

Abstract
Matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) mass spectrometry (MS) imaging has been used for rapid phenotyping of enzymatic activities, but is mainly limited to single-step conversions. Herein we report a label-free method for high-throughput engineering of multistep biochemical reactions based on optically guided MALDI-ToF MS analysis of bacterial colonies. The bacterial cells provide containment of multiple enzymes and access to substrates and cofactors via metabolism. Automated MALDI-ToF MS acquisition from randomly distributed colonies simplifies procedures to prepare strain libraries without liquid handling. MALDI-ToF MS profiling was utilized to screen both substrate and enzyme libraries for natural product biosynthesis. Computational algorithms were developed to process and visualize the resulting mass spectral data sets. For analogues of the peptidic antibiotic plantazolicin, multivariate analyses by t-distributed stochastic neighbor embedding were used to group similar spectra for rapid identification of nonisobaric variants. After MALDI-ToF MS screening, follow-up analyses using high-resolution MS and tandem MS were readily performed on the same sample target. Separately, relative ion intensities of rhamnolipid congeners with various lipid moieties were evaluated to engineer enzymatic specificity. The glycolipid profiles of each colony were overlaid with optical images to facilitate the recovery of desirable mutants. For both the antibiotic and rhamnolipid cases, large populations of colonies were rapidly surveyed at the molecular level, providing information-rich insights not easily obtained with traditional screening assays. Utilizing standard microbiological techniques with routine microscopy and MALDI-ToF MS instruments, this simple yet effective workflow is applicable for a wide range of screening campaigns targeting multistep enzymatic reactions.
Introduction
Mass spectrometry (MS) has become an invaluable analytical technique, in part because it offers label-free analyses of target molecules. Matrix-assisted laser desorption/ionization time-of-flight (MALDI-ToF) MS is particularly well suited for the rapid inspection of a large number of biological samples because of its simple sample preparation, high salt tolerance, and wide coverage of diverse biomolecules.1,2 Accordingly, MALDI mass spectrometry imaging (MSI) has been increasingly applied for the rapid profiling of enzymatic reactions.3−8 However, MALDI MSI uses fixed raster steps for sampling, which requires high-density deposition of reaction components in a regular array.6−8 Although up to three enzymes with no cross-activities have been multiplexed in MSI screening,7,9−11 it is technically demanding to colocalize multiple enzymes, requiring advanced liquid handling, such as acoustic deposition,7 to increase the throughput. Compared with multiplexed enzyme assays, there are further challenges associated with screening multistep reactions, such as enzyme complex formation, temporal and stoichiometric control to avoid intermediate build-up, and the need to fine-tune reaction conditions to be compatible with multiple enzymes. This may explain the limited use of MSI-based screening of multistep biochemical reactions. To engineer a multistep reaction, modified intermediates must be accepted at each step of the catalytic sequence to obtain a final product. Engineering an individual step in isolation ignores possible downstream effects, and MSI screening platforms primarily designed for single-step enzymatic reactions may be ill-suited for engineering multistep pathways. Multistep biosynthesis is vital for the production of many important molecules, including fuels, fine chemicals, and pharmaceuticals,12,13 driving the need to develop improved methods to screen multistep enzymatic reactions.
In this work, we apply optically guided MALDI-ToF MS to engineer multistep enzymatic reactions via high-throughput, direct profiling of microbial colonies. Using microbial cells as reaction vessels, a set of enzymes are encoded as a single biosynthetic pathway on a DNA vector. Routine molecular biology enables mutagenesis, delivery, and expression of multiple enzymes encapsulated in a single cell. Similarly, living microbial colonies have been utilized to express a single heterologous enzyme for biocatalyst screening using desorption electrospray ionization (DESI) MS,14 which is an ambient pressure liquid extraction-ionization technique particularly suitable for real-time monitoring of living microbial cells.15−17 In addition, cell growth and metabolism in colonies facilitate analyte accumulation, which may help to eliminate the current requirement of analyte immobilization/capture for optimal sensitivity in MSI screening.8 MALDI MSI has been successfully utilized to study the spatial heterogeneity of microbial metabolism in biofilms,18−20 multispecies cocultures,21−24 and host–pathogen interactions.25 The approach has not been applied to screen libraries of microbial colonies, which are randomly distributed and widely spaced on agar media when prepared using standard techniques. MSI of such sparse objects is inefficient as most acquisitions occur on the spaces between colonies because of the fixed raster steps.14 As beneficial mutations are generally rare, a large number of mutants are often screened to isolate desirable variants.12,26,27 Advanced liquid handling systems may be used to deposit colonies into defined patterns for MSI screening,6−8 but they are costly and time-consuming. Our alternative approach utilizes microscopy images coupled with feature detection in image analyses to program the MALDI-ToF MS acquisition for rapid “colony picking” (Figure 1). The use of optically guided MS profiling for bacterial colonies is an extension of the methodology developed for single-cell MS analysis.28−31
Figure 1.
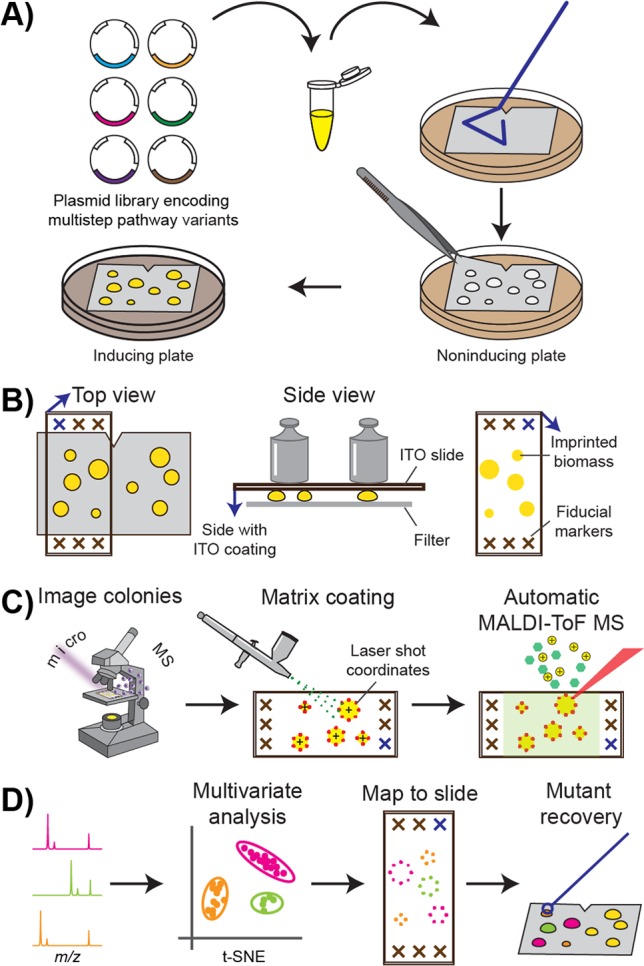
Optically guided MALDI-ToF MS screening. (A) Strain library preparation. (B) Imprinting of a colony biomass onto indium tin oxide (ITO)-coated glass slides (blue arrows indicate the coating side). (C) Generation of laser coordinates for automated MALDI-ToF MS profiling using machine vision on microscopic images. (D) Multivariate analysis and visualization of resulting mass spectra data sets.
