

Abstract
Acetylcholine signaling is essential for cognitive functioning and blocks inflammation. To maintain homeostasis, cholinergic signaling is subjected to multi‐leveled and bidirectional regulation by both proteins and non‐coding microRNAs (‘CholinomiRs’). CholinomiRs coordinate the cognitive and inflammatory aspects of cholinergic signaling by targeting major cholinergic transcripts including the acetylcholine hydrolyzing enzyme acetylcholinesterase (AChE). Notably, AChE inhibitors are the only currently approved line of treatment for Alzheimer's disease patients. Since cholinergic signaling blocks neuroinflammation which is inherent to Alzheimer's disease, genomic changes modifying AChE's properties and its susceptibility to inhibitors and/or to CholinomiRs regulation may affect the levels and properties of inflammasome components such as NLRP3. This calls for genomic‐based medicine approaches based on genotyping of both coding and non‐coding single nucleotide polymorphisms (SNPs) in the genes involved in cholinergic signaling. An example is a SNP in a recognition element for the primate‐specific microRNA‐608 within the 3′ untranslated region of the AChE transcript. Carriers of the minor allele of that SNP present massively elevated brain AChE levels, increased trait anxiety and inflammation, accompanied by perturbed CholinomiR‐608 regulatory networks and elevated prefrontal activity under exposure to stressful insults. Several additional SNPs in the AChE and other cholinergic genes await further studies, and might likewise involve different CholinomiRs and pathways including those modulating the initiation and progression of neurodegenerative diseases. CholinomiRs regulation of the cholinergic system thus merits in‐depth interrogation and is likely to lead to personalized medicine approaches for achieving better homeostasis in health and disease.
This is an article for the special issue XVth International Symposium on Cholinergic Mechanisms.

Keywords: acetylcholinesterase (AChE), Alzheimer's disease, genetics, neuroinflammation, single nucleotide polymorphisms (SNPs)
Abbreviations used
- ACh
acetylcholine
- AChE
acetylcholinesterase
- AD
Alzheimer's disease
- ADHD
attention Deficit–Hyperactivity Disorder
- BChE
butyrylcholinesterase
- ChE‐Is
Cholinesterase inhibitors
- CholinomiRs
non‐coding microRNA regulators of ACh signaling
- CHRN
nicotinic Cholinergic Receptor
- IBS‐D
diarrheal Predominant Irritable Bowel Syndrome
- IL
interleukin
- LPS
lipopolysaccharide
- miR
microRNA
- PD
Parkinson's Disease
- PPARγ
peroxisomal proliferation receptor gamma
- PTSD/S
post‐traumatic stress disorder/symptoms
- SLITRK1
slit and Trk‐like 1
- SNPs
single nucleotide polymorphisms
- TZD
thiazolidinedione
- UTR
untranslated region
Acetylcholine signaling supports cognition and blocks inflammation
Acetylcholine (ACh) signaling is best characterized as a neurotransmitter‐derived signaling pathway. Within the nervous system, ACh operates both as a neurotransmitter and as a neuromodulator, and in the periphery, it conveys messages of neurons or other ACh‐producing cells to their parasympathetic effectors (Picciotto et al. 2012; Soreq 2015). Cholinergic signaling at large, and the ACh hydrolyzing enzyme AChE specifically, is simultaneously involved in central cognitive processes such as learning, memory, and stress responses and in activating the parasympathetic system and mediating both neuromuscular and anti‐inflammatory responses (Soreq 2015). In brain neurons, ACh signaling elevates under anxiety (Graef et al. 2011) and suppresses inflammation (Rosas‐Ballina et al. 2011). At neuromuscular junctions of skeletal muscles, ACh determines channel opening (Fambrough 1979); in peripheral cells including pancreatic alpha cells (Rodriguez‐Diaz et al. 2011), placenta cells (Wessler et al. 2001), thrombocytes (Schedel et al. 2011) and lymphocytes (Kawashima and Fujii 2004; Olofsson et al. 2016), non‐neuronal ACh controls numerous signaling processes. The diverse roles of ACh, and the need to achieve homeostasis predict complex regulatory processes over its levels.
Recently, microRNA(miR) regulation emerges as a new level of control for ACh signaling (Simon et al. 2008; Hanin et al. 2014; Nadorp and Soreq 2014). MiRs are short (~22 nucleotides), non‐coding RNAs that regulate various molecular pathways by post‐transcriptional gene silencing (Bartel 2009; Krol et al. 2010). Each miR may target several mRNAs via interacting with short ‘seed’ motifs, often in specific locations on their 3′‐untranslated region (3′‐UTR) and can rapidly and effectively modulate entire pathways in a rheostat‐like manner(Chen et al. 2004). MiRs are hence particularly suitable for controlling the rapidly adjustable physiology of the parasympathetic system, and could modulate both the neuronal and immune functions of ACh by controlling its production and destruction (Shaked et al. 2009). However, the great majority of current miR studies focus on the interaction and silencing activities of one miR and one selected target, whereas individual mRNA transcripts may be silenced by many different miRs (Boudreau et al. 2014; Hsu et al. 2014). This complexity provides checks and balances ensuring that silencing would work efficiently, while implying that many miRs may share part of their targets. Therefore, when a particular miR interacts with one specific target, its availability for interaction with other targets may be reduced; and inversely, when one mRNA interacts with a specific miR, the probability of that mRNA to be silenced by other miRs would likely be reduced, albeit in a cell‐ and tissue‐specific manner. Very little is still known about the effects of such competition on basic physiological processes, although some studies from recent years have started unveiling the context dependence of this complexity (Hanin and Soreq 2011; Khella et al. 2013; Bracken et al. 2014; Wojtowicz et al. 2016); and we believe that the parasympathetic system is especially suitable to test this concept of complexity and appreciate its impact.
Bidirectional cholinergic signaling regulators include ‘CholinomiRs’
We have previously designated those miRs which regulate key cholinergic proteins ‘CholinomiRs’ (Nadorp and Soreq 2014). CholinomiRs extend regulatory functions over various cholinergic transcripts including nicotinic (Simon et al. 2008) and muscarinic (Scarr et al. 2013) receptors, as well as ACh packaging and degrading enzymes (Hanin and Soreq 2011). Many of these sequences are shared predicted targets of several miRs (Nadorp and Soreq 2014). This implies the existence of a common regulatory mechanism over cholinergic signaling at both pre‐ and post‐synaptic sites, keeping it in balance. Notably, many of the CholinomiRs are primate‐specific and their levels are altered in various conditions, including inflammation, anxiety, and neurodegeneration.
