

Key Points
Thrombomodulin deficiency in adult mice induces a fatal coagulopathy caused by the lack of cofactor function for activation of protein C.
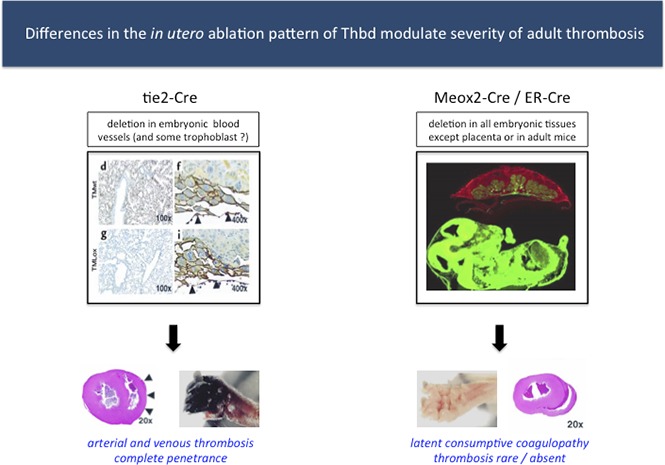
The severity of thrombosis after birth is modulated by in utero thrombomodulin expression in extraembryonic tissues.
Abstract
Thrombomodulin (Thbd) exerts pleiotropic effects on blood coagulation, fibrinolysis, and complement system activity by facilitating the thrombin-mediated activation of protein C and thrombin-activatable fibrinolysis inhibitor and may have additional thrombin- and protein C (pC)-independent functions. In mice, complete Thbd deficiency causes embryonic death due to defective placental development. In this study, we used tissue-selective and temporally controlled Thbd gene ablation to examine the function of Thbd in adult mice. Selective preservation of Thbd function in the extraembryonic ectoderm and primitive endoderm via the Meox2Cre-transgene enabled normal intrauterine development of Thbd-deficient (Thbd−/−) mice to term. Half of the Thbd−/− offspring expired perinatally due to thrombohemorrhagic lesions. Surviving Thbd−/− animals only rarely developed overt thrombotic lesions, exhibited low-grade compensated consumptive coagulopathy, and yet exhibited marked, sudden-onset mortality. A corresponding pathology was seen in mice in which the Thbd gene was ablated after reaching adulthood. Supplementation of activated PC by transgenic expression of a partially Thbd-independent murine pC zymogen prevented the pathologies of Thbd−/− mice. However, Thbd−/− females expressing the PC transgene exhibited pregnancy-induced morbidity and mortality with near-complete penetrance. These findings suggest that Thbd function in nonendothelial embryonic tissues of the placenta and yolk sac affects through as-yet-unknown mechanisms the penetrance and severity of thrombosis after birth and provide novel opportunities to study the role of the natural Thbd-pC pathway in adult mice and during pregnancy.
Visual Abstract
Introduction
Thrombomodulin (Thbd) is a multifunctional transmembrane thrombin receptor expressed on endothelial, hematopoietic, placental trophoblast, and various other cells. Thbd inhibits thrombin’s prothrombotic activity. In a complex with Thbd, thrombin’s substrate selectivity is redirected toward activation of protein C (PC), the key effector protease of the cytoprotective and anticoagulant PC pathway,1,2 and activation of thrombin-activatable fibrinolysis inhibitor.3 In addition, Thbd exerts thrombin- and PC-independent functions to regulate complement activity and inflammatory signaling.4,5
In humans, missense mutations in the Thbd gene have been linked to various diseases, including atypical hemolytic uremic syndrome,6 arterial,7-10 and, to a lesser extent, venous thrombotic disease11-13 and recently to a bleeding disorder through a mutation resulting in increased plasma levels of soluble Thbd.14,15 Lower expression of Thbd in the placenta has been reported in preeclampsia pregnancies.16 To our knowledge, no complete loss of function mutations have been described in humans.
In mice, complete Thbd deficiency is incompatible with normal embryonic development, secondary to an early block in placental development.17 The expression of Thbd by fetal placental trophoblast cells appears necessary to counteract coagulation and platelet activation at the fetomaternal interface, which interferes with the placental labyrinth layer formation through as-yet-unknown mechanisms.18 Selective retention of Thbd expression in trophoblast cells by tissue engineering19 or via Tie2Cre-mediated, endothelial-specific ablation of the Thbd gene20 both bypassed the early placental defect, but later resulted in a partial second block in intrauterine development. Live-born mice with endothelial-restricted Thbd deficiency succumb to pronounced consumptive coagulopathy with spontaneous thrombosis, with complete penetrance and uniform manifestation of disease within 3 to 5 weeks after birth. Mice carrying a Glu404Pro mutation that partially abolished the cofactor activity of Thbd for the activation of PC showed only a latent prothrombotic phenotype, but exhibited various defects in response to inflammatory challenge with lipopolysaccharide, additional genetic thrombotic risk factors, or radiation exposure.21-23
In the current study, we applied tissue-selective and temporally regulated Thbd gene ablation and genetic complementation of PC activation to further examine the role of Thbd in development and adult physiology in mice.
Methods
Animals
ERCre mice (Gt(ROSA)26Sortm9(cre/ESR1)Arte; stock no. 10471, Taconic, Albany, NY),24 Meox2Cre mice (B6.129S4-Meox2tm1(cre)Sor/J; stock no. 003755, The Jackson Laboratory, Bar Harbor, ME),25 and the dual-color Cre-reporter strain mT/mG (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J; stock no. 007676, The Jackson Laboratory)26 were obtained from commercial providers. ThbdloxP, ThbdloxP-Tie2Cre, and Thbd+/− mice have been described previously.20,27 Transgenic mice expressing the murine D168F/N173K PC variant under the control of the liver-selective transthyretin (TTR) promoter were generated by site-directed mutagenesis of mouse PC complementary DNA and inserting the variant complementary DNA into a Stu1 restriction site in exon 2 of a 3-kb fragment of the TTR gene containing the TTR promoter, exon 1, and exon 2, as previously described for generation of the human APCHI transgene. Transgenic mice were obtained via pronuclear injection of the vector-free transgene DNA into C57Bl6 oocytes. All strains were maintained on a C57Bl6/N background in the specific pathogen-free facility of the Translational Biomedical Research Center of the Medical College of Wisconsin. Experiments were conducted in adherence with National Institutes of Health guidelines on the use of laboratory animals and were approved by the Medical College of Wisconsin’s Institutional Animal Care and Use Committee.