Engineering multistep enzymatic pathways may modify the structures and/or quantities of the reaction products. These changes can be reflected in the mass spectra, such as mass shifts in nonisomerization reactions, or differences in the relative ion intensities of congeners as a result of altered enzymatic specificities. The mass spectra obtained from the microbial library screening provide a large, information-rich data set, which requires computational tools to extract, interpret, and visualize the most relevant signals to aid mutant recovery. The diverse molecular profiles can be surveyed with targeted, multivariate clustering if the molecular weight information on the desirable products is available. Alternatively, for discovery efforts, nontargeted clustering can group colonies exhibiting similar spectra without a priori knowledge. For researchers with limited MS experience, it is desirable to be able to visualize the screening data in a manner similar to classical, colorimetric assays. Given the wide application of multistep enzymatic reactions, data analysis pipelines tailored for diverse engineering objectives are needed.
Research on natural products (NPs) advances fundamental biochemistry and provides a valuable source of chemical matter for drug discovery.32,33 Many NP molecules synthesized via secondary metabolism contain complex chemical modifications created via a number of enzymatic reactions. NP analogues are widely applied in mode-of-action, substrate tolerance, and structure–activity relationship studies.34 NP variants are also engineered to develop compounds with improved medicinal properties.34 However, the lack of high-throughput methods for structure-based screening remains a main bottleneck in this research area.34,35
Here, we utilized MALDI-ToF MS profiling of microbial colonies for rapid screening of NP analogue libraries. We first characterized the substrate tolerance of a five-enzyme pathway synthesizing the antibiotic plantazolicin (PZN) from a ribosomal precursor peptide. We then applied MALDI-ToF MS screening in directed protein evolution to alter the congener compositions of rhamnolipids (RLs) produced from a two-enzyme pathway. Custom sampling and analysis algorithms were developed for each system, with the former focusing on the structural variations of analogues and the latter on the relative abundance of target congeners. We demonstrated the successful application of optically guided MALDI-ToF MS profiling in both examples, resulting in the discovery of new compounds and isolation of enzymes with the desirable chemical selectivity.
Results
Workflow Development
The workflow for high-throughput MALDI-based characterization of bacterial colonies consists of the creation of a strain library, optically guided MALDI-ToF MS profiling, and data analyses and visualization (Figure 1). Recombinant variants of a multienzymatic pathway are constructed as plasmid DNA libraries, which are used to transform a production host such as Escherichia coli (Figure 1A). The transformants are plated on a filter membrane for straightforward manipulation of many colonies simultaneously,36 such as exchanging culture media or imprinting onto MALDI targets. Microbial cells are initially cultivated on noninducing agar media to obtain individual colonies and then transferred onto inducing plates to initiate enzyme expression and target molecule production (Figure 1A). Each clonal population contains a single variant of the multistep pathway. Separation of the induction phase from the growth phase allows accumulation of sufficient biomass before cellular resources are diverted to target enzyme production, and minimizes uneven growth among mutant colonies if the final products affect cellular fitness.
For MALDI-ToF MS analyses, colonies are imprinted on conductive, indium tin oxide (ITO)-coated glass slides (Figure 1B). Relative to agar bacterial cultures mounted directly onto a MALDI target, profiling from imprinted biomass on a uniform target surface obtains better ion signal for cell-associated compounds.37 The use of transparent MALDI targets allows acquisition of optical images prior to matrix application using an artist’s airbrush (Figure 1C). To aid automated colony finding, whole-slide images are acquired to locate the etched fiducials and imprinted colonies in bright-field and autofluorescence images (Figure 1C). At least 16 fiducial markers were etched directly on the ITO-coated slide in the area surrounding the imprint region (Figure 1B) using a diamond-tipped pen. A target accuracy of ∼20 μm during MALDI-ToF MS profiling can be achieved following fiducial training.38 A Python platform for image-guided MS analyses, microMS,38 is used to generate MALDI laser coordinates for automatic colony profiling (Figure 1C). Laser shots are patterned around the peripheries of the imprinted colonies for optimal sensitivity, as described below.
The resulting mass spectra are processed using multivariate statistical analyses. For a strain library producing analogues of a target compound, t-distributed stochastic neighbor embedding (t-SNE)39 was utilized to visualize spectral similarity and identify nonisobaric variants (Figure 1D). t-SNE is a dimensionality reduction method, similar to principle component analysis (PCA), for the visualization of high-dimensional data sets.39 While PCA is commonly utilized to separate dissimilar data within a low-dimensional map, t-SNE also keeps similar data close together. As such, we found clearer grouping of spectra within a t-SNE plot than a PCA score plot. The observed grouping is advantageous for spectral data with multiple classes, such as those acquired from a compound analogue library. Alternatively, when screening a strain library producing the same set of products at different ratios, ion intensities at select m/z values are extracted to calculate the total and relative abundances.
In both cases, high-dimensional data are reduced to low-dimensional variates to reflect a target phenotype, such as spectral classifications or congener ratios. The reduced data sets are then visualized by overlaying optical images with false color markers to produce straightforward readouts similar to colorimetric assays (Figure 1D). Notably, as m/z values in the MALDI-ToF mass spectra are utilized in the computational analyses to link mass spectral signals to target compounds, analyte assignments are further confirmed using high-resolution MS and tandem MS (MS/MS). These analyses can be performed subsequent to high-throughput profiling, as more than 60% of the analytes remain on the target even after exhaustive MALDI-ToF MS measurements.40 This two-tiered approach allows rapid survey of the whole library, which helps to identify colonies of interest for slower but more informative MS analyses. Finally, select colonies are recovered for further characterization, such as DNA sequencing or liquid fermentation (Figure 1D).
