

Abstract
In blood coagulation, thrombin converts fibrinogen into fibrin monomers that polymerize into a clot network. Thrombin also activates Factor XIII by cleaving the R37-G38 peptide bond of the Activation Peptide (AP) segment. The resultant transglutaminase introduces covalent crosslinks into the fibrin clot. A strategy to modify clot architecture would be to design FXIII AP sequences that are easier or more difficult to be thrombin-cleaved thus controlling initiation of crosslinking. To aid in this design process, FXIII V34X (28–41) activation peptides were kinetically ranked for cleavage by wild-type thrombin and several anticoagulant mutants. Thrombin-catalyzed hydrolysis of aromatic FXIII F34, W34, and Y34 APs was compared with V34 and L34. Cardioprotective FXIII L34 remained the variant most readily cleaved by wild-type thrombin. The potent anticoagulant thrombins W215A and W215A/E217A (missing a key substrate platform for binding fibrinogen) were best able to hydrolyze FXIII F34 and W34 APs. Thrombin I174A and L99A could effectively accommodate FXIII W34 and Y34 APs yielding kinetic parameters comparable to FXIII AP L34 with wild-type thrombin. None of the aromatic FXIII V34X APs could be hydrolyzed by thrombin Y60aA. FXIII F34 and W34 are promising candidates for FXIII – anticoagulant thrombin systems that could permit FXIII-catalyzed crosslinking in the presence of reduced fibrin formation. By contrast, FXIII Y34 with thrombin (Y60aA or W215A/E217A) could help assure that both fibrin clot formation and protein crosslinking are hindered. Regulating the activation of FXIII is predicted to be a strategy for helping to control fibrin clot architecture and its neighboring environments.
Keywords: Factor XIII, transglutaminase, thrombin, serine protease, kinetics, coagulation
Graphical abstract
1. Introduction
In the latter stages of the blood coagulation cascade, the proteins Fibrinogen (Fbg), Factor XIII (FXIII), and thrombin participate in the formation of a blood clot and define its subsequent architecture [1–6]. Thrombin cleaves off the N-terminal portions of the Aα and Bβ chains of fibrinogen (AαBβγ)2 to release fibrinopeptides A and B. The resultant fibrin monomers polymerize linearly and laterally to create an ordered fibrin network [1, 2]. Simultaneously, thrombin activates FXIII by hydrolyzing the R37-G38 peptide bond of the Activation Peptide (AP) segment [5, 7, 8]. Activated FXIII is then responsible for catalyzing the formation of γ-glutamyl-ε-lysyl crosslinks within the fibrin network thereby generating a more mechanically and proteolytically resistant clot [5, 9]. Besides cleaving fibrinogen and FXIII, thrombin contributes to anticoagulation by activating Protein C in the presence of thrombomodulin [3, 10]. Thrombin also activates protease activated receptors (PARs) that control initiation of platelet aggregation and secretion [3, 11, 12].
Thrombin is a sodium binding, serine protease whose catalytic residues include S195, H57, and D102 [3, 13]. Thrombin employs a series of surface loops to control substrate specificity. In addition, the enzyme utilizes two anion binding exosites (ABE I and II) to direct the binding of substrates and supporting ligands [14]. The 60-insertion or β-insertion loop (Y60a-K60f) of thrombin limits substrate access to the catalytic site and includes two key residues Y60a and W60d (Footnote 1) (Fig 1) [13, 15]. The extended active site region contains two nonpolar residues L99 and I174 that assist in accommodating individual substrate residues [13]. Thrombin residue W215 serves as a key substrate binding platform within the aryl binding site [13]. When W215 is replaced with an alanine (W215A), thrombin catalyzed hydrolysis of fibrinogen becomes greatly hindered whereas the activation of Protein C still occurs [16, 17]. As a result, the anticoagulant functions of thrombin become more dominant. The double mutant W215A/E217A (WE) exhibits a collapsed active site cleft and generates an even better anticoagulant thrombin [18, 19].
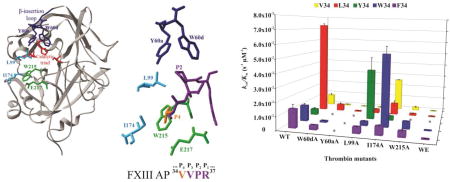
Figure 1.

X-ray crystal structure of thrombin highlighting key residues that surround the bound structure of FXIII AP (34–37). A) Ribbon diagram of thrombin (gray) showing selected residues as colored sticks (PDB ID: 1DE7). Residues include the catalytic triad (red), Y60a and W60d (blue), L99 and I174 (cyan), W215 and E217 (green). B) The enzyme-bound FXIII AP segment (34V orange, 35VPR37 purple) surrounded by thrombin residues that were mutated to alanines for the kinetic studies (PDB ID: 1DE7).
A critical substrate that thrombin must accommodate is the FXIII AP segment [5, 9]. The V34 at the P4 position is located just a few residues N-terminal to the FXIII R37-G38 (P1-P1') cleavage site (Table 1) [5, 9, 20]. The X-ray crystal structures of the zymogen FXIII A2 dimer show that each AP segment straddles across the dimer interface and protects against premature access to the opposite FXIII catalytic site [21, 22]. The free FXIII AP 1–37 segment is even more flexible with little defined secondary structure [23, 24]. To be cleaved by thrombin, the FXIII AP must interact appropriately with the enzyme subsites of this serine protease. Previous kinetic and NMR studies demonstrated that the P5-P1 residues (33GVVPR37) of FXIII AP (28–41) are most critical for targeting the thrombin active site region [25]. (Footnote 2) Of particular note are the P4 and P2 positions of FXIII AP that interact with the S4 and S2 subsites on thrombin [15, 20]. Interestingly, the electron density for this AP region is often not resolved in FXIII A2 crystal structures thus suggesting that this region is highly flexible [21, 22]. In a physiological environment, the location and orientation of FXIII AP (1–37) in intact FXIII A may still contribute to accessibility and optimal binding properties at the thrombin active site.
Table 1.
Substrate sequences that target the thrombin active site a
| P9…………P4……P1… | |
|---|---|
| Factor XIII (28–41) V34 AP | 28TVELQGVVPRGVNL41 |
| Factor XIII (28–41) L34 AP | 28TVELQGLVPRGVNL41 |
| Factor XIII (28–41) F34 AP | 28TVELQGFVPRGVNL41 |
| Factor XIII (28–41) W34AP | 28TVELQGWVPRGVNL41 |
| Factor XIII (28–41) Y34 AP | 28TVELQGYVPRGVNL41 |
| Factor XIII (28–41) P34 AP | 28TVELQGPVPRGVNL41 |
| Fibrinogen Aα (7–20) | 7DFLAEGGGVRGPRV20 |
| Thrombin Receptor PAR1 (32–45) | 32KATNATLDPRSFLL45 |
| Thrombin Receptor PAR4 (38–51) | 38STPSILPAPRGYPG51 |
Sequences of human FXIII, fibrinogen Aα and Thrombin Receptors (PAR1 and PAR4) are displayed. The residues are aligned along the P4 position.
The common polymorphism FXIII L34 is found in about 25% of the population and its presence has been associated with decreases in myocardial infarction and coronary artery disease particularly when fibrinogen concentrations are elevated to health risk levels [26–32]. Interestingly, FXIII L34 is activated more readily by thrombin than the more common FXIII V34 [27, 33–35]. Kinetic studies with synthetic peptides derived from FXIII AP (28–41) revealed that the extra methylene group in FXIII AP L34 (28–41) generates a thrombin substrate with a strong enhancement in kcat [33, 36]. NMR studies indicated that L34 and P36 promote stabilizing P4 to P2 interactions that better anchor the AP segment onto the thrombin active site surface and promote hydrolysis [37]. Additional kinetic studies further demonstrated the critical role that the 34 (P4) position can play in controlling thrombin-catalyzed hydrolysis of the FXIII (28–41) AP segment [25, 38–40]. Results with recombinant full length FXIII variants involving these same AP residues support this proposal [41, 42].