Immunohistochemistry
Tissues were fixed in 10% formalin and embedded in paraffin. Five-micrometer sections were cut and counterstained with Gil’s hematoxylin and eosin or Masson’s trichome (Vector Laboratories, Burlingame, CA). Immunohistochemistry was performed by using the Vectastain Elite ABC and VIP staining kits (Vector Laboratories). Goat anti-mouse Thbd (AF3894) or rat anti-mouse Thbd 201B, biotin-conjugated anti-goat immunoglobulin G (B7014), and biotin-conjugated anti-sheep immunoglobulin G (A3415) were from R&D Systems (Minneapolis, MN). Digital images were captured on a Nikon Eclipse E600 microscope equipped with a Diagnostic Instruments Color Mosaic 11.2 digital camera or a Hamamatsu NanoZoomer slide scanner. Fluorescence microscopy was conducted on a fluorescent slide scanner (VS120) from Olympus.
Measurement of plasma markers, platelets, and hematology
Plasma fibrinogen, thrombin-antithrombin, and fibrin degradation products (d-dimer) in citrated blood samples obtained from venipuncture of the inferior vena cava or from the submandibular vein were measured as described.20 Mouse activated protein C (aPC) and PC were assayed in citrated plasma containing 10 mM benzamidine-hydrogen chloride essentially as previously described28 by immunocapture of aPC or PC with maB1587 or maB1582 (gift from Charles T. Esmon, Oklahoma City, OK), respectively, followed by detection of PC with goat anti-mouse PC polyclonal antibody and measurement of aPC activity with a Spectrozyme PCa substrate. Levels of a selection of cytokines were measured in citrated plasma by using the 13-plex LEGENDplex Mouse Inflammation Panel following the manufacturer’s instructions (BioLegend, San Diego, CA). Differential hematologic profiles were acquired on a Vet ABC Hematology Analyzer (scil animal care company, Gurnee, IL).
Platelet activation status and abundance of platelet-leukocyte aggregates were determined by flow cytometry of citrated whole blood diluted in 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid in phosphate-buffered saline by using a LSRII instrument (BD Biosciences) and analyzed with FlowJo software version 9.7 (FlowJo, Ashland, OR). Platelets were gated as a Ter119-negative population (Ter119-BV421; BioLegend) as well as by size in log scale for forward and side scatter plots and characterized for expression of activated integrin αIIbβ3 and P-selectin (JON/A phycoerythrin-conjugated, Wug.E9 fluorescein isothiocyanate, Emfret Analytics, Würzburg, Germany). Platelets that stained positive for both JON/A-phycoerythrin and Wug.E9-fluorescein isothiocyanate were considered activated. Leukocyte-platelet aggregates were characterized as CD45/P-selectinPOS or CD45/Wug.E9POS (CD45.2-Alexa Fluor 700/allophycocyanin-cyanin fluorochrome Cy7; BioLegend).
FeCl3-induced in vivo thrombosis and calibrated automated thrombography
Ferric chloride (FeCl3)-induced thrombus formation in the carotid artery was performed as previously described29,30 with modifications. After isoflurane inhalation anesthesia, the right carotid was isolated bluntly from surrounding tissues, including the vagal nerve, and a plastic sheath was placed underneath. A 1 × 2–mm filter paper saturated with distilled water containing 5% weight-to-volume FeCl3 was placed on the vessel for 3 minutes. After flushing the area with saline, a Doppler flow probe (model 0.5 VB, Transonic Systems, Ithaca, NY) was placed around the vessel. The signal was recorded continuously by using a perivascular flow module (T402/TS420, Transonic Systems) and LabChart 7 software (ADInstruments, Colorado Springs, CO). Time to occlusion was measured from the moment the filter paper was removed. A decrease in flow back to baseline level that remained stable for 3 minutes was considered an occlusion. Recording was continued for 30 minutes or until an occlusion occurred. The rate of occlusion was estimated from the slope of the percent flow decrease over the time between the moments where the flow reached 85% and 25% of the initial stable flow value after placing the probe. For calibrated automated thrombography, blood was collected from the inferior vena cava by using a 25-gauge needle into a 1:10 volume of 3.8% citrate/0.5 mg/mL corn trypsin inhibitor. Thrombograms were acquired as described previously31 after activation with 0.6 pm tissue factor and 24 µM phospholipid mixture (Dade Innovin, Siemens).
Evans blue assay
Vascular permeability was measured as described previously.32 Sixty minutes after IV infusion of Evans blue-albumin (25 mg/kg of body weight), a blood sample was drawn for measurement of plasma dye concentration, animals were perfused with 30 mL of warmed saline, and organs were dissected. Evans blue content in blood and organs was determined by imaging on an Odyssey infrared imager (LI-COR Biosciences, Lincoln NE) with Odyssey Application software, version 3.0.
Statistical analysis
Parametric data are presented as means and standard deviations, with differences tested with Student t test. Nonparametric data are presented as medians and interquartile ranges, with differences tested with a Mann-Whitney U test. Given the time-to-event type of data in the in vivo thrombosis model, differences in occlusion times were tested with a log-rank test, where nonocclusion was regarded as censored data at 30 minutes. Differences in longitudinal body weight data were determined by using a regression model with genotype and age as fixed effects and individual mouse as a random effect.
Results
Temporally controlled Thbd gene ablation in adult mice
To examine the effect of Thbd knockout in fully developed mice, ThbdloxP/loxP mice were crossed with a mouse strain expressing the estrogen receptor-Cre-recombinase fusion protein under the transcriptional control of the ROSA26 locus (Gt(ROSA)26Sortm9(cre/ESR1)Arte (ERCre)).24 Thbd gene ablation was induced in 8- to 14-week old ERCre-ThbdloxP/loxP mice by daily intraperitoneal infusion of 1 mg tamoxifen (Sigma Aldrich, St. Louis, MO) in 100 µl corn oil for 4 days. Two mice died within the first week after tamoxifen treatment. Remaining animals stayed symptom-free over the first 10 weeks after induction. Immunohistochemistry and western blot analyses, conducted 4 weeks after tamoxifen induction, documented near-complete absence of Thbd antigen in most tissues except for the lungs, where 11 of 14 mice retained clearly detectable Thbd antigen (Figure 1A-C). A survey of tissue sections from various internal organs did not reveal gross pathological changes or the presence of large, obstructive blood clots. Around 10 weeks after tamoxifen treatment, animals developed variable pathologies, including visible cachexia and decreased movement/hunched posture (2 of 21 mice), eye matting followed by rapid loss/degeneration of the affected eye (7 of 21 mice), and edematic swelling and purple discoloration of distal tail segments (13 of 21 mice) and/or hind limb digits (2 of 21 mice). Overall, 15 of 21 mice were removed from the study on developing these pathologies or spontaneously expired without prior grossly observable indications (n = 6) within 20 weeks after tamoxifen treatment (Figure 1D). No lethality was observed in a cohort of 16 noninduced ERCre-ThbdloxP/loxP mice up to 34 weeks of age (latest time point observed).