Substrate Libraries of a Peptidic NP
We first applied the workflow to survey the structural diversity of PZN 1 analogues, created using a substrate library. Ribosomally synthesized and post-translationally modified peptides (RiPPs) form a major class of NPs.41,42 As the product is synthesized from a ribosomal peptide, product variants can be generated by mutagenesis of the precursor gene.43,44 PZN 1 is a member of one RiPP subclass termed the linear azol(in)e-containing peptides. During biosynthesis of this subclass, a trimeric heterocycle synthetase converts select cysteine (C), serine (S), and threonine (T) residues in the C-terminal (core) region of the precursor peptide to thiazol(in)e and (methyl)oxazol(in)e heterocycles (Scheme S1). PZN 1 is naturally produced by Bacillus velezensis FZB4245 and exhibits remarkable antibacterial selectivity against Bacillus anthracis,(46) the causative agent of anthrax. We previously achieved heterologous production of PZN 1 in E. coli using a fosmid bearing the corresponding biosynthetic gene cluster.47 Analogues of PZN 1 were also created by site-directed mutagenesis of the precursor peptide gene, followed by liquid cultivation and methanol extraction before MS analyses.47 For successful synthesis of an analogue of PZN 1, a mutant precursor peptide must be accepted as a substrate by multiple enzymes of the biosynthetic pathway, including the cyclodehydratase, dehydrogenase, leader peptidase, and methyltransferase (Scheme S1).47
To apply optically guided MALDI-ToF MS screening to E. coli colonies producing PZN 1 analogues, we targeted two noncyclized positions, I7 and I8 (Scheme 1, red; the numbering for original core residues is given in Scheme S1), where mutations are relatively tolerated by the native biosynthetic machinery.47 Site-saturation mutagenesis was performed using degenerate codon (NNK)-containing primers. Polyclonal plasmid DNA was used to transform competent E. coli cells harboring a refactored version of the PZN cluster, where native Bacillus promoters were replaced with a strong T7 promoter to enhance production. Isopropyl β-d-1-thiogalactopyranoside (IPTG) was used to induce production of PZN 1 on M9 medium containing acetate as the sole carbon source.
Scheme 1. Structure of PZN 1.

For I7 and I8 libraries, 352 and 393 colonies were screened, respectively, achieving a > 99.9% probability of full coverage of the NNK libraries.48 Following the analysis workflow, we first performed t-SNE analyses for unsupervised clustering of the resulting mass spectra, utilizing each binned m/z value to evaluate population heterogeneity and variance. We manually examined each spectral class for tentative PZN peaks, and found all the base peaks (Figure S1) were consistent with single-residual-mutation analogues with “wild-type-like” modifications: nine azole rings, one azoline ring, leader peptidolysis N-terminal to arginine R1, and N-terminal dimethylation (Scheme 1).47 A targeted clustering (Figure 2) was then performed using the predicted m/z values of these analogues (see SI “Multivariate data analysis” for details) (Figure S1).
Figure 2.
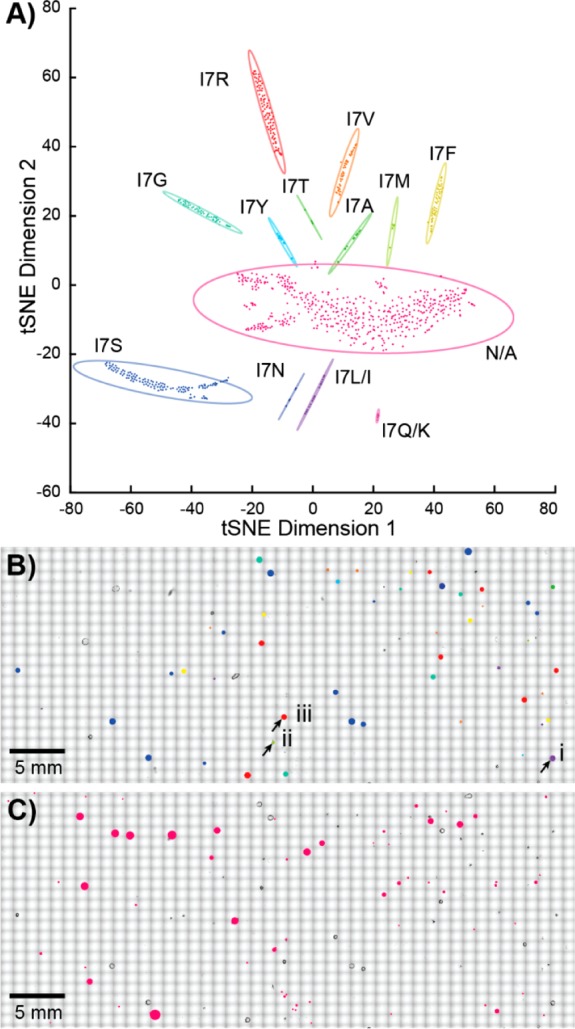
Multivariate analysis of PZN analogues. (A) Visualization with targeted t-SNE clustering of the I7 library from a single experiment. Each point corresponds to a single mass spectrum, with each cluster surrounded by a 95% confidence ellipsoid. The N/A cluster contains spectra without observable peptide signals. The position of (B) each mutant or (C) N/A colony is mapped onto the optical image to aid mutant recovery. The three colonies highlighted in panel B are displayed in more detail in Figure 3.
From the MALDI-ToF MS data alone, we observed 12 and 9 variant classes of PZN 1 for the libraries of I7 (Figure 2) and I8 (Figure S1), respectively. However, further analyses are required to differentiate the single-residual mutations between glutamine (Q) and lysine (K), as well as between leucine (L) and isoleucine (I). It is challenging to distinguish the 0.036 Da mass difference between the Q and K mutations, as an m/z tolerance of ±0.25 Da was employed to assign residue substitutions of PZN analogues from the MALDI-ToF MS data. Such mass accuracy results from the limited mass resolution of a ToF mass analyzer (∼10 000 in a m/z window of 1100–1600 for PZN molecules), and uneven sample heights of imprinted biomass that affect the time-of-flight of target ions and reduce mass accuracy. As noted above, after MALDI-ToF MS profiling, enough of the analytes remain on the targets,40 allowing follow-up analyses. High-resolution MALDI-Fourier transform ion cyclotron resonance (FT-ICR) MS analyses were performed on selected colonies of interest, and unambiguously differentiated between Q and K mutations, revealing both K and Q substitutions at I7, but only Q at I8 (Figure S1). The high-resolution MS data also corroborated the predicted molecular formulas of other PZN analogues with <1 ppm mass errors (Figure S1)
On the other hand, as the PZN analogues with I and L substitutions are isomers with no mass differences, DNA sequencing is necessary to differentiate these two mutants (Figure S2). Colonies belonging to each class were inoculated in liquid cultures for plasmid isolation and DNA sequencing. Each colony sequenced presented mutations consistent with predicted and observed mass shifts in the base peaks assuming full maturation (Figure S1). In this study, all 13 previously isolated PZN analogues with single residue mutations at I7 or I8 were detected,47 as well as 10 previously unreported variants (Figure S1). Select analogues with sufficient residual analytes on the sample target were subjected to in situ ion identification with MS/MS (Figures S2–S5). The tandem mass spectra suggested “wild-type-like” modifications (Scheme 1) for all examined base peaks (Figures S2–S5). The detection of unreported PZN analogues in this study validates the improved methodology. Production of PZN 1 was increased through pathway refactoring and growth medium optimization, enabling observation of PZN variants not previously detected due to insufficient amounts. Also, enhanced production allowed detection of PZN 1 analogues directly from single colonies. This improvement was leveraged by optically guided MALDI-ToF MS to substantially increase analytical throughput, allowing more comprehensive codons (NNK versus NNC47) for mutagenesis while retaining high probabilities of full library coverage.