Fibrin clot structure is dependent upon fibrinogen levels, rates of fibrinopeptide cleavage, and FXIII activity [6, 43–46]. Clot architecture could be further controlled by designing FXIII AP sequences that are easier or more difficult to be cleaved by thrombin [27, 41]. As a result, FXIIIa catalyzed crosslinking could be initiated earlier or later in the fibrin clot assembly process. Moreover, specific FXIII mutants could be matched with thrombin species that have either procoagulant or anticoagulant properties [3, 4, 19, 40]. To design such FXIII variants, more information is needed on how the FXIII AP segment interacts with specific regions of wild-type thrombin and its potential therapeutic mutants.
Wild-type and mutant thrombin studies have already been carried out with FXIII AP (28–41) peptides containing V34, L34, P34, or F34 [36, 40]. The FXIII P34 variant allowed platelet PAR4 character to be introduced into the FXIII AP sequence (Table 1) [47]. Earlier NMR studies had revealed that the FXIII F34 provided the same beneficial P4-P2 interactions with the thrombin surface as L34 [39]. Importantly, the FXIII F34 AP was demonstrated to be the first FXIII-based substrate that exhibited little change in Km upon loss of thrombin residue W215 [40]. An aromatic residue at the P4 position was proposed to be an effective strategy for promoting FXIII activation in the presence of an anticoagulant thrombin.
Based on the promising features of FXIII F34 AP, two additional aromatic residues at the P4 position were explored. With FXIII W34 AP (28–41), the ability to accommodate the bulkier indole ring could be probed and thus help map out the S4 site on thrombin. FXIII Y34 AP (28–41) incorporated a more polar phenol group that was predicted to have difficulties handling the hydrophobic β-insertion loop and the W215 platform environment. Hydrolysis of FXIII W34 and Y34 APs (28–41) were examined with WT thrombin and the mutants W60dA, Y60aA, L99A, I174A, W215A, and WE. The kinetic effects were compared with FXIII V34, L34, and F34 [36, 40]. Results reveal that FXIII W34 and F34 are promising candidates for creating therapeutic FXIII – thrombin mutant systems that contain some crosslinking ability in an environment of decreased fibrin(ogen) clotting. A combination of Factor XIII Y34 and an anticoagulant thrombin would hinder both fibrinogen clotting and protein crosslinking.
2. Materials and Methods
2.1. Synthetic Peptides
Peptides derived from human FXIII activation peptide (AP) residues 28–41 were synthesized by New England Peptide (Gardner, MA). The amino acid sequences of the peptides included: FXIII (28–41) V34 AP, Ac-TVELQGVVPRGVNL-NH2; FXIII (28–41) L34 AP, Ac-TVELQGLVPRGVNL-NH2, FXIII (28–41) F34 AP, Ac-TVELQGFVPRGVNL- NH2; FXIII (28–41) W34 AP, Ac-TVELQGWVPRGVNL-NH2; and FXIII (28–41) Y34 AP, Ac-TVELQGYVPRGVNL-NH2. The purity of the peptides were determined by analytical reversed phase HPLC and the peptide m/z values were confirmed using matrix assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry. Peptides were soluble in deionized water to 7mM. The concentrations of the peptides in solution were determined by quantitative amino acid analysis (AAA Service Laboratory, Damascus, OR).
2.2. Thrombin Preparation
All human recombinant thrombin was generously provided by Dr. Enrico Di Cera and Ms. Leslie Pelc, Saint Louis University, St. Louis, MO. These thrombin species were expressed and purified as described previously [48]. The thrombin variants examined in this project included wild-type (WT), the single alanine substitutions W60dA, Y60aA, L99A, I174A, and W215A, and the double substitution W215A/E217A (WE). Thrombin concentrations were determined from absorbance measurements at E280nm0.1% = 1.83 mL/(mg cm) using MW = 36,500 g/mol.
2.3. Kinetics Procedure
The HPLC-based kinetic assay of Trumbo and Maurer was employed [33]. Briefly, a solution of peptide in assay buffer (50 mM H3PO4, 100mM NaCl, 0.1% PEG, pH 7.4) was heated to 25 °C in a heat block. The final concentrations of the FXIII APs spanned the concentration range of 75–2500 µM. Hydrolysis of each Factor XIII AP was initiated with the addition of human thrombin (WT or mutant). The thrombin concentrations were between 6 nM and 1 µM depending on the particular FXIII AP and thrombin mutant being examined.
An aliquot of the reaction mixture was removed at regular intervals and quenched with 12.5% H3PO4. Thrombin concentrations and reaction time points were selected such that less than 17% of the FXIII V34X APs were hydrolyzed in 30 min. Peptide peaks were separated by RP-HPLC using a Waters X-Bridge C18 Column on a Waters 2695 or a Waters 600 series HPLC system. For thrombin-catalyzed hydrolysis of FXIII (28–41) F34 and Y34 AP, substrate and product peaks were separated using an acetonitrile based gradient of [15% CH3CN, 0.09% trifluoroacetic acid in dI water] to [50% CH3CN, 0.09% trifluoroacetic acid in dI water]. A methanol based gradient of [17% methanol, 0.09% trifluoroacetic acid in dI water] to [40% methanol, 0.09% trifluoroacetic acid in dI water] was employed for reactions involving FXIII W34 AP.
After the time points for the thrombin-catalyzed reactions were run on the HPLC columns, the FXIII AP (28–37) product peaks were integrated. The peak areas were converted to concentration using product calibration curves for each FXIII activation peptide. The slopes of product concentration versus time plots were used to determine the initial velocities (in µM/s) for the different thrombin-catalyzed reactions. The results represent experiments which were done in triplicate. Individual kinetic constants were determined following nonlinear regression analysis fits to the equation V = Vmax/(1 + Km/[S]) using Sigma Plot (Jandel Scientific) or Kaleidagraph (Synergy). The kcat values were then calculated knowing the final thrombin concentrations employed in the assays.
2.4. Expression of Full-Length Recombinant FXIII Variants
A pGEX plasmid vector encoding GST-tagged FXIII-A V34 was kindly provided by Dr. Helen Philippou, Dr. Robert Ariens, and Dr. Kerrie Smith from the University of Leeds, UK [49]. Single amino acid V34X substitutions (L, F, W, or Y) were introduced using the QuikChange II Mutagenesis kit (Agilent Technologies). The expression vectors for the FXIII-A V34X variants were transformed into E. coli BL21 (DE3) Gold cells. An auto-induction approach was employed for the expression of full length FXIII-A variants [50]. Cells were harvested by centrifugation, washed from the remaining culture medium, and stored in pellets at −20°C. The pellets were resuspended in phosphate buffered saline and lysed. The cell lysate was cleared by centrifugation and then treated with 1% streptomycin. After centrifugation and filtration through a 0.22 µm membrane filter, the cleared supernatant was applied to a GST-trap column (GE Healthcare, USA) using an AKTA Prime FPLC system. FXIII-A protein was cleaved from the GST-tag by in-column digestion overnight with PreScission protease. The protein was eluted from the column with phosphate buffered saline. Molar concentration was determined from absorbance readings at 280 nm using ε = 125710 M−1cm−1 for L34, F34, and Y34 variants (calculated using Protparam tool, www.expasy.org). An ε = 131210 M−1cm−1 was applied for FXIII-A W34.
2.5. Gel Electrophoresis Assay to Follow Thrombin Cleavage of FXIII
Thrombin-catalyzed cleavage of FXIII APs was monitored using SDS-PAGE. 1 µM FXIII V34X variant was incubated with 30 nM of WT recombinant human thrombin at 37°C. At different time points (1, 2, 5, 10, 15, 20, 25, and 30 min), aliquots were withdrawn and supplemented with 6 µM of the thrombin active site inhibitor PPACK (D-Phe-Pro-Arg chloromethyl ketone). Samples were then mixed with reducing loading buffer and subjected to electrophoresis using 8% polyacrylamide gels. Gels were stained with Coomassie Blue. Cleavage process was monitored by densitometric analysis using GelAnalyzer 2010 (www.gelanalyzer.com).
3. Results
The HPLC based assay proved to be a successful strategy for characterizing the ability of wild-type and mutant thrombins (W60dA, Y60aA, L99A, I174A, W215A, and W215A/E217A [WE]) to hydrolyze a series of Factor XIII AP segments (28–41) containing V34X substitutions. Km, kcat, and kcat/Km for the different thrombin and FXIII V34X AP pairs are shown in Table 2. Bar chart comparisons across the peptide and thrombin series are displayed in Fig 2 and 3.