CholinomiR regulation over the various transcript variants of AChE, a central regulator of cholinergic signaling, is of clinical, pharmacological, and physiological significance. The ‘synaptic’ AChE‐S (or AChE‐T) is active in the synapse, whereas both the soluble variant AChE‐R and the erythrocyte‐expressed AChE‐E (AChE‐H) are active in the periphery (Soreq and Seidman 2001). For example, miR‐186 is an evolutionarily conserved predicted regulator of the both, the anxiety‐induced AChE‐R and the peripheral butyrylcholinesterase (BChE), which shares much of its protein sequence with AChE and hydrolyzes ACh as well. BChE's expression is elevated in the intestine of mice exposed to stressful conditions, predicting limited cholinergic blockade of intestinal inflammation (Nadorp and Soreq 2014). This may possibly be relevant for the prodromal stages of Parkinson's Disease (PD), which likely involve gastrointestinal symptoms that are part of the disease pathogenesis (Fasano et al. 2015). The unique entity of central nervous system inflammation is often referred to as neuroinflammation, and the cholinergic involvement in its physiological aspects (Filiou et al. 2014), is also relevant for other mental and neurodegenerative diseases. Thus, miR‐132 targets AChE in both the brain and the periphery, its levels decrease sharply in Alzheimer's disease (AD) and the disruption of its regulation causes behavioral and learning‐related phenomena (Shaltiel et al. 2013), as well as exacerbated inflammation (Shaked et al. 2009). Hence, CholinomiRs are key controllers of the interface between cholinergic signaling, neuroinflammation, and neurodegeneration.
AChE inhibitors potentially serve as genomics‐based modulators of AD therapeutics
Neuroinflammation is involved in AD and many other neurodegenerative conditions such as PD (Simchovitz et al. 2016), frontotemporal dementia, amyotrophic lateral sclerosis, Huntington's Disease and more (Heneka et al. 2014). The diminished risk of AD among users of non‐steroidal anti‐inflammatory drugs (in t' veld et al. 2001; Wang et al. 2015) attests to its importance, but steroids do not exert a similar effect; which calls for a closer inspection of the neuroinflammatory pathway in AD (Heneka et al. 2015a,c; Wang et al. 2015). In this context, AChE inhibitors (ChE‐Is) are currently the only effective line of treatment for AD patients (Birks 2006). ChE‐Is may delay the consistent ACh depletion because of cholinergic cell death (McGeer et al. 1984), decreasing the excessive activation of microglia that promotes neuroinflammation‐mediated damage in response to the formation of Aβ plaques (Heppner et al. 2015). Some of the non‐steroidal anti‐inflammatory drugs found to decrease the risk for AD were also identified as agonists of peroxisomal proliferation receptor gamma (PPARγ), a target of the thiazolidinedione class of antidiabetics (Lehmann et al. 1997). Strikingly, activation of PPARγ was protective in AD model mice (Heneka et al. 2005), activation of all PPARs was even more protective (Kummer et al. 2015), and thiazolidinediones were protective against dementia in human patients (Heneka et al. 2015b). Of note, PPARγ is repressed in the liver indirectly by down‐regulation of miR‐132 (Mann et al. 2010), which is down‐regulated in AD (Lau et al. 2013), and importantly is also a regulator of AChE (Shaked et al. 2009). It is hence tempting to speculate that the failure of endogenous PPARγ to protect against AD without pharmacological aid involves CholinomiRs contribution (Fig. 1).
Figure 1.
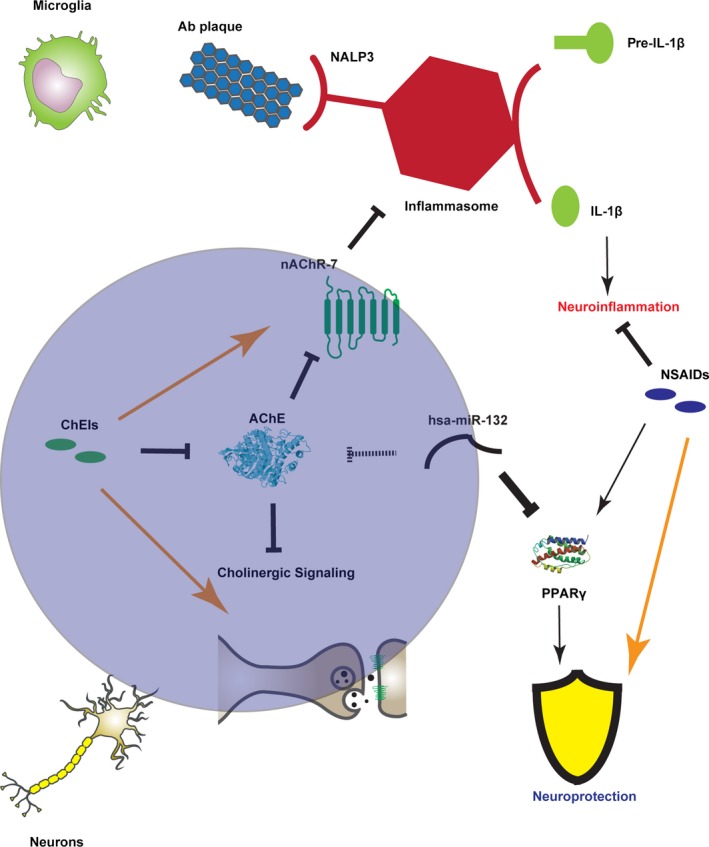
The network of Alzheimer's disease (AD), neuroinflammation, and the cholinergic system. Alzheimer's Disease‐related processes occur in both microglia (upper panel) and neurons (lower panel); The cholinergic system and ChE‐I treatment might modulate both these aspects, as does the treatment with certain non‐steroidal anti‐inflammatory drugs (NSAIDs). Microglia are activated by Aβ plaques through the NALP3 receptor of the inflammasome. The activated inflammasome then promotes the cleavage of pre‐IL1β to mature IL1β, which induces neuroinflammation. The nicotinic receptor inhibits inflammasome formation; Therefore, ChE‐Is, which increase cholinergic signaling, might inhibit the inflammasome as well. NSAIDs, on the other hand, directly inhibit neuroinflammation by their anti‐inflammatory action. In cholinergic neurons, synaptic transmission initiates signaling processes, which are altered in AD and might be potentiated by ChE‐Is. Finally, NSAIDs involvement in neuronal activity might be mediated by neuroprotective peroxisomal proliferation receptor gamma (PPARγ), which is activated by NSAIDs and inhibited by miR‐132, which also targets acetylcholinesterase (AChE) and is down‐regulated in AD.
Inflammatory reaction to Aβ entails the release of Interleukin‐1β (IL‐1β), a primary pro‐inflammatory cytokine activated via cleavage by caspase‐1, a part of the inflammasome (Halle et al. 2008). The inflammasome is a soluble cytoplasmic complex of several proteins mediating the production and effect of pro‐inflammatory cytokines (Martinon et al. 2002), activated by the NALP3 protein as a reaction to toxins, ATP (Mariathasan et al. 2006) and also fibrillar Aβ (Halle et al. 2008). The inflammasome activation in AD is reflected as higher caspase activity in the post‐mortem brains of AD patients and mild cognitively impaired volunteers, compared to cognitively spared age‐matched controls (Saresella et al. 2016). Furthermore, mice lacking the NLRP3 gene coding to the NALP3 protein, as well as mice lacking caspase‐1, show less cognitive deterioration when carrying familial AD mutations, indicating that the inflammasome is causally involved in the pathogenicity itself (Heneka et al. 2013). Neuroinflammation and its regulation are hence key elements in the initiation and progression of AD.