In addition to spectral classes containing base peaks matching predicted m/z values of PZN analogues, a class exhibiting a low signal-to-noise ratio or chemical background was also observed (Figure 2). This spectral class likely resulted either from (1) mutations not tolerated by the biosynthetic machinery or reducing analogue production below our detection limit, (2) the UAG stop codon contained in the degenerate NNK codon, (3) artifacts during optical image acquisition such as dust, and/or (4) problems targeting irregularly shaped colony imprints (Figure 3B). The first two possibilities were not further investigated given the consistency between current and previous results.47 The latter two can be alleviated by more vigilant colony finding and target patterning. In particular, we found the best sensitivity was obtained when directing the MALDI laser to the peripheries of the imprinted biomass (Figure 3). Direct sampling on the imprinted biomass often yielded mass spectra with a low signal-to-noise ratio (Figure 3B), possibly due to insufficient matrix-to-analyte mixing and nonideal matrix crystallization,37,49 poor laser focus, or inefficient ion transfer from the elevated sample heights.
Figure 3.
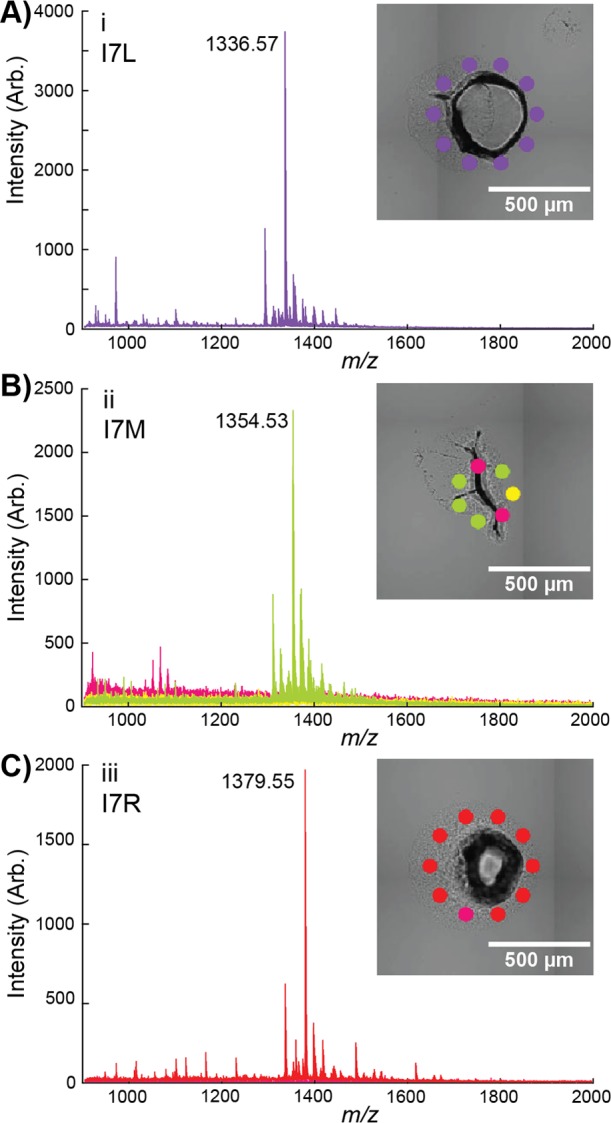
Detailed view of annotated colonies (i, ii and iii) from Figure 2 shown in panels (A–C) respectively. The color of each cluster corresponds to the t-SNE plot in Figure 2. Average spectra of each cluster for the colony are displayed with the base peak labeled. Mutations were confirmed by DNA sequencing (Figure S1).
Enzyme Libraries in Biosynthesis of RL 5
Next, we sought to quantify changes in the relative abundance of the reaction products (RLs 5a–d) due to modified enzymatic specificities. We focused on a two-step biochemical pathway for RL synthesis (Scheme 2). Initially discovered from Pseudomonas aeruginosa,50 RLs are a class of biosurfactants that have been extensively studied for their potential application in enhanced oil recovery, biodegradation, and bioremediation.51,52 To form monorhamnolipids (mono-RLs, RL 5), RhlB (rhamnosyltransferase 1 chain B) catalyzes the condensation of 3 and 4. Different variants of 3 are synthesized by RhlA (rhamnosyltransferase 1 chain A) using 2 of varying chain lengths and degrees of unsaturation,53 contributing to the structural diversity of RL lipid moieties in nature.54
Scheme 2. Biosynthesis of RL 5.
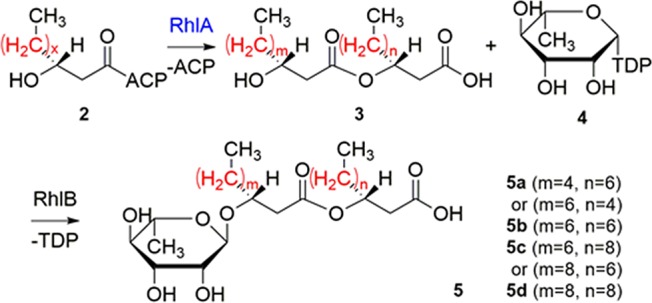
The most abundant RL species produced by P. aeruginosa and other bacteria consist of β-hydroxydecanoyl-β-hydroxydecanoate as the fatty acyl moiety, as in RL 5b, which is attributed to the role of RhlA as a “molecular ruler” with a high preference toward β-hydroxydecanoyl-ACP (2, x = 6) in vitro.53 As fatty acyl chain lengths affect the physiochemical and biological properties of RLs,50,55 it is desirable to produce RLs with custom congener compositions for specific applications. Previous screening assays relied on the link between antimicrobial activity and RL mixture composition, and suffered from low chemical specificity.55 Here we sought to directly measure the relative abundance of different RL congeners using MALDI-ToF MS in high throughput.