Table 2.
Kinetic Constants for Hydrolysis of FXIII (28–41) F34, W34, Y34, V34, and L34 Activation Peptides with Different Thrombin (IIa) Mutantsa. L34 is a Common Polymorphism
| FXIII (28–41) F34 AP | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
| WT IIab | 442 ± 93 | 6.27 ± 0.54 | 1.4×10−2 ± 3.0×10−3 |
| W60dA IIab | 435 ± 98 | 1.46 ± 0.15 | 3.4×10−3 ± 8.0×10−4 |
| Y60aA IIab | NA | NA | NA |
| L99A IIab | 570 ± 125 | 2.71 ± 0.3 | 4.8×10−3 ± 1.2×10−4 |
| I174A IIa | 920 ± 88 | 6.08 ± 0.25 | 6.6×10−3 ± 6.9×10−4 |
| W215A IIb | 437 ± 81 | 1.97 ± 0.14 | 4.5×10−3 ± 9.0×10−4 |
| WE (W215A/E217A) IIab | 190 ± 43 | 0.07 ± 0.007 | 3.6×10−4 ± 8.0×10−5 |
| FXIII (28–41) W34 AP | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
| WT IIa | 637 ± 56 | 7.62 ± 0.3 | 1.2×10−2 ± 1.2×10−3 |
| W60dA IIa | 976 ± 89 | 1.21 ± 0.05 | 1.2×10−3 ± 1.2×10−4 |
| Y60aA IIa | NA | NA | NA |
| L99A IIa | 719 ± 97 | 2.3 ± 0.17 | 3.2×10−3 ± 4.9×10−4 |
| I174A IIa | 256 ± 24 | 13.9 ± 0.52 | 5.4×10−2 ± 5.5×10−3 |
| W215A IIa | 1030 ± 57 | 5.76 ± 0.14 | 5.6×10−3 ± 3.4×10−4 |
| WE (W215A/E217A) IIa | 1281 ± 161 | 0.35 ± 0.03 | 2.7×10−4 ± 3.5×10−5 |
| FXIII (28–41) Y34 AP | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
| WT IIa | 484 ± 88 | 2.23 ± 0.18 | 4.6×10−3 ± 9.0×10−4 |
| W60dA IIa | NA | NA | NA |
| Y60aA IIa | NA | NA | NA |
| L99A IIa | 347 ± 80 | 13.5 ± 1.4 | 3.9×10−2 ± 9.8×10−3 |
| I174A IIa | 349 ± 120 | 1.17 ± 0.16 | 3.3×10−3 ± 2.0×10−3 |
| W215A IIa | NA | NA | NA |
| WE (W215A/E217A) IIa | NA | NA | NA |
| FXIII (28–41) V34 AP | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
| WT IIac | 298 ± 42 | 2.57 ± 0.005 | 8.6×10−3 ± 1.0×10−3 |
| W60dA IIab | 1375 ± 422 | 0.86 ± 0.046 | 6.3×10−4 ± 1.9×10−4 |
| Y60aA IIab | 678 ± 120 | 0.048 ± 0.0032 | 7.1×10−5 ± 1.3×10−5 |
| L99A IIac | 2074 ± 414 | 0.89 ± 0.07 | 4.3×10−4 ± 1.0×10−4 |
| I174A IIac | 204 ± 36 | 5.37 ± 0.01 | 2.6×10−2 ± 1.0×10−4 |
| W215A IIac | 2374 ± 372 | 5.53 ± 0.017 | 2.3×10−3 ± 7.0×10−4 |
| WE (W215A/E217A) IIac | 745 ± 98 | 0.117 ± 0.008 | 1.6×10−4 ± 2.0×10−5 |
| FXIII (28–41) L34 AP [common polymorphism] | Km (µM) | kcat (s−1) | kcat/Km (µM−1 s−1) |
| WT IIac | 315 ± 42 | 22.9 ± 0.003 | 7.3×10−2 ± 1.2×10−3 |
| W60dA IIab | 1000 ± 201 | 5.07 ± 0.021 | 5.1×10−3 ± 1.0×10−3 |
| Y60aA IIab | 696 ± 116 | 0.37 ± 0.012 | 5.3×10−4 ± 1.2×10−4 |
| L99A IIac | 1930 ± 354 | 5.94 ± 0.02 | 3.1×10−3 ± 2.0×10−4 |
| I174A IIc | 1109 ± 247 | 10.2 ± 0.04 | 9.1×10−3 ± 2.0×10−3 |
| W215A IIac | 3619 ± 470 | 5.31 ± 0.002 | 1.5×10−3 ± 2.0×10−4 |
| WE (W215A/E217A) IIac | 142 ± 28 | 0.052 ± 0.003 | 3.7×10−4 ± 1.0×10−4 |
The results shown here represent averages of at least three independent experiments. Kinetic values were calculated using nonlinear regression analysis methods using SigmaPlot or Kaleidagraph. The error values correspond to standard error of the mean (SEM).
Jadhav, M. et al. [40]
Isetti, G. et al [36]
Figure 2.

The kinetic constants Km and kcat for hydrolysis of FXIII V34X AP (28–41) peptides by WT and mutant thrombins. A) A comparison of the Km values for FXIII V34 AP (yellow), L34 AP (red), F34 AP (purple), W34 AP (light blue), and Y34 AP (green) in the presence of WT thrombin and the single site mutants W60dA, Y60aA, L99A, I174A, W215A, and WE. B) A comparison of the kcat values for FXIII V34 AP (yellow), L34 AP (red), F34 AP (purple), W34 AP (light blue), and Y34 AP (green) in the presence of WT thrombin and the single site mutants W60dA, Y60aA, L99A, I174A, W215A, and WE. The individual kinetic parameters were derived from an HPLC assay described in Materials and Methods. Results represent data from at least three independent trials, and error bars correspond to standard error of the mean.
Figure 3.

Three dimensional representation of kcat/Km values as a function of thrombin mutant and FXIII V34X AP. The thrombin species examined include WT and the single site mutants W60dA, Y60aA, L99A, I174A, W215A, and WE. The different FXIII V34X AP (28–41) peptides consisted of FXIII V34 AP (yellow), L34 AP (red), F34 AP (purple), W34 AP (light blue), and Y34 AP (green). The individual kinetic parameters were derived from an HPLC assay described in Materials and Methods. The * symbols correspond to peptide-thrombin combinations whose rates of hydrolysis were too low to be characterized. Results represent data from at least three independent trials, and error bars correspond to standard error of the mean.
3.1. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by WT Thrombin
Prior kinetic studies revealed that FXIII L34 AP was a better substrate for WT thrombin than FXIII V34 AP (Table 2 and Figure 2, 3) due to a 9-fold higher kcat [36]. The aromatic residue in FXIII F34 AP resulted in a 1.5-fold increase in Km relative to FXIII V34 AP; however, the kcat value increased 2.4-fold over the FXIII V34 AP sequence [36, 40]. In the current work, the bulkier tryptophan indole group found in FXIII W34 AP led to a 2-fold increase in Km relative to FXIII V34 AP. Similar to FXIII F34 AP, the W34 exhibited a 3-fold increase in kcat relative to FXIII V34 AP. The FXIII Y34 AP exhibited a Km value comparable to that of FXIII F34 AP, but the kcat value was reduced back down to the value of FXIII V34 AP. Overall, the kcat/Km values were best for FXIII L34 AP followed by F34 and W34. Kinetic improvements relative to FXIII V34 AP were primarily due to increases in the kcat values.
