Corporation, Novo Nordisk, Inc, Sanofi, and Takeda and has received research support from and acted as an author for Sanofi. MHa has no disclosures. VM has received research support from and acted as an author for Eli Lilly and Company, Johnson & Johnson, Merck, Novo Nordisk, Inc, Sanofi, and USV. MHo has served on advisory panels and acted as an author for AstraZeneca Pharmaceuticals LP and Novartis Pharmaceuticals Corporation; has received research support from and acted as an author for Eli Lilly and Company, Novartis Pharmaceuticals Corporation and Sanofi; has served on a speaker bureau and acted as an author for Boehringer Ingelheim Pharmaceuticals, Inc., Eli Lilly and Company, Merck Sharp & Dohme Limited and Sanofi; and is a stock/shareholder in and acted as an author for Satiogen Pharmaceuticals, Inc.
AUTHOR CONTRIBUTIONS
RCB, with others, conceived this post hoc analysis, reviewed the data, planned further analysis, and wrote and revised the manuscript. LB reviewed the data analyses and participated in writing and revising the manuscript. MA participated in reviewing the data and revising the manuscript. RB reviewed the data analyses and participated in writing and revising the manuscript. PG reviewed the data analyses and participated in writing and revising the manuscript. MHa reviewed the data analyses and participated in writing and revising the manuscript. VM participated in writing and revising the manuscript. MHo reviewed the data analyses and contributed to writing and revising the manuscript.
PRIOR PUBLICATION OF DATA
These data have been presented in part at the 74th Scientific Sessions of the American Diabetes Association, San Francisco, CA, USA (June 13‐17, 2014).
Supporting information
Supplementary Table 1. Patient demographics and baseline characteristics in the unmatched population
Supplementary Table 2. Efficacy outcomes in the unmatched population
Supplementary Table 3. Pearson correlation analysis of baseline HOMA‐β index measurements with patient clinical characteristics in the unmatched population
Supplementary Table 4. Composite endpoints in the unmatched population
ACKNOWLEDGEMENTS
The authors would like to thank Jay Lin, PhD, Novosys Health, for his statistical support. Editorial assistance was provided by Christina Holleywood, PhD, of Caudex and was funded by Sanofi.
Bonadonna RC, Blonde L, Antsiferov M, et al. Lixisenatide as add‐on treatment among patients with different β‐cell function levels as assessed by HOMA‐β index. Diabetes Metab Res Rev. 2017;33:e2897 https://doi.org/10.1002/dmrr.2897
REFERENCES
- 1. Fletcher EL, Phipps JA, Wilkinson‐Berka JL. Dysfunction of retinal neurons and glia during diabetes. Clin Exp Optom. 2005;88(3):132‐145. [DOI] [PubMed] [Google Scholar]
- 2. Luitse MJ, Biessels GJ, Rutten GE, Kappelle LJ. Diabetes, hyperglycaemia, and acute ischaemic stroke. Lancet Neurol. 2012;11(3):261‐271. [DOI] [PubMed] [Google Scholar]
- 3. Powell DW, Kenagy DN, Zheng S, et al. Associations between structural and functional changes to the kidney in diabetic humans and mice. Life Sci. 2013;93(7):257‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yudkin JS, Oswald GA. Hyperglycaemia, diabetes and myocardial infarction. Diabet Med. 1987;4(1):13‐18. [DOI] [PubMed] [Google Scholar]
- 5. Monnier L, Lapinski H, Colette C. Contributions of fasting and postprandial plasma glucose increments to the overall diurnal hyperglycemia of type 2 diabetic patients: variations with increasing levels of HbA(1c). Diabetes Care. 2003;26(3):881‐885. [DOI] [PubMed] [Google Scholar]