We first examined the composition of the mono-RL 5 mixtures produced from recombinant E. coli colonies using optically guided MALDI-ToF MS profiling. Heterologous production of mono-RL 5 in E. coli was achieved by coexpression of the wild-type rhlA and rhlB genes of P. aeruginosa, as previously reported56 (denoted as WT). Following the screening workflow (Figure 1), eight peaks in the MALDI mass spectra were tentatively assigned as Na+ and K+ adduct ions of RL 5a–d, based on mass matching (Figure S6A) and comparison of MS/MS results with previous reports18,19 (Figure S6B). We utilized the ion intensities of the RL 5a–d peaks in the MALDI mass spectra to estimate relative congener abundance, and the results provided a good approximation of the RL 5b percentile when compared with the MALDI-ToF MS or liquid chromatography (LC)–MS/MS quantification of organic solvent extracts from ∼100 WT colonies (Figure S7, white bars). For the percentiles of RL 5a, 5c and 5d, however, different results were observed among the MALDI MS of colonies, MALDI MS of extracts, and LC–MS of extracts (Figure S7). Such discrepancies may result from two sources. The variations between the MALDI MS and LC–MS results of the same extract (Figure S7) suggest that congener ionization efficiency may vary in different mass analyzers. Also, the relative transfer efficiency of RL 5a–d during imprinting may differ from that during solvent extraction. Therefore, we devised a two-step screening strategy, where MALDI-ToF MS screening served as the first approximation of relative congener content in high throughput, followed by LC–MS quantification on liquid cultures of identified mutant strains without solvent extraction. Liquid cultures were utilized as a confirmation as they are more similar to industrial fermentation conditions than colonies on agar media.
We then applied directed protein evolution to RhlA to engineer the relative abundance of RL 5a–d in mono-RL production. Random mutations were introduced in the WT rhlA gene using error-prone polymerase chain reaction (PCR).57,58 The PCR product was inserted into a plasmid harboring a WT rhlB gene, and the resulting DNA library was used to transform the E. coli cells. Following the MALDI-ToF MS screening workflow (Figure 1), the resulting data sets were visualized by overlaying the optical image with a bubble chart to provide a rapid assessment of the variance in relative abundance and total production of RL molecules (Figure 4). Each circle has a radius determined by the log-base 10 intensity of the sum of all RL peaks. The color is determined by the relative abundance of RL 5b. Compared with the WT strain (Figure 4A), the strain library in the first round of mutagenesis (denoted as R1) exhibited increased diversity in terms of both the total intensities of RL ions and relative percentiles of RL 5b (Figure 4B). These results agree with the description of RhlA as the “molecular ruler” of RL lipid moiety synthesis.53 From R1 (3085 colonies were screened), variant strains producing RL 5b at larger fractions relative to WT were recovered with the visual aid of bubble charts (red circles). After plasmid isolation and retransformation into a fresh strain background, two mutant strains (R1#6 and R1#15) were confirmed to produce significantly larger proportions of RL 5b than WT in liquid cultures (Figure 5). R1#6 and R1#15 harbor a single amino acid mutation of V10I and A64V, respectively (Table S1).
Figure 4.
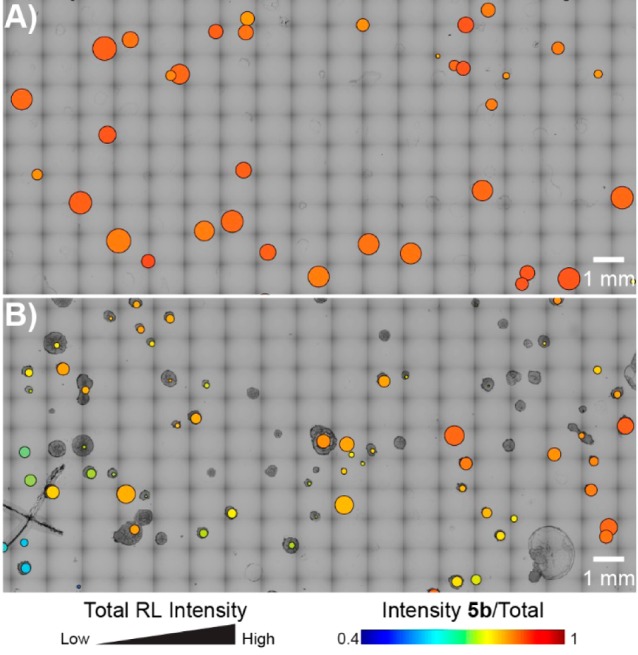
Visualization of MALDI-ToF MS screening results of mono-RL-producing E. coli colonies. (A) WT. (B) Strain library in the first round of mutagenesis (R1). For each circle overlaying the corresponding colony, the radius scales with the log-base 10 intensity of the sum of all RL peaks, and the false color scales with the relative abundance of RL 5b. Only a small, representative region is shown.
Figure 5.
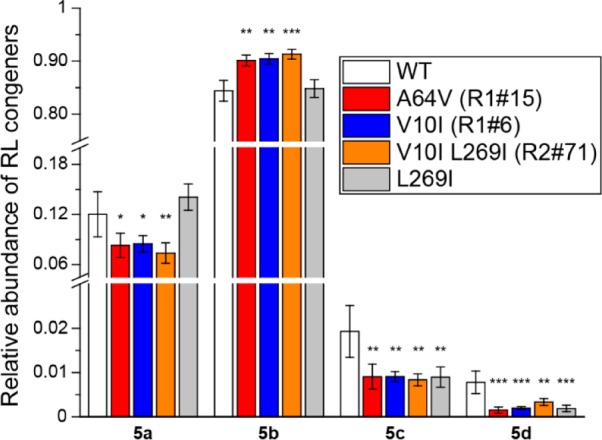
Comparison of RL production in liquid cultures between WT and isolated mutant strains quantified using LC–MS/MS in multiple reaction monitoring mode. Error bars indicate the standard deviations of biological triplicates. Significant differences were determined between WT and mutants using an independent two-tailed, two-sample t-test for equal sample sizes and equal variance. Significance levels: * p < 0.05; ** p < 0.01; *** p < 0.001.