Figure 1.

Tamoxifen-induced systemic thrombomodulin ablation in adult mice. (A-B) Residual expression of Thbd antigen detected in the lungs by western blot analysis of tissue lysate (A) and on histological sections (B). Numbers in panels A and B identify individual animals. (C) Detection of Thbd protein by immunohistochemistry in various tissues of tamoxifen-treated ERCreThbdloxP mice. Brown staining indicates Thbd protein detected by horseradish peroxidase–coupled anti-Thbd antibody. Hematoxylin counterstain. Red bars indicate 50 μm. In several animals, Thbd antigen was virtually undetectable. (D) Kaplan-Meier survival plot of ERCreThbdloxP mice after tamoxifen treatment at 8 to 10 weeks. Difference in survival between groups is significant (Mantel-Cox log-rank analysis). Ctrl, non–tamoxifen-treated mice.
These results show that partial and in some cases near-complete systemic ablation of Thbd leads to mortality in adult mice. Notably, in marked contrast to the severe thrombotic diathesis described in mice with endothelial cell–restricted Thbd deficiency (Tie2Cre-ThbdloxP),20 the phenotypic penetrance of thrombosis remained largely limited to the tails of these animals.
Thbd expression restricted to the placenta is sufficient for intrauterine development
To explore whether the relatively mild phenotype of tamoxifen-treated ERCre-ThbdloxP mice, as compared with mice with constitutive endothelial Thbd-deficiency, was due to residual Thbd expression (observed in the lungs), we generated a mouse model of complete and constitutive Thbd deficiency. The expression of Cre recombinase under the control of the endogenous Meox2 gene promoter mediates ablation of loxP-flanked genes in virtually all embryonic tissues, but preserves normal gene function in placental derivatives of trophectoderm and primitive endoderm.25 Crosses between female ThbdloxP/loxP mice and male Meox2Cre-Thbd+/− (compound heterozygotes for the Meox2Cre-knockin allele and the constitutive loss-of-function Thbd-null allele) produced slightly less than half of the expected yield of Meox2Cre-Thbddel/lacZ (hereafter termed Thbd-null) offspring at weaning age (3 weeks), but normal representation of full-term Thbd-null embryos (Table 1), indicating that selective loss of Thbd-null neonates occurred between birth and weaning age. Seven of 12 carcasses recovered the morning after births were Thbd-null neonates with overt hemorrhagic lesions (Figure 2A). Analysis of Meox2Cre expression in the Cre-recombinase mT/mG reporter mouse strain26 at day 12 of embryonic development confirmed ubiquitous recombinase activity in the embryo proper and fetal blood vessels in the placenta, but a lack thereof in all extraembryonic fetal trophoblast cells (Figure 2B). Accordingly, Thbd antigen expression at embryonic day 15.5 was preserved in trophoblast derivatives, but undetectable in the embryo proper and in fetal endothelium in the placenta (Figure 2C-D).
Table 1.
Intrauterine survival of Meox2Cre-Thbd-null mice
| Mating: male Meox2Cre-Thbd+/– × female ThbdloxP/loxP | |||
|---|---|---|---|
| Offspring genotype | Male | Female | Total |
| E 15-18 | — | — | |
| ThbdloxP/lacZ Meox2Cre | — | — | 14 |
| Thbd+/lacZ Meox2Cre | — | — | 11 |
| ThbdloxP/lacZ | — | — | 11 |
| Thbd+/lox | — | — | 7 |
| 3 weeks | |||
| ThbdloxP/lacZ Meox2Cre | 13 | 8 | 21* |
| Thbd+/lacZ Meox2Cre | 28 | 31 | 59 |
| ThbdloxP/lacZ | 33 | 34 | 67 |
| Thbd+/lox | 33 | 28 | 61 |
Genotype frequencies at 15 to 18 days postcoitum and at 3 weeks postpartum, statistically tested against expected frequencies. Findings indicate selective loss of Meox2Cre-Thbd-null mice between the 2 time points.
–, allele deleted; +, wild-type allele; E, embryonic day; lox, loxP-flanked gene.
P < .05 (χ2 analysis).
Figure 2.

Constitutive Thbd ablation with selective preservation in the placenta in Meox2Cre-ThbdloxP mice. (A) Thbd-deficient neonates found dead. Arrows indicate hemorrhagic lesions. (B) Photomicrograph of a flash-frozen section prepared from a day 15.5 embryo expressing the Meox2Cre-gene and the mT/mG recombinase reporter gene: red (mT) fluorescence indicates the nonrecombined reporter gene expressed in the placenta, green fluorescence indicates Cre-mediated activation of the mG reporter in the embryo proper, chorionic plate, and fetal blood vessels in the placenta, but lack thereof in all extraembryonic fetal trophoblast cells. (C-D) Immunohistochemical detection of Thbd antigen (brown staining; hematoxylin counterstain) in the day 12 embryo and placenta of ThbdloxP mice (in panel C, Thbd is detected in the embryo and placenta) and Meox2Cre-ThbdloxP mice (in panel D, Thbd is present in the placenta, but not the embryo). Scale bars, 1 mm.
This showed that the Meox2Cre-mediated loss of Thbd function was compatible with normal intrauterine development, but caused perinatal death of approximately half of the mutant neonates, likely due to birth trauma–induced thrombotic hemorrhaging.