The anti‐inflammatory cholinergic pathway is an active controller of neuroinflammation. Vagus nerve signaling through the alpha‐7 nicotinic receptor inhibits the activation of the inflammasome (Lu et al. 2014); and nitric oxide‐mediated central muscarinic activity is anti‐inflammatory as well (Pavlov et al. 2009), suggesting co‐amplified anti‐inflammatory role for ChE‐Is in AD treatment. Correspondingly, ChE‐I treatment reduces cytokine levels in AD patients (Reale et al. 2004). Nevertheless, the vast variability in response to ChE‐Is among AD patients (Birks 2006) contrasts this potential robustness of ChE‐Is against various aspects of AD, even when considering general pharmacokinetic‐related inter‐individual differences, such as polymorphisms in the CYP2D6 gene (Xiao et al. 2016). This raises the question of whether other inter‐individual genomic differences, for example, changes in the 3′‐UTR regions interacting with CholinomiRs can be responsible for the variable efficacy of ChE‐Is (Martinelli‐Boneschi et al. 2013). AChE, therefore, presents an attractive target for studying the personalized genetic aspects in the junction between the cognitive and neuroinflammatory pathways of AD pathology.
Both coding and non‐coding mutations in cholinergic genes modify signaling
The personalized medicine implications of inter‐individual genomic differences represent a prominent trend in modern medicine (Hamburg and Collins 2010). Specifically, relatively common genomic variations that are not necessarily disease‐causing but rather disease and treatment modifying, rapidly become a focus of intensive research. Single nucleotide polymorphisms (SNPs) constitute a large portion of these changes, with some of those existing in cholinergic genes and their regulators (Fig. 2). For example, a coding SNP in the neuronal nicotinic cholinergic receptor α‐ 5 subunit (CHRNA5) changes aspartate to asparagine and associates with increased risk of chronic obstructive pulmonary disorder (Cui et al. 2014), and coding SNPs in the CHRNB4 gene modify receptor affinity to ACh, with consequent implications on cognition and addiction (Liang et al. 2005). Also, an aspartate to glycine substitution in the BChE gene reduces the capacity for ACh hydrolysis by the BChE protein, such that carriers of this substitution are more susceptible to side‐effects of ChE‐Is and cholinergic‐related general anesthetics (Loewenstein‐Lichtenstein et al. 1995). Similarly, a common variant in the SLC5A7 gene, coding for the Sodium/Choline cotransporter protein, associates with an increased rate of attention deficit–hyperactivity disorder (English et al. 2009), whereas an alanine to threonine substitution in SLC5A7 associates with better response to ChE‐Is treatment in AD (Harold et al. 2006; Lee et al. 2015). Individual variability in the cholinergic network genes may hence be pivotal for the individual variability in the efficacy of AD therapeutics.
Figure 2.
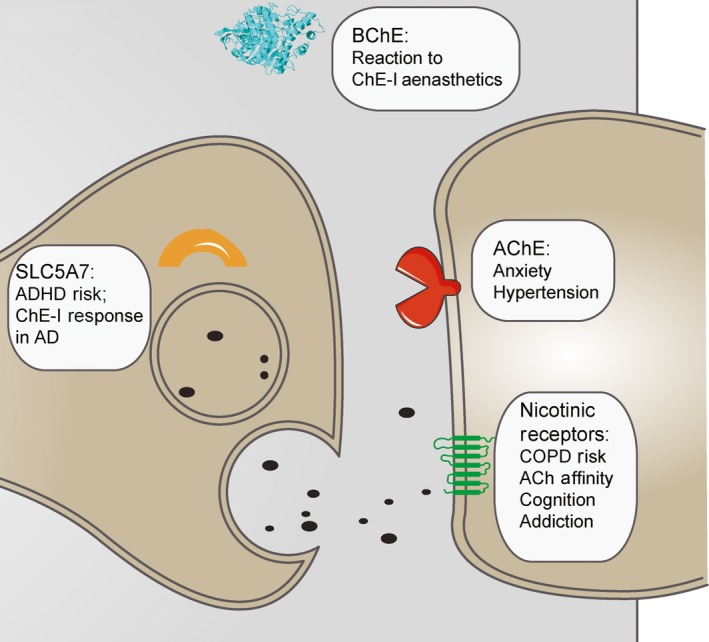
Single nucleotide polymorphisms (SNPs) in Coding and non‐coding regions in cholinergic genes and their reported impact. Cholinergic transcripts include, but are not limited to SLC5A7, responsible for choline transport into the cell [SNPs described in – (English et al. 2009; Harold et al. 2006; Lee et al. 2015)]; Acetylcholinesterase, responsible for central degradation of ACh (Hasin et al. 2004; Lin et al. 2016) (Hanin et al. 2014); Butyrylcholinesterase, responsible for peripheral degradation of ACh (Loewenstein‐Lichtenstein et al. 1995); and several nicotinic and muscarinic receptors, responsible for signal transmission across the synapse (Cui et al. 2014; Liang et al. 2005; Luo et al. 2005; Schlaepfer et al. 2008; Wang et al. 2004; Mobascher et al. 2016; Russo et al. 2015). These are all major players in the cholinergic signaling pathway, and may well be affected by miR‐interfering SNPs in both their coding and non‐coding regions. These effects modulate protein activity and regulation and may induce significant systemic impact, as reported in the cited references.
Several synonymous SNPs in ACh receptor genes have been associated with other brain‐mediated phenotypes. SNPs in CHRNA4 associate with smoking, PD symptoms (Zhang et al. 2015), and electroencephalography (EEG)‐measured reaction to stimuli (Mobascher et al. 2016). SNPs in non‐coding intron domains of the CHRNA4/A5/B3 cluster associate with early tobacco and alcohol initiation (Schlaepfer et al. 2008); intron SNPs in CHRNB4 associate with alcohol and drug dependence, as well as with affective disorders (Wang et al. 2004; Luo et al. 2005); and intron SNPs in the CHRNA7 gene regulate patients’ response to ChE‐Is treatment in AD (Russo et al. 2015). SNPs in cholinergic genes can therefore have a large impact on systemic and cognitive function, potentially also effecting the variability in individual responses to AD treatment, whether they reside in protein‐coding or non‐coding regions of these transcripts.