- 6. Riddle M, Umpierrez G, DiGenio A, Zhou R, Rosenstock J. Contributions of basal and postprandial hyperglycemia over a wide range of A1C levels before and after treatment intensification in type 2 diabetes. Diabetes Care. 2011;34(12):2508‐2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta‐cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):32‐42. [DOI] [PubMed] [Google Scholar]
- 8. Levy J, Atkinson AB, Bell PM, McCance DR, Hadden DR. Beta‐cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10‐year follow‐up of the Belfast diet study. Diabet Med. 1998;15(4):290‐296. [DOI] [PubMed] [Google Scholar]
- 9. American Diabetes Association . Standards of medical care in diabetes—2013. Diabetes Care. 2013;36(Suppl 1):S11‐S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Garber AJ, Abrahamson MJ, Barzilay JI, et al. American association of clinical endocrinologists' comprehensive diabetes management algorithm 2013 consensus statement – executive summary. Endocr Pract. 2013;19(3):536‐557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient‐centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35(6):1364‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ryden L, Standl E, Bartnik M, et al. Guidelines on diabetes, pre‐diabetes, and cardiovascular diseases: executive summary. The task force on diabetes and cardiovascular diseases of the European Society of Cardiology (ESC) and of the European Association for the Study of Diabetes (EASD). Eur Heart J. 2007;28(1):88‐136. [DOI] [PubMed] [Google Scholar]
- 13. Meier JJ. GLP‐1 receptor agonists for individualized treatment of type 2 diabetes mellitus. Nat Rev Endocrinol. 2012;8(12):728‐742. [DOI] [PubMed] [Google Scholar]
- 14. Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP‐1 receptor activated insulin secretion from pancreatic beta‐cells: mechanism and glucose dependence. Diabetes Obes Metab. 2013;15(1):15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ahrén B, Leguizamo DA, Miossec P, Saubadu S, Aronson R. Efficacy and safety of lixisenatide once‐daily morning or evening injections in type 2 diabetes inadequately controlled on metformin (GetGoal‐M). Diabetes Care. 2013;36(9):2543‐2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fonseca VA, Alvarado‐Ruiz R, Raccah D, Boka G, Miossec P, Gerich JE. Efficacy and safety of the once‐daily GLP‐1 receptor agonist lixisenatide in monotherapy: a randomized, double‐blind, placebo‐controlled trial in patients with type 2 diabetes (GetGoal‐mono). Diabetes Care. 2012;35(6):1225‐1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Riddle MC, Aronson R, Home P, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled by established basal insulin: a 24‐week, randomized, placebo‐controlled comparison (GetGoal‐L). Diabetes Care. 2013;36(9):2489‐2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Riddle MC, Forst T, Aronson R, et al. Adding once‐daily lixisenatide for type 2 diabetes inadequately controlled with newly initiated and continuously titrated basal insulin glargine: a 24‐week, randomized, placebo‐controlled study (GetGoal‐Duo 1). Diabetes Care. 2013;36(9):2497‐2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seino Y, Min KW, Niemoeller E, Takami A. Randomized, double‐blind, placebo‐controlled trial of the once‐daily GLP‐1 receptor agonist lixisenatide in Asian patients with type 2 diabetes insufficiently controlled on basal insulin with or without a sulfonylurea (GetGoal‐L‐Asia). Diabetes Obes Metab. 2012;14(10):910‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rosenstock J, Hanefeld M, Shamanna P, et al. Beneficial effects of once‐daily lixisenatide on overall and postprandial glycemic levels without significant excess of hypoglycemia in type 2 diabetes inadequately controlled on a sulfonylurea with or without metformin (GetGoal‐S). J Diabetes Complications. 2014;28(3):386‐392. [DOI] [PubMed] [Google Scholar]