Morbidity of adult mice with complete Thbd deficiency
Thbd-null mice that survived the perinatal period were smaller than their Thbd-expressing littermates, but showed normal weight gain (Figure 3A-B). Quantitation of residual Thbd messenger RNA expression in 6 different organs of 6 individual Thbd-null mice by quantitative reverse transcription polymerase chain reaction indicated on average a ≥94% reduction of organ-wide Thbd messenger RNA levels, with 1 animal showing substantial expression in 1 organ only (the heart; data not shown). Immunohistochemical analyses at age 10 to 16 weeks confirmed the persistent absence of Thbd antigen in micro- and macrovascular endothelium in all tissues analyzed (Figure 3C); 1 of 10 animals analyzed exhibited isolated patches of microvascular Thbd expression in the ventricular wall of the heart. Beginning at ∼4 weeks of age, the Thbd-null mice expired spontaneously or reached humane end points, with only 2 of 28 mice surviving to week 40 (Figure 3D). Eighteen of 28 animals developed externally observable thrombotic lesions (ie, purple discoloration, edematic swelling, and necrosis of distal tail segments [17 of 28 mice], or hind limb digits [1 of 28 mice], uni- or bilateral eye degeneration [7 of 28 mice], or a combination thereof [6 of 28 mice]). Postmortem inspection and autopsies of spontaneously expired animals were unremarkable, except for severe gastrointestinal bleeding as the putative cause of death in 1 animal. Histology of postmortem lungs, hearts, livers, and brains of 5 animals recovered within 12 hours after death remained inconclusive with respect to the cause of death. Histological surveys of internal organs from surviving 10- to 20-week-old Thbd-null mice (n = 6) did not reveal evidence of widespread thrombotic organ damage (ie, tissue necrosis, fibrotic tissue remodeling, or vascular occlusions). One animal, analyzed 1 day after the onset of eye matting, exhibited extensive infiltration of neutrophils into the anterior eye chamber, consistent with anterior uveitis (Figure 3E).
Figure 3.

Adult phenotype of Meox2Cre-Thbd-null mice. (A) Smaller body size of Thbd-null mice. (B) Thbd-null mice had significantly lower body weight at all time points measured (n ≥ 16 for each time point), but showed near-normal growth as compared with Thbd-expressing littermates (n ≥ 92 for each time point). (C) Histological immunostaining of different organs at 10 to 16 weeks of age showing persistent absence of Thbd from the endothelium in different vascular beds. (D) Kaplan-Meier survival plot of live-born Meox2Cre-Thbd-null mice (n = 28), as compared with Thbd-expressing littermates of various genotypes. (E) Masson-Trichrome–stained section of affected eye in a Meox2Cre-Thbd-null mouse showing leukocyte infiltration into the anterior eye chamber (left); normal eye histology in a ThbdloxP/+ (1 loxP-flanked but nondeleted Thbd allele and 1 wild-type Thbd allele) (right); hematoxylin and eosin stain.
Genetic background does not affect severity or onset of thrombosis in mice with endothelial cell–restricted Thbd deficiency
The above observations showed that complete loss of Thbd-function (Meox2Cre-ThbdloxP; C57Bl6 background) did not aggravate, but paralleled the gross phenotype seen in mice with temporally controlled Thbd gene ablation in adulthood (ERCre-ThbdloxP; C57Bl6 background). Both phenotypes were, however, much less pronounced than those of mice with endothelial-restricted Thbd-deficiency (Tie2Cre-ThbdloxP; mixed 129/Sv-C57Bl6-FVB genetic background), as reported previously.20 To determine whether these differences could be attributed to genetic background, a small cohort of mice with endothelial cell–restricted Thbd deficiency was re-created on an inbred C57Bl6 background (≥15 backcrosses). Intrauterine embryonic lethality persisted (∼50%), and 6 out of 7 live-born Tie2Cre-ThbdloxP mice developed severe thrombosis of their hind limbs and other external lesions within 4 to 9 weeks, reproducing earlier observations.
Hemostatic status of adult mice with complete Thbd deficiency
Plasma levels of d-dimer and thrombin-antithrombin complex (TAT) appeared elevated in Thbd-null mice as compared with controls, but the difference did not reach statistical significance (P = .056 and P = .24 respectively; see Table 2 for all hemostatic parameters). Normalized fibrinogen levels were decreased. Platelet, hemoglobin, and white blood cell counts were within the normal range. Except for elevated levels of interleukin-6 (IL-6), cytokine plasma levels were indistinguishable from Thbd-expressing littermates. Platelets from Thbd-null mice did not exhibit elevated surface expression of P-selectin/activated GPαIIbβ3 or an increased abundance of circulating platelet-leukocyte aggregates. FeCl3-induced injury to the exposed carotid artery was used to measure the effect of Thbd deficiency on an artificial prothrombotic stimulus. Thbd-null mice exhibited a borderline significant delay in the time to occlusion with a corresponding, but nonsignificant decrease in the rate of occlusion. In calibrated automated thrombography assays of platelet-poor plasma samples, Thbd-null mice showed diminished endogenous thrombin potential (P = .003), prolonged time to peak thrombin generation (P = .017), and diminished peak thrombin generation (P = .013) (Figure 4A-B). Lung vascular permeability, measured by Evan’s blue extravasation, was elevated in the lungs of Thbd-null mice, but not in the kidneys, brains, hearts, or livers (Figure 4C-D).
Table 2.
Hemostatic status of Meox2Cre-Thbd-null mice with and without expression of the murine PC transgene, as compared with controls
| Control | Thbd-null | Thbd-null hmPC+ | hmPC+ | |
|---|---|---|---|---|
| d-dimer, median (IQR), ng/mL | 95 (86-139) (n = 15) | 135 (130-159) (P = .056) (n = 8) | 97 (81-145) (n.s.) (n = 8) | 95 (89-124) (n.s.) (n = 9) |
| TAT, median (IQR), μg/L | 27 (15-81) (n = 10) | 74 (41-119) (n.s.) (n = 4) | 14 (13-17) (n.s.) (n = 8) | 23 (10-52) (n.s.) (n = 9) |
| Fibrinogen, mean ± SD, % | 100 ± 16 (n = 5) | 51 ± 32 (p 0.011) (n = 6) | — | — |
| IL-6, median (IQR), pg/mL | 19 (14-27) (n = 14) | 162 (60-369) (P = .0011) (n = 8) | 244 (69-320) (P = .0016) (n = 12) | 17 (13-88) (n.s.) (n = 9) |
| Platelets, mean ± SD, × 109/L | 279 ± 81 (n = 12) | 223 ± 62 (n.s.) (n = 11) | — | — |
| Hemoglobin, mean ± SD, g/dL | 13.0 ± 0.7 (n = 12) | 11.7 ± 0.5 (n.s.) (n = 11) | — | — |
| White blood cells, mean ± SD, × 109/L | 6.3 ± 0.8 (n = 12) | 8.6 ± 1.3 (n.s.) (n = 11) | — | — |
| Activated platelets, median (IQR), % P-selectin+ αIIbβ3+ | 0.030 (0.009-0.25) (n = 5) | 0.065 (0.040-0.077) (n.s.) (n = 5) | — | — |
| Platelet leukocyte aggregates, median (IQR), % CD45/P-selectin+ or CD45/αIIbβ3+ | 10.9 (9.4-11.0) (n = 5) | 16.0 (12.2-19.0) (n.s.) (n = 5) | — | — |
| Carotid injury thrombosis model | ||||
| Time to occlusion, median (IQR), s | 189 (163-203) (n = 8) | 538 (394-899) (P = 0.0413) (n = 3) | 226 (160-355) (n.s.) (n = 8) | 276 (238-392) (P < 0.001) (n = 7) |
Controls comprised Thbd wild-type and ThbdloxP/+ C57black mice. Normally distributed parameters are presented as means and standard deviations; non–normally distributed parameters are presented as medians and IQRs.