3.2. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W60dA Thrombin
W60d exists in the center of the thrombin 60-insertion loop and is part of the S2 enzyme subsite (Fig 1) [13, 15]. In prior work on FXIII V34 and L34 AP, the Km values increased 3–5 fold while kcat values decreased 3–5 fold [40]. There was an overall 14-fold decrease in kcat/Km relative to WT thrombin. FXIII F34 AP binding to W60dA thrombin could handle the loss of the W60d residue generating a Km similar to that of WT thrombin [40]. By contrast, the ability to be hydrolyzed had been compromised leading to a 4-fold decrease in kcat. Overall, there was a 4-fold decrease in kcat/Km relative to wild-type thrombin. In the current work on FXIII W34 AP, the Km increased 1.5-fold and the kcat decreased 6-fold relative to WT thrombin. As a result, the kcat/Km value for hydrolysis of FXIII W34 AP by W60dA thrombin was 10-fold lower than by WT thrombin. FXIII Y34 AP hydrolysis rates were too low at the thrombin stock concentrations available for this project, thus kinetic constants could not be determined.
3.3. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by Y60aA Thrombin
Thrombin residue Y60a (part of the S2 enzyme subsite) is positioned at the beginning of the thrombin 60-insertion loop and helps control entrance into the thrombin active site region (Fig 1) [13, 15]. Prior results revealed that hydrolysis of FXIII V34 and L34 AP both led to 2-fold increases in Km accompanied by 54 to 61-fold decreases in kcat [40]. Unlike the studies with W60dA thrombin, FXIII F34 AP could not be hydrolyzed by Y60aA thrombin at the enzyme concentrations available [40]. In the current project, the same difficulties were observed with FXIII W34 AP and Y34 AP. All three aromatic FXIII AP V34X varians (F, W, and Y) could not handle replacement of thrombin Y60a with the smaller A residue. Individual kinetic values could not be determined.
3.4. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by L99A Thrombin
L99 is also a member of the S2 enzyme subsite that contributes to the thrombin apolar binding site (Fig 1) [13, 15]. Thrombin residues L99 and Y60a both function to help encompass incoming substrates. In prior work with L99A thrombin, FXIII V34 and L34 AP exhibited 6–7-fold increases in Km and 3–4 fold decreases in kcat relative to WT thrombin [36]. The stronger kcat value for L34 allowed this FXIII AP to still exhibit a 7-fold advantage in kcat /Km relative to V34. Unlike the Y60aA thrombin studies, the FXIII F34 AP could be accommodated by the L99A mutant with only a 1.3-fold increase in Km and 2-fold decrease in kcat relative to WT thrombin [40]. The kcat/Km value for FXIII F34 AP in the presence of thrombin L99A was 10-fold higher than FXIII V34 AP. For the current work, FXIII W34 AP and thrombin L99A exhibited no improvement in Km over WT thrombin, and similar to FXIII F34 AP, there was a 3-fold decrease in kcat. Still, the kcat/Km value for FXIII W34 AP in the presence of thrombin L99A was 7-fold higher than with FXIII V34 AP. Curiously, FXIII V34Y AP could finally be examined with a thrombin mutant. The Km value in the presence of L99A thrombin was comparable to WT thrombin and the kcat value exhibited a surprising 6-fold improvement over WT thrombin. Overall, L99A thrombin was best at hydrolyzing FXIII V34Y AP. There was a 8.5-fold increase in kcat/Km relative to FXIII W34AP in the presence of L99A thrombin and a 90-fold increase relative to FXIII V34AP.
3.5. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by I174A Thrombin
Extending more into the apolar binding site, the I174 residue borders the S4 pocket of thrombin (Fig 1) [13, 15]. Prior work revealed that with FXIII V34 AP, thrombin I174A leads to a 1.5-fold increase in Km and a 2-fold increase in kcat resulting in a 2.7-fold increase in kcat/Km relative to WT thrombin [36]. By contrast, hydrolysis of FXIII L34 AP by I174A thrombin exhibits a 3.5-fold increase in Km and a 2-fold decrease in kcat leading to an 8-fold decrease in kcat/Km [36]. The influence of thrombin I174 on other FXIII V34X substitutions had not been examined before. In the current work, FXIII V34F AP also underwent a 2-fold increase in Km but no change in kcat. FXIII V34Y exhibited a 1.4-fold decrease in Km and a 2-fold decrease in kcat. FXIII V34W AP was the best substrate of the I174A thrombin series revealing a 2.4-fold decrease in Km and a 2-fold increase in kcat producing a 4.5-fold increase in kcat/Km. The kcat/Km value for FXIII W34 AP was 2-fold increased relative to FXIII V34 AP in the presence of I174A thrombin.
3.6. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W215A Thrombin
The indole ring of W215 provides a thrombin platform to support such critical substrates as the Fbg Aα chain (Fig 1) [13, 15, 51]. Moreover, the W215 must be in a correct orientation to help maintain an open serine protease active site region [18, 52]. Because of these features, there was strong interest in assessing how well FXIII V34X APs could be accommodated by a thrombin W215A mutant. Prior studies had revealed that FXIII V34 and L34 AP were both less efficiently hydrolyzed by W215A thrombin than FXIII F34 AP [36, 40]. FXIII V34 and L34 AP exhibited 8 to 11-fold increases in Km with W215A thrombin and now their kcat values were similar. By contrast, FXIII F34 exhibited little change in Km relative to WT thrombin and the kcat value underwent a 3-fold decrease [40]. As a result, the kcat/Km values were reduced 4-fold for FXIII V34 AP, 49-fold for FXIII L34 AP, and 3-fold for FXIII F34 AP relative to WT thrombin. Overall, FXIII F34 AP was the best substrate sequence for W215A thrombin because the Km value was preserved and there was a smaller decrease in kcat [40].
In the current work, the effects of the indole ring of W and the phenol of Y were compared to that of the smaller F. Unlike FXIII F34 AP, the FXIII W34 AP with its bulkier indole side chain experienced a 1.6-fold increase in Km relative to WT thrombin. Moreover, there was a 3-fold improvement in kcat for FXIII W34 AP relative to F34 AP. As a result, FXIII APs W34, V34, and L34 all have similar kcat values. The kcat/Km values for FXIII APs F34 and W34 were found to be comparable with F34 providing a better Km and W34 a stronger kcat. Individual kinetic parameters could not be determined for hydrolysis of FXIII Y34 by thrombin W215A.
3.7. Kinetics Involving FXIII (28–41) V34X AP Hydrolyzed by W215A/ E217A (WE) Thrombin
X-ray crystallography has revealed that replacement of W215 and E217 (S4 enzyme subsite) with alanines leads to collapse of the thrombin active site cleft (Fig 1) [18]. This double mutant known as the WE thrombin has shown much promise as a therapeutic anticoagulant [53–56]. Earlier studies revealed that WE thrombin led to a 2.5-fold increase in Km for FXIII V34 AP and 2-fold decreases in Km for both L34 and F34 relative to WT thrombin [36, 40]. Decreases in kcat were 22-fold for V34, 90-fold for F34, and 440-fold for L34 relative to WT thrombin. The overall kcat/Km values were similar for FXIII APs L34 and F34 in the presence of WE thrombin [36, 40].
In the current work, the Km for FXIII W34 AP with WE thrombin was comparable to that of W215A thrombin with both exhibiting a 2-fold decrease over WT thrombin. Similar to FXIII V34 AP, the kcat was reduced 22-fold for FXIII W34 AP and WE thrombin. When examining kcat/Km values of FXIII V34, L34, F34, W34 APs, the L34 and F34 were the best of the series and V34 the worst. No significant hydrolysis of FXIII Y34 AP was observed in the presence of WE thrombin.
3.8. Thrombin Cleavage of Full Length FXIII V34X Variants
Studies with synthetic peptides have provided valuable clues about how different FXIII V34X residues can be accommodated by WT thrombin and its selective active site region mutants. Verifying that full length FXIII V34X proteins will generate comparable results is important. FXIII V34, L34, F34, W34, and Y34 were successfully expressed in E. coli and purified by affinity chromatography. FXIII V34X variants at a concentration of 1 µM were individually subjected to cleavage by 30 nM thrombin. The hydrolysis reactions were carried out at 37°C and halted at 1, 2, 5, 10, 15, 20, 25, and 30 min. Thrombin cleavage at R37-G38 leads to the loss of a 4 kDa fragment, and as seen in Figure 4, the uncleaved and cleaved FXIII A-subunits can be separated by SDS-PAGE. As predicted from synthetic peptide studies, FXIII L34 underwent hydrolysis at the fastest rate with 60% cleavage occurring by 5 min and full cleavage by 20 min. Cleavage of FXIII V34 started to be visible at 2 min and became 50% cleaved by 10 min. There was almost full cleavage of FXIII V34 by 30 minutes. Rates of cleavage for the other FXIII variants followed the trend of F34 > W34 ≈ Y34. These gel electrophoresis studies confirm that Activation Peptide cleavage can be monitored over time with intact FXIII proteins and that differences can be detected. More detailed HPLC kinetic studies could be carried out with full length FXIII variants [27, 57]. However, the FXIII AP (28–41) sequences have some distinct advantages. These smaller synthetic AP substrates can be obtained at much higher yields and importantly, they produce better resolved HPLC peaks for kinetic data analysis. Moreover, the synthetic peptides can be used more readily in assays designed to determine individual kinetic constants [33]. The synthetic peptide studies thus remain a highly effective approach for comparing individual kinetic constants for a series of variant FXIII Activation Peptide sequences.