- 21. Kapitza C, Forst T, Coester HV, Poitiers F, Ruus P, Hincelin‐Mery A. Pharmacodynamic characteristics of lixisenatide once daily versus liraglutide once daily in patients with type 2 diabetes insufficiently controlled on metformin. Diabetes Obes Metab. 2013;15(7):642‐649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meier JJ, Rosenstock J, Hincelin‐Mery A, et al. Contrasting effects of lixisenatide and liraglutide on post‐prandial glycemic control, gastric emptying, and safety parameters in patients with type 2 diabetes on optimized insulin glargine with or without metformin: a randomized, open‐label trial. Diabetes Care. 2015;38(7):1263‐1273. [DOI] [PubMed] [Google Scholar]
- 23. Pinget M, Goldenberg R, Niemoeller E, Muehlen‐Bartmer I, Guo H, Aronson R. Efficacy and safety of lixisenatide once daily versus placebo in type 2 diabetes insufficiently controlled on pioglitazone (GetGoal‐P). Diabetes Obes Metab. 2013;15(11):1000‐1007. [DOI] [PubMed] [Google Scholar]
- 24. Yu Pan C, Han P, Liu X, et al. Lixisenatide treatment improves glycaemic control in Asian patients with type 2 diabetes mellitus inadequately controlled on metformin with or without sulfonylurea: a randomized, double‐blind, placebo‐controlled, 24‐week trial (GetGoal‐M‐Asia). Diabetes Metab Res Rev. 2014;30(8):726‐735. [DOI] [PubMed] [Google Scholar]
- 25. Lorenz M, Pfeiffer C, Steinstrasser A, et al. Effects of lixisenatide once daily on gastric emptying in type 2 diabetes – relationship to postprandial glycemia. Regul Pept. 2013;185:C1‐C8. [DOI] [PubMed] [Google Scholar]
- 26. Moore MC, Werner U, Smith MS, Farmer TD, Cherrington AD. Effect of the glucagon‐like peptide‐1 receptor agonist lixisenatide on postprandial hepatic glucose metabolism in the conscious dog. Am J Physiol Endocrinol Metab. 2013;305(12):E1473‐E1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nauck MA, Niedereichholz U, Ettler R, et al. Glucagon‐like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. 1997;273(5 Pt 1):E981‐E988. [DOI] [PubMed] [Google Scholar]
- 28. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28(7):412‐419. [DOI] [PubMed] [Google Scholar]
- 29. Hermans MP, Levy JC, Morris RJ, Turner RC. Comparison of tests of beta‐cell function across a range of glucose tolerance from normal to diabetes. Diabetes. 1999;48(9):1779‐1786. [DOI] [PubMed] [Google Scholar]
- 30. Levy JC, Rudenski A, Burnett M, Knight R, Matthews DR, Turner RC. Simple empirical assessment of beta‐cell function by a constant infusion of glucose test in normal and type 2 (non‐insulin‐dependent) diabetic subjects. Diabetologia. 1991;34(7):488‐499. [DOI] [PubMed] [Google Scholar]
- 31. Stumvoll M, Mitrakou A, Pimenta W, et al. Use of the oral glucose tolerance test to assess insulin release and insulin sensitivity. Diabetes Care. 2000;23(3):295‐301. [DOI] [PubMed] [Google Scholar]
- 32. Wallace TM, Levy JC, Matthews DR. An increase in insulin sensitivity and basal beta‐cell function in diabetic subjects treated with pioglitazone in a placebo‐controlled randomized study. Diabet Med. 2004;21(6):568‐576. [DOI] [PubMed] [Google Scholar]
- 33. Ludvik B, Nolan JJ, Baloga J, Sacks D, Olefsky J. Effect of obesity on insulin resistance in normal subjects and patients with NIDDM. Diabetes. 1995;44(9):1121‐1125. [DOI] [PubMed] [Google Scholar]
- 34. Duman BS, Turkoglu C, Gunay D, Cagatay P, Demiroglu C, Buyukdevrim AS. The interrelationship between insulin secretion and action in type 2 diabetes mellitus with different degrees of obesity: evidence supporting central obesity. Diabetes Nutr Metab. 2003;16(4):243‐250. [PubMed] [Google Scholar]