hmPC+, mice expressing the PC transgene; IQR, interquartile range; n.s., nonsignificant; SD, standard deviation.
Figure 4.

Thrombin generation and vascular permeability in Meox2Cre-Thbd-null mice. (A) Calibrated automated thrombinography of plasma from control mice (ThbdloxP/+, orange, n = 4) and Thbd-deficient mice (Thbd−/−; blue, n = 4). (B) Thbd-null mice displayed prolonged lag time and time to peak thrombin generation (ttPeak), reduced endogenous thrombin potential (ETP), and peak thrombin generation (Peak). (C-D) Evans blue extravasation assay showing increased vascular permeability in the lungs of Thbd-null mice compared with controls ThbdloxP/loxP; n = 5 per group). (D) Data shown in panel C were generated by quantitative scanning of fluorescence (at 720 nm) of whole-organ lung tissue.
Transgenic aPC supplementation is sufficient to rescue Meox2Cre-Thbd-null mice
To determine whether the pathology of Thbd-null mice was due to impaired PC activation, we generated a transgenic mouse strain (mAPCHI mice) expressing a D168F/N173K variant of the murine PC zymogen. This is the mouse analog of the hyperactivatable human D167F/D172K PC variant, which exhibits enhanced activation in the absence of Thbd.33 The murine PC transgene fully substituted for the function of endogenous PC, because mAPCHI-PC−/− transgenic mice lacking endogenous PC were viable and able to reproduce. Transgene expression resulted in two- and threefold increased plasma levels of murine PC and aPC, respectively (Figure 5A-B) and prolonged the time to occlusion in the FeCl3 carotid injury model (Table 2). The D168F/N173K PC transgene was introduced into Thbd-null mice. This genetic supplementation of aPC prevented the perinatal lethality, ameliorated the growth retardation (Figure 5C), enabled symptom-free, long-term survival of the majority of both Meox2Cre- and ERCre-Thbd-null mice (Figure 5D), and normalized median plasma levels of TAT, plasma d-dimer, and the time to occlusion in the FeCl3 carotid injury model (Table 2). It did not restore vascular barrier function in the lungs (Figure 4C-D) or suppress the augmented plasma IL-6 levels seen in Thbd-null mice (Table 2).
Figure 5.

Genetic supplementation of APC by PC transgene expression. (A-B) The D168F/N173K protein C transgene (mAPCHI) increased plasma levels of PC (A) and activated PC (B) as measured in mice with normal Thbd gene expression. To assess the expression of the mAPCHI transgene in the absence of endogenous aPC, PC-deficient mice with and without the transgene were added as additional experimental groups. Transgene-derived D168F/N173K PC expression was determined in APCHI mice lacking endogenous PC (PC−/−). Heterozygous PC-deficient mice (PC+/−) were included as controls. (C) The transgene partially restored the body weight of Thbd-null mice (for each time point: Thbd-null, n ≥ 24; mAPCHI Thbd-null, n ≥ 16; Thbd-expressing littermates, n ≥ 92). (D) D168F/N173K PC supplementation reduced mortality of tamoxifen-induced ERCre-ThbdloxP mice as well as of constitutively Thbd-deficient Meox2Cre-Thbd-null mice. (E) Pregnant Thbd-null mAPCHI mice showed pregnancy-induced bleeding, as shown by hemosiderin depositions in the lungs (left; original magnification ×4, hematoxylin and eosin stain) and hemorrhage in the uterus (right).
Pregnancy-induced mortality of Thbd-null females expressing the PC transgene
Thirteen pregnancies from timed matings of female Meox2Cre-Thbd-null mice carrying the mAPCHI transgene with wild-type males (embryonic placenta expresses Thbd) ended either in stillbirth (n = 2), death of the mother shortly after delivery of 1 or 2 live pups (n = 2), or death or euthanasia mandated by severe distress of the mother between day 11 postcoitum and term (n = 6). Three females that had gained substantial weight and were visibly pregnant lost their pregnancies without showing distress, and 1 of the latter expelled tissue fragments identified as placental remnants at 34 days postcoitum. Necropsy of 5 affected females revealed grossly visible hemorrhage in the uterus (n = 3), lungs (n = 1), or kidney (n = 1) and histological evidence of hemosiderin deposition and erythrocyte extravasation in the lungs (n = 3; Figure 5E). No such pathologies nor reduced fecundity were observed in wild-type mice expressing the mAPCHI transgene, mitigating against the detrimental effects of augmented aPC levels alone.
Discussion
By using tissue-selective and temporally controlled gene ablation, we have generated novel mouse models with a disrupted Thbd-PC pathway. In the first model (ERCre-ThbdloxP mice), tamoxifen treatment led to near-complete loss of Thbd expression in adult mice. In the second model (Meox2Cre-Thbd-null mice), the Thbd gene was ablated in all embryonic tissues during early development, with preservation in extraembryonic lineages.