Figure 4.

SDS-PAGE analysis of thrombin cleavage of FXIII activation peptide by thrombin. A) 1 µM FXIII V34X variant was incubated with 30 nM recombinant thrombin at 37°C. At defined time points, the aliquots were removed, quenched with PPACK and then reducing loading buffer, and finally quickly frozen at −80°C. Samples were later subjected to electrophoresis on 8% gels and stained for proteins using Coomassie blue. Intact FXIII appears at 83kDa whereas cleavage of the Activation Peptide (1–37) results in a protein band at 79kDa. B) Cleavage progress for FXIII L34 (red), V34 (yellow), F34 (purple), Y34 (green), and W34 (blue) was monitored by densitometric analysis using GelAnalyzer 2010 (www.gelanalyzer.com) for the time points 1, 2, 5, 10, 15, 20, 25, and 30 min
4. Discussion
Thrombin catalyzed hydrolysis of the FXIII Activation Peptide segment is a key step in generating an active Factor XIII that can covalently crosslink the fibrin clot network and other physiological substrates [5, 9]. The common polymorphism FXIII L34 utilizes its extra methylene group to produce an AP segment that is better oriented for thrombin-catalyzed hydrolysis than FXIII V34 [27, 33, 37]. There is now interest in kinetically characterizing other FXIII AP V34X (28–41) peptides that interact with WT and mutant thrombins [36, 40]. Specific sets of thrombin and FXIII V34X variants may later be used to regulate fibrin clot formation and extent of FXIII-catalyzed substrate crosslinking.
4.1. Interactions between WT Thrombin and FXIII AP (28–41) V34, L34, F34, W34, and Y34
Activation Peptides with F34, W34, and Y34 substitutions were used to explore how well the extended thrombin active site region could accommodate three different aromatic residues at the P4 position. FXIII APs F34, W34, and Y34 could still foster binding interactions with the thrombin surface and the turnover numbers were at least as good as those of FXIII V34 AP (Fig 2, 3). Based on their kcat values, FXIII AP (28–41) sequences containing W34 and F34 are proposed to be better oriented for thrombin catalyzed hydrolysis than V34 and Y34. The common polymorphism L34 still remains the dominant FXIII AP substrate for WT thrombin. Detailed thrombin cleavage studies can be carried out with synthetic peptides and further supported by work with full-length FXIII V34X proteins (Fig 4). Experiments with full-length proteins revealed that FXIII L34 is cleaved at the fastest rate followed by V > F > (W ≈ Y). These results are mostly in accord with the synthetic peptide studies although the FXIII W34 was cleaved more slowly than expected. All full-length FXIII species were examined at the same single, initial concentration. Having the weakest Km of the V34X series, the overall cleavage rate of the W34 variant may have been limited by its low concentration. A greater extent of thrombin-catalyzed cleavage would have occurred with a higher concentration of the FXIII V34W substrate. From reviewing all the hydrolysis data, the fast cleaving FXIII L34 would be poised to introduce crosslinking earlier in the fibrin clot assembly process than the aromatic FXIII V34X candidates.
4.2. Interactions involving thrombin W60dA and thrombin Y60aA
The thrombin residue W60d is located in the center of the 60-insertion loop and is known to play a vital role in positioning Fbg Aα, Fbg Bβ, and the thrombin receptor PAR4 within the thrombin active site surface [19, 48]. Kinetic studies revealed that FXIII F34 AP was the best of the V34X sequences for promoting binding interactions with W60dA thrombin. The phenyl side chain of FXIII F34 may find a better way to interact with this new thrombin environment than V34, L34, or the bulkier indole of FXIII W34 AP. Kinetic constants could not be determined for FXIII Y34 AP. The Y34 polar phenol may cause a steric clash with the mutated 60-insertion loop.
In contrast to W60d, the thrombin residue Y60a is situated at the start of the 60-insertion loop and reported to influence the binding of Fbg Aα, hirudin, and thrombomodulin [19, 58, 59]. X-ray crystallography has shown that the FXIII V34 side chain borders thrombin Y60a [15]. Thrombin Y60aA could not hydrolyze any of the FXIII AP segments containing F34, W34, or Y34. Aromatic V34X peptides are proposed to benefit from binding at or near thrombin Y60a, but not A60a. From reviewing the kinetic data, thrombin W60dA is an example of a mutant that could modestly hinder cleavage of fibrinogen but still allow for FXIII F34 or W34 activation and thus crosslinking activity. Alternatively, thrombin Y60aA could function to greatly hinder cleavage of fibrinogen and also cleavage of FXIII AP segments with an aromatic V34X substitution.
4.3. Interactions involving thrombin L99A and thrombin I174A
Thrombin residues L99 and Y60a both function to help cage the P2 residue of thrombin substrates (Fig 1) [13, 15]. It is already known that FXIII P36 at the P2 position interacts with thrombin L99, Y60aA, and W215 [15]. Unlike Y60aA, the FXIII F34, W34, and Y34 AP segments could each be hydrolyzed by L99A thrombin further supporting the proposal that the FXIII AP aromatic rings are oriented more toward the thrombin 60-insertion loop than the S2 region near thrombin L99. Unexpectedly, L99A thrombin was best able to hydrolyze FXIII Y34 AP relative to the other V34X APs examined (Fig 2, 3). The expansion of the thrombin S2 site upon the L99A substitution must generate a better environment to accommodate the FXIII Y34 side chain and encourage an orientation that promotes thrombin cleavage of the R37-G38 peptide bond. Of the FXIII V34X APs, FXIII V34 was the worst of the series. Surprisingly, the altered S2 subsite may no longer provide an appropriate surface for binding the smaller-sized FXIII V34.
The thrombin I174 residue contributes to the S4 subsite on this protease [13]. Thrombin I174 helps surround the F8 residue of Fbg Aα and plays a supporting role in accommodating the P4 and P5 positions of FXIII AP (28–37) (Figure 1) [15, 19, 51]. Kinetic studies with I174A thrombin helped to detect unique differences between F34, W34, and Y34. The loss of the aliphatic I174 side chain caused the FXIII F34 AP to only experience a decrease in binding interactions. Reactions with FXIII Y34 AP revealed, in addition, a lower substrate to product turnover. The hydroxyl group of the phenol may cause polar repulsions with the thrombin surface that ultimately contribute to a non-optimal orientation for R37-G38 hydrolysis. Intriguingly, FXIII W34 AP exhibited improvements in both binding interactions and turnover. The larger space provided by the thrombin A174 is proposed to be most effectively utilized by the bulkier indole group of FXIII W34.
Hydrolysis of FXIII W34 AP by I174A thrombin produced, for the first time, kcat/Km values that approach those of WT thrombin with the strong substrate FXIII L34 AP (Fig 2, 3). FXIII W34 AP and I174A thrombin could be a promising therapeutic combination. I174A thrombin is already known to decrease the kcat/Km value for fibrinopeptide A release by 22-fold [19]. Fibrin clot formation could be hindered but FXIII W34 AP cleavage could be at a rate comparable to FXIII L34 AP. Not far behind in kinetic effectiveness would be the enzyme-substrate pair composed of thrombin L99A and FXIII Y34 AP. L99A thrombin is however not as effective in hindering FpA cleavage as I174A thrombin [19]. There is only a 2-fold decrease in kcat/Km value for FpA release by L99A thrombin. From an enzyme design perspective, the thrombin substitutions L99A or I174A could also be helpful for modifying the characteristics of the S2 and S4 enzyme subsites. These alanine mutations might be utilized to help a more potent anticoagulant thrombin better accommodate certain FXIII AP residues within these subsites.