CholinomiR‐target interactions as potential modulators of neuroinflammation in AD
How SNPs in the non‐coding regions of cholinergic genes exert global systemic effects and increase the risk for certain pathologies is largely unclear, but a recent study demonstrated one possible mechanism. This study predicted that SNPs which modify miR binding to the 3′‐UTR of their target genes may initiate a domino‐like cascade involving several layers of regulation. Publicly available SNP databases revealed 250 SNPs in the 3′‐UTRs of miR target genes (Saunders et al. 2007); however, those SNPs do not necessarily lie in the region interacting with the ‘seed’ region (Hariharan et al. 2009). Nevertheless, some of these SNPs may interfere with miR/target interactions and thus affect the expression of their targets. This might be because of a modified secondary structure near the miR‐binding site, which changes the accessibility of that miR to its binding site (Kertesz et al. 2007). Examples include a SNP in the 3′ ‐UTR of the human ATF1 gene that associates with essential hypertension because of weakened binding by miR‐1283, leading to the target ATF1 transcript up‐regulation in peripheral blood (Yang et al. 2015). A variant of the aldosterone synthase gene Cyp112, in which an A>G SNP interrupts miR‐590‐3p regulation, is also associated with essential hypertension in Chinese and German cohorts (Xiao et al. 2015).
MiR‐interacting SNPs are also of relevance to higher cognitive functioning and CNS‐related disorders, as is seen for a variant in the 3‐’UTR of the Slit and Trk‐like 1 (SLITRK1) gene which strengthens an existing miR‐189 target site. The SLITRK1 gene is expressed at high levels in the brain and supports neurite growth, and this SNP is involved in Tourette's syndrome (TS) and attention deficit–hyperactivity disorder (Abelson et al. 2005). In PD, a 3′ ‐UTR SNP in the fibroblast growth factor 20 (FGF20) gene was identified as a risk factor for the disease by disrupting a binding site for miR‐433, increasing translation of FGF20 which is associated with PD (Wang et al. 2008). Both strengthening and weakening of miR/target interactions may therefore be physiologically relevant.
At the inflammatory level, diarrheal predominant irritable bowel syndrome (IBS‐D) is generally associated with dysfunctions in the serotoninergic system. A 76G>A variant in the 3′ ‐UTR of the serotonin receptor type 3 subunit gene (HTR3A) showed a strong association with female IBS‐D by affecting binding of miR‐510 to the HTR3E 3′‐UTR, resulting in high expression of the receptor subunit (Kapeller et al. 2008). Strikingly, genes involved in neurodegeneration were shown to have higher prospects of carrying SNPs with a potential to modulate miR binding (Saba et al. 2014). These novel findings make SNPs which potentially disrupt miR‐binding sites in 3′‐UTRs of cholinergic genes promising candidates for research. Supporting this notion, recent work described below has shown their relevance and the major complexity that arises from the disruption of large miR networks, compatible with the dual paradigm of single miR/many targets; single target/many miRs. The genomics‐affected interactions between miRs and their targets are therefore of special importance to therapeutic efforts aimed at addressing the various aspects of neurodegenerative diseases, including cognitive decline and inflammation.
A SNP interrupting CholinomiR regulation has wide systemic effects
The complex nature of miR regulation over a network of transcripts is particularly evident in the case of miR‐608 and its disrupted regulation of AChE by the rs17228616 SNP (Fig. 3). The minor allele of this SNP associates with highly elevated AChE activity in the brains of homozygous carriers. Also, this is accompanied by reduced levels of other hsa‐miR‐608 targets including CDC42, CD44, and IL‐6 (Hanin et al. 2014). Systemic effects of this SNP include elevated anxiety and blood pressure, as well as relative resilience to post‐traumatic stress symptoms (Lin et al. 2016). Reduced CDC42 levels were anxiogenic in mice, and reports by others suggest that reduced levels of the pro‐inflammatory IL‐6 might attenuate post‐traumatic stress disorder‐related symptoms (Gill et al. 2009; Wilson et al. 2013) and modulate inflammation (Zimmerman et al. 2012; Gill et al. 2013). Down‐regulation of IL‐6 might also explain why despite excessive AChE levels, which might limit the cholinergic anti‐inflammatory pathway, SNP carriers present only slightly increased inflammatory markers (Hanin et al. 2014). Skewing homeostasis by tilting a single miR/target interaction is therefore shown as a possible mechanism for the initiation of a complex phenotype because of dual changes in the level of the single affected target as well as in the multiple other targets of that miR and the interactions between these targets.
Figure 3.
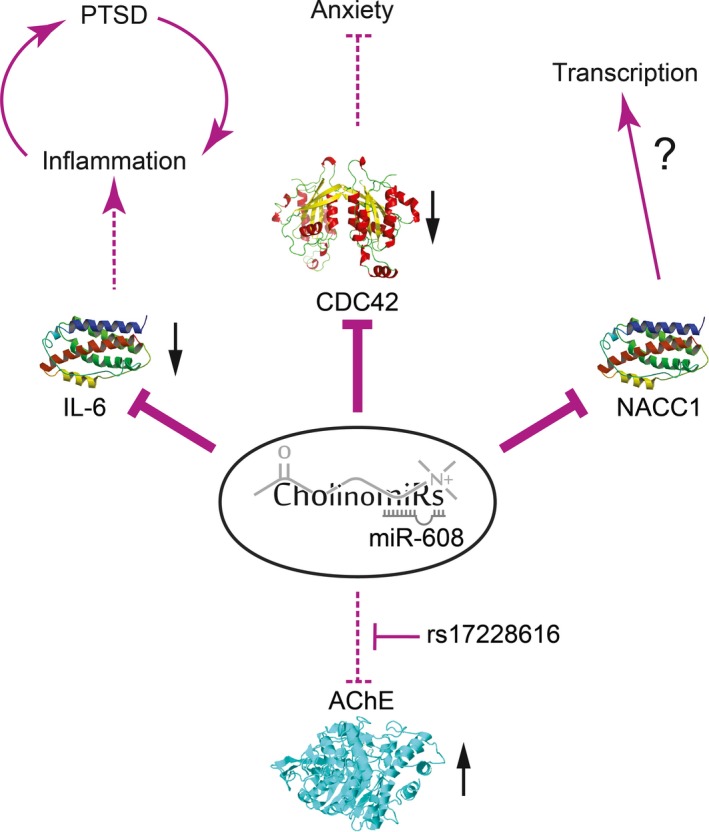
AChE single nucleotide polymorphisms (SNP)‐induced propagation of imbalanced miR‐608 network. As previously reported (Hanin et al. 2014; Lin et al. 2016), a SNP in the 3’‐UTR of AChE diminishes miR‐608 binding and regulatory effect, causing both significant elevation of brain acetylcholinesterase (AChE) activity and down‐regulation of other miR‐608 targets attributed to ‘unemployed’ miR‐608 molecules. These include the anxiety‐inhibiting CDC42, the inflammatory‐promoting IL6, and the neuronal transcription factor NACC1, with possibly complex effects over brain transcriptome and activity. Thick arrows represent an increase in positive or negative regulation, whereas stroked arrows represent diminished regulation.