- 35. Meier JJ, Bonadonna RC. Role of reduced beta‐cell mass versus impaired beta‐cell function in the pathogenesis of type 2 diabetes. Diabetes Care. 2013;36(Suppl 2):S113‐S119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pfeifer MA, Halter JB, Porte D Jr. Insulin secretion in diabetes mellitus. Am J Med. 1981;70(3):579‐588. [DOI] [PubMed] [Google Scholar]
- 37. Ward WK, Bolgiano DC, McKnight B, Halter JB, Porte D Jr. Diminished B cell secretory capacity in patients with noninsulin‐dependent diabetes mellitus. J Clin Invest. 1984;74(4):1318‐1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Freemantle N, Balkau B, Home PD. A propensity score matched comparison of different insulin regimens 1 year after beginning insulin in people with type 2 diabetes. Diabetes Obes Metab. 2013;15(12):1120‐1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang L, Wei W, Miao R, Xie L, Baser O. Real‐world outcomes of US employees with type 2 diabetes mellitus treated with insulin glargine or neutral protamine Hagedorn insulin: a comparative retrospective database study. BMJ Open. 2013;3: e002348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vora J. Combining incretin‐based therapies with insulin: realizing the potential in type 2 diabetes. Diabetes Care. 2013;36(Suppl 2):S226‐S232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gupta V. Glucagon‐like peptide‐1 analogues: an overview. Indian J Endocrinol Metab. 2013;17(3):413‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garber AJ. Incretin effects on beta‐cell function, replication, and mass: the human perspective. Diabetes Care. 2011;34(Suppl 2):S258‐S263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cai X, Han X, Luo Y, Ji L. Efficacy of dipeptidyl‐peptidase‐4 inhibitors and impact on beta‐cell function in Asian and Caucasian type 2 diabetes mellitus patients: a meta‐analysis. J Diabetes. 2015;7(3):347‐359. [DOI] [PubMed] [Google Scholar]
- 44. Li J, Tian H, Li Q, et al. Improvement of insulin sensitivity and beta‐cell function by nateglinide and repaglinide in type 2 diabetic patients – a randomized controlled double‐blind and double‐dummy multicentre clinical trial. Diabetes Obes Metab. 2007;9(4):558‐565. [DOI] [PubMed] [Google Scholar]
- 45. U.K. Prospective Diabetes Study Group . U.K. Prospective diabetes study 16. Overview of 6 years' therapy of type II diabetes: a progressive disease. Diabetes. 1995;44(11):1249‐1258. [PubMed] [Google Scholar]
- 46. Gautier JF, Sauvanet JP. Efficacy of saxagliptin as an add‐on to oral monotherapy in the phase 3 clinical development program: predictive factors of the treatment response in type 2 diabetes. Ann Endocrinol (Paris). 2011;72(4):287‐295. [DOI] [PubMed] [Google Scholar]
- 47. Jones AG, McDonald TJ, Shields BM, et al. Markers of beta‐cell failure predict poor glycemic response to GLP‐1 receptor agonist therapy in type 2 diabetes. Diabetes Care. 2016;39(2):250‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wallace TM, Levy JC, Matthews DR. Use and abuse of HOMA modeling. Diabetes Care. 2004;27(6):1487‐1495. [DOI] [PubMed] [Google Scholar]
- 49. Rudenski AS, Matthews DR, Levy JC, Turner RC. Understanding "insulin resistance": both glucose resistance and insulin resistance are required to model human diabetes. Metabolism. 1991;40(9):908‐917. [DOI] [PubMed] [Google Scholar]
- 50. Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427‐2443. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1. Patient demographics and baseline characteristics in the unmatched population
Supplementary Table 2. Efficacy outcomes in the unmatched population
Supplementary Table 3. Pearson correlation analysis of baseline HOMA‐β index measurements with patient clinical characteristics in the unmatched population
Supplementary Table 4. Composite endpoints in the unmatched population