These approaches led to new observations that contrast with previous outcomes of endothelium-selective Thbd gene ablation in Tie2Cre-ThbdloxP mice.20 The Meox2Cre-Thbd-null model confirmed that placenta-selective preservation of Thbd is sufficient to overcome the developmental arrest at embryonic day 8.5 that is consistently seen in completely Thbd-deficient mice,17 which is in line with observations in Tie2Cre-ThbdloxP mice.20 However, the latter revealed a secondary developmental block that resulted in the partial loss of the Thbd-deficient embryos at midterm and was associated with an increased prevalence of placental thrombosis. The subset of Tie2Cre-ThbdloxP mice that escaped intrauterine developmental failure showed little pathology during the perinatal period, but developed severe fatal thrombosis of their extremities and internal organs thereafter. In contrast, first, the Meox2Cre-Thbd-null mice showed normal development to term. Second, about half of Meox2Cre-Thbd-null mice died shortly after birth, likely due to birth-induced thrombohemorrhagic complications. Third, in Thbd-null mice that survived the neonatal period, macroscopically evident thrombosis remained largely confined to the tail, and we were unable to document overt thrombotic damage to internal organs, which is a hallmark of the thrombotic phenotype in the Tie2Cre model.
Tie2Cre-ThbdloxP mice clearly displayed a severe consumptive coagulopathy (that partially normalized over time) with decreases in platelet counts and fibrinogen levels and increased TAT and d-dimer levels along with signs of hemorrhage and thrombosis. These abnormalities were much less pronounced in Meox2Cre-Thbd-null mice, reflected in a somewhat retarded in vivo clot formation, reduced ex vivo thrombin generation, nonsignificantly elevated TAT levels, elevated levels of d-dimer, a moderate decline in fibrinogen levels, and normal platelet counts. Taken together, these findings are consistent with a chronic but mild consumption of clotting factors that resembles compensated disseminated intravascular coagulopathy, where continuous exposure to tissue factor leads to a state of consumption of clotting factors with normalization of platelet levels in some prolonged cases.34 Although this might explain the underlying cause of morbidity in Thbd-deficient mice, the proximal cause of death in individual animals proved difficult to document. Other than the common thrombotic pathologies in the tail and, in rare cases, the hind limbs, we did not find evidence for recurring thrombotic events in live animals or indications of massive thrombotic events in critical organs, such as the kidney, heart, lung or brain. The single identified cause of death was massive gastrointestinal bleeding, suggesting that at least 1 animal death was associated with a transition from compensated to uncompensated disseminated intravascular coagulation.35
The absence of a secondary development block in Meox2Cre-Thbd-null mice as well as the less severe phenotypic penetrance (in comparison with Tie2Cre-ThbdloxP mice) were unexpected. Because Meox2Cre-mediated Thbd gene disruption eliminates Thbd in all embryonic tissue, including endothelium and hematopoietic cells targeted by the Tie2Cre transgene, Meox2Cre-Thbd-null mice were predicted to develop the same or even more severe phenotype. Having ruled out differences in genetic background or residual Thbd expression in the endothelial cells of MeoxCre2-Thbd-null mice, the underlying cause of such differences remains unknown at present. It is possible that insertion of the Tie2Cre transgene alters the function of some unknown modifier gene, or that the heterozygous Meox2 gene inactivation itself (due to insertion of the Cre transgene into the Meox2 allele) ameliorates the prothrombotic phenotype. Indeed, Meox2 is involved in myogenesis, and a recent study has identified the skeletal muscle protein myosin as a potent actor in the coagulation system.36 Alternatively, the discrepancy could be caused by differences in the developmentally regulated expression patterns of the Tie2- and Meox2-Cre transgenes. Cre expression, controlled by the endogenous Meox2 promoter, is strictly limited to the epiblast beginning as early as embryonic day 5, thereby preserving Thbd expression in all extraembryonic lineages, including visceral endoderm, parietal endoderm, and all trophoblast subpopulations. In contrast, expression of endogenous Tie2/Tek has been documented in trophoblast derivatives, including giant trophoblast cells of the day 9.5 mouse conceptus,37 possibly resulting in partial ablation of Thbd gene function in trophoblast cells of Tie2Cre-ThbdloxP mice. Disruption of gene function in placental trophoblast cells can indeed exert downstream defects in cardiovascular or hematopoietic development in mice.38-41 Likewise, abnormal placental development has been correlated with subsequent cardiovascular disease in humans (reviewed in Burton et al42). The current animal models provide an experimentally accessible opportunity for testing the hypothesis that the phenotypic penetrance of spontaneous, overt thrombosis in adult Thbd-deficient mice is in part governed by placental abnormalities caused by the absence of Thbd from trophoblast cells.
Transgenic supplementation of aPC prevented peri- and postnatal mortality of Thbd-null mice, prevented tail necrosis, normalized coagulation markers, normalized ex vivo thrombin generation, and corrected the time to thrombus formation in the carotid injury model. These outcomes imply that the pathologies of Thbd-null mice are explained by the loss of Thbd’s anticoagulant cofactor function for PC activation. This conclusion is further supported by earlier observations that animals infused with a PC-blocking antibody developed similar eye pathologies associated with thrombus formation in the cephalic vein around the eyeball and lacrimal glands,43 as described in this article for Thbd-null mice, whereas transgenic aPC supplementation suppresses this phenotype. Notably, IL-6 levels were not normalized by the PC transgene, indicating persistent production of this cytokine by immune cells and/or by the endothelium. IL-6 elaboration in the absence of a concurrent elevation of additional biomarkers of inflammation (IL-1, γ-interferon, and TNF-α) militates against a systemic proinflammatory response to thrombotic organ damage as a major factor contributing to the morbidity of Thbd-null mice.