The future prospects of genomics‐based cholinergic‐targeted medicine
The physical proximity of individual SNPs on a single transcript may affect the potency of the relevant miRs to suppress their targets, because of competition on the binding interactions. In this context, several of the identified SNPs in the AChE gene (Hasin et al. 2004) reside close to each other in the 3′‐UTR region, such that interaction with one miR might possibly interfere with other miR/AChE interactions. Of note are two specific SNPs – rs17228602 and rs1799806 (Fig. 4). Rs17228602 resides in the recognition element for miR‐125b, which predictably targets both the soluble AChE‐R variant (suppression of which would reduce ACh destruction) and the vesicular acetylcholine transporter (suppression of which would inversely reduce ACh production) (Nadorp and Soreq 2014). This indicates homeostatic bidirectional impact of miR‐125b on cholinergic signaling. Intriguingly, miR‐125b is down‐regulated in response to bacterial lipopolysaccharide exposure, possibly through the activation of NFκB (Tili et al. 2007). This creates a seemingly self‐controlled mechanism, by which NFkB suppresses hsa‐miR‐125b, which in turn re‐balances cholinergic signaling, keeping the inflammatory response to anxiety under continuous control. However, deficient miR‐125b/AChE interaction in carriers of the rs17228602 SNP might impair the capacity for keeping homeostatic cholinergic signaling under both stress and inflammation, leading to a vicious anxio‐inflammatory cycle (Zimmerman et al. 2012).
Figure 4.
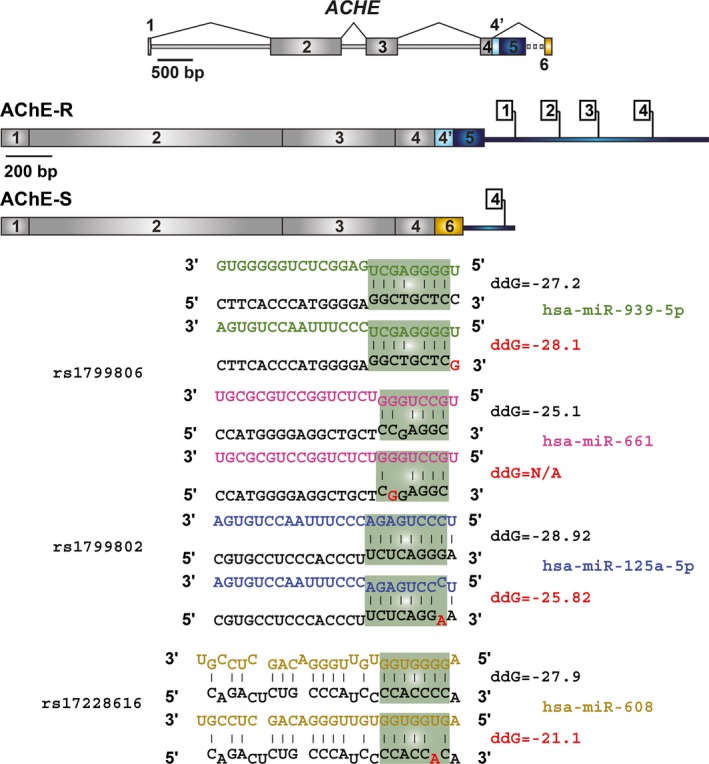
Four single nucleotide polymorphisms (SNPs) in predicted miR‐binding sites of the AChE 3′‐UTR. Shown are common SNPs in the 3′‐UTR of acetylcholinesterase (AChE) splice variants according to the 1000 genome project, and the bioinformatics prediction of the effect of three of them on binding of miRs (Kertesz et al. 2007). The presented ddG values predict the difference in free energy between the bound and unbound conditions of the miR and mRNA. For example, rs1799806 would slightly enhance miR‐939‐5p binding, but reduce miR‐661 binding; similar effects are observed for rs1799802 and miR‐125b and for rs17228616 and miR‐608 (the latter prediction was experimentally validated) (Hanin et al. 2014).
In addition to its direct impact on AChE transcripts, the rs17228602 SNP might leave ‘unemployed’ miR‐125b chains that fail to bind AChE mRNA, similarly to the rs17228616 miR‐608/AChE‐interfering SNP (Hanin et al. 2014). These chains will be free to target other transcripts carrying the same ‘seed’ domain, decreasing the levels of other miR‐125b targets which would likely impair synaptic plasticity (Edbauer et al. 2010). The anxiety phenotype of mice lacking the AChE‐3′‐UTR (Shaltiel et al. 2013), where miR‐125b would fail to exert its effect, is compatible with this prediction. Interestingly, miR‐608 and miR‐125b interact with closely positioned sites on the AChE 3′‐UTR and share some targets and pathways, indicating a synergistic function of the SNPs interrupting their AChE binding; for example, miR‐608 regulates IL‐6 (Hanin et al. 2014), whereas miR‐125b regulates its receptor, IL‐6R (Gong et al. 2013), implying a synergistic anti‐inflammatory effect of the two miRs and an amplified inflammatory phenotype in co‐carriers of the two SNPs. However, this is not always the case. For example, miR‐608 down‐regulates the anxiolytic Rho GTPase CDC42 (Zhang et al. 2016), which would predictably elevate anxiety; whereas, miR‐125b predictably targets the learning‐associated NR2A, the levels of which decrease upon miR‐125b over‐expression (Edbauer et al. 2010), with a seemingly inverse predicted outcome. In this context, the two SNPs may counterbalance each other's impact. Thus, interrogating the outcome of SNP interruptions of brain targets is rather complicated and probably depends on many different parameters.
The complex bidirectional effects of AChE 3′‐UTR SNPs comprise only part of the genomic variability in this one gene. Along with other SNPs in the AChE gene, these SNPs may provide a useful example for studying the personalized outcome of different SNPs in the non‐coding domains of brain‐expressed genes. Additional examples include the common rs1799806 SNP [minor allele frequency 26.9% (Auton et al. 2015)], which localizes at both the coding region of the erythrocyte AChE‐E variant, and at the non‐coding 3′‐UTR region of the AChE‐R protein. This SNP abolishes a predicted recognition element for miR‐661, which targets the pregnancy‐related histocompatibility complex variant HLA‐G (Castelli et al. 2009). Additionally, this SNP might enhance the potency of a putative binding site for miR‐939‐5p, which regulates nitric oxide production by suppressing the translation of the inducible nitric oxide synthase gene (Guo et al. 2012). Different SNPs in the AChE 3′‐UTR might therefore dysregulate diverse miRs and implement multi‐leveled imbalances in those processes that are regulated by these miRs. This, in turn raises the question of potential inter‐ and intra‐haplotype interactions between these SNPs, both at the mechanistic and the population level, which awaits further research.
Of note, AChE SNPs might skew the balance between different cholinergic signaling pathways, including but not limited to AD‐related events. This provides a possible explanation for the vast variability in AD patients’ reactions to ChE‐Is. However, AChE is only one member of the cholinergic signaling pathway, and many other modulators of this system should be added to this equation, such as the nicotinic and muscarinic ACh receptors, the vesicular ACh transporter and choline acetyltransferase (Soreq 2015). Therefore, treatment with ChE‐Is might only be effective in patients with particular genotypes, in whom it may limit some of the pathology‐related and delay the cognitive decline, as was originally intended; however, in others, existing SNPs may interfere with this treatment's impact. Interrogating the genomic profiles of patients who are responsive and unresponsive to ChE‐Is, with special attention to the 3′‐UTR of the AChE gene may provide answers to this and related questions. Also, the involvement of AChE in other phenotypes suggests similar considerations for other medical conditions, providing an opportunity to develop individual, genome‐based therapy strategies.