4.4. Interactions involving thrombin W215A and WE
The thrombin mutant W215A has already been proven to be a promising anticoagulant enzyme that exhibits greatly hindered cleavage of the Fbg Aα chain (500-fold decrease in kcat/Km) but still allows for Protein C activation [17, 19]. With the W215A substitution, the Fbg Aα residue F8 loses a key stabilizing side chain interaction from W215 within the thrombin active site region (Fig 1) [51]. An even more potent anticoagulant thrombin involves the W215A/E217A (WE) substitution that contains a collapsed primary specificity cleft [18]. With this double mutant, there is 1900-fold decrease in kcat/Km for FpA cleavage [17, 19]. The WE thrombin has been successfully tested as a therapeutic anticoagulant in mice and in non-human primates [53–56].
According to X-ray crystal structures, the backbones of FXIII AP V34 and L34 are both supported by thrombin W215A (Figure 1) [15]. With this thrombin mutant, binding interactions for FXIII V34 and L34 are dramatically reduced along with loss of the strong turnover benefit of L34 [36]. As with W60dA, FXIII F34 AP was the best at preserving binding interactions with W215A thrombin [40]. FXIII W34 AP was better at maintaining the turnover number. The kcat/Km values for FXIII F34 and W34 AP were comparable. WE thrombin studies resulted in the lowest kcat/Km values; however, ranking trends similar to those of W215A thrombin were still observed. Unexpectedly, difficulties were encountered when trying to kinetically characterize hydrolysis of FXIII Y34 AP by thrombin W215A. Decreased velocities observed at higher peptide substrate concentrations suggest the FXIII Y34 residue may be causing polar repulsion directed toward the thrombin 60-insertion loop region. Proceeding to the more potent anticoagulant WE thrombin, no cleavage of FXIII Y34 AP was observed. FXIII Y34 AP must rely on a well-positioned thrombin W215 platform to help assure that the phenol ring finds an appropriate binding environment within the thrombin active site region.
Based on their individual kinetic constants, FXIII F34 AP and W34 AP are both examples of sequences that may still be accommodated by anticoagulant thrombins W215A and WE. With such mutant thrombin – FXIII combinations, Fbg Aα cleavage would be greatly reduced but FXIII activation would still prevail. As a result, other physiological substrates could still be crosslinked by FXIII. These protein combinations may permit crosslinking activities in an environment with diminished fibrin clot formation. Unlike W34 and F34, FXIII Y34 remains the best variant of the series for assuring minimal FXIII activation. With W215A and WE thrombin, clot formation and FXIII-catalyzed crosslinking activity would definitely be hindered.
4.5. Conclusions
A series of FXIII V34X (28–41) activation peptides was kinetically characterized for cleavage by WT and mutant thrombins. FXIII L34 AP remained the variant most readily cleaved by WT thrombin and would be expected to initiate crosslinking activities the earliest of the V34X AP series (Fig 3). FXIII F34 and W34 AP showed much promise as FXIII species that could still be activated in the presence of the potent anticoagulant thrombins W215A and WE. In a localized therapeutic environment, fibrin clotting would be greatly hindered but crosslinking of other physiological substrates would still be permitted. Intriguingly, the I174A and L99A thrombin mutants helped to improve hydrolysis of FXIII W34 and Y34 APs leading to kinetic properties approaching those of FXIII L34 AP with WT thrombin (Fig 3). Such substitutions could be used to generate milder anticoagulant thrombin – FXIII combinations. Alternatively, I174A and L99A could be used to further optimize the thrombin binding environments for different FXIII AP residues. Interestingly, none of the aromatic FXIII AP residues could be effectively hydrolyzed by Y60aA thrombin. Moreover, W215A and WE thrombin had the greatest difficulties cleaving FXIII Y34 AP (Fig 3). Such mutant FXIII-thrombin combinations would hinder fibrin clot formation and FXIII-catalyzed crosslinking.
To achieve different FXIII-related goals, mutant FXIII A and thrombin species could be introduced into a localized environment. The amounts applied would be sufficient to out compete endogenous counterpart levels for a therapeutic period of time. Recombinant WT FXIII A2 has already been used successfully as a prophylaxis in FXIII A-deficient patients [60–62]. The administered FXIII A becomes readily complexed with endogenous FXIII B-subunits. These B-subunits mediate FXIII interactions with fibrinogen and protect FXIII A from premature degradation [60, 61]. Additional control studies could assess how combinations of mutant FXIII A2B2 V34X and mutant thrombin species function in FXIII-deficient plasma. From the current work, it is already encouraging to learn that there are full-length FXIII A2 V34X variants that can be cleaved by WT thrombin with rankings comparable to those of FXIII V34X (28–41) AP. Overall, our kinetic results on hydrolysis of FXIII V34X activation peptides by WT and mutant thrombins reveal novel strategies to regulate FXIII activation and thus its ability to initiate crosslinking reactions. These approaches may be used to further control fibrin blood clot structure and its surrounding architectures.
Highlights.
Thrombin activates Factor XIII by cleaving R37-G38 bond of the Activation Peptide
A strategy to modify fibrin clots is to design FXIII APs that are easier or more difficult to cleave.
Combinations of FXIII APs and mutant thrombins were screened for cleavage ability.
FXIII F34 and W34 are predicted to permit crosslinking in presence of reduced fibrin.
Acknowledgments
The authors thank Dr. Enrico Di Cera and Ms Leslie Pelc (Saint Louis University) for generously supplying the recombinant thrombins. Plasmid DNA for GST-tagged FXIII-A V34 was kindly provided by Dr. Helen Philippou, Dr. Kerrie Smith, and Dr. Robert Ariëns (University of Leeds). We also appreciate further tips for optimizing FXIII expression levels from Dr. Jeffrey Keillor and Dr. A. Mulani (University of Ottawa). The authors thank K. Mouapi, R. Billur, and L. Wagner for critical discussions related to this project. W.G. acknowledges undergraduate fellowships from the University of Louisville Summer Research Opportunity (SROP) and the Institute of Molecular Diversity and Drug Design (IMD3). K.L. acknowledges undergraduate summer research support from the NSF-REU program (CHE-1156861). S.Z and C.R. received undergraduate summer research funding from NIH R15 grant HL120068. This research project was supported by grants from the National Institutes of Health (R01 HL68440 and R15 HL120068).
Abbreviations
- FXIII
blood coagulation Factor XIII
- FXIII AP
Factor XIII Activation Peptide
- ABE-I
anion-binding exosite-I
- ABE-II
anion-binding exosite-II
- Fbg
fibrinogen
- PAR
protease activated receptor
- PPACK
D-Phe-Pro-Arg chloromethyl ketone
- WE thrombin
W215A/E217A thrombin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Amino acid residues of thrombin are designated by single letter abbreviations and chymotrypsin numbering is employed. This numbering scheme requires accommodations for the thrombin insertion loops. As a result, Y60a corresponds to the tyrosine in the first residue position of the 60- or β-insertion loop. W60d corresponds to the tryptophan at the fourth or “d” position of this same loop.
The P nomenclature system is used to assign the individual amino acid positions on the substrate peptides. The scissile bond is designated by P1-P1′. The substrate amino acids N-terminal of the hydrolysis site are labeled P2, P3, P4, etc. whereas those that are C-terminal are labeled P2′, P3′, P4′, etc. Likewise, the S nomenclature is used to assign positions on the enzyme. S1 accepts the P1 residue from the peptide substrate and so on.