Thbd-null females that expressed the mAPCHI transgene nevertheless developed marked pregnancy-induced mortality and morbidity and had a severely reduced capacity to produce live litter. The pregnancy-associated morbidity of completely Thbd-deficient females (carrying heterozygous Thbd-knockout embryos) is in stark contrast to the intrauterine death of completely Thbd-deficient mice carried by mothers with heterozygous Thbd deficiency.17,44 The latter was associated with a failure of Thbd-null embryos to initiate differentiation of the placenta, but had no effect on the fecundity or well-being of the mother. The same is true when embryos with a defective Thbd cofactor function for PC activation are carried by heterozygous factor V Leiden females (with aPC resistance).18 Conversely, in the current animal model, pregnancy results in maternal death that is likely caused by major hemorrhage, most prominently in the uterus and lungs. Although 2 Thbd-null-mAPCHI females gave birth to viable offspring (and subsequently died), embryos and fetuses recovered from morbid females were found arrested in various stages of development, indicating that Thbd deficiency of the mother interfered with fetal development before compromising maternal survival. The precise etiology of maternal death remains to be fully resolved. Potential triggers may include: the pregnancy-associated shift in hemostasis, which occurs physiologically in both humans and mice45,46; the hemostatic challenge associated with placental/uterine vascular remodeling; or high levels of (transgenic) aPC combined with a profibrinolytic state secondary to the loss of Thbd-dependent, thrombin-activatable fibrinolysis inhibitor activation. A local inflammatory reaction of the mother’s immune system against the paternal Thbd neoantigen expressed on the placenta might further contribute. The presence of hemorrhagic lesions not only in the reproductive tract, but also in remote organs of the mother suggests that these pregnancy-specific risk factors combine to trigger a systemic response reminiscent of overt consumptive coagulopathy that is associated with pregnancy-related diseases, such as stillbirth and preeclampsia/hemolysis, elevated liver enzymes, low platelets syndrome.47
Acknowledgments
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants HL117132, HL133348 (H.W.), and HL112873 (R.S.) and by the Ziegler Family Chair for Research (H.W.).
Footnotes
Presented in abstract form at the 58th annual meeting of the American Society of Hematology, San Diego, CA, 3-6 December 2016.
Authorship
Contribution: T.E.v.M., H.-P.H.L., S.B., I.H., J.M., R.S., C.S.K., and H.W. performed experiments and data analysis; M. Zogg and J.M. maintained the mouse colonies; M. Zogg and H.W. designed breeding strategies and genetic constructs; M. Zhan, Q.Y., and R.S. measured intrauterine survival and analyzed embryonic expression of Thbd/Cre-recombinase; J.F., S.K., and H.W. generated the transgenic mice; T.E.v.M. and H.W. wrote the manuscript; H.W. designed the study; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for H.-P.H.L. is ANZAC Research Institute, Concord Repatriation General Hospital, Concord, NSW, Australia.
Correspondence: Hartmut Weiler, Blood Research Institute, BloodCenter of Wisconsin, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: hartmut.weiler@bcw.edu.
References
- 1.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109(8):3161-3172. [DOI] [PubMed] [Google Scholar]
- 2.Morser J. Thrombomodulin links coagulation to inflammation and immunity. Curr Drug Targets. 2012;13(3):421-431. [DOI] [PubMed] [Google Scholar]
- 3.Plug T, Meijers JCM. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J Thromb Haemost. 2016;14(4):633-644. [DOI] [PubMed] [Google Scholar]
- 4.Abeyama K, Stern DM, Ito Y, et al. . The N-terminal domain of thrombomodulin sequesters high-mobility group-B1 protein, a novel antiinflammatory mechanism. J Clin Invest. 2005;115(5):1267-1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Van de Wouwer M, Plaisance S, De Vriese A, et al. . The lectin-like domain of thrombomodulin interferes with complement activation and protects against arthritis. J Thromb Haemost. 2006;4(8):1813-1824. [DOI] [PubMed] [Google Scholar]
- 6.Delvaeye M, Noris M, De Vriese A, et al. . Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(4):345-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu KK, Aleksic N, Ahn C, Boerwinkle E, Folsom AR, Juneja H; Atherosclerosis Risk in Communities Study (ARIC) Investigators. Thrombomodulin Ala455Val polymorphism and risk of coronary heart disease. Circulation. 2001;103(10):1386-1389. [DOI] [PubMed] [Google Scholar]
- 8.Wang H, Dong P. Thrombomodulin -33G/A and Ala455Val polymorphisms are associated with the risk of coronary artery disease: a meta-analysis including 12 584 patients. Coron Artery Dis. 2015;26(1):72-77. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Jin J, Tan S. Association of Thrombomodulin Gene Polymorphisms with Susceptibility to Atherosclerotic Diseases: A Meta-Analysis. Ann Hum Genet. 2016;80(3):172-181. [DOI] [PubMed] [Google Scholar]
- 10.Auro K, Komulainen K, Alanne M, et al. . Thrombomodulin gene polymorphisms and haplotypes and the risk of cardiovascular events: a prospective follow-up study. Arterioscler Thromb Vasc Biol. 2006;26(4):942-947. [DOI] [PubMed] [Google Scholar]
- 11.Sugiyama S, Hirota H, Kimura R, et al. . Haplotype of thrombomodulin gene associated with plasma thrombomodulin level and deep vein thrombosis in the Japanese population. Thromb Res. 2007;119(1):35-43. [DOI] [PubMed] [Google Scholar]
- 12.Le Flem L, Mennen L, Aubry ML, et al. . Thrombomodulin promoter mutations, venous thrombosis, and varicose veins. Arterioscler Thromb Vasc Biol. 2001;21(3):445-451. [DOI] [PubMed] [Google Scholar]
- 13.Heit JA, Petterson TM, Owen WG, Burke JP, DE Andrade M, Melton LJ III. Thrombomodulin gene polymorphisms or haplotypes as potential risk factors for venous thromboembolism: a population-based case-control study. J Thromb Haemost. 2005;3(4):710-717. [DOI] [PubMed] [Google Scholar]
- 14.Langdown J, Luddington RJ, Huntington JA, Baglin TP. A hereditary bleeding disorder resulting from a premature stop codon in thrombomodulin (p.Cys537Stop). Blood. 2014;124(12):1951-1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dargaud Y, Scoazec JY, Wielders SJH, et al. . Characterization of an autosomal dominant bleeding disorder caused by a thrombomodulin mutation. Blood. 2015;125(9):1497-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner RJ, Bloemenkamp KWM, Bruijn JA, Baelde HJ. Loss of thrombomodulin in placental dysfunction in preeclampsia. Arterioscler Thromb Vasc Biol. 2016;36(4):728-735. [DOI] [PubMed] [Google Scholar]
- 17.Healy AM, Rayburn HB, Rosenberg RD, Weiler H. Absence of the blood-clotting regulator thrombomodulin causes embryonic lethality in mice before development of a functional cardiovascular system. Proc Natl Acad Sci USA. 1995;92(3):850-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sood R, Zogg M, Westrick RJ, et al. . Fetal gene defects precipitate platelet-mediated pregnancy failure in factor V Leiden mothers. J Exp Med. 2007;204(5):1049-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Isermann B, Hendrickson SB, Hutley K, Wing M, Weiler H. Tissue-restricted expression of thrombomodulin in the placenta rescues thrombomodulin-deficient mice from early lethality and reveals a secondary developmental block. Development. 2001;128(6):827-838. [DOI] [PubMed] [Google Scholar]
- 20.Isermann B, Hendrickson SB, Zogg M, et al. . Endothelium-specific loss of murine thrombomodulin disrupts the protein C anticoagulant pathway and causes juvenile-onset thrombosis. J Clin Invest. 2001;108(4):537-546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiler H, Lindner V, Kerlin B, et al. . Characterization of a mouse model for thrombomodulin deficiency. Arterioscler Thromb Vasc Biol. 2001;21(9):1531-1537. [DOI] [PubMed] [Google Scholar]
- 22.Pathak R, Shao L, Ghosh SP, et al. . Thrombomodulin contributes to gamma tocotrienol-mediated lethality protection and hematopoietic cell recovery in irradiated mice. PLoS One. 2015;10(4):e0122511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geiger H, Pawar SA, Kerschen EJ, et al. . Pharmacological targeting of the thrombomodulin-activated protein C pathway mitigates radiation toxicity. Nat Med. 2012;18(7):1123-1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seibler J, Zevnik B, Küter-Luks B, et al. . Rapid generation of inducible mouse mutants. Nucleic Acids Res. 2003;31(4):e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tallquist MD, Soriano P. Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis. 2000;26(2):113-115. [DOI] [PubMed] [Google Scholar]
- 26.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593-605. [DOI] [PubMed] [Google Scholar]
- 27.Weiler-Guettler H, Aird WC, Husain M, Rayburn H, Rosenberg RD. Targeting of transgene expression to the vascular endothelium of mice by homologous recombination at the thrombomodulin locus. Circ Res. 1996;78(2):180-187. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Zheng X, Gu J, et al. . Overexpressing endothelial cell protein C receptor alters the hemostatic balance and protects mice from endotoxin. J Thromb Haemost. 2005;3(7):1351-1359. [DOI] [PubMed] [Google Scholar]
- 29.Kerlin B, Cooley BC, Isermann BH, et al. . Cause-effect relation between hyperfibrinogenemia and vascular disease. Blood. 2004;103(5):1728-1734. [DOI] [PubMed] [Google Scholar]
- 30.Fay WP, Parker AC, Ansari MN, Zheng X, Ginsburg D. Vitronectin inhibits the thrombotic response to arterial injury in mice. Blood. 1999;93(6):1825-1830. [PubMed] [Google Scholar]
- 31.Hemker HC, Giesen P, Al Dieri R, et al. . Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb. 2003;33(1):4-15. [DOI] [PubMed] [Google Scholar]
- 32.von Drygalski A, Furlan-Freguia C, Mosnier LO, Yegneswaran S, Ruf W, Griffin JH. Infrared fluorescence for vascular barrier breach in vivo--a novel method for quantitation of albumin efflux. Thromb Haemost. 2012;108(5):981-991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Isermann B, Vinnikov IA, Madhusudhan T, et al. . Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13(11):1349-1358. [DOI] [PubMed] [Google Scholar]
- 34.Taylor FB Jr, Wada H, Kinasewitz G. Description of compensated and uncompensated disseminated intravascular coagulation (DIC) responses (non-overt and overt DIC) in baboon models of intravenous and intraperitoneal Escherichia coli sepsis and in the human model of endotoxemia: toward a better definition of DIC. Crit Care Med. 2000;28(9 Suppl):S12-S19. [DOI] [PubMed] [Google Scholar]
- 35.Bick RL. Disseminated intravascular coagulation: a review of etiology, pathophysiology, diagnosis, and management: guidelines for care. Clin Appl Thromb Hemost. 2002;8(1):1-31. [DOI] [PubMed] [Google Scholar]
- 36.Deguchi H, Sinha RK, Marchese P, et al. . Prothrombotic skeletal muscle myosin directly enhances prothrombin activation by binding factors Xa and Va. Blood. 2016;128(14):1870-1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abbott BD, Buckalew AR. Placental defects in ARNT-knockout conceptus correlate with localized decreases in VEGF-R2, Ang-1, and Tie-2. Dev Dyn. 2000;219(4):526-538. [DOI] [PubMed] [Google Scholar]
- 38.Maruyama EO, Lin H, Chiu S-Y, Yu H-MI, Porter GA, Hsu W. Extraembryonic but not embryonic SUMO-specific protease 2 is required for heart development. Sci Rep. 2016;6:20999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garcia-Gonzalez MA, Outeda P, Zhou Q, et al. . Pkd1 and Pkd2 are required for normal placental development. PLoS One. 2010;5(9):e12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wenzel PL, Wu L, de Bruin A, et al. . Rb is critical in a mammalian tissue stem cell population. Genes Dev. 2007;21(1):85-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu L, de Bruin A, Saavedra HI, et al. . Extra-embryonic function of Rb is essential for embryonic development and viability. Nature. 2003;421(6926):942-947. [DOI] [PubMed] [Google Scholar]
- 42.Burton GJ, Fowden AL, Thornburg KL. Placental origins of chronic disease. Physiol Rev. 2016;96(4):1509-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kurosawa-Ohsawa K, Kimura M, Kume-Iwaki A, Tanaka T, Tanaka S, Anti-Protein C. Anti-protein C monoclonal antibody induces thrombus in mice. Blood. 1990;75(11):2156-2163. [PubMed] [Google Scholar]
- 44.Isermann B, Sood R, Pawlinski R, et al. . The thrombomodulin-protein C system is essential for the maintenance of pregnancy. Nat Med. 2003;9(3):331-337. [DOI] [PubMed] [Google Scholar]
- 45.Bleker SM, Coppens M, Middeldorp S. Sex, thrombosis and inherited thrombophilia. Blood Rev. 2014;28(3):123-133. [DOI] [PubMed] [Google Scholar]
- 46.Tchaikovski SN, van Vlijmen BJM, Cleuren AC, et al. . Pregnancy-associated changes in the hemostatic system in wild-type and factor V Leiden mice. J Thromb Haemost. 2009;7(2):312-318. [DOI] [PubMed] [Google Scholar]
- 47.Erez O, Mastrolia SA, Thachil J. Disseminated intravascular coagulation in pregnancy: insights in pathophysiology, diagnosis and management. Am J Obstet Gynecol. 2015;213(4):452-463. [DOI] [PubMed] [Google Scholar]