Acknowledgments and conflict of interest disclosure
Work performed by A.S and H.S was supported by The Israel Science Foundation (Grant No. 817/13) and the Israeli Ministry of Science and Technology Grant Number 53140. MTH was supported by a grant from the German Research Council (DFG KFO177, TP8) and by the EU's Seventh Framework Programme InMIND.
Michael Heneka is an editor with the Journal of Neurochemistry.Other than that, the authors declare they have no conflict of interests.
This is an article for the special issue XVth International Symposium on Cholinergic Mechanisms.
References
- Abelson J. F., Kwan K. Y., O'Roak B. J. et al (2005) Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310, 317–320. [DOI] [PubMed] [Google Scholar]
- Auton A., Brooks L. D., Durbin R. M. et al (2015) A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel D. P. (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birks J. (2006) Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst. Rev. CD005593 DOI: 10.1002/14651858.CD005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R. L., Jiang P., Gilmore B. L., Spengler R. M., Tirabassi R., Nelson J. A., Ross C. A., Xing Y. and Davidson B. L. (2014) Transcriptome‐wide discovery of microRNA binding sites in human brain. Neuron 81, 294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken C. P., Li X., Wright J. A. et al (2014) Genome‐wide identification of miR‐200 targets reveals a regulatory network controlling cell invasion. EMBO J. 33, 2040–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelli E. C., Moreau P., Oya e Chiromatzo A., Mendes‐Junior C. T., Veiga‐Castelli L. C., Yaghi L., Giuliatti S., Carosella E. D. and Donadi E. A. (2009) In silico analysis of microRNAS targeting the HLA‐G 3’ untranslated region alleles and haplotypes. Hum. Immunol., 70, 1020–1025. [DOI] [PubMed] [Google Scholar]
- Chen C. Z., Li L., Lodish H. F. and Bartel D. P. (2004) MicroRNAs modulate hematopoietic lineage differentiation. Science 303, 83–86. [DOI] [PubMed] [Google Scholar]
- Cui K., Ge X. and Ma H. (2014) Four SNPs in the CHRNA3/5 alpha‐neuronal nicotinic acetylcholine receptor subunit locus are associated with COPD risk based on meta‐analyses. PLoS ONE 9, e102324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edbauer D., Neilson J. R., Foster K. A. et al (2010) Regulation of synaptic structure and function by FMRP‐associated microRNAs miR‐125b and miR‐132. Neuron 65, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English B. A., Hahn M. K., Gizer I. R., Mazei‐Robison M., Steele A., Kurnik D. M., Stein M. A., Waldman I. D. and Blakely R. D. (2009) Choline transporter gene variation is associated with attention‐deficit hyperactivity disorder. J. Neurodev. Disord. 1, 252–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fambrough D. M. (1979) Control of acetylcholine receptors in skeletal muscle. Physiol. Rev. 59, 165–227. [DOI] [PubMed] [Google Scholar]
- Fasano A., Visanji N. P., Liu L. W., Lang A. E. and Pfeiffer R. F. (2015) Gastrointestinal dysfunction in Parkinson's disease. Lancet Neurol. 14, 625–639. [DOI] [PubMed] [Google Scholar]
- Filiou M. D., Arefin A. S., Moscato P. and Graeber M. B. (2014) ‘Neuroinflammation’ differs categorically from inflammation: transcriptomes of Alzheimer's disease, Parkinson's disease, schizophrenia and inflammatory diseases compared. Neurogenetics 15, 201–212. [DOI] [PubMed] [Google Scholar]
- Gill J. M., Saligan L., Woods S. and Page G. (2009) PTSD is associated with an excess of inflammatory immune activities. Perspect Psychiatr. Care 45, 262–277. [DOI] [PubMed] [Google Scholar]
- Gill J. M., Saligan L., Lee H., Rotolo S. and Szanton S. (2013) Women in recovery from PTSD have similar inflammation and quality of life as non‐traumatized controls. J. Psychosom. Res. 74, 301–306. [DOI] [PubMed] [Google Scholar]
- Gong J., Zhang J. P., Li B., Zeng C., You K., Chen M. X., Yuan Y. and Zhuang S. M. (2013) MicroRNA‐125b promotes apoptosis by regulating the expression of Mcl‐1, Bcl‐w and IL‐6R. Oncogene 32, 3071–3079. [DOI] [PubMed] [Google Scholar]
- Graef S., Schonknecht P., Sabri O. and Hegerl U. (2011) Cholinergic receptor subtypes and their role in cognition, emotion, and vigilance control: an overview of preclinical and clinical findings. Psychopharmacology 215, 205–229. [DOI] [PubMed] [Google Scholar]
- Guo Z., Shao L., Zheng L., Du Q., Li P., John B. and Geller D. A. (2012) miRNA‐939 regulates human inducible nitric oxide synthase posttranscriptional gene expression in human hepatocytes. Proc. Natl Acad. Sci. USA 109, 5826–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A., Hornung V., Petzold G. C. et al (2008) The NALP3 inflammasome is involved in the innate immune response to amyloid‐beta. Nat. Immunol. 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburg M. A. and Collins F. S. (2010) The path to personalized medicine. N. Engl. J. Med. 363, 301–304. [DOI] [PubMed] [Google Scholar]
- Hanin G. and Soreq H. (2011) Cholinesterase‐targeting microRNAs identified in silico affect specific biological processes. Front Mol. Neurosci. 4, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanin G., Shenhar‐Tsarfaty S., Yayon N. et al (2014) Competing targets of microRNA‐608 affect anxiety and hypertension. Hum. Mol. Genet. 23, 4569–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariharan M., Scaria V. and Brahmachari S. K. (2009) dbSMR: a novel resource of genome‐wide SNPs affecting microRNA mediated regulation. BMC Bioinformatics 10, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harold D., Macgregor S., Patterson C. E. et al (2006) A single nucleotide polymorphism in CHAT influences response to acetylcholinesterase inhibitors in Alzheimer's disease. Pharmacogenet. Genomics 16, 75–77. [DOI] [PubMed] [Google Scholar]
- Hasin Y., Avidan N., Bercovich D., Korczyn A., Silman I., Beckmann J. S. and Sussman J. L. (2004) A paradigm for single nucleotide polymorphism analysis: the case of the acetylcholinesterase gene. Hum. Mutat. 24, 408–416. [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Sastre M., Dumitrescu‐Ozimek L. et al (2005) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1‐42 levels in APPV717I transgenic mice. Brain 128, 1442–1453. [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Kummer M. P., Stutz A. et al (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., Kummer M. P. and Latz E. (2014) Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 14, 463–477. [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Carson M. J., El Khoury J. et al (2015a) Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka M. T., Fink A. and Doblhammer G. (2015b) Effect of pioglitazone medication on the incidence of dementia. Ann. Neurol. 78, 284–294. [DOI] [PubMed] [Google Scholar]
- Heneka M. T., Golenbock D. T. and Latz E. (2015c) Innate immunity in Alzheimer's disease. Nat. Immunol. 16, 229–236. [DOI] [PubMed] [Google Scholar]
- Heppner F. L., Ransohoff R. M. and Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358–372. [DOI] [PubMed] [Google Scholar]
- Hsu S. D., Tseng Y. T., Shrestha S. et al (2014) miRTarBase update 2014: an information resource for experimentally validated miRNA‐target interactions. Nucleic Acids Res. 42, D78–D85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- in t’ Veld B. A., Ruitenberg A., Hofman A., Launer L. J., vanDuijn C. M. , Stijnen T., Breteler M. M. and Stricker B. H. (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N. Engl. J. Med., 345, 1515–1521. [DOI] [PubMed] [Google Scholar]
- Kapeller J., Houghton L. A., Monnikes H. et al (2008) First evidence for an association of a functional variant in the microRNA‐510 target site of the serotonin receptor‐type 3E gene with diarrhea predominant irritable bowel syndrome. Hum. Mol. Genet. 17, 2967–2977. [DOI] [PubMed] [Google Scholar]
- Kawashima K. and Fujii T. (2004) Expression of non‐neuronal acetylcholine in lymphocytes and its contribution to the regulation of immune function. Front Biosci. 9, 2063–2085. [DOI] [PubMed] [Google Scholar]
- Kertesz M., Iovino N., Unnerstall U., Gaul U. and Segal E. (2007) The role of site accessibility in microRNA target recognition. Nat. Genet. 39, 1278–1284. [DOI] [PubMed] [Google Scholar]
- Khella H. W., Bakhet M., Allo G. et al (2013) miR‐192, miR‐194 and miR‐215: a convergent microRNA network suppressing tumor progression in renal cell carcinoma. Carcinogenesis 34, 2231–2239. [DOI] [PubMed] [Google Scholar]
- Krol J., Loedige I. and Filipowicz W. (2010) The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610. [DOI] [PubMed] [Google Scholar]
- Kummer M. P., Schwarzenberger R., Sayah‐Jeanne S., Dubernet M., Walczak R., Hum D. W., Schwartz S., Axt D. and Heneka M. T. (2015) Pan‐PPAR modulation effectively protects APP/PS1 mice from amyloid deposition and cognitive deficits. Mol. Neurobiol. 51, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau P., Bossers K., Janky R. et al (2013) Alteration of the microRNA network during the progression of Alzheimer's disease. EMBO Mol. Med. 5, 1613–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. U., Lee J. H., Lee D. Y. et al (2015) The effect of choline acetyltransferase genotype on Donepezil treatment response in patients with Alzheimer's disease. Clin. Psychopharmacol. Neurosci. 13, 168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann J. M., Lenhard J. M., Oliver B. B., Ringold G. M. and Kliewer S. A. (1997) Peroxisome proliferator‐activated receptors alpha and gamma are activated by indomethacin and other non‐steroidal anti‐inflammatory drugs. J. Biol. Chem. 272, 3406–3410. [DOI] [PubMed] [Google Scholar]
- Liang Y., Salas R., Marubio L., Bercovich D., De Biasi M., Beaudet A. L. and Dani J. A. (2005) Functional polymorphisms in the human beta4 subunit of nicotinic acetylcholine receptors. Neurogenetics 6, 37–44. [DOI] [PubMed] [Google Scholar]
- Lin T., Simchovitz A., Shenhar‐Tsarfaty S. et al (2016) Intensified vmPFC surveillance over PTSS under perturbed microRNA‐608/AChE interaction. Transl. Psychiatry 6, e801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewenstein‐Lichtenstein Y., Schwarz M., Glick D., Norgaard‐Pedersen B., Zakut H. and Soreq H. (1995) Genetic predisposition to adverse consequences of anti‐cholinesterases in ‘atypical’ BCHE carriers. Nat. Med. 1, 1082–1085. [DOI] [PubMed] [Google Scholar]
- Lu B., Kwan K., Levine Y. A. et al (2014) alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol. Med. 20, 350–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X., Kranzler H. R., Zuo L., Wang S., Blumberg H. P. and Gelernter J. (2005) CHRM2 gene predisposes to alcohol dependence, drug dependence and affective disorders: results from an extended case‐control structured association study. Hum. Mol. Genet. 14, 2421–2434. [DOI] [PubMed] [Google Scholar]
- Mann J., Chu D. C., Maxwell A., Oakley F., Zhu N. L., Tsukamoto H. and Mann D. A. (2010) MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology, 138, 705–714, 714 e701‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariathasan S., Weiss D. S., Newton K. et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440, 228–232. [DOI] [PubMed] [Google Scholar]
- Martinelli‐Boneschi F., Giacalone G., Magnani G. et al (2013) Pharmacogenomics in Alzheimer's disease: a genome‐wide association study of response to cholinesterase inhibitors. Neurobiol. Aging 34, 1711, e1717–1713. [DOI] [PubMed] [Google Scholar]
- Martinon F., Burns K. and Tschopp J. (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL‐beta. Mol. Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- McGeer P. L., McGeer E. G., Suzuki J., Dolman C. E. and Nagai T. (1984) Aging, Alzheimer's disease, and the cholinergic system of the basal forebrain. Neurology 34, 741–745. [DOI] [PubMed] [Google Scholar]
- Mobascher A., Diaz‐Lacava A., Wagner M. et al (2016) Association of common polymorphisms in the nicotinic acetylcholine receptor Alpha4 subunit gene with an electrophysiological endophenotype in a large population‐based sample. PLoS ONE 11, e0152984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadorp B. and Soreq H. (2014) Predicted overlapping microRNA regulators of acetylcholine packaging and degradation in neuroinflammation‐related disorders. Front Mol. Neurosci. 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson P. S., Steinberg B. E., Sobbi R. et al (2016) Blood pressure regulation by CD4 + lymphocytes expressing choline acetyltransferase. Nat. Biotechnol. 34, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov V. A., Parrish W. R., Rosas‐Ballina M., Ochani M., Puerta M., Ochani K., Chavan S., Al‐Abed Y. and Tracey K. J. (2009) Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti‐inflammatory pathway. Brain Behav. Immun. 23, 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto M. R., Higley M. J. and Mineur Y. S. (2012) Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reale M., Iarlori C., Gambi F. et al (2004) Treatment with an acetylcholinesterase inhibitor in Alzheimer patients modulates the expression and production of the pro‐inflammatory and anti‐inflammatory cytokines. J. Neuroimmunol. 148, 162–171. [DOI] [PubMed] [Google Scholar]
- Rodriguez‐Diaz R., Dando R., Jacques‐Silva M. C. et al (2011) Alpha cells secrete acetylcholine as a non‐neuronal paracrine signal priming beta cell function in humans. Nat. Med. 17, 888–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas‐Ballina M., Olofsson P. S., Ochani M. et al (2011) Acetylcholine‐synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo P., Kisialiou A., Moroni R., Prinzi G. and Fini M. (2015) Effect of genetic polymorphisms (SNPs) in CHRNA7 gene on response to acetylcholinesterase inhibitors (AChEI) in patients with Alzheimer's disease. Curr. Drug Targets. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Saba R., Medina S. J. and Booth S. A. (2014) A functional SNP catalog of overlapping miRNA‐binding sites in genes implicated in prion disease and other neurodegenerative disorders. Hum. Mutat. 35, 1233–1248. [DOI] [PubMed] [Google Scholar]
- Saresella M., La Rosa F., Piancone F. et al (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer's disease. Mol. Neurodegener. 11, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders M. A., Liang H. and Li W. H. (2007) Human polymorphism at microRNAs and microRNA target sites. Proc. Natl Acad. Sci. USA 104, 3300–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarr E., Craig J. M., Cairns M. J. et al (2013) Decreased cortical muscarinic M1 receptors in schizophrenia are associated with changes in gene promoter methylation, mRNA and gene targeting microRNA. Transl. Psychiatry 3, e230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedel A., Thornton S., Schloss P., Kluter H. and Bugert P. (2011) Human platelets express functional alpha7‐nicotinic acetylcholine receptors. Arterioscler. Thromb. Vasc. Biol. 31, 928–934. [DOI] [PubMed] [Google Scholar]
- Schlaepfer I. R., Hoft N. R., Collins A. C. et al (2008) The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol. Psychiatry 63, 1039–1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I., Meerson A., Wolf Y., Avni R., Greenberg D., Gilboa‐Geffen A. and Soreq H. (2009) MicroRNA‐132 potentiates cholinergic anti‐inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973. [DOI] [PubMed] [Google Scholar]
- Shaltiel G., Hanan M., Wolf Y., Barbash S., Kovalev E., Shoham S. and Soreq H. (2013) Hippocampal microRNA‐132 mediates stress‐inducible cognitive deficits through its acetylcholinesterase target. Brain Struct. Funct. 218, 59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simchovitz A., Soreq L. and Soreq H. (2016) Transcriptome profiling in Parkinson's leukocytes: from early diagnostics to neuroimmune therapeutic prospects. Curr. Opin. Pharmacol. 26, 102–109. [DOI] [PubMed] [Google Scholar]
- Simon D. J., Madison J. M., Conery A. L., Thompson‐Peer K. L., Soskis M., Ruvkun G. B., Kaplan J. M. and Kim J. K. (2008) The microRNA miR‐1 regulates a MEF‐2‐dependent retrograde signal at neuromuscular junctions. Cell 133, 903–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soreq H. (2015) Checks and balances on cholinergic signaling in brain and body function. Trends Neurosci. 38, 448–458. [DOI] [PubMed] [Google Scholar]
- Soreq H. and Seidman S. (2001) Acetylcholinesterase–new roles for an old actor. Nat. Rev. Neurosci. 2, 294–302. [DOI] [PubMed] [Google Scholar]
- Tili E., Michaille J. J., Cimino A. et al (2007) Modulation of miR‐155 and miR‐125b levels following lipopolysaccharide/TNF‐alpha stimulation and their possible roles in regulating the response to endotoxin shock. J. Immunol. 179, 5082–5089. [DOI] [PubMed] [Google Scholar]
- Wang J. C., Hinrichs A. L., Stock H. et al (2004) Evidence of common and specific genetic effects: association of the muscarinic acetylcholine receptor M2 (CHRM2) gene with alcohol dependence and major depressive syndrome. Hum. Mol. Genet. 13, 1903–1911. [DOI] [PubMed] [Google Scholar]
- Wang G., van der Walt J. M., Mayhew G., Li Y. J., Zuchner S., Scott W. K., Martin E. R. and Vance J. M. (2008) Variation in the miRNA‐433 binding site of FGF20 confers risk for Parkinson disease by overexpression of alpha‐synuclein. Am. J. Hum. Genet. 82, 283–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Tan L., Wang H. F., Tan C. C., Meng X. F., Wang C., Tang S. W. and Yu J. T. (2015) Anti‐inflammatory drugs and risk of Alzheimer's disease: an updated systematic review and meta‐analysis. J. Alzheimers Dis. 44, 385–396. [DOI] [PubMed] [Google Scholar]
- Wessler I., Roth E., Deutsch C., Brockerhoff P., Bittinger F., Kirkpatrick C. J. and Kilbinger H. (2001) Release of non‐neuronal acetylcholine from the isolated human placenta is mediated by organic cation transporters. Br. J. Pharmacol. 134, 951–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson C. B., McLaughlin L. D., Nair A., Ebenezer P. J., Dange R. and Francis J. (2013) Inflammation and oxidative stress are elevated in the brain, blood, and adrenal glands during the progression of post‐traumatic stress disorder in a predator exposure animal model. PLoS ONE 8, e76146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtowicz E. E., Lechman E. R., Hermans K. G. et al (2016) Ectopic miR‐125a expression induces long‐term repopulating stem cell capacity in mouse and human hematopoietic progenitors. Cell Stem Cell. 19, 383–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B., Gu S. M., Li M. J. et al (2015) Rare SNP rs12731181 in the miR‐590‐3p target site of the prostaglandin F2alpha receptor gene confers risk for essential hypertension in the han chinese population. Arterioscler. Thromb. Vasc. Biol. 35, 1687–1695. [DOI] [PubMed] [Google Scholar]
- Xiao T., Jiao B., Zhang W., Tang B. and Shen L. (2016) Effect of the CYP2D6 and APOE polymorphisms on the efficacy of Donepezil in patients with Alzheimer's disease: a systematic review and meta‐analysis. CNS Drugs. 30, 899–907 [DOI] [PubMed] [Google Scholar]
- Yang S., Gao Y., Liu G., Li J., Shi K., Du B., Si D. and Yang P. (2015) The human ATF1 rs11169571 polymorphism increases essential hypertension risk through modifying miRNA binding. FEBS Lett. 589, 2087–2093. [DOI] [PubMed] [Google Scholar]
- Zhang L. M., Zhang X. P., Chen Y. Q. and Ye W. (2015) Association of CHRNA4 gene rs1044396 and rs1044397 polymorphisms with Parkinson's disease symptoms and smoking. Genet. Mol. Res. 14, 5112–5122. [DOI] [PubMed] [Google Scholar]
- Zhang X., Li Q., Wang L., Liu Z. J. and Zhong Y. (2016) Cdc42‐Dependent Forgetting Regulates Repetition Effect in Prolonging Memory Retention. Cell Rep. 16, 817–825. [DOI] [PubMed] [Google Scholar]
- Zimmerman G., Shaltiel G., Barbash S. et al (2012) Post‐traumatic anxiety associates with failure of the innate immune receptor TLR9 to evade the pro‐inflammatory NFkappaB pathway. Transl. Psychiatry 2, e78. [DOI] [PMC free article] [PubMed] [Google Scholar]