

Abstract
Hematopoietic stem cell (HSC) transplantation can restore a new functional hematopoietic system in recipients in cases where the recipient’s own system is not functional, or for example, leukemic. However, the number of available donor HSCs is sometimes too low for successful transplantation. Expansion of HSCs and thus, HSC self-renewal ex vivo would greatly improve transplantation therapy in the clinic. In vivo, HSCs expand significantly in the niche, but establishing protocols that result in HSC expansion ex vivo remains challenging. In this review, we discuss current knowledge of niche biology, the intrinsic regulators of HSC self-renewal in vivo, and introduce novel niche-informed strategies of HSC expansion ex vivo.
Significance of Ex Vivo HSC Expansion
Hematopoietic stem cells (HSCs) sustain blood-cell formation in a process called hematopoiesis (see Glossary, Box 1). This is achieved by their ability to regenerate themselves long-term, which is referred to as self-renewal activity, and through their ability to differentiate into cells of all mature blood lineages. Human HSCs are rare cells (~0.01%) primarily found in bone marrow (BM) in adults [1]. Hematopoietic stem cell transplantation (HSCT) can restore a new functional hematopoietic system and blood cell production in recipients [2–4]. It is used in the clinic to treat leukemia and other cancers, as well as bone marrow failure syndromes, and in gene therapy settings. The source of HSCs for a transplant is either a patient’s own HSCs (autologous transplant) or HSCs from a human leukocyte antigen (HLA) matched donor in an allograft transplant setting [5, 6]. For HSCT, HSCs from BM or umbilical cord blood (UCB) or HSCs in blood, mobilized to blood by the cytokine granulocyte-colony stimulating factor (G-CSF), can be used. Infused donor HSCs then home to and engraft in discrete BM niches to reconstitute the blood system of the recipient [2]. Currently, autologous transplants have survival rates exceeding 80%, while the success rate for allogeneic transplants at five years fluctuates between 30% and 70%, based in part, on the initial donor match [5]. The number of HSCs transplanted correlates with successful engraftment and patient survival. For a successful HSCT, high numbers of CD34+ cells (i.e. 3–4 × 106/kg of human body weight) are required [6, 7] and thus, the numbers of HSCs in a given graft may not be sufficient to allow transplantation to proceed. Thus, protocols that result in expansion of HSCs ex vivo would be a highly desirable tool to further increase positive outcomes in the clinic. Understanding mechanisms of HSC self-renewal in vivo in depth should be a pre-requisite for the development of successful protocols to expand HSCs ex vivo for therapeutic applications.
Box-1. Mammalian Hematopoiesis and HSCs.
The hematopoietic system is one of the best-studied adult stem cell systems in humans and rodents:
Functionally, HSCs are defined as cells that give rise to long-term multi-lineage engraftment that persists for at least 20 weeks after primary and secondary transplantation [171].
Multipotent progenitors can generate all major hematopoietic lineages in transplantation assays in lethally irradiated recipients but fail to engraft long-term. Long-term reconstitution of hematopoiesis in a transplant setting can be achieved by a single long-term (LT)-HSC [3, 4, 32].
Recent research has been able to phenotypically define murine long-term HSCs:
(LT-HSCs) as Lin−IL-7α−Sca-1+c-Kit+Flt3−CD34−CD150+CD48−, progenitors including short-term HSCs (ST-HSCs) as Lin−IL-7Rα−Sca-1+c-Kit+Flt3 CD34+CD150+ CD48−,
and multipotent progenitors (MPPs) as Lin−IL-7Rα−Sca-1+c-Kit+Flt3low-highCD34+ [4, 32, 172–174].
Human long-term HSCs and MPPs have been phenotypically defined as being:
HSC self-renewal is regulated by a complex interplay of intrinsic factors such as transcription factors, cell cycle status and metabolic pathways, as well as extrinsically, by both the local and the systemic environment. The local environment in the BM is referred to as stem cell niche [8, 9]. It is believed that signals from the niche are critical for the regulation of HSC self-renewal as well as for differentiation decisions [1, 8, 10, 11]. In recent years, numerous cellular constituents of the murine BM niche and committed hematopoietic progeny have been investigated that interact either directly or indirectly with HSCs and which might contribute to the regulation of HSC self-renewal and differentiation [9, 12–24]. As such, experiments usually impair genetically or pharmacologically one type of cell niche to then analyze the changes in HSC phenotype; however, much remains unknown regarding the mechanisms that regulate the complex interplay among the distinct types of stromal elements under native conditions. HSCs expand in numbers in vivo within their niche environment. Theoretically, the number of HSCs in the niche is determined by the frequency of symmetric cell divisions that lead to the generation of two stem cells or two progenitor cells, relative to the frequency of asymmetric cell divisions that posit a balance between HSC and daughter cell generation [25]. HSCs generally remain quiescent in the BM niche, while diverse stimuli that trigger loss of quiescence cause robust entry into the cell cycle, and induce proliferation often associated with stress, DNA-damage and apoptosis [26, 27]. Ex vivo expansion will thus require approaches that result in symmetric stem cell divisions [25] and hence, HSC self-renewal without further differentiation and apoptosis. Mammalian HSCs undergo symmetric cell divisions in vivo during development [25] and in adulthood. For example, using mice where HSCs were labeled with a dye ‘diluting’ HSCs following division (label-retaining HSCs (LR-HSCs), murine HSC were found to complete four symmetric self-renewal divisions in vivo before re-entering a state of dormancy [28]; and yet, persistent inflammatory signaling can disturb HSC dormancy, resulting in HSC exhaustion [29]. Because adult HSCs have been shown to undergo self-renewal/expansion following chemotherapy, radiation challenge or transplantation, thus replenishing the hematopoietic niche [4, 30, 31], it may be possible to achieve HSC expansion ex vivo, once we improve our understanding of the HSC-intrinsic and niche-dependent mechanisms that are responsible for HSC expansion in vivo. We review below the most recent knowledge on mechanisms of HSC self-renewal, placing a particular focus on the contribution of the HSC niche.
HSC Localization within the Niche
Adult HSCs reside in specific BM locations with unique environments known as niches. A large set of data have revealed that there is vast heterogeneity of niches for HSCs within the BM (recently reviewed in [22]). Niches for HSCs comprise endosteal niches and vascular niches further divided into arteriolar as well as sinusoidal components [9, 18–24, 32]. Deeply quiescent (dormant) HSCs are believed to localize around arterioles and closer to the endosteum in the mouse BM, while activated HSCs – which are significantly more abundant than dormant HSCs—are thought to reside in the vicinity of sinusoids [33–36]. Indeed, recent studies have revealed that most murine HSCs are present in perivascular locations in close contact with either sinusoids or arterioles [20, 23, 36, 37]. Indeed, such new and detailed knowledge on niche architecture and on the association/proximity of HSCs to non-HSC niche cells has been based on high-resolution imaging studies in mouse BM (particularly from femur) [12, 16, 21, 33, 34, 38, 39]. Thus, the role of the HSC niche as a critical contributor to the regulation of HSC cell self-renewal [1] has been confirmed in mouse models where niche cells and/or distinct soluble growth factors have been genetically modified; the cell types and factors within the niche contributing specifically to HSC self-renewal in vivo are detailed below.
Endosteal and Vascular Niches
Both the endosteal niche and the vascular (arteriolar and sinusoidal) niches have been recognized as regulators of HSC self-renewal as well as HSC function, based in part on the cellular composition and soluble components found in both of these niches. For example, multiple heterologous mouse cell types including endosteal osteoblasts [19, 33, 39], sinusoidal blood vessels and leptin receptor-positive (Lepr+) perivascular stromal cells [23, 32], CXCL12-abundant reticular (CAR) cells [40, 41], nestin+ mesenchymal stem cells [18], non-myelinating Schwann cells [16], regulatory T cells (Treg) [38] and megakaryocytes [12, 42] have been shown to locate in close proximity to murine HSCs in vivo. The underlying assumption has been that proximity serves as a surrogate marker for the relative importance of the regulatory influence of particular types of stromal cells on HSCs [12, 16, 21, 33, 34, 38, 39]. However, there are also multiple examples in which cells that are, on average, relatively distant from HSCs within the BM influence HSCs, such as osteoblasts [43]. Osteoblasts have been reported to enable a 3–4 fold expansion of human long-term culture-initiating cells (LTC-ICs) in vitro [44, 45], suggesting that stem cell self-renewal can be supported by osteoblast-derived factors. When parathyroid hormone (PTH/PTHrP) receptors (PPRs) were specifically introduced into murine osteoblasts, they produced a high level of the Notch ligand Jagged1 [19]. This caused a significant increase in the number of osteoblasts, which in turn resulted in an increase in the number of HSCs in vivo [19]. Similarly, mice with a conditional inactivation of BMP receptor type IA (Bmpr1a) exhibited an increase in N-cadherin+ osteoblasts, which also resulted in an increase in the number of HSCs in vivo [9]. However, the tissue-specific promoters used in these studies [9, 19] could also target perivascular osteoblastic progenitors and not only osteoblasts. Moreover, genetic manipulations of mature osteoblasts have not resulted in altered HSC function [46–48]. Taken together, a controversial debate still exists regarding the role mature osteoblasts play in the putative regulation of HSCs. Accordingly, because mice devoid of N-cadherin in hematopoietic and stromal cells or osteoblastic lineages have not led to changes in HSC numbers in vivo, the potential role for N-cadherin signaling in the regulation of HSC expansion in vivo also remains controversial [49, 50]. Osteoclasts in turn have been found to be dispensable for HSC maintenance and might thus not be involved in regulating murine HSC self-renewal in vivo [51].
Notch ligand signaling, such as via Jagged-1 in murine endothelial cells (i.e. vascular niche) has been found to support HSC self-renewal and prevent HSC exhaustion, both in vivo and in serum-free co-culture assays [52, 53]. BM macrophages (osteomacs) have also been reported to maintain retention of murine HSC in the BM through direct contact of perivascular cells with HSCs [14, 17]. Furthermore, the depletion of neutrophils in mouse BM can increase the number of CAR cells, and subsequently, CXCL12 levels, reducing the size and function of the hematopoietic niche; and, neutrophil clearance by macrophages can promote hematopoietic stem and progenitor cell (HSPC) mobilization in the circulation [54]. Indeed, research over the last couple of years has provided a more comprehensive characterization of the cellular composition of the murine HSC niche and the components that might influence HSC self-renewal in vivo with respect to both endosteal and vascular niches. Furthermore, multiple new types of niche cells have been linked to novel putative mechanisms of HSC regulation in terms of number and function within the BM (Key Figure, Figure 1, see also [8, 55]). However, further studies are required to finely dissect the relative and specific contribution of this panoply of niche cell types in HSC self-renewal, and the functional significance in the interdependence of these signaling pathways.
Key Figure, Figure 1. Mammalian Bone Marrow HSC Niche.
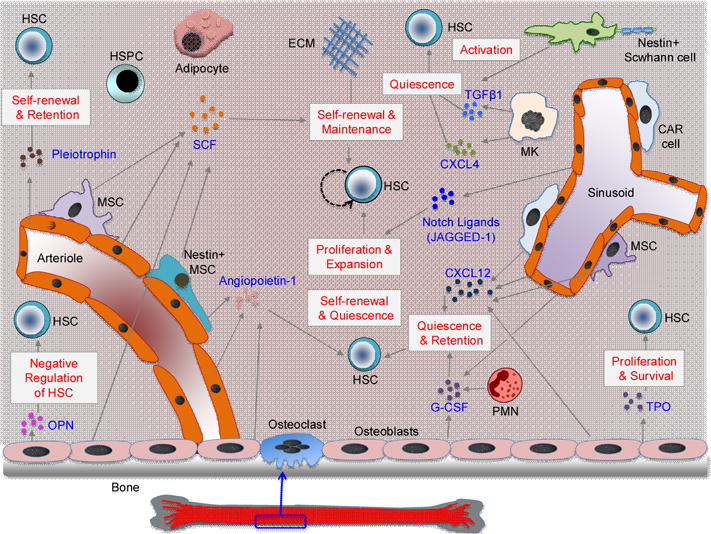
The diagram shows the cellular composition and cytokines/growth factors that can impact HSC self-renewal and function in the bone marrow (BM) niche. Recent research has identified the role of diverse BM niche cells and HSC progeny including osteoblasts, nestin+ mesenchymal stem cells, CAR cells, non-myelinating Schwann cells, BM endothelial cells and adipocytes, macrophages, megakaryocytes and neutrophils (PMN) in HSC self-renewal, differentiation and function. Niche cells also produce/release several cytokines/growth factors, such as SCF, TPO, TGF-β1, CXCL-4, CXCL-12, G-CSF, OPN, notch ligands, angiopoietin 1 and pleiotrophin to regulate HSC self-renewal, maintenance, survival, retention and function. The extra-cellular matrix (ECM) can also regulate HSC self-renewal and maintenance. SCF, stem cell factor; TPO, thrombopoietin; TGF-β1, transforming growth factor beta 1;CXCL-4, CXC chemokine ligand 4; CXCL-12, CXC chemokine ligand 12; G-CSF, Granulocyte-colony stimulating factor; OPN, osteopontin.
Cytokine Secretion by Niche Cells
Niche cells secrete cytokines and growth factors which regulate HSC self-renewal and differentiation in vivo (table 1). Osteoblast-derived cytokines, including osteopontin (OPN), angiopoietin -1, -3, thrombopoietin (TPO), granulocyte colony-stimulating factor (G-CSF), stem cell factor (SCF) and CXC-chemokine ligand 12 (CXCL12 or SDF-1) have been reported to regulate murine HSC self-renewal [30, 41, 44, 56–61]. Niche-expressed SCF or kit-ligand, TPO and their receptors on HSCs (c-Kit and c-Mpl) are well studied with respect to their role in murine HSC expansion [23, 57, 62–65]. Osteoblasts and endothelial cells release SCF, while TPO is mainly released by osteoblasts. Both c-Kit and c-Mpl are expressed on highly purified HSCs and genetic deletion of TPO or c-Mpl leads to a reduction of the number of murine HSCs [64, 66]. Leptin receptor positive (LepR+) perivascular and endothelial cells are another major component of the HSC niche and are the primary sources of CXCL12, SCF, pleiotrophin and Notch ligands (such as Jagged1) implicated in HSC regulation [20, 23, 61]. The puttaive role for angiopoietin-1 in HSC self-renewal – a cytokine well known for its role in in endothelial cell remodeling – is controversially discussed, and a recent report suggests that it might not directly influence HSC function [67]. Murine BM sinusoidal endothelial cells (BMECs) of the vascular niche secrete pleiotrophin (PTN) which may positively regulate HSC self-renewal [68, 69]. Specifically, PTN-deficient mice harbor decreased numbers of HSCs in the BM along with impaired hematopoietic regeneration [68], while PTN can promote in vitro expansion of long-term repopulating HSCs, both from mouse and human umbilical cord blood [69]. Moreover, PTN-induced HSC expansion could be blocked by inhibition of Notch activation through γ-secretase [69]. Another study has further implicated Notch signaling in HSC regulation by showing in serum/cytokine-free co-culture systems, that BMECs, secreting Notch ligands, could enhance in vitro HSC self-renewal [52].
Table 1.
Soluble Factors in the Bone Marrow Niche Affecting Mammalian HSCs
| Soluble Factor | Secreted by Niche cells | Impact on HSC | References |
|---|---|---|---|
| SCF | Osteoblasts, Endothelial cells, MSCs and Nestin+ MSCs | Induces HSC maintenance and self-renewal | [19, 23] |
| TPO | Osteoblasts | Enhances HSC self-renewal and survival | [57, 107, 146] |
| G-CSF | Osteoblasts, Endothelial cells, Neutrophils | Induces retention and quiescence | [17, 44] |
| CXCL-12 | Osteoblasts, Endothelial cells, CAR cells, MSCs, LepR+ Perivascular cells | Positive regulator of self-renewal, retention and function | [20, 40, 61] |
| CXCL-4 | Megakaryocytes | Inhibits self-renewal and induces quiescence | [12] |
| Pleiotrophin | Sinusoidal endothelial cells, LepR+ perivascular cells | Enhances self-renewal and BM retention | [68] |
| Osteopontin | Osteoblastic lineage | Negative regulation of HSC | [56] |
| Angiopoietin-1 | Osteoblasts, Osteoprogenitors, Endothelial cells, Nestin+ MSCs | Induces self-renewal, and survival | [58–60] |
| TGF-β1 | Nestin+ Schwann cells, Megakaryocytes | Maintenance of HSC quiescence, inhibition of cell cycle activity | [16, 42] |
| Notch Ligand Jagged-1 | Osteoblasts, Endothelial cells, LepR+ perivascular cells | Supports HSC self-renewal and prevents exhaustion | [19, 52, 53] |
Recent mouse studies also suggest an inhibitory role for peri-sinusoidal megakaryocytes (MKs) in HSCs expansion in the BM; for instance, depletion of MKs in the BM can cause HSC expansion due to loss of HSC quiescence [12, 42]. Furthermore, MKs can secrete the chemokine CXCL4/Cxcl4 and genetic depletion of Cxcl4 in MKs has resulted in increased numbers of murine HSCs, while Cxcl4 administration in mice can reduce HSC numbers in vivo, presumably via increased quiescence [12]. In addition, deletion of Tgfb1 in MKs increased HSC activation and proliferation, and conversely, activation of TGF-β1 signaling in MKs has resulted in HSC quiescence [42]; indeed, injection of TGF-β1 in MK-ablated mice restored HSC quiescence and inhibited self-renewal [42]. Also, nonmyelinating Nestin+ Schwann cells have been found to activate TGF-β1-mediated inhibition of HSC self-renewal in mice [16]. Finally, murine BM adipocytes can also secrete soluble factors that inhibit HSCs self-renewal [70], with recent reports suggesting that adipocytes can support murine HSCs in-vitro, while not exhibiting any effects on HSCs in vivo [71]. Taken together, data indicate that various cytokines and growth factors derived from the BM niche are able to regulate HSC self-renewal and differentiation (table 1), but further functional characterization will be required.
Wnt Signaling and HSC Self-Renewal
Wnt signaling is known to act in a very context-dependent manner and might also be involved in regulating murine as well as human HSC self-renewal (reviewed in [72]). Expression of constitutively active β-catenin, a component of the canonical Wnt pathway, resulted in enhanced murine HSC self-renewal [73] and accordingly, Wnt3A proteins have been shown to increase murine HSC self-renewal ex vivo [74]. Mice lacking Wnt3a die prenatally and deficiency of Wnt3a has been found to impair HSC self-renewal, as evidenced by reduced reconstitution capacity of fetal liver HSCs [75]. Moreover, exogenous Wnt3a has been shown to cause reduced murine HSC proliferation relative to cells treated with TPO, but can lead to higher long-term reconstitution, suggesting an enhanced ability for self-HSC renewal [76]. Others have reported that disrupted secretion of Wnt ligands by genetic deletion of the Porcn factor – essential for Wnt secretion [77] – or upon deletion of β-catenin and γ-catenin does not affect adult murine hematopoiesis [78–80]; consequently, this may likely imply a context-dependent action of Wnt-signaling in hematopoiesis, but has not yet been elucidated. Specifically, HSCs from β-catenin-deficient mice have shown normal HSC counts, but yet exhibit impaired long-term growth and maintenance or support of BCR-ABL-induced chronic myelogenous leukemia (CML) [81] In other studies, constitutive β-catenin activation resulted in enforced cell cycle entry and subsequent exhaustion of murine HSCs, with induction of multilineage differentiation in vivo [82, 83]. In a compound genetic mouse model of Pten deletion and β-catenin activation in HSPCs, the number of HSCs was increased, though they exhibited defects in differentiation [84]. Additional studies revealed that stabilization of beta-catenin in stromal cells promote maintenance and self-renewal of HSCs in a contact-dependent manner, whereas direct stabilization in hematopoietic cells caused loss of HSCs [85]. Another mouse study using serial transplantation assays reported an increase in cell cycling, but a decline in HSC function; upon expression of the pan-inhibitor of canonical Wnt signaling, Dickkopf1 (Dkk1) in the niche (driven by an osteoblast-specific promoter (Col1α2.3), caused inhibition of Wnt signaling in HSCs [86]. Recently Dkk-1 was also found to promote murine hematopoietic regeneration in response to irradiation, acting both directly on stem cells to regulate reactive oxygen species (ROS) levels as well as on niche cells to regulate EGF levels via paracrine cross-talk between BM osteolineage cells and endothelial cells [87]. Genetic deficiencies of Flamingo (Fmi) or Frizzled (Fz) 8, members of non-canonical Wnt signaling cascade, have been found to reduce the frequency of murine HSCs in vivo [34]. In this study, Fmi regulated the distribution of Fz8 at the cell-cell interface between HSCs and N-cadherin(+) osteoblasts, as the non-canonical Wnt signaling initiated by Fz8 suppressed the Ca(2+)-NFAT- IFNγ pathway, antagonizing canonical Wnt signaling [34]; this resulted in maintenance of quiescent long-term HSCs in the niche. Consistent with such observations, ex vivo cultivation of HSCs with non-canonical Wnt5A proteins increased the HSC repopulation potential in murine transplantation experiments [88]. The role of Wnt/β-catenin signaling with respect to murine HSC self-renewal and differentiation, while already investigated to a great extent, remains complex and controversial and is likely dependent on variables such as genetic dosage and context dependency [72, 89]. Nevertheless, the data overall suggest a distinct positive role of canonical Wnt signaling initiated by the niche to mediate HSC self-renewal [90]. However, the role of Wnt proteins and Wnt regulatory factors in the stem cell niche remain to be investigated in greater detail in order to elucidate the potential of modulating Wnt signaling to achieve ex-vivo human HSC expansion.
Metabolic Regulation of HSC
HSCs also exhibit a stringent regulation of their hypoxic status [91, 92] and their metabolic [93] and mitochondrial profiles [94] (Figure 2).
Figure 2. Metabolic Regulation of Mammalian HSCs.
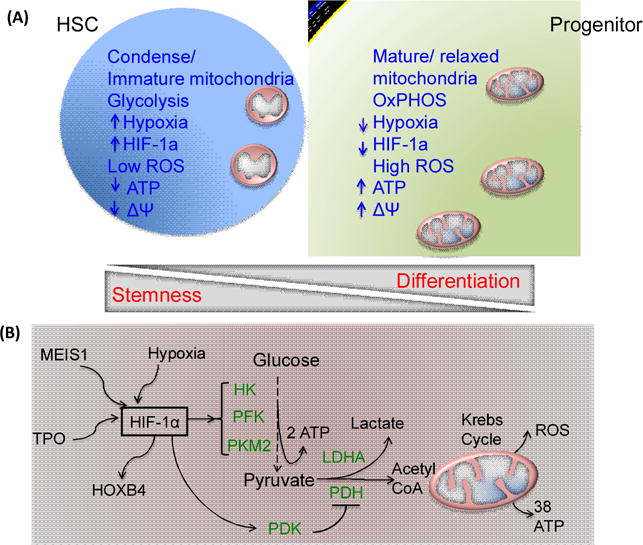
(A) Hematopoietic stem cells (HSCs) exhibit condense and immature mitochondria, low metabolic status and high glycolytic activity, as suggested by low ATP, low ROS and low membrane potential (ΔΨ) which uphold stemness of the HSCs, in contrast to progenitors and more differentiated cells that exhibit high mitochondrial activity and utilize oxidative phosphorylation (OXPHOS). Furthermore, stabilized hypoxia-inducible factor 1α (HIF1α) in HSCs can support self-renewal and stemness potential. (B) Mechanisms of metabolic regulation of HSC function. Under hypoxic conditions, cytokine TPO and MEIS1 can stabilize HIF1α to promote glycolysis by regulating glycolytic pathway enzymes including HK, hexokinase; LDHA, lactate dehydrogenase A; PFK, phosphofructokinase 2; PKM2, pyruvate kinase M2. HIF1α can also controls HOXB4 and pyruvate dehydrogenase kinase (PDK) activation that prevents pyruvate oxidation by suppressing the pyruvate dehydrogenase (PDH) complex, inhibiting oxidative phosphorylation and leading to maintenance of HSC quiescence and stemness. ROS, Reactive Oxygen Species: MEIS1, Meis Homeobox 1; TPO, thrombopoietin; HIF, hypoxia-inducible factor; HOXB4, Homeobox B4; PDK, pyruvate dehydrogenase kinase;
Recent research has identified regulatory pathways and probable links between HSC metabolism, mitochondrial function, energy demands and their role in regulating HSC quiescence and self-renewal. These pathways might possibly serve as additional novel targets for HSC expansion ex vivo. For example, the Lkb1 tumor suppressor is a kinase that functions upstream of AMP-activated protein kinase (AMPK). Deletion of Lkb1 in mice causes rapid HSC depletion due to loss of quiescence leading to pancytopenia [95–97]. Lkb1-deficient HSCs exhibit reduced mitochondrial membrane potential, alterations in lipid and nucleotide metabolism, and depletion of cellular ATP [95–97]. Furthermore, transcriptome analyses have identified decreased gene expression of the peroxisome proliferator-activating receptor (Ppar) mediated metabolic pathway in Lkb1-deficient murine HSCs in contrast to WT cells [95]. In addition, studies demonstrated a novel role for the promyelocytic leukemia (PML)-driven PPAR-δ- (fatty acid oxidation) FAO pathway in murine HSC self-renewal through regulation of cell-division symmetry, with the PML–PPAR-δ–FAO pathway being able to control the mode of HSC division [98, 99]. Indeed, loss of PPAR-δ or pharmacological inhibition of mitochondrial FAO induced loss of HSC self-renewal and loss of symmetric cell division; thus, symmetric differentiation commitment was implicated as the prevailing mode of HSC maintenance[99]. The symmetric differentiation mode of HSC division was further confirmed in murine experiments where ex vivo daughter cells from the first HSC division were transplanted into recipient animals to assess HSC function [99].
A role for metabolic regulation of HSC self-renewal was investigated in animal models where glucose intake was altered, or where HSCs harboring a genetic deletion of an enzyme involved in glycolysis were analyzed [100–102]. Specifically, using a Zebrafish embryo-to-adult transplantation model, a transient elevation in glucose levels in fish was found to accelerate the induction of functional HSCs from hemogenic endothelium, as identified from various murine HSC-reporter lines in contrast to control embryos following glucose exposure (Tg(runx1P1:eGFP), Tg(cmyb:eGFP), and Tg(CD41:eGFP)) [100]. Mechanistically, elevated glucose increased mitochondrial ROS which induced expression of hypoxia inducible factor-1α (Hif1α); this in turn, led to an increased HSC number, while pharmacological inhibition of ROS, mitochondrial ROS, and Hif1α using N-acetylcysteine, MitoQ or Dimethyloxallyl glycine (DMOG) led to a decrease in HSC numbers [100]. Murine HSCs in the BM niche have been thought to utilize glycolysis rather than mitochondrial oxidative phosphorylation as they show low mitochondrial respiration and high glycolytic flux [94]; this suggests a unique metabolic requirement for HSCs which might enable these cells to adapt to low oxygen tension in the BM niche [94]. Murine HSCs also exhibit a higher pyruvate kinase activity compared to progenitors and more differentiated BM cells through a pyruvate dehydrogenase kinase (Pdk)-dependent mechanism [93]. A dependency of HSC on glycolysis has also been reported in cases where enzymes involved in aerobic glycolysis have been genetically deleted in mice. For instance, deletion of lactate dehydrogenase A (Ldha) was reported to block the number and function of both murine HSCs and progenitors upon secondary BM transplantation [102]. Others have found that by blocking glutaminolysis with 6-diazo-5-oxo-L-norleucine (DON) in vitro, or in vivo in mice, that erythroid specification of human and murine HSCs requires glutamine-dependent de novo nucleotide biosynthesis[101]. Furthermore, supplementation with nucleosides rescued erythropoiesis [101]. This suggests a broader regulatory input of metabolic pathways in terms of glycolysis and glutaminolysis on HSC self-renewal and differentiation [101]. In addition, starvation-induced metabolic stress in murine HSCs appears to be reduced by active autophagy resulting in improved HSC maintenance [103], but the role of autophagy in regulating HSC self-renewal requires further investigation. Finally, stem cell divisions can result in an asymmetric allocation of mitochondria to one daughter cell versus another; daughter cells receiving aged mitochondria will differentiate, while daughter cells receiving low amounts of mitochondria maintain stemness in human mammary stem-like cells [104] (Figure 3). These data suggest that HSC self-renewal is metabolically fine-tuned, at least with respect to glycolysis. These novel findings might open up alternate avenues to explore the potential of enhancing ex vivo human HSC self-renewal.
Figure 3. Metabolic Regulation of Mammalian HSC Self-Renewal.
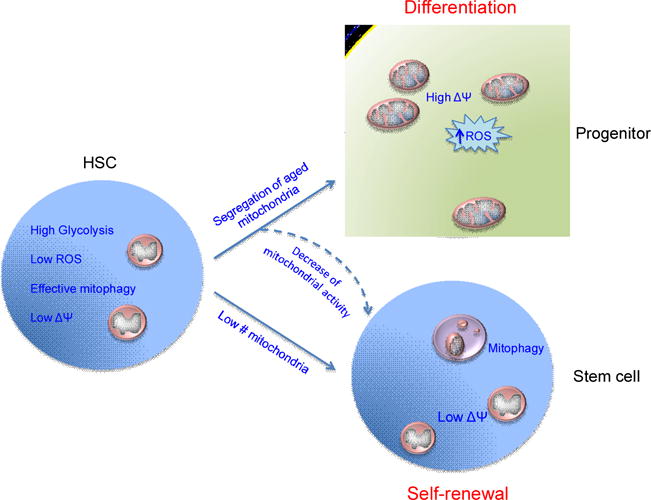
HSCs maintain low metabolic status and high glycolytic activity with low ROS and low membrane potential (ΔΨ) that helps to maintain stemness, during steady state or stress conditions. HSCs divide to produce stem and progenitor cells. Recent research has suggested that daughter cells fated to perform as progenitors through differentiation exhibit high mitochondrial activity with increased mitochondrial numbers, high membrane potential and utilize oxidative phosphorylation to produce more ROS, while daughter cells that receive low mitochondrial activity fate to perform as stem cells through self-renewal decisions. Furthermore, pharmacological modulation of mitochondrial activity using uncoupling agents or mitophagy can lead to increased stem cell self-renewal decisions.
Mitochondria, Hypoxia and ROS
Mitochondria are indispensable for energy generation. Mammalian HSCs exhibit low mitochondrial content and mitochondrial potential with reduced rates of oxygen consumption and low ATP content but higher lactate production [93, 94] (Figure 2); this suggests that HSC utilize glycolysis rather than oxidative phosphorylation. Moreover, mice devoid of Ptpmt1−/− (mitochondrial phosphatase) have shown a 40-fold increase in the number of HSCs in the BM relative to wild type animals, which has been attributed to defective HSC differentiation [105]. Consequently, it is possible that mitochondrial bioenergetics may be directly involved in the mode of HSC division. These studies further imply a distinct mitochondrial activity profile in HSCs relative to more differentiated cells – a process which may be necessary to meet the energy demands of HSCs upon activation, and to favor self-renewal over differentiation [93, 94, 105].
A prior study demonstrated that HSCs, aside from residing in hypoxic niches, can also exhibit intracellular hypoxia, expressing a stabilized form of the transcription factor Hif1α within mouse BM HSCs [92]. Under normoxia, Hif1α is hydroxylated by O2-dependent prolyl hydroxylases, followed by von Hippel-Lindau protein (VHL) and E3 ubiquitin ligase-derived Hif1α degradation [106]. However, during hypoxic conditions, Hif1α can be stabilized upon suppression of Hif1α prolyl hydroxylation [11, 106]. Stabilized Hif1α associates with Hif1β to form a transcription factor that activates the promoters of multiple glycolytic genes [11]. By contrast, Hif1α-deficient mice exhibit loss of HSC cell cycle quiescence and reduced HSC numbers upon stress, indicating that hypoxia/Hif1α-dependent regulation of HSC quiescence and self-renewal may occur [92]. A positive role of HIF-1α in HSC self-renewal has been further supported in vitro in human hematopoietic cell lines or murine primitive cells treated with either SCF [63] or TPO [107], exhibiting more stabilized levels of HIF-1α than cells without treatment. Another study also reported that HSPCs in murine femur BM, could maintain a hypoxic profile cell-intrinsically, regardless of their localization in the vicinity of vascular structures or of their cell cycle status, as evidenced from imaging cytometry revealing HIF-1α expression and reduced pimonidazole levels, (a surrogate marker for hypoxia) [39]. However, as the pimonidazole adduct is insensitive to re-oxygenation [108], further studies will be necessary to confirm the extent to which hypoxia within HSCs can be directly correlated to a hypoxic niche. Together, a niche-regulated as well as a cell-intrinsic hypoxic status have been implicated in HSC maintenance in-vivo and might be exploited during ex-vivo expansion protocols.
From another angle, mitochondrial aerobic metabolism is the main source of ROS generation in HSCs [109]. Ablation of the Polycomb repressor protein (Bmi1) in mice has led to defects in stem cell self-renewal and been mechanistically linked to impaired mitochondrial function, reduced ATP generation and increased intracellular ROS levels [110]. Indeed, ROS, at low levels, can also function in signaling [111]. Quiescent HSCs exhibit a low level of ROS that contributes to higher self-renewal potential and long-term stemness, while a higher level of ROS within HSCs or in the niche can result in loss of HSC from differentiation, proliferation or apoptosis [112–114]. Consequently, mice that lack components of the ROS regulatory system frequently present with a loss of HSC self-renewal [113–115]. For example, Ataxia telangiectasia mutated deficient (Atm−/−) mice have shown progressive BM failure (HSC), as evidenced by from failed BM reconstitution and a decline in hematocrit levels in old mice relative to young, which has been attributed to elevated ROS levels in HSCs [115], which in addition, lead to p38 activation [116]. Moroever, forkhead transcription factors (FoxO) that act downstream of the Pten/PI3K/Akt pathway and are activated in response to ROS, have been found to be critical for HSC self-renewal in mice, specifically for the in vivo maintenance of the HSC pool that depends on self-renewal [113, 114]. Indeed, FoxO1/3/4 triple knockout (KO) mice exhibited a reduced HSC pool size, and HSCs exhibited defective long-term repopulation as well as increased cell cycling and apoptosis associated with high ROS [114]. Similarly, the BM of FoxO3a KO mice exhibited decreased HSC repopulation potential, as well as defective maintenance of quiescence associated with elevated ROS, leading to p38 activation [113]. In this study. the antioxidant N-acetyl-L cysteine decreased p38 activation while inhibition of p38 restored the colony-forming capacity of FoxO3a KO Lin−Sca-1+c-Kit+ (LSK) cells, at-least in vitro [113]. These data support the notion that appropriate levels of ROS and antioxidant enzyme activity may be critical for the regulation of HSC quiescence, self-renewal and differentiation [117, 118]. Thus, it is possible that the pharmacological modulation of ROS concentrations as well as of signaling pathways regulated by ROS in HSCs might facilitate HSC expansion ex vivo, though this remains to be tested.
HSC Expansion Ex Vivo: Status and Perspective
HSCs undergo massive expansion in numbers in vivo during the process of hematopoietic reconstitution after stress, such as from an infection, lipopolysaccharide challenge, chemotherapy, radiation or transplantation [4, 30, 31], but which still cannot be recapitulated by ex vivo expansion approaches. Successful protocols for the expansion of both HSCs as well as hematopoietic progenitor cells ex vivo are warranted in the clinic, as higher numbers of progenitors and HSCs in transplants provide more short-term progeny required for better cell survival and at the same time generate more robust long-term reconstitution [119]. HSC ex vivo expansion efforts are primarily based on protocols to expand both murine LSK cells, (containing stem and progenitor cells) and human CD34+ cells (also containing both stem and progenitor cell populations). High-throughput screening of chemical compound libraries (see below) has resulted in a few successful attempts towards human HSC expansion ex-vivo [120, 121] (table 2). In addition, protocols for bioengineering HSC niches using extra-cellular matrix components and 3D cultures have been established for human and mouse HSC expansion. In this section, we discuss these recent findings and other niche-informed approaches for HSC expansion ex vivo that aim to conserve the functional and molecular characteristics of HSCs.
Table 2.
Successful Protocols of HSC Expansion Ex-Vivo
| Factors | Cells tested | Species | Supplement | Culture period | Assay | Fold expansion | References |
|---|---|---|---|---|---|---|---|
| Cytokine derived expansion | |||||||
| Cytokines | CB CD34+38− | H | SCF, Flt3L, G-CSF, IL-3, IL-6 | 4 days | CFU & CRU (HSC frequency) | 15 fold CFU/4 fold chimera | [128] |
| Cytokines | CB CD34+38− | H | flt-3, SCF, IL-3, Il-6 and G-CSF | 5–8 days | CFU, LTC-IC, CRU (HSC frequency) | 100-fold CFU/4-fold LTC-IC, 2-fold CRU | [131] |
| Angiopoietin | SP CD45+ Sca-1+ | M | SCF, TPO, FGF-1, IGF-2 | 10 days | CRU (HSC frequency) | 24–30-fold | [130] |
| Pleiotrophin | CD34− LSK, CB CD34+ 38− | M & H | SCF, Flt3L, TPO | 7 days | CRU frequency/LT engraftment | 4 fold CRU/10 fold chimera | [69] |
| Virus mediated overexpression, TFs, etc | |||||||
| HoxB4 | CB CD34+ | H | Co-culture on MS-5 mouse stromal cells | 4–5 weeks | LTC-IC assay, reconstitution analysis | 20 fold LTC-ICs/2.5 fold Long term repopulation | [145] |
| Fbxw7 | LSK | M | SCF, TPO | 10 days | Competitive reconstitution analysis | >2 fold Long term repopulation | [147] |
| Dppa5 | CD34–48− LSK | M | SCF, TPO | 14 days | Competitive reconstitution analysis | 6–10 fold | [154] |
| Hypoxia, ROS and metabolic modulations | |||||||
| PDH inhibitor [1-aminoethylphos phinic acid (1-AA) | CD34-Flts-LSK | M | SF-O3 medium 1.0% BSA, Serum free, SCF, TPO | 2–4 Weeks | CFU/Competitive reconstitution analysis | 2 fold CFUs/5 fold LT repopulation | [93] |
| Mitochondrial phosphatase Ptpmt1 inhibitor, alexidine dihydrochloride (AD) | LSK & In vivo treatment CD34+ 38− |
M H |
SCF, TPO, Flk-3 | 7 days | CFU/and LT Chimera 34+38− number/CFU |
2 fold CFUs/3–5 fold LT repopulation 2 fold number/2 fold CFUs |
[150] |
| GSK-3β inhibitor, CHIR99021 | LSK & CB CD34+ LSK Flk− |
M & H M |
With Rapamycin in cytokine free X-VIVO medium SCF, TPO & insulin |
7 days 14 days |
HSC frequency/LT engraftment Competitive reconstitution analysis |
10–20 fold HSC number, 2–5 fold LT repopulation 100 fold number, 2–10 fold LT repopulation |
[135] [84] |
| High Throughput Chemical screens | |||||||
| SR-1 | CD34+ MPB UBC | H | SCF, Flt3L, TPO, IL-6 | 7–21 days | Number/CFU/Competitive reconstitution analysis | 65 fold CFUs/17 fold enhanced chimera | [120] |
| UM171 | CD34+ MPB UBC | H | SCF, Flt3L, TPO | 7–21 days | Number/CFU/Competitive reconstitution analysis | >100 fold LT-HSC/35 fold enhanced chimera | [121] |
CB, cord blood; SCF, stem cell factor; TPO, thrombopoietin; TGF-β1, transforming growth factor beta 1; CXCL-4, CXC chemokine ligand 4; CXCL-12, CXC chemokine ligand 12; G-CSF, granulocyte-colony stimulating factor; IL-3, interleukin 3; OPN, osteopontin; Flt3L, FMS-like tyrosine kinase 3 ligand; FGF-1, fibroblast growth factor 1; IGF-2, Insulin-like growth factor 2; MS-5, murine MS-5 stromal cell line; SP, side population; Fbxw7, F-box and WD repeat domain containing 7, E3 ubiquitin protein ligase; Dppa5, developmental pluripotency associated 5; SR-1, StemRegenin1; MPB, mobilized peripheral blood; UCB, umbilical cord blood; CFU, colony forming unit; CRU, competitive repopulating unit; LT, long-term; LTC-IC, long-term culture initiating cell; TFs, transcription factors.
High-throughput Screening of Compounds for Expansion
A major break-through in ex vivo expansion of HSCs was achieved by the Cooke laboratory [120]. They utilized high-throughput technology for an unbiased screen to search for factors that could expand human HSCs ex vivo, using a library of 100,000 small molecules and serum-free expansion medium containing TPO, SCF, Flt3L and IL-6 [120]. A purine derivative, StemRegenin 1 (SR1), was found to promote a 50-fold ex vivo expansion of human cord blood-derived CD34+ cells and a 17-fold increase in the number of human HSCs engrafting long-term in immunodeficient mice [120]. SR1 antagonizes the aryl hydrocarbon receptor (AHR) [120]. Recently, a clinical study using SR-1 demonstrated a remarkable early neutrophil and platelet recovery and better engraftments in human recipients that received umbilical cord blood CD34+ cells treated for 15 days ex vivo with SR-1 compared to recovery in recipients that received equal “start” numbers of CD34+ cells from the same unit, but not expanded [122]. A recent study from the Sauvageau laboratory showed that a pyrimidoindole derivative called UM171 induces human HSC self-renewal and ex vivo expansion in an AhR-independent manner, given that the expression of AhR targets AhRR and CYP1B1 remained un-altered upon UM171 treatment [121]. A library of 5280 low-molecular weight compounds and 300 analogs were screened to identify UM171. UM171 resulted in a better expansion of more primitive human CD34+CD45RA cells from mobilized peripheral blood (mPB) than SR1[121]. Consequently, UM171 and SR1 may represent promising chemical compounds for ex vivo expansion of human HSCs. Of note, they do not act on murine HSCs [120, 121]. However, the mechanisms of SR-1 or UM171-mediated HSC self-renewal and differentiation block are not known. We speculate that given their putative role in determining the mode of HSC division, these might interfere with either the regulation of ROS, or the mitochondrial or metabolic function that allows HSC self-renewal and expansion.
Reliance on Cytokines and Growth Factors
Cytokines were among the first drugs tested for HSC ex vivo expansion (table 2). As mentioned, numerous cytokines have been shown to influence murine HSC numbers at least in vivo [1, 58, 64–66, 69, 123–130]. Several cytokines singly, or in combination, have been investigated for their effect on murine and human HSC cultures and expansion ex vivo; however, only a maximum of 2–4 fold expansion of murine and human HSCs with long-term repopulation potential has been achieved (see also [126]). One study reported a modest increase of a 4- and 10-fold in the number of human cord blood (CB) CD34+CD38− cells and colony forming units (CFUs), respectively, as well as a 2- to 4-fold increase in SCID-repopulating cells (SRC) frequency in NOD/SCID mice after 4 days (d) of culture with cytokines (SCF, Flt3L, G-CSF, IL-3, IL-6) [128]. However, after 9 d of culture, despite further increase in the total number of CD34+ cells, the reconstitution ability was lost [128]. In another study, human CB CD34+CD38− cells in serum-free medium containing Flt-3, SCF, IL-3, IL-6 and G-CSF for 5–8 d resulted in robust 100-fold CFU expansion, 4-fold LTC-IC, and 2-fold increase in the competitive repopulating unit (CRU) [131].
Overall, these cytokines and their combinations can maintain HSCs and progenitors and most likely protect them against apoptosis during ex vivo proliferation, as evidenced from flow cytometric assays [66, 132], though only resulting in a modest expansion of human HSC, albeit a multilog expansion of progenitor cells [126, 132]. Thus, additional factors are clearly necessary for successful expansion of human HSCs ex vivo. Newly identified factors in stroma-conditioned medium, such as nerve growth factor and collagen 1, have resulted in a better expansion of murine HSCs compared to the “standard” cocktail listed above [133]. This suggests that the stroma still harbors additional yet-to-be-determined factors that alone, or in combination, might result in significant ex vivo expansion of HSCs, and representing an exciting novel area of research to improve HSCs expansion ex vivo.
Several laboratories have also tested the activation of Wnt signaling for stem cell expansion. The Reya laboratory reported an 8–80-fold expansion of functional murine HSCs upon short-term culture in serum-free medium supplemented with low concentrations of cytokines (SLF, Flt-3L, IL-6), transducing HSCs with constitutively active β-catenin through up-regulation of the self-renewal gene homeobox B4 (HoxB4) [73]. Moreover, short-term pretreatment of cells with a GSK-3β inhibitor (6-bromoindirubin 3′-oxime or BIO) that activates β-catenin was demonstrated to enhance the engraftment of ex vivo-expanded human cord blood CD34+ HSCs in murine xenograft models [134]. Another study reported that a pharmacological and thus reversible activation of both Wnt/β-catenin and PI3K/Akt signaling in HSCs using another type of GSK-3β inhibitor (CHIR99021) in combination with cytokines (SCF, TPO) and insulin significantly expanded (~100 fold) the number of HSCs [84] after 14 d′ cultivation of murine HSCs in serum-free medium, suggesting that cooperation between these pathways might be beneficial for HSC self-renewal and expansion [84]. Cultivation of human CD34+ cells as well as murine LSK cells in cytokine-free medium for 7 d in the presence of rapamycin (inhibition of the mammalian target of rapamycin (mTOR) pathway) and CHIR99021 (activation of canonical Wnt) resulted in the maintenance in the number of human and murine HSCs, as confirmed in serial transplantation assays [135]. Also, when provided in excess, prostaglandin E2 (PGE2) – which modifies the Wnt signaling cascade at the level of β-catenin degradation [136] – has been shown to result in an increase in HSC numbers in zebrafish and mouse embryos [137]. Treating HSCs with PGE2 increased the number of human CFUs in vitro, and enhanced the engraftment of unfractionated and human cord blood CD34+ HSPCs upon xenotransplantation [138], which may supports a role for Wnt-signaling in HSC expansion. Another study used an automatic fed-batch media dilution approach to control inhibitory feedback signals during culture of human cord blood HSPCs; this led to an 11-fold expansion of SCID repopulating cells with self-renewing, multilineage repopulating ability [139], implying a critical role for inhibitory feedback loops in mitigating HSCs expansion ex-vivo.
Regulation of ROS, Antioxidants and Hypoxia
Cytokines such as GM-CSF, IL-3, SCF, and TPO have been shown to increase murine and human HSC proliferation through a rapid increase in the level of ROS in quiescent cells [118]. Specifically, elevation of ROS have induced HSC-specific phosphorylation of p38 MAPK upon culture in serum-free media supplemented with cytokines including SCF, IL-3 and EPO, while antioxidant treatment or inhibition of p38 MAPK ex vivo has rescued ROS-induced defects in HSC repopulating capacity, preventing exhaustion of murine HSCs in serial transplantation experiments [116]. These data suggest that p38 MAPK or ROS inhibition in ex vivo cultures might be able to contribute to HSC expansion. In addition, a role of hypoxia and fine-tuned regulation of HIF-1α stabilization in HSC maintenance have been established by both biochemical and genetic approaches [91, 92, 140, 141]. For instance, mouse BM cells cultured under hypoxia have shown 5-fold higher day-14 spleen colony-formation efficiency as well as enhanced radio-protection ability than under normoxic conditions [140], suggesting better maintenance and expansion of HSCs. Upon cultivation under hypoxic conditions, murine HSCs have also been shown to accumulate at the G0 stage of the cell division cycle which results in an increase in HSCs with long-term engraftment potential relative to non-hypoxic conditions [141]. Furthermore, hypoxia induces Hif1α dependent expression of the cell cycle regulators p21, p27 and p57 in murine HSCs [141]. Isolating and manipulating murine BM and human cord blood under strict hypoxic conditions in vitro has demonstrated a higher number of HSCs recovered from the BM under these conditions [142], while a brief exposure to ambient oxygen was found to decrease the number of HSCs upon BM harvest, through an extraphysiologic oxygen shock/stress (EPHOSS) mechanism [142]. Together, it is therefore possible that maintaining a strict hypoxic environment might be beneficial for ex vivo expansion of HSCs but robust validation of this hypothesis is still warranted.
Retrovirus-Mediated Introduction of Stem Cell Regulators and Reprogramming
Multiple approaches have been reported for ex vivo HSC expansion based on retrovirus-mediated expression of HSC maintenance or expansion genes. For example, overexpression of HoxB4 expands murine HSCs approximately 40–1000 fold in vitro and in vivo respectively [126, 143, 144] without stem cell transformation. Human cord blood CD34+ HSCs have been expanded approximately 2.5-fold using a HoxB4 fusion protein expressed by the stromal cell line MS-5 [145]. A challenge remains as the HoxB4 protein is unstable in culture when provided extrinsically, as in the previous approach. TPO also positively regulates HoxB4 expression in murine and human hematopoietic cell lines [146], which might explain in part, the beneficial effect of TPO on HSC maintenance ex vivo, although this remains speculative. The expression of the ubiquitin-ligase, F-box and WD-40 domain protein 7 (Fbxw7) that mediates degradation of cell-cycle activators in HSCs is up-regulated by hypoxia [147]. As such, overexpression of Fbxw7 in murine LSK cells has been reported to cause >2 fold higher reconstitution capacity during ex vivo culture by maintaining HSC quiescence through a reduction in expression of c-Myc, Notch1 and mTOR [147]. While the transduction of HSCs with the above mentioned regulators expand HSCs to small but distinct levels, there is a risk of insertion-mediated oncogene activation (as with all transgenic approaches), which will most likely preclude the application of these current approaches in the clinic.
Recently, two studies have described the generation of functional HSCs via reprogramming from adult endothelium and human pluripotent stem cells [148, 149], introducing novel exciting technologies into the field of HSCs generation. Adult mouse endothelial cells were fully reprogrammed to hematopoietic stem cells (rEC-HSCs) through transient ectopic expression of four transcription-factors (Fosb, Gfi1, Runx1, and Spi1; FGRS) and vascular-niche-derived angiocrine factors from a feeder layer [148]. This fidning was interesting because rEC-HSCs presented a transcriptome profile and long-term self-renewal capacity similar to those found in adult HSCs [148]. Furthermore, multi-lineage reconstitution was achieved in both primary and secondary bone marrow transplantation settings [148]. In another study, human pluripotent stem cells were first directed towards the hemogenic endothelium using chemical signals, and seven transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1, and SPI1) pushed the hemogenic endothelium towards a blood-stem-cell state that provided in vivo multilineage reconstitution in a xenograft mouse model [149]. These findings provide significant advancement in generating HSCs that do not involve altering the mode of division of existing HSCs, but rather, involve generating from “scratch” unlimited HSC numbers. Nevertheless, the translational potential of these approaches and newly generated cells currently remains limited due to the use of reprogramming factors bearing oncogenic potential.
Targeting Metabolic Pathways
HSC metabolism has also been targeted to investigate HSC expansion, but with mixed success. When glycolysis was favored in murine HSCs using a pyruvate dehydrogenase (PDH) inhibitor [1-aminoethylphosphinic acid (1-AA)], the cycling and colony growth of HSCs was suppressed during ex-vivo culture, while HSC frequency and reconstitution ability was maintained even after 4 weeks of culture [93]. Alexidine dihydrochloride (AD) inhibits mitochondrial phosphatase Ptpmt1 [150] and can shift mitochondrial aerobic metabolism to glycolysis through AMPK [150]. Thus, treatment of murine LSK cells with AD under normoxic conditions increased their transplantation efficiency about 3-fold in competitive transplant settings relative to untreated control cells [150]. Consequently, it will be interesting to test if there is a synergistic effect of hypoxia and treatment with AD with respect to HSC expansion. Recent reports demonstrate that chemical uncoupling of the electron transport chain, which decreases the mitochondrial activity, resulted in increased murine HSC self-renewal under ex-vivo culture conditions generally causing rapid differentiation [151]. In general, active mitophagy appears to be a mechanism necessary for directing HSCs towards self-renewal from differentiation (at least in mice) [152] (Figure 3). Indeed, murine HSCs have been demonstrated to exhibit high mitophagy function via the PPAR-FAO pathway, preferentially undergoing symmetric divisions to self-renew. Accordingly, GW501516, a PPAR-FAO agonist, can enhance LTC-IC frequency via mitophagy activation in human HSCs [152]. Altogether, these studies suggest that targeting the “metabolic switch” to enhance HSC glycolysis during an ex vivo culture might potentially enhance ex vivo self-renewal and perhaps even HSC expansion, a hypothesis awaiting robust validation.
Targeting ER Stress Pathways
HSCs can encounter diverse types of stress such as elevated ROS and DNA damage, but also endoplasmic reticulum (ER)-dependent stress stemming from the unfolded protein response (UPR). Indeed, recent work demonstrates that an overall appropriate response to ER stress from unfolded proteins can support HSC maintenance, self-renewal and expansion [153–155]. For example, human HSCs, but not progenitors, are highly predisposed to undergo apoptosis through a PERK-mediated UPR to ER stress, while overexpression of the co-chaperone ERDJ4 (that increases ER protein folding) has been found to enhance human HSC engraftment in a mouse xenograft model [153]. Moreover, in contrast to BM HSCs, fetal liver (FL) HSCs undergo a very rapid expansion in vivo, and despite increased protein synthesis rate, they do not exhibit ER stress [155]. Instead, taurocholic acid, the major maternal and fetal liver bile acid (BA) form has been shown to serve as a chemical chaperone that can inhibit protein aggregation and support HSC growth in mice [155]. Such recently identified chaperones might thus comprise a novel class of compounds to be tested in ex vivo expansion approaches for adult HSCs. Developmental pluripotency-associated 5 (Dppa5), an RNA binding protein, is highly enriched in HSCs [154]. Murine HSCs that ectopically expressed Dppa5 have been reported to robustly increase their reconstitution potential in transplantation experiments, reducing (ER) stress and apoptosis during ex vivo culture for 14 days [154]. Correspondingly, tauroursodeoxycholic acid (TUDCA), a chemical chaperone that reduces ER stress, was shown to enhance HSC engraftment approximately 5-fold in this mouse model [154]. In general, these studies could indicate that minimizing stress to the ER might potentially contribute to successful HSC expansion ex vivo.
Extra-cellular Matrix and Niche Engineering
Niches in the BM provide, besides soluble factors, specific extracellular matrix (ECM) components and structural 3D architectures [8, 156, 157]. Several polymeric biomaterial substrates that mimic the structure of the ECM have been explored with respect to their function to enhance HSC expansion. Diverse ECM substrates including polyethylene terephthalate (PET), tissue culture polystyrene (TCPS) and polyether sulfone (PES) (BOX 2) have failed to enhance HSCs expansion ex vivo [158]. However, fibronectin-coated PET has shown elevated expansion in the number of human HSCs ex vivo in contrast to unmodified biomaterials [159, 160]. Similarly, aminated-PES substrates and cytokines (SCF, Flt3, TPO and IL-3) have been reported to support a 3- to 4-fold expansion of human CD34+ HSCs derived from umbilical cord blood when compared to tissue culture polystyrene [161, 162]. Moreover, cultivation of human HSCs with BM mesenchymal stem cells (MSCs) in the absence of additional cytokines has resulted in a 5- to 7-fold increase in the number of LTC-ICs, when compared to HSCs cultivated in the absence of MSCs [163]. MSCs might thus provide niche components, including soluble cytokines, that support HSC expansion ex-vivo.
BOX 2. Extracellular Matrix Modeling and HSC Niche Reconstitution.
The extracellular matrix (ECM) consists of, among others, collagen, fibronectin, dystroglycan, heparin sulfate, proteoglycans, osteopontin and laminin [56, 158, 159, 169, 176] and can bind adhesion molecules such as integrins, on HSCs. Several polymeric biomaterial substrates such as polyethylene terephthalate (PET), tissue culture polystyrene (TCPS) and polyether sulfone (PES) fibers exhibit the advantage of a defined composition, surface chemistry, and toxicity profile. Recent evidence further suggests that substrate elasticity can influence self-renewal versus differentiation outcomes of murine and human HSC divisions ex vivo [156, 164, 165]. For example, the tropoelastin substrate can enhance HSC self-renewal through mechanotransduction machinery; blockage of mechanotransduction using myosin II inhibition abrogated tropoelastin-induced expansion effects [165]. Currently, the field focuses on developing 3D biomaterials of low-density with open-cell foam structure scaffolds and distinct levels of elasticity, using stromal cells to support HSC expansion and which could be adopted as analogs of the trabecular bone [170, 176]. Novel approaches in the area of 3D ECM research for stem cell expansion also include microfluidic trap devices for capturing individual HSCs to perform post-culture single-cell analysis [177].
Various studies have demonstrated that elasticity, dimensionality and topography of the matrix positively influences HSC proliferation and expansion [156, 164, 165]. Specifically, cultivation of mouse or human primitive hematopoietic cells on a tropoelastin substrate has led to a 2 to 3-fold expansion of HSCs compared to cultivation on bare tissue culture plates due to changes in substrate elasticity [165]. 3D collagen-coated porous reticulated polyvinyl formal (PVF) resin scaffolds with low oxygen have also led to murine HSC expansion over three weeks in the presence of BM stromal cells without exogenous cytokines [166]. Furthermore, 3D PVF resin scaffolds that produced an oxygen gradient, as opposed to a constant hypoxic environment, mimicked key features of marrow physiology [91] leading to 3-fold higher expansion of primitive CD34+ cells under a 3D setting, in contrast to a 2D culture systems [167]. Accordingly, 3D culture systems (e.g., nonwoven porous carriers, macroporous collagen carriers and porous microspheres such as PET and collagen) have resulted in a 3-fold increase in human HSC self-renewal when compared to 2D cultures, which was further enhanced 7-fold in the presence of TPO and Flt-3 ligand supplementation [168]. In addition, a fibronectin-immobilized 3D PET scaffold has led to a remarkable 100-fold expansion of human HSCs [160]. A high immuno-phenotypic expansion (1014 vs 106 input cells) of cord blood CD34+ HSCs was also observed with fibrin scaffolds in the presence of human umbilical cord (UC)-MSCs and cytokine supplementation [SCF, TPO, FGF-1, angiopoietin like-5 (Angptl-5), insulin-like growth factor binding-protein 2 (IGFBP2), and heparin] following 14 d of culture, which was mirrored by a high long-term reconstitution ability (58.5%) in murine xenotransplantation models [169].
The Blau laboratory utilized a hydrogel microwell array for rapid analysis of murine HSC proliferation kinetics which correlated well with subsequent serial long-term blood reconstitution in mice in vivo [76]. In such assays, Wnt3a resulted in slow HSC proliferation compared to several other tested proteins such as TPO and IL-11, which led to higher long-term reconstitution, suggesting that Wnt3a might potentially enhance HSC self-renewal, while TPO and IL-11 induced robust proliferation as well as differentiation [76]. Others have described a bone marrow-on-a-chip platform to replicate murine BM niche-like analogs for HSC in vitro cultures [170]. To generate an artificial niche, they combined demineralized bone powder and BMP2/4 into a collagen scaffold, which was subcutaneously transplanted into mice to form into a bone encased marrow compartment containing hematopoietic cells. This engineered bone marrow (eBM), when used in a microfluidic device ex vivo, retained the number of HSCs after one week of culture [170]. This system may represent a promising novel platform for screening diverse drugs for ex-vivo HSC expansion in an in vivo-like artificial niche setting. Collectively, the data suggest that novel ECM and niche engineering approaches in the presence of stromal cells might support HSC self-renewal and expansion ex vivo (Box 2).
Concluding Remarks
Over the last decade, several novel studies have suggested that HSC expansion ex vivo might actually be feasible. This is based on a better understanding of HSC-intrinsic as well as niche-specific factors regulating HSC self-renewal in vivo, and which have led to novel putative strategies to expand HSCs ex vivo. Significant advancements in HSC expansion have also been made recently in high-throughput screening approaches of low-molecular weight compound libraries. Ultimately, a combination of 3D scaffolds mimicking the niche 3D architecture, mixed with cytokines/chemokines and stromal cells under the right oxygen and metabolic conditions might be a solid option for achieving robust expansion of adult HSCs ex vivo. However, many mechanisms of HSC regulation in the niche remain poorly understood (see outstanding questions and box 3) and extensive and robust validation of these platforms will be required as a first essential step for clinical translation. Nevertheless, various modalities, strategies and methodologies will undoubtedly emerge for HSC expansion in the near future, and it will be quite exciting to follow such advances in stem cell research.
Outstanding Questions Box.
What is the relative contribution of cellular (stem cell intrinsic) and niche factors (stem cell extrinisic) towards the regulation of HSC self-renewal?
What is the role of a hypoxic environment on HSC expansion in vivo, and what is the best model to use ex vivo?
Can ROS/stress accumulation usually associated with HSC proliferation ex vivo be efficiently targeted/suppressed?
Can we harvest novel knowledge on HSC metabolism for HSC expansion strategies?
Do novel compounds such as SR1 and UM171 that result in HSC expansion also regulate HSC metabolism? if yes, how?
Which molecular mechanisms are triggered by engineered 3D extra-cellular matrix environments to modulate HSC self-renewal ex vivo?
Box 3. Clinician’s Corner.
A recent clinical trial of ex-vivo expanded umbilical cord blood CD34+ cells used SR-1 and demonstrated better engraftment and improved early recovery of leukocytes, suggesting that ex-vivo expansion of HSCs might be potentially achievable [120–122].
Similarly, co-cultures of cord blood CD34+ cells with mesenchymal stromal cells has led to expansion of CD34+ cells by a median factor of 30.1, improving time to neutrophil engraftment at 15 days, as compared to 24 days in recipients who received un-manipulated cord blood CD34+ cells [178].
Growing evidence indicates that targeting metabolism/mitophagy and cellular stress for HSC expansion might potentially lead to successful HSC expansion approaches for transplantation therapies in the future [150, 152].
Trends.
Hematopoietic stem cell niches have been defined in more detail with respect to their cellular composition over the last couple of years. Molecular characterization of these niches has been successfully initiated.
Molecular pathways that govern the mode of division of HSCs are being discovered.
HSC intrinsic mechanisms, such as metabolic pathways regulating HSC self-renewal can be modulated by the environment and/or niche factors.
High-throughput screening of chemical compound libraries has resulted in a few successful attempts towards HSC expansion ex-vivo.
Attempts to engineer stem cell niches ex vivo in 3D matrix culture systems are promising.
Acknowledgments
S.K. is currently supported by a Ramalingaswami fellowship, India. H.G. is supported by NIH grants HL076604, DK077762, AG040118, and the DFG SFB 1074 and 1149, the BMBF and the Excellence program of the Baden-Württemberg Foundation.
Glossary
- Allograft transplant
Stem cells from an HLA matched donor are transplanted
- Asymmetric Cell Division
leads to the generation of two cells with different potentials: a daughter stem cell and a daughter progenitor cell
- Ataxia telangiectasia mutated deficient (Atm−/−) mice
Atm regulates reprogramming efficiency and genomic stability, Atm−/− mice exhibit pancytopenia, BM failure and HSC exhaustion
- Autologous transplant
A person’s own stem cells are collected in advance and transplanted to herself/himself after chemotherapy or radiation therapy
- Autophagy/Mitophagy
Process that degrades/destructs dysfunctional components of the cytoplasm (autophagy) or dysfunctional mitochondria (mitophagy) in lysosomes
- CD34+ cells
human cells expressing CD34 which include both stem and progenitor cell populations
- Cell-intrinsic
property of cells governed/regulated by signaling/factors within, but not through the niche environment
- Competitive transplant settings
transplantation of donor HSCs or BM cells in the presence of genetically trackable congenic competitor BM cells
- Differentiation
decision to generate progenitors from stem cells necessary to produce mature blood cells
- Endosteal
HSC niche in close association to a bone surface
- HSC exhaustion
a state of turnover, being ‘used up’; HSCs can undergo exhaustion due to a high demand of reconstitution under stress or serial transplant settings.
- Extracellular Matrix (ECM)
The ECM includes among other elements, collagen, fibronectin, dystroglycan, heparin sulfate, proteoglycans, osteopontin and laminins
- Hemogenic endothelium
a subset of endothelial cells with the potential to differentiate into hematopoietic cells
- Hematopoiesis
The process of blood cell formation from HSCs
- HSC expansion
The process of increasing the number of HSCs
- HSC niche
A specific BM environment that provides cellular, chemical and molecular constituents and contributes to the regulation of HSC survival, self-renewal and differentiation
- Hematopoietic stem cell transplantation (HSCT)
A procedure to replenish the blood system of a recipient by providing a sufficient number of new HSCs cells from a donor
- Human leukocyte antigen (HLA)
encodes the major histocompatibility complex (MHC) proteins in humans and functions as a determinant of transplant rejections
- Infused donor HSCs
intravenously injected HSCs from a donor into a recipient to reconstitute hematopoietic system
- Long-term (LT)-HSC
give rise to multi-lineage engraftment post-transplantation for a time-frame of at least 20 weeks. LT-HSCs are phenotypically characterized as Lin−IL-7α−Sca-1+c-Kit+Flt3−CD34−CD150+CD48− cells
- Long-term culture-initiating cells (LTC-ICs)
Primitive hematopoietic cells capable of initiating and sustaining in vitro cultures for >5 weeks, including colony forming cells (CFCs) or cobblestone area forming cells
- LSK cells
distinct fraction of murine hematopoietic stem and progenitor cells in BM, characterized as Lin−Sca-1+c-Kit+ based on surface marker expression
- mTOR pathway
controls nutrient sensing, metabolism and mitogenic signals to regulate cell quiescence, proliferation, cell survival, and longevity; important for PI3K, AKT and insulin signal transduction pathways
- NOD/SCID mice
Nonobese diabetic (NOD)-severe combined immunodeficiency (SCID) mice present with impaired T and B cell lymphocyte and NK cells. They can accept allogeneic and xenogeneic grafts and are thus an excellent model system to study human cell transplantation and engraftment (xenotransplants)
- Pancytopenia
reduction in the number of all three blood cell types: red blood cells, white blood cells and platelets
- Pimonidazole
nontoxic exogenous 2-nitroimidazole small compound that forms adducts with thiol groups in hypoxic environments and works as an effective and nontoxic hypoxia marker
- Quiescence
state of being inactive or dormant in the cell division cycle
- Radio-protection ability
the capacity to confer long-term survival after lethal irradiation (e.g. mice)
- Serial transplantation assay
regarded as the gold standard assay to determine HSC function in vivo. Serial (multiple, consecutive, up to 6) transplantations (e.g. in mice), test the ability of HSCs to undergo self-renewal in vivo
- SCID-repopulating cells (SRC)
Human HSCs capable of long-term reconstitution in immunodeficient mice (xenotransplant approach)
- Self-renewal
cell division producing two daughter stem cells
- Sinusoids
Small blood vessel capillaries of irregular tubular space for blood passage within the BM. HSCs can reside near the sinusoid networks that present a sinusoidal niche
- Symmetric Cell Division
leads to the generation of two similar types of daughter cells: either two stem or tow progenitor cells
- Transplantation assay
well-established assay to measure multi-lineage reconstitution and self-renewal potential of hematopoietic stem and progenitor cells in irradiated recipient mice in vivo.
- Xenografts
A transplantation setting where a tissue graft or organ transplant is from a donor of a different species than the recipient, e.g. human stem cell transplantation into mice. Generally, immunodeficient mice, i.e. SCID, NOD/SCID or NOD/SCID/Ycnull (NSG) mice are used as recipients in human-mouse xenograft models
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Seita J, Weissman IL. Hematopoietic stem cell: self-renewal versus differentiation. Wiley Interdiscip Rev Syst Biol Med. 2010;2(6):640–53. doi: 10.1002/wsbm.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Copelan EA. Hematopoietic stem-cell transplantation. N Engl J Med. 2006;354(17):1813–26. doi: 10.1056/NEJMra052638. [DOI] [PubMed] [Google Scholar]
- 3.Notta F, et al. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333(6039):218–21. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 4.Osawa M, et al. Long-term lymphohematopoietic reconstitution by a single CD34− low/negative hematopoietic stem cell. Science. 1996;273(5272):242–5. doi: 10.1126/science.273.5272.242. [DOI] [PubMed] [Google Scholar]
- 5.Brunstein CG, et al. Allogeneic hematopoietic cell transplantation for hematologic malignancy: relative risks and benefits of double umbilical cord blood. Blood. 2010;116(22):4693–9. doi: 10.1182/blood-2010-05-285304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Al-Anazi KA. Autologous Hematopoietic Stem Cell Transplantation for Multiple Myeloma without Cryopreservation. Bone Marrow Res. 2012;2012:917361. doi: 10.1155/2012/917361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Remberger M, et al. Effect of Total Nucleated and CD34(+) Cell Dose on Outcome after Allogeneic Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2015;21(5):889–93. doi: 10.1016/j.bbmt.2015.01.025. [DOI] [PubMed] [Google Scholar]
- 8.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505(7483):327–34. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425(6960):836–41. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 10.Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. 2014;20(8):833–46. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ito K, Suda T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat Rev Mol Cell Biol. 2014;15(4):243–56. doi: 10.1038/nrm3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bruns I, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. 2014;20(11):1315–20. doi: 10.1038/nm.3707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimoto D, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow A, et al. CD169(+) macrophages provide a niche promoting erythropoiesis under homeostasis and stress. Nat Med. 2013;19(4):429–36. doi: 10.1038/nm.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Winkler IG, et al. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat Med. 2012;18(11):1651–7. doi: 10.1038/nm.2969. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki S, et al. Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. 2011;147(5):1146–58. doi: 10.1016/j.cell.2011.09.053. [DOI] [PubMed] [Google Scholar]
- 17.Winkler IG, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. 2010;116(23):4815–28. doi: 10.1182/blood-2009-11-253534. [DOI] [PubMed] [Google Scholar]
- 18.Mendez-Ferrer S, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Calvi LM, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–6. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 20.Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495(7440):231–5. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Acar M, et al. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526(7571):126–30. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Birbrair A, Frenette PS. Niche heterogeneity in the bone marrow. Ann N Y Acad Sci. 2016 doi: 10.1111/nyas.13016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding L, et al. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481(7382):457–62. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itkin T, et al. Distinct bone marrow blood vessels differentially regulate haematopoiesis. Nature. 2016;532(7599):323–8. doi: 10.1038/nature17624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068–74. doi: 10.1038/nature04956. [DOI] [PubMed] [Google Scholar]
- 26.Walter D, et al. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature. 2015 doi: 10.1038/nature14131. [DOI] [PubMed] [Google Scholar]
- 27.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–29. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 28.Bernitz JM, et al. Hematopoietic Stem Cells Count and Remember Self-Renewal Divisions. Cell. 2016;167(5):1296–1309 e10. doi: 10.1016/j.cell.2016.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matatall KA, et al. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep. 2016;17(10):2584–2595. doi: 10.1016/j.celrep.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iscove NN, Nawa K. Hematopoietic stem cells expand during serial transplantation in vivo without apparent exhaustion. Curr Biol. 1997;7(10):805–8. doi: 10.1016/s0960-9822(06)00341-1. [DOI] [PubMed] [Google Scholar]
- 31.Takizawa H, et al. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. J Exp Med. 2011;208(2):273–84. doi: 10.1084/jem.20101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiel MJ, et al. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–21. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, et al. Detection of functional haematopoietic stem cell niche using real-time imaging. Nature. 2009;457(7225):97–101. doi: 10.1038/nature07639. [DOI] [PubMed] [Google Scholar]
- 34.Sugimura R, et al. Noncanonical Wnt signaling maintains hematopoietic stem cells in the niche. Cell. 2012;150(2):351–65. doi: 10.1016/j.cell.2012.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ehninger A, Trumpp A. The bone marrow stem cell niche grows up: mesenchymal stem cells and macrophages move in. J Exp Med. 2011;208(3):421–8. doi: 10.1084/jem.20110132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kunisaki Y, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502(7473):637–43. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Asada N, et al. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat Cell Biol. 2017;19(3):214–223. doi: 10.1038/ncb3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fujisaki J, et al. In vivo imaging of Treg cells providing immune privilege to the haematopoietic stem-cell niche. Nature. 2011;474(7350):216–9. doi: 10.1038/nature10160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nombela-Arrieta C, et al. Quantitative imaging of haematopoietic stem and progenitor cell localization and hypoxic status in the bone marrow microenvironment. Nat Cell Biol. 2013;15(5):533–43. doi: 10.1038/ncb2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Omatsu Y, et al. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity. 2010;33(3):387–99. doi: 10.1016/j.immuni.2010.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Sugiyama T, et al. Maintenance of the hematopoietic stem cell pool by CXCL12− CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25(6):977–88. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- 42.Zhao M, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. 2014;20(11):1321–6. doi: 10.1038/nm.3706. [DOI] [PubMed] [Google Scholar]
- 43.Guidi N, et al. Osteopontin attenuates aging-associated phenotypes of hematopoietic stem cells. EMBO J. 2017;36(10):1463. doi: 10.15252/embj.201796968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taichman RS, Emerson SG. Human osteoblasts support hematopoiesis through the production of granulocyte colony-stimulating factor. J Exp Med. 1994;179(5):167782. doi: 10.1084/jem.179.5.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taichman RS, et al. Human osteoblasts support human hematopoietic progenitor cells in vitro bone marrow cultures. Blood. 1996;87(2):518–24. [PubMed] [Google Scholar]
- 46.Kiel MJ, et al. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell. 2007;1(2):20417. doi: 10.1016/j.stem.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Ma YD, et al. Defects in osteoblast function but no changes in long-term repopulating potential of hematopoietic stem cells in a mouse chronic inflammatory arthritis model. Blood. 2009;114(20):4402–10. doi: 10.1182/blood-2008-12-196311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu J, et al. Osteoblasts support B-lymphocyte commitment and differentiation from hematopoietic stem cells. Blood. 2007;109(9):3706–12. doi: 10.1182/blood-2006-08-041384. [DOI] [PubMed] [Google Scholar]
- 49.Bromberg O, et al. Osteoblastic N-cadherin is not required for microenvironmental support and regulation of hematopoietic stem and progenitor cells. Blood. 2012;120(2):303–13. doi: 10.1182/blood-2011-09-377853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kiel MJ, et al. Hematopoietic stem cells do not depend on N-cadherin to regulate their maintenance. Cell Stem Cell. 2009;4(2):170–9. doi: 10.1016/j.stem.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miyamoto K, et al. Osteoclasts are dispensable for hematopoietic stem cell maintenance and mobilization. J Exp Med. 2011;208(11):2175–81. doi: 10.1084/jem.20101890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butler JM, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6(3):251–64. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Poulos MG, et al. Endothelial Jagged-1 is necessary for homeostatic and regenerative hematopoiesis. Cell Rep. 2013;4(5):1022–34. doi: 10.1016/j.celrep.2013.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Casanova-Acebes M, et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell. 2013;153(5):1025–35. doi: 10.1016/j.cell.2013.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015 doi: 10.1182/blood-2014-09-570192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stier S, et al. Osteopontin is a hematopoietic stem cell niche component that negatively regulates stem cell pool size. J Exp Med. 2005;201(11):1781–91. doi: 10.1084/jem.20041992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yoshihara H, et al. Thrombopoietin/MPL signaling regulates hematopoietic stem cell quiescence and interaction with the osteoblastic niche. Cell Stem Cell. 2007;1(6):68597. doi: 10.1016/j.stem.2007.10.020. [DOI] [PubMed] [Google Scholar]
- 58.Arai F, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. 2004;118(2):149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 59.Sacchetti B, et al. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131(2):324–36. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 60.Zheng J, et al. Angiopoietin-like protein 3 supports the activity of hematopoietic stem cells in the bone marrow niche. Blood. 2011;117(2):470–9. doi: 10.1182/blood-2010-06-291716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Greenbaum A, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495(7440):227–30. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zsebo KM, et al. Stem cell factor is encoded at the Sl locus of the mouse and is the ligand for the c-kit tyrosine kinase receptor. Cell. 1990;63(1):213–24. doi: 10.1016/0092-8674(90)90302-u. [DOI] [PubMed] [Google Scholar]
- 63.Pedersen M, et al. Stem cell factor induces HIF-1alpha at normoxia in hematopoietic cells. Biochem Biophys Res Commun. 2008;377(1):98–103. doi: 10.1016/j.bbrc.2008.09.102. [DOI] [PubMed] [Google Scholar]
- 64.Kimura S, et al. Hematopoietic stem cell deficiencies in mice lacking c-Mpl, the receptor for thrombopoietin. Proc Natl Acad Sci U S A. 1998;95(3):1195–200. doi: 10.1073/pnas.95.3.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yagi M, et al. Sustained ex vivo expansion of hematopoietic stem cells mediated by thrombopoietin. Proc Natl Acad Sci U S A. 1999;96(14):8126–31. doi: 10.1073/pnas.96.14.8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Domen J, Weissman IL. Hematopoietic stem cells need two signals to prevent apoptosis; BCL-2 can provide one of these, Kitl/c-Kit signaling the other. J Exp Med. 2000;192(12):1707–18. doi: 10.1084/jem.192.12.1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou BO, et al. Hematopoietic stem and progenitor cells regulate the regeneration of their niche by secreting Angiopoietin-1. Elife. 2015;4:e05521. doi: 10.7554/eLife.05521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Himburg HA, et al. Pleiotrophin regulates the retention and self-renewal of hematopoietic stem cells in the bone marrow vascular niche. Cell Rep. 2012;2(4):964–75. doi: 10.1016/j.celrep.2012.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Himburg HA, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16(4):475–82. doi: 10.1038/nm.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Naveiras O, et al. Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature. 2009;460(7252):259–63. doi: 10.1038/nature08099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Spindler TJ, et al. Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev. 2014;23(4):434–41. doi: 10.1089/scd.2013.0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell. 2009;4(1):27–36. doi: 10.1016/j.stem.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Reya T, et al. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature. 2003;423(6938):409–14. doi: 10.1038/nature01593. [DOI] [PubMed] [Google Scholar]
- 74.Willert K, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature. 2003;423(6938):448–52. doi: 10.1038/nature01611. [DOI] [PubMed] [Google Scholar]
- 75.Luis TC, et al. Wnt3a deficiency irreversibly impairs hematopoietic stem cell self-renewal and leads to defects in progenitor cell differentiation. Blood. 2009;113(3):546–54. doi: 10.1182/blood-2008-06-163774. [DOI] [PubMed] [Google Scholar]
- 76.Lutolf MP, et al. Perturbation of single hematopoietic stem cell fates in artificial niches. Integr Biol (Camb) 2009;1(1):59–69. doi: 10.1039/b815718a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kabiri Z, et al. Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood. 2015;126(9):1086–94. doi: 10.1182/blood-2014-09-598540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koch U, et al. Simultaneous loss of beta- and gamma-catenin does not perturb hematopoiesis or lymphopoiesis. Blood. 2008;111(1):160–4. doi: 10.1182/blood-2007-07-099754. [DOI] [PubMed] [Google Scholar]
- 79.Cobas M, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199(2):221–9. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeannet G, et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008;111(1):142–9. doi: 10.1182/blood-2007-07-102558. [DOI] [PubMed] [Google Scholar]
- 81.Zhao C, et al. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007;12(6):528–41. doi: 10.1016/j.ccr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Scheller M, et al. Hematopoietic stem cell and multilineage defects generated by constitutive beta-catenin activation. Nat Immunol. 2006;7(10):1037–47. doi: 10.1038/ni1387. [DOI] [PubMed] [Google Scholar]
- 83.Kirstetter P, et al. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7(10):1048–56. doi: 10.1038/ni1381. [DOI] [PubMed] [Google Scholar]
- 84.Perry JM, et al. Cooperation between both Wnt/{beta}-catenin and PTEN/PI3K/Akt signaling promotes primitive hematopoietic stem cell self-renewal and expansion. Genes Dev. 2011;25(18):1928–42. doi: 10.1101/gad.17421911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim JA, et al. Identification of a stroma-mediated Wnt/beta-catenin signal promoting self-renewal of hematopoietic stem cells in the stem cell niche. Stem Cells. 2009;27(6):1318–29. doi: 10.1002/stem.52. [DOI] [PubMed] [Google Scholar]
- 86.Fleming HE, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2(3):274–83. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Himburg HA, et al. Dickkopf-1 promotes hematopoietic regeneration via direct and niche-mediated mechanisms. Nat Med. 2017;23(1):91–99. doi: 10.1038/nm.4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nemeth MJ, et al. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc Natl Acad Sci U S A. 2007;104(39):15436–41. doi: 10.1073/pnas.0704747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Staal FJ, et al. Caught in a Wnt storm: Complexities of Wnt signaling in hematopoiesis. Exp Hematol. 2016;44(6):451–7. doi: 10.1016/j.exphem.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 90.Luis TC, et al. Canonical wnt signaling regulates hematopoiesis in a dosage-dependent fashion. Cell Stem Cell. 2011;9(4):345–56. doi: 10.1016/j.stem.2011.07.017. [DOI] [PubMed] [Google Scholar]
- 91.Parmar K, et al. Distribution of hematopoietic stem cells in the bone marrow according to regional hypoxia. Proc Natl Acad Sci U S A. 2007;104(13):5431–6. doi: 10.1073/pnas.0701152104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Takubo K, et al. Regulation of the HIF-1alpha level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7(3):391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 93.Takubo K, et al. Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell. 2013;12(1):4961. doi: 10.1016/j.stem.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Simsek T, et al. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell. 2010;7(3):380–90. doi: 10.1016/j.stem.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gan B, et al. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 2010;468(7324):701–4. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gurumurthy S, et al. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 2010;468(7324):659–63. doi: 10.1038/nature09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakada D, et al. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 2010;468(7324):653–8. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ito K, et al. PML targeting eradicates quiescent leukaemia-initiating cells. Nature. 2008;453(7198):1072–8. doi: 10.1038/nature07016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ito K, et al. A PML-PPAR-delta pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat Med. 2012;18(9):1350–8. doi: 10.1038/nm.2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harris JM, et al. Glucose metabolism impacts the spatiotemporal onset and magnitude of HSC induction in vivo. Blood. 2013;121(13):2483–93. doi: 10.1182/blood-2012-12-471201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Oburoglu L, et al. Glucose and glutamine metabolism regulate human hematopoietic stem cell lineage specification. Cell Stem Cell. 2014;15(2):169–84. doi: 10.1016/j.stem.2014.06.002. [DOI] [PubMed] [Google Scholar]
- 102.Wang YH, et al. Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell. 2014;158(6):1309–23. doi: 10.1016/j.cell.2014.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Warr MR, et al. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature. 2013;494(7437):323–7. doi: 10.1038/nature11895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Katajisto P, et al. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015;348(6232):340–3. doi: 10.1126/science.1260384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yu WM, et al. Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation. Cell Stem Cell. 2013;12(1):62–74. doi: 10.1016/j.stem.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 107.Kirito K, et al. Thrombopoietin enhances expression of vascular endothelial growth factor (VEGF) in primitive hematopoietic cells through induction of HIF-1alpha. Blood. 2005;105(11):4258–63. doi: 10.1182/blood-2004-07-2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ragnum HB, et al. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br J Cancer. 2015;112(2):382–90. doi: 10.1038/bjc.2014.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Finkel T. Signal transduction by mitochondrial oxidants. J Biol Chem. 2012;287(7):4434–40. doi: 10.1074/jbc.R111.271999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Liu J, et al. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature. 2009;459(7245):387–92. doi: 10.1038/nature08040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Urao N, Ushio-Fukai M. Redox regulation of stem/progenitor cells and bone marrow niche. Free Radic Biol Med. 2013;54:26–39. doi: 10.1016/j.freeradbiomed.2012.10.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jang YY, Sharkis SJ. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood. 2007;110(8):3056–63. doi: 10.1182/blood-2007-05-087759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miyamoto K, et al. Foxo3a is essential for maintenance of the hematopoietic stem cell pool. Cell Stem Cell. 2007;1(1):101–12. doi: 10.1016/j.stem.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 114.Tothova Z, et al. FoxOs are critical mediators of hematopoietic stem cell resistance to physiologic oxidative stress. Cell. 2007;128(2):325–39. doi: 10.1016/j.cell.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 115.Ito K, et al. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature. 2004;431(7011):997–1002. doi: 10.1038/nature02989. [DOI] [PubMed] [Google Scholar]
- 116.Ito K, et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med. 2006;12(4):446–51. doi: 10.1038/nm1388. [DOI] [PubMed] [Google Scholar]
- 117.Juntilla MM, et al. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood. 2010;115(20):4030–8. doi: 10.1182/blood-2009-09-241000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sattler M, et al. Hematopoietic growth factors signal through the formation of reactive oxygen species. Blood. 1999;93(9):2928–35. [PubMed] [Google Scholar]
- 119.Martin PS, et al. Infused total nucleated cell dose is a better predictor of transplant outcomes than CD34+ cell number in reduced-intensity mobilized peripheral blood allogeneic hematopoietic cell transplantation. Haematologica. 2016;101(4):499–505. doi: 10.3324/haematol.2015.134841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Boitano AE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329(5997):1345–8. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fares I, et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014;345(6203):1509–12. doi: 10.1126/science.1256337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wagner JE, Jr, et al. Phase I/II Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell. 2016;18(1):144–55. doi: 10.1016/j.stem.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yonemura Y, et al. In vitro expansion of hematopoietic progenitors and maintenance of stem cells: comparison between FLT3/FLK-2 ligand and KIT ligand. Blood. 1997;89(6):1915–21. [PubMed] [Google Scholar]
- 124.Nishinakamura R, et al. Hematopoiesis in mice lacking the entire granulocyte-macrophage colony-stimulating factor/interleukin-3/interleukin-5 functions. Blood. 1996;88(7):2458–64. [PubMed] [Google Scholar]
- 125.Qian H, et al. Critical role of thrombopoietin in maintaining adult quiescent hematopoietic stem cells. Cell Stem Cell. 2007;1(6):671–84. doi: 10.1016/j.stem.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 126.Sauvageau G, et al. In vitro and in vivo expansion of hematopoietic stem cells. Oncogene. 2004;23(43):7223–32. doi: 10.1038/sj.onc.1207942. [DOI] [PubMed] [Google Scholar]
- 127.de Haan G, et al. In vitro generation of long-term repopulating hematopoietic stem cells by fibroblast growth factor-1. Dev Cell. 2003;4(2):241–51. doi: 10.1016/s1534-5807(03)00018-2. [DOI] [PubMed] [Google Scholar]
- 128.Bhatia M, et al. Quantitative analysis reveals expansion of human hematopoietic repopulating cells after short-term ex vivo culture. J Exp Med. 1997;186(4):61924. doi: 10.1084/jem.186.4.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Takizawa H, et al. Ex vivo expansion of hematopoietic stem cells: mission accomplished? Swiss Med Wkly. 2011;141:w13316. doi: 10.4414/smw.2011.13316. [DOI] [PubMed] [Google Scholar]
- 130.Zhang CC, et al. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006;12(2):240–5. doi: 10.1038/nm1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Conneally E, et al. Expansion in vitro of transplantable human cord blood stem cells demonstrated using a quantitative assay of their lympho-myeloid repopulating activity in nonobese diabetic-scid/scid mice. Proc Natl Acad Sci U S A. 1997;94(18):9836–41. doi: 10.1073/pnas.94.18.9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhang CC, Lodish HF. Cytokines regulating hematopoietic stem cell function. Curr Opin Hematol. 2008;15(4):307–11. doi: 10.1097/MOH.0b013e3283007db5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wohrer S, et al. Distinct stromal cell factor combinations can separately control hematopoietic stem cell survival, proliferation, and self-renewal. Cell Rep. 2014;7(6):1956–67. doi: 10.1016/j.celrep.2014.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ko KH, et al. GSK-3beta inhibition promotes engraftment of ex vivo-expanded hematopoietic stem cells and modulates gene expression. Stem Cells. 2011;29(1):108–18. doi: 10.1002/stem.551. [DOI] [PubMed] [Google Scholar]
- 135.Huang J, et al. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012;18(12):1778–85. doi: 10.1038/nm.2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Goessling W, et al. Genetic interaction of PGE2 and Wnt signaling regulates developmental specification of stem cells and regeneration. Cell. 2009;136(6):1136–47. doi: 10.1016/j.cell.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447(7147):1007–11. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Goessling W, et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8(4):445–58. doi: 10.1016/j.stem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Csaszar E, et al. Rapid expansion of human hematopoietic stem cells by automated control of inhibitory feedback signaling. Cell Stem Cell. 2012;10(2):218–29. doi: 10.1016/j.stem.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 140.Cipolleschi MG, et al. The role of hypoxia in the maintenance of hematopoietic stem cells. Blood. 1993;82(7):2031–7. [PubMed] [Google Scholar]
- 141.Eliasson P, et al. Hypoxia mediates low cell-cycle activity and increases the proportion of long-term-reconstituting hematopoietic stem cells during in vitro culture. Exp Hematol. 2010;38(4):301–310 e2. doi: 10.1016/j.exphem.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 142.Mantel CR, et al. Enhancing Hematopoietic Stem Cell Transplantation Efficacy by Mitigating Oxygen Shock. Cell. 2015;161(7):1553–65. doi: 10.1016/j.cell.2015.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Antonchuk J, et al. HOXB4-induced expansion of adult hematopoietic stem cells ex vivo. Cell. 2002;109(1):39–45. doi: 10.1016/s0092-8674(02)00697-9. [DOI] [PubMed] [Google Scholar]
- 144.Sauvageau G, et al. Overexpression of HOXB4 in hematopoietic cells causes the selective expansion of more primitive populations in vitro and in vivo. Genes Dev. 1995;9(14):1753–65. doi: 10.1101/gad.9.14.1753. [DOI] [PubMed] [Google Scholar]
- 145.Amsellem S, et al. Ex vivo expansion of human hematopoietic stem cells by direct delivery of the HOXB4 homeoprotein. Nat Med. 2003;9(11):1423–7. doi: 10.1038/nm953. [DOI] [PubMed] [Google Scholar]
- 146.Kirito K, et al. Thrombopoietin stimulates Hoxb4 expression: an explanation for the favorable effects of TPO on hematopoietic stem cells. Blood. 2003;102(9):3172–8. doi: 10.1182/blood-2003-03-0944. [DOI] [PubMed] [Google Scholar]
- 147.Iriuchishima H, et al. Ex vivo maintenance of hematopoietic stem cells by quiescence induction through Fbxw7α overexpression. Blood. 2011;117(8):2373–7. doi: 10.1182/blood-2010-07-294801. [DOI] [PubMed] [Google Scholar]
- 148.Lis R, et al. Conversion of adult endothelium to immunocompetent haematopoietic stem cells. Nature. 2017;545(7655):439–445. doi: 10.1038/nature22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sugimura R, et