The mutated rhlA gene from the R1#6 strain was subjected to another round of mutagenesis to further increase the relative abundance of RL 5b. However, the majority of the recovered strains from the second round of screening (R2, 3649 colonies were screened) were found to contain no additional mutations relative to the parent R1#6. To understand the challenge in isolating mutants with larger RL 5b portions, RL production of WT, R1, and R2 was compared in the same batch, and the relative abundance of RL 5b on the population level was observed in the sequence of R2 < R1 < WT (Figure S8). These observations suggest that as the WT RhlA exhibits relatively high selectivity,53 most random mutations tend to decrease rather than enhance its catalytic specificity.26 After retransformation, one mutant strain (R2#71) was isolated bearing a single amino acid mutation (L269I, Table S1) that reduced the proportions of RL 5c and RL 5d in liquid cultures compared with WT, but failed to further enhance the relative abundance of RL 5b relative to the parent R1#6 (Figure 5). Future investigation is needed to elucidate the mechanisms on why selected mutations confer distinct observed phenotypes.
Currently, ∼1000 colonies can be screened per slide with an MS sampling rate of 1–2.5 s (2000–5000 MALDI laser shots) per colony. With regard to sample preparation, the total time and cost to analyze one colony takes approximately 5 s and $0.0065 using the optically guided approach, compared with 15 s and $0.86 using a liquid-handing robot (see SI “Comparison with liquid-handling approaches” for details). This throughput is sufficient for screening variant libraries of small sizes (<104), such as those created in this study via site-saturation mutagenesis at a single residue or error-prone PCR at a low mutation rate. But the approach becomes limiting when screening larger populations. The throughput may be further improved by several approaches. First, it currently takes ∼1 h to acquire both bright-field and fluorescence images of a 25 mm × 75 mm ITO-coated glass target using a 10× microscopic objective lens. With more advanced machine vision, a single bright-field image at lower resolution or even a cell phone image could be utilized for both fiducial and colony finding, potentially reducing total analysis time to <2 s per colony. Second, the current upper limit of colony density (2000–3000 per 100 mm Petri dish analyzed on two ITO slides) is largely to ensure reliable picking of individual clones.59 To further improve the throughput, it is necessary to plate variant strains at higher densities, shorten growth time to minimize overlapping clones, and utilize robotic mutant recovery of smaller colonies.
In the current workflow, colonies are transferred onto MALDI targets using imprinting, which is suitable for detection of cell-associated compounds. As organic solvents are used for airbrush spray coating of matrix solutions, intracellular molecules are extracted, incorporated into matrix crystals, and detected together with secreted molecules. However, the sample preparation procedure can be modified and optimized to specific target analyte classes. For example, matrix application by sublimation offers a solvent-free preparation to minimize detection of intracellular metabolites. Alternatively, the geometry of MALDI laser shots is easily modified to examine the interior regions of imprinted biomass to enhance detection of intracellular metabolites (provided there is sufficient solvent extraction and matrix crystallization on top of the biomass). Finally, colonies grown on thin agar may be mounted directly on a MALDI target to acquire mass spectra from areas surrounding colonies to improve detection of secreted compounds.49
Conclusions
We have developed an integrated workflow for high-throughput screening of multistep enzymatic reactions in bacterial colonies. Traditional screening methods are either limited to photometrically active molecules and labeled surrogates, or require low-throughput chromatographic separation.12 MS provides a label-free, highly sensitive platform for monitoring products, reactants, and byproducts with high accuracy. Incorporating machine vision and automatic target patterning greatly improves MS acquisition efficiency over traditional MSI assays, especially for randomly distributed colonies. The resulting mass spectra data sets may be subjected to multivariate clustering or reduced into univariate plots to quickly assess and select mutants with desirable phenotypes. Moreover, the capacity to rapidly survey the molecular contents of a whole library provides new biological insights, such as the overall substrate tolerance of a biosynthetic pathway and the trade-off between phenotypic gain and further evolvability during the course of directed protein evolution. Additional improvements may be achieved through increased throughput, automatic imprinting/matrix coating to enhance sample uniformity, and derivatization of analytes with poor native MALDI-ToF MS sensitivity. The optically guided approach may also be incorporated when profiling randomly distributed colonies using other surface analysis approaches, such as DESI,15−17 which provide complementary molecular coverage and analytical capabilities relative to MALDI-ToF MS, as in real-time monitoring of living cells. Given its simplicity and effectiveness, this workflow should be applicable to a wide range of multistep enzyme reactions and facilitate high-throughput screening of microbial libraries.
Acknowledgments
We gratefully acknowledge financial support from the National Institutes of Health (GM077596 to H.Z., AI113219 to J.V.S., and GM097142 to D.A.M.). J.V.S. also acknowledges NSF CHE 16-067915. T.S. acknowledges postdoctoral fellowship support from the Carl R. Woese Institute for Genomic Biology (UIUC). T.J.C. acknowledges support from an NSF Graduate Research Fellowship Program, the Springborn Fellowship, and the Training Program at the Chemistry-Biology Interface (T32 GM070421). We thank Prof. Joshua D. Shrout at the University of Notre Dame for kindly providing P. aeruginosa genomic DNA. We thank Elizabeth Neumann for the help with microscopy and FT-ICR analyses, Sage J.B. Dunham for initiating the rhamnolipid engineering project, and Adam J. DiCaprio with the construction of PZN analogue libraries. We also thank Dr. Benjamin Bowen at the Lawrence Berkeley National Lab for the help with mass spectrum deposition at OpenMSI.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b04641.