References
- 1.Weisel JW, Litvinov RI. Mechanisms of fibrin polymerization and clinical implications. Blood. 2013;121:1712–1719. doi: 10.1182/blood-2012-09-306639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weisel JW. Fibrinogen and fibrin. Advances in protein chemistry. 2005;70:247–299. doi: 10.1016/S0065-3233(05)70008-5. [DOI] [PubMed] [Google Scholar]
- 3.Di Cera E. Thrombin. Molecular aspects of medicine. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Cera E. Thrombin as an anticoagulant. Progress in molecular biology and translational science. 2011;99:145–184. doi: 10.1016/B978-0-12-385504-6.00004-X. [DOI] [PubMed] [Google Scholar]
- 5.Muszbek L, Bereczky Z, Bagoly Z, Komaromi I, Katona E. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiological reviews. 2011;91:931–972. doi: 10.1152/physrev.00016.2010. [DOI] [PubMed] [Google Scholar]
- 6.Ariens RA. Fibrin(ogen) and thrombotic disease. Journal of thrombosis and haemostasis : JTH. 2013;11(Suppl 1):294–305. doi: 10.1111/jth.12229. [DOI] [PubMed] [Google Scholar]
- 7.Takagi T, Doolittle RF. Amino acid sequence studies on factor XIII and the peptide released during its activation by thrombin. Biochemistry. 1974;13:750–756. doi: 10.1021/bi00701a018. [DOI] [PubMed] [Google Scholar]
- 8.Hornyak TJ, Bishop PD, Shafer JA. Alpha-thrombin-catalyzed activation of human platelet factor XIII: relationship between proteolysis and factor XIIIa activity. Biochemistry. 1989;28:7326–7332. doi: 10.1021/bi00444a027. [DOI] [PubMed] [Google Scholar]
- 9.Schroeder V, Kohler HP. Factor XIII: Structure and Function. Seminars in thrombosis and hemostasis. 2016;42:422–428. doi: 10.1055/s-0036-1571341. [DOI] [PubMed] [Google Scholar]
- 10.Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Seminars in thrombosis and hemostasis. 2006;32(Suppl 1):49–60. doi: 10.1055/s-2006-939554. [DOI] [PubMed] [Google Scholar]
- 11.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. Journal of thrombosis and haemostasis : JTH. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 12.Nieman MT. Protease-activated receptors in hemostasis. Blood. 2016;128:169–177. doi: 10.1182/blood-2015-11-636472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bode W, Turk D, Karshikov A. The refined 1.9-A X-ray crystal structure of D-Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis, overall structure, electrostatic properties, detailed active-site geometry, and structure-function relationships. Protein science : a publication of the Protein Society. 1992;1:426–471. doi: 10.1002/pro.5560010402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bock PE, Panizzi P, Verhamme IM. Exosites in the substrate specificity of blood coagulation reactions. Journal of thrombosis and haemostasis : JTH. 2007;5(Suppl 1):81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadasivan C, Yee VC. Interaction of the factor XIII activation peptide with alpha - thrombin. Crystal structure of its enzyme-substrate analog complex. The Journal of biological chemistry. 2000;275:36942–36948. doi: 10.1074/jbc.M006076200. [DOI] [PubMed] [Google Scholar]
- 16.Arosio D, Ayala YM, Di Cera E. Mutation of W215 compromises thrombin cleavage of fibrinogen, but not of PAR-1 or protein C. Biochemistry. 2000;39:8095–8101. doi: 10.1021/bi0006215. [DOI] [PubMed] [Google Scholar]
- 17.Cantwell AM, Di Cera E. Rational design of a potent anticoagulant thrombin. The Journal of biological chemistry. 2000;275:39827–39830. doi: 10.1074/jbc.C000751200. [DOI] [PubMed] [Google Scholar]
- 18.Pineda AO, Chen ZW, Caccia S, Cantwell AM, Savvides SN, Waksman G, Mathews FS, Di Cera E. The anticoagulant thrombin mutant W215A/E217A has a collapsed primary specificity pocket. The Journal of biological chemistry. 2004;279:39824–39828. doi: 10.1074/jbc.M407272200. [DOI] [PubMed] [Google Scholar]
- 19.Marino F, Pelc LA, Vogt A, Gandhi PS, Di Cera E. Engineering thrombin for selective specificity toward protein C and PAR1. The Journal of biological chemistry. 2010;285:19145–19152. doi: 10.1074/jbc.M110.119875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schechter I, Berger A. On the active site of proteases. 3. Mapping the active site of papain; specific peptide inhibitors of papain. Biochemical and biophysical research communications. 1968;32:898–902. doi: 10.1016/0006-291x(68)90326-4. [DOI] [PubMed] [Google Scholar]
- 21.Yee VC, Pedersen LC, Le Trong I, Bishop PD, Stenkamp RE, Teller DC. Three-dimensional structure of a transglutaminase: human blood coagulation factor XIII. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:7296–7300. doi: 10.1073/pnas.91.15.7296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weiss MS, Metzner HJ, Hilgenfeld R. Two non-proline cis peptide bonds may be important for factor XIII function. FEBS letters. 1998;423:291–296. doi: 10.1016/s0014-5793(98)00098-2. [DOI] [PubMed] [Google Scholar]
- 23.Schroeder V, Vuissoz JM, Caflisch A, Kohler HP. Factor XIII activation peptide is released into plasma upon cleavage by thrombin and shows a different structure compared to its bound form. Thrombosis and haemostasis. 2007;97:890–898. [PubMed] [Google Scholar]
- 24.Ortner E, Schroeder V, Walser R, Zerbe O, Kohler HP. Sensitive and selective detection of free FXIII activation peptide: a potential marker of acute thrombotic events. Blood. 2010;115:5089–5096. doi: 10.1182/blood-2009-11-253062. [DOI] [PubMed] [Google Scholar]
- 25.Isetti G, Maurer MC. Thrombin activity is unaltered by N-terminal truncation of factor XIII activation peptides. Biochemistry. 2004;43:4150–4159. doi: 10.1021/bi049796v. [DOI] [PubMed] [Google Scholar]
- 26.Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood. 2002;100:743–754. doi: 10.1182/blood.v100.3.743. [DOI] [PubMed] [Google Scholar]
- 27.Ariens RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000;96:988–995. [PubMed] [Google Scholar]
- 28.Lim BC, Ariens RA, Carter AM, Weisel JW, Grant PJ. Genetic regulation of fibrin structure and function: complex gene-environment interactions may modulate vascular risk. Lancet. 2003;361:1424–1431. doi: 10.1016/S0140-6736(03)13135-2. [DOI] [PubMed] [Google Scholar]
- 29.Kohler HP, Ariens RA, Whitaker P, Grant PJ. A common coding polymorphism in the FXIII A-subunit gene (FXIIIVal34Leu) affects cross-linking activity. Thrombosis and haemostasis. 1998;80:704. [PubMed] [Google Scholar]
- 30.Kohler HP, Stickland MH, Ossei-Gerning N, Carter A, Mikkola H, Grant PJ. Association of a common polymorphism in the factor XIII gene with myocardial infarction. Thrombosis and haemostasis. 1998;79:8–13. [PubMed] [Google Scholar]
- 31.Chen F, Qiao Q, Xu P, Fan B, Chen Z. Effect of factor XIII-A Val34Leu polymorphism on myocardial infarction risk: a meta-analysis. Clinical and applied thrombosis/hemostasis : official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis. 2014;20:783–792. doi: 10.1177/1076029613504130. [DOI] [PubMed] [Google Scholar]
- 32.Voko Z, Bereczky Z, Katona E, Adany R, Muszbek L. Factor XIII Val34Leu variant protects against coronary artery disease. A meta-analysis. Thrombosis and haemostasis. 2007;97:458–463. [PubMed] [Google Scholar]
- 33.Trumbo TA, Maurer MC. Examining thrombin hydrolysis of the factor XIII activation peptide segment leads to a proposal for explaining the cardioprotective effects observed with the factor XIII V34L mutation. The Journal of biological chemistry. 2000;275:20627–20631. doi: 10.1074/jbc.M000209200. [DOI] [PubMed] [Google Scholar]
- 34.Balogh I, Szoke G, Karpati L, Wartiovaara U, Katona E, Komaromi I, Haramura G, Pfliegler G, Mikkola H, Muszbek L. Val34Leu polymorphism of plasma factor XIII: biochemistry and epidemiology in familial thrombophilia. Blood. 2000;96:2479–2486. [PubMed] [Google Scholar]
- 35.Wartiovaara U, Mikkola H, Szoke G, Haramura G, Karpati L, Balogh I, Lassila R, Muszbek L, Palotie A. Effect of Val34Leu polymorphism on the activation of the coagulation factor XIII-A. Thrombosis and haemostasis. 2000;84:595–600. [PubMed] [Google Scholar]
- 36.Isetti G, Maurer MC. Employing mutants to study thrombin residues responsible for factor XIII activation peptide recognition: a kinetic study. Biochemistry. 2007;46:2444–2452. doi: 10.1021/bi0622120. [DOI] [PubMed] [Google Scholar]
- 37.Trumbo TA, Maurer MC. Thrombin hydrolysis of V29F and V34L mutants of factor XIII (28–41) reveals roles of the P(9) and P(4) positions in factor XIII activation. Biochemistry. 2002;41:2859–2868. doi: 10.1021/bi0157823. [DOI] [PubMed] [Google Scholar]
- 38.Trumbo TA, Maurer MC. V34I and V34A substitutions within the factor XIII activation peptide segment (28–41) affect interactions with the thrombin active site. Thrombosis and haemostasis. 2003;89:647–653. [PubMed] [Google Scholar]
- 39.Isetti G, Maurer MC. Probing thrombin's ability to accommodate a V34F substitution within the factor XIII activation peptide segment (28–41) The journal of peptide research : official journal of the American Peptide Society. 2004;63:241–252. doi: 10.1111/j.1399-3011.2004.00132.x. [DOI] [PubMed] [Google Scholar]
- 40.Jadhav MA, Lucas RC, Goldsberry WN, Maurer MC. Design of Factor XIII V34X activation peptides to control ability to interact with thrombin mutants. Biochimica et biophysica acta. 2011;1814:1955–1963. doi: 10.1016/j.bbapap.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duval C, Ali M, Chaudhry WW, Ridger VC, Ariens RA, Philippou H. Factor XIII A-Subunit V34L Variant Affects Thrombus Cross-Linking in a Murine Model of Thrombosis. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:308–316. doi: 10.1161/ATVBAHA.115.306695. [DOI] [PubMed] [Google Scholar]
- 42.Andersen MD, Kjalke M, Bang S, Lautrup-Larsen I, Becker P, Andersen AS, Olsen OH, Stennicke HR. Coagulation factor XIII variants with altered thrombin activation rates. Biological chemistry. 2009;390:1279–1283. doi: 10.1515/BC.2009.142. [DOI] [PubMed] [Google Scholar]
- 43.Ryan EA, Mockros LF, Weisel JW, Lorand L. Structural origins of fibrin clot rheology. Biophysical journal. 1999;77:2813–2826. doi: 10.1016/S0006-3495(99)77113-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wolberg AS. Thrombin generation and fibrin clot structure. Blood reviews. 2007;21:131–142. doi: 10.1016/j.blre.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Hethershaw EL, Cilia La Corte AL, Duval C, Ali M, Grant PJ, Ariens RA, Philippou H. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. Journal of thrombosis and haemostasis : JTH. 2014;12:197–205. doi: 10.1111/jth.12455. [DOI] [PubMed] [Google Scholar]
- 46.Byrnes JR, Wolberg AS. Newly-Recognized Roles of Factor XIII in Thrombosis. Seminars in thrombosis and hemostasis. 2016;42:445–454. doi: 10.1055/s-0036-1571343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cleary DB, Trumbo TA, Maurer MC. Protease-activated receptor 4-like peptides bind to thrombin through an optimized interaction with the enzyme active site surface. Archives of biochemistry and biophysics. 2002;403:179–188. doi: 10.1016/s0003-9861(02)00220-5. [DOI] [PubMed] [Google Scholar]
- 48.Ayala YM, Cantwell AM, Rose T, Bush LA, Arosio D, Di Cera E. Molecular mapping of thrombin-receptor interactions. Proteins. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- 49.Smith KA, Adamson PJ, Pease RJ, Brown JM, Balmforth AJ, Cordell PA, Ariens RA, Philippou H, Grant PJ. Interactions between factor XIII and the alphaC region of fibrinogen. Blood. 2011;117:3460–3468. doi: 10.1182/blood-2010-10-313601. [DOI] [PubMed] [Google Scholar]
- 50.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein expression and purification. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Martin PD, Robertson W, Turk D, Huber R, Bode W, Edwards BF. The structure of residues 7–16 of the A alpha-chain of human fibrinogen bound to bovine thrombin at 2.3-A resolution. The Journal of biological chemistry. 1992;267:7911–7920. [PubMed] [Google Scholar]
- 52.Gandhi PS, Page MJ, Chen Z, Bush-Pelc L, Di Cera E. Mechanism of the anticoagulant activity of thrombin mutant W215A/E217A. The Journal of biological chemistry. 2009;284:24098–24105. doi: 10.1074/jbc.M109.025403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gruber A, Cantwell AM, Di Cera E, Hanson SR. The thrombin mutant W215A/E217A shows safe and potent anticoagulant and antithrombotic effects in vivo. The Journal of biological chemistry. 2002;277:27581–27584. doi: 10.1074/jbc.C200237200. [DOI] [PubMed] [Google Scholar]
- 54.Gruber A, Fernandez JA, Bush L, Marzec U, Griffin JH, Hanson SR, E DIC. Limited generation of activated protein C during infusion of the protein C activator thrombin analog W215A/E217A in primates. Journal of thrombosis and haemostasis : JTH. 2006;4:392–397. doi: 10.1111/j.1538-7836.2006.01760.x. [DOI] [PubMed] [Google Scholar]
- 55.Feistritzer C, Schuepbach RA, Mosnier LO, Bush LA, Di Cera E, Griffin JH, Riewald M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. The Journal of biological chemistry. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- 56.Wood DC, Pelc LA, Pozzi N, Wallisch M, Verbout NG, Tucker EI, Gruber A, Di Cera E. WEDGE: an anticoagulant thrombin mutant produced by autoactivation. Journal of thrombosis and haemostasis : JTH. 2015;13:111–114. doi: 10.1111/jth.12774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Philippou H, Rance J, Myles T, Hall SW, Ariens RA, Grant PJ, Leung L, Lane DA. Roles of low specificity and cofactor interaction sites on thrombin during factor XIII activation. Competition for cofactor sites on thrombin determines its fate. The Journal of biological chemistry. 2003;278:32020–32026. doi: 10.1074/jbc.M305364200. [DOI] [PubMed] [Google Scholar]
- 58.Mengwasser KE, Bush LA, Shih P, Cantwell AM, Di Cera E. Hirudin binding reveals key determinants of thrombin allostery. The Journal of biological chemistry. 2005;280:26997–27003. doi: 10.1074/jbc.M502678200. [DOI] [PubMed] [Google Scholar]
- 59.Xu H, Bush LA, Pineda AO, Caccia S, Di Cera E. Thrombomodulin changes the molecular surface of interaction and the rate of complex formation between thrombin and protein C. The Journal of biological chemistry. 2005;280:7956–7961. doi: 10.1074/jbc.M412869200. [DOI] [PubMed] [Google Scholar]
- 60.Lovejoy AE, Reynolds TC, Visich JE, Butine MD, Young G, Belvedere MA, Blain RC, Pederson SM, Ishak LM, Nugent DJ. Safety and pharmacokinetics of recombinant factor XIII-A2 administration in patients with congenital factor XIII deficiency. Blood. 2006;108:57–62. doi: 10.1182/blood-2005-02-0788. [DOI] [PubMed] [Google Scholar]
- 61.Inbal A, Oldenburg J, Carcao M, Rosholm A, Tehranchi R, Nugent D. Recombinant factor XIII: a safe and novel treatment for congenital factor XIII deficiency. Blood. 2012;119:5111–5117. doi: 10.1182/blood-2011-10-386045. [DOI] [PubMed] [Google Scholar]
- 62.Carcao M, Fukutake K, Inbal A, Kerlin B, Lassila R, Oldenburg J, Garly ML, Nugent D. Developing the First Recombinant Factor XIII for Congenital Factor XIII Deficiency: Clinical Challenges and Successes. Seminars in thrombosis and hemostasis. 2017;43:59–68. doi: 10.1055/s-0036-1585076. [DOI] [PubMed] [Google Scholar]