Experimental details on the DNA and strain construction, the acquisition, processing and visualization of MALDI-ToF MS screening data sets, Table S1, Scheme S1, and Figures S1–S8 (PDF)
Author Contributions
¶ T.S., B.L., and T.J.C. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Bothner B.; Chavez R.; Wei J.; Strupp C.; Phung Q.; Schneemann A.; Siuzdak G. J. Biol. Chem. 2000, 275, 13455–13459. 10.1074/jbc.275.18.13455. [DOI] [PubMed] [Google Scholar]
- Greis K. D. Mass Spectrom. Rev. 2007, 26, 324–339. 10.1002/mas.20127. [DOI] [PubMed] [Google Scholar]
- Northen T. R.; Lee J.-C.; Hoang L.; Raymond J.; Hwang D.-R.; Yannone S. M.; Wong C.-H.; Siuzdak G. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 3678–3683. 10.1073/pnas.0712332105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban L.; Pettit N.; Li L.; Stuparu A. D.; Cai L.; Chen W.; Guan W.; Han W.; Wang P. G.; Mrksich M. Nat. Chem. Biol. 2012, 8, 769–773. 10.1038/nchembio.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heins R. A.; Cheng X.; Nath S.; Deng K.; Bowen B. P.; Chivian D. C.; Datta S.; Friedland G. D.; D’Haeseleer P.; Wu D.; Tran-Gyamfi M.; Scullin C. S.; Singh S.; Shi W.; Hamilton M. G.; Bendall M. L.; Sczyrba A.; Thompson J.; Feldman T.; Guenther J. M.; Gladden J. M.; Cheng J. F.; Adams P. D.; Rubin E. M.; Simmons B. A.; Sale K. L.; Northen T. R.; Deutsch S. ACS Chem. Biol. 2014, 9, 2082–2091. 10.1021/cb500244v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurard-Levin Z. A.; Scholle M. D.; Eisenberg A. H.; Mrksich M. ACS Comb. Sci. 2011, 13, 347–350. 10.1021/co2000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greving M.; Cheng X.; Reindl W.; Bowen B.; Deng K.; Louie K.; Nyman M.; Cohen J.; Singh A.; Simmons B.; Adams P.; Siuzdak G.; Northen T. Anal. Bioanal. Chem. 2012, 403, 707–711. 10.1007/s00216-012-5908-8. [DOI] [PubMed] [Google Scholar]
- de Rond T.; Danielewicz M.; Northen T. Curr. Opin. Biotechnol. 2015, 31, 1–9. 10.1016/j.copbio.2014.07.008. [DOI] [PubMed] [Google Scholar]
- Smith A. M. E.; Brennan J. D. ChemBioChem 2014, 15, 587–594. 10.1002/cbic.201300739. [DOI] [PubMed] [Google Scholar]
- Rathore R.; Pribil P.; Corr J. J.; Seibel W. L.; Evdokimov A.; Greis K. D. J. Biomol. Screening 2010, 15, 1001–1007. 10.1177/1087057110363824. [DOI] [PubMed] [Google Scholar]
- Reindl W.; Deng K.; Gladden J. M.; Cheng G.; Wong A.; Singer S. W.; Singh S.; Lee J. C.; Yao C. H.; Hazen T. C.; Singh A. K.; Simmons B. A.; Adams P. D.; Northen T. R. Energy Environ. Sci. 2011, 4, 2884–2893. 10.1039/c1ee01112j. [DOI] [Google Scholar]
- Dietrich J. A.; McKee A. E.; Keasling J. D. Annu. Rev. Biochem. 2010, 79, 563–590. 10.1146/annurev-biochem-062608-095938. [DOI] [PubMed] [Google Scholar]
- Du J.; Shao Z.; Zhao H. J. Ind. Microbiol. Biotechnol. 2011, 38, 873–890. 10.1007/s10295-011-0970-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.; Parmeggiani F.; Jones E. A.; Claude E.; Hussain S. A.; Turner N. J.; Flitsch S. L.; Barran P. E. J. Am. Chem. Soc. 2017, 139, 1408–1411. 10.1021/jacs.6b12165. [DOI] [PubMed] [Google Scholar]
- Song Y.; Talaty N.; Tao W. A.; Pan Z.; Cooks R. G. Chem. Commun. (Cambridge, U. K.) 2007, 61–63. 10.1039/B615724F. [DOI] [PubMed] [Google Scholar]
- Song Y.; Talaty N.; Datsenko K.; Wanner B. L.; Cooks R. G. Analyst 2009, 134, 838–841. 10.1039/b900069k. [DOI] [PubMed] [Google Scholar]
- Roach P. J.; Laskin J.; Laskin A. Analyst 2010, 135, 2233–2236. 10.1039/c0an00312c. [DOI] [PubMed] [Google Scholar]
- Masyuko R. N.; Lanni E. J.; Driscoll C. M.; Shrout J. D.; Sweedler J. V.; Bohn P. W. Analyst 2014, 139, 5700–5708. 10.1039/C4AN00435C. [DOI] [PubMed] [Google Scholar]
- Lanni E. J.; Masyuko R. N.; Driscoll C. M.; Aerts J. T.; Shrout J. D.; Bohn P. W.; Sweedler J. V. Anal. Chem. 2014, 86, 9139–9145. 10.1021/ac5020222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si T.; Li B.; Zhang K.; Xu Y.; Zhao H.; Sweedler J. V. J. Proteome Res. 2016, 15, 1955–1962. 10.1021/acs.jproteome.6b00127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. L.; Xu Y.; Straight P.; Dorrestein P. C. Nat. Chem. Biol. 2009, 5, 885–887. 10.1038/nchembio.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham S. J.; Ellis J. F.; Li B.; Sweedler J. V. Acc. Chem. Res. 2017, 50, 96–104. 10.1021/acs.accounts.6b00503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watrous J. D.; Dorrestein P. C. Nat. Rev. Microbiol. 2011, 9, 683–694. 10.1038/nrmicro2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakeman C. A.; Moore J. L.; Noto M. J.; Zhang Y.; Singleton M. D.; Prentice B. M.; Gilston B. A.; Doster R. S.; Gaddy J. A.; Chazin W. J.; Caprioli R. M.; Skaar E. P. Nat. Commun. 2016, 7, 11951. 10.1038/ncomms11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zackular J. P.; Moore J. L.; Jordan A. T.; Juttukonda L. J.; Noto M. J.; Nicholson M. R.; Crews J. D.; Semler M. W.; Zhang Y.; Ware L. B.; Washington M. K.; Chazin W. J.; Caprioli R. M.; Skaar E. P. Nat. Med. 2016, 22, 1330–1334. 10.1038/nm.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero P. A.; Arnold F. H. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. Nat. Rev. Drug Discovery 2011, 10, 188–195. 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- Ong T. H.; Kissick D. J.; Jansson E. T.; Comi T. J.; Romanova E. V.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2015, 87, 7036–7042. 10.1021/acs.analchem.5b01557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansson E. T.; Comi T. J.; Rubakhin S. S.; Sweedler J. V. ACS Chem. Biol. 2016, 11, 2588–2595. 10.1021/acschembio.6b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do T. D.; Comi T. J.; Dunham S. J.; Rubakhin S. S.; Sweedler J. V. Anal. Chem. 2017, 89, 3078–3086. 10.1021/acs.analchem.6b04819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi T. J.; Do T. D.; Rubakhin S. S.; Sweedler J. V. J. Am. Chem. Soc. 2017, 139, 3920–3929. 10.1021/jacs.6b12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues T.; Reker D.; Schneider P.; Schneider G. Nat. Chem. 2016, 8, 531–541. 10.1038/nchem.2479. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. J. Nat. Prod. 2012, 75, 311–335. 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E.; Moore B. S.; Yoon Y. J. Nat. Chem. Biol. 2015, 11, 649–659. 10.1038/nchembio.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans B. S.; Chen Y.; Metcalf W. W.; Zhao H.; Kelleher N. L. Chem. Biol. 2011, 18, 601–607. 10.1016/j.chembiol.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornvik T.; Dahlroth S. L.; Magnusdottir A.; Herman M. D.; Knaust R.; Ekberg M.; Nordlund P. Nat. Methods 2005, 2, 507–509. 10.1038/nmeth767. [DOI] [PubMed] [Google Scholar]
- Watrous J.; Hendricks N.; Meehan M.; Dorrestein P. C. Anal. Chem. 2010, 82, 1598–1600. 10.1021/ac9027388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi T. J.; Neumann E. K.; Do T. D.; Sweedler J. V. J. Am. Soc. Mass Spectrom. 2017, 28, 1919. 10.1007/s13361-017-1704-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L.; Hinton G. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Page J. S.; Sweedler J. V. Anal. Chem. 2002, 74, 6200–6204. 10.1021/ac025898k. [DOI] [PubMed] [Google Scholar]
- Skinnider M. A.; Johnston C. W.; Edgar R. E.; Dejong C. A.; Merwin N. J.; Rees P. N.; Magarvey N. A. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E6343–E6351. 10.1073/pnas.1609014113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnison P. G.; Bibb M. J.; Bierbaum G.; Bowers A. A.; Bugni T. S.; Bulaj G.; Camarero J. A.; Campopiano D. J.; Challis G. L.; Clardy J.; Cotter P. D.; Craik D. J.; Dawson M.; Dittmann E.; Donadio S.; Dorrestein P. C.; Entian K. D.; Fischbach M. A.; Garavelli J. S.; Goransson U.; Gruber C. W.; Haft D. H.; Hemscheidt T. K.; Hertweck C.; Hill C.; Horswill A. R.; Jaspars M.; Kelly W. L.; Klinman J. P.; Kuipers O. P.; Link A. J.; Liu W.; Marahiel M. A.; Mitchell D. A.; Moll G. N.; Moore B. S.; Muller R.; Nair S. K.; Nes I. F.; Norris G. E.; Olivera B. M.; Onaka H.; Patchett M. L.; Piel J.; Reaney M. J.; Rebuffat S.; Ross R. P.; Sahl H. G.; Schmidt E. W.; Selsted M. E.; Severinov K.; Shen B.; Sivonen K.; Smith L.; Stein T.; Sussmuth R. D.; Tagg J. R.; Tang G. L.; Truman A. W.; Vederas J. C.; Walsh C. T.; Walton J. D.; Wenzel S. C.; Willey J. M.; van der Donk W. A. Nat. Prod. Rep. 2013, 30, 108–160. 10.1039/C2NP20085F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young T. S.; Dorrestein P. C.; Walsh C. T. Chem. Biol. 2012, 19, 1600–1610. 10.1016/j.chembiol.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffner D. E.; Schmidt E. W.; Heemstra J. R. ACS Synth. Biol. 2015, 4, 482–492. 10.1021/sb500267d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz R.; Molohon K. J.; Nachtigall J.; Vater J.; Markley A. L.; Sussmuth R. D.; Mitchell D. A.; Borriss R. J. Bacteriol. 2011, 193, 215–224. 10.1128/JB.00784-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molohon K. J.; Blair P. M.; Park S.; Doroghazi J. R.; Maxson T.; Hershfield J. R.; Flatt K. M.; Schroeder N. E.; Ha T.; Mitchell D. A. ACS Infect. Dis. 2016, 2, 207–220. 10.1021/acsinfecdis.5b00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deane C. D.; Melby J. O.; Molohon K. J.; Susarrey A. R.; Mitchell D. A. ACS Chem. Biol. 2013, 8, 1998–2008. 10.1021/cb4003392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nov Y. Appl. Environ. Microbiol. 2012, 78, 258–262. 10.1128/AEM.06265-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Y.; Phelan V. V.; Simkovsky R.; Watrous J. D.; Trial R. M.; Fleming T. C.; Wenter R.; Moore B. S.; Golden S. S.; Pogliano K.; Dorrestein P. C. J. Bacteriol. 2012, 194, 6023–6028. 10.1128/JB.00823-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe J.; Bauer J.; Andra J.; Schromm A. B.; Ernst M.; Rossle M.; Zahringer U.; Rademann J.; Brandenburg K. FEBS J. 2006, 273, 5101–5112. 10.1111/j.1742-4658.2006.05507.x. [DOI] [PubMed] [Google Scholar]
- Dobler L.; Vilela L. F.; Almeida R. V.; Neves B. C. New Biotechnol. 2016, 33, 123–135. 10.1016/j.nbt.2015.09.005. [DOI] [PubMed] [Google Scholar]
- Muller M. M.; Kugler J. H.; Henkel M.; Gerlitzki M.; Hormann B.; Pohnlein M.; Syldatk C.; Hausmann R. J. Biotechnol. 2012, 162, 366–380. 10.1016/j.jbiotec.2012.05.022. [DOI] [PubMed] [Google Scholar]
- Zhu K.; Rock C. O. J. Bacteriol. 2008, 190, 3147–3154. 10.1128/JB.00080-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdel-Mawgoud A. M.; Lepine F.; Deziel E. Appl. Microbiol. Biotechnol. 2010, 86, 1323–1336. 10.1007/s00253-010-2498-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L.; Liu P.; Peng Y.; Lin J.; Wang Q.; Ma Y. J. Appl. Microbiol. 2014, 117, 139–150. 10.1111/jam.12515. [DOI] [PubMed] [Google Scholar]
- Cabrera-Valladares N.; Richardson A. P.; Olvera C.; Trevino L. G.; Deziel E.; Lepine F.; Soberon-Chavez G. Appl. Microbiol. Biotechnol. 2006, 73, 187–194. 10.1007/s00253-006-0468-5. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Chockalingam K.; Chen Z. Curr. Opin. Biotechnol. 2002, 13, 104–110. 10.1016/S0958-1669(02)00291-4. [DOI] [PubMed] [Google Scholar]
- Wang M.; Si T.; Zhao H. Bioresour. Technol. 2012, 115, 117–125. 10.1016/j.biortech.2012.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N. J. In Enzyme Assays; Wiley-VCH Verlag GmbH & Co. KGaA, 2006; pp 137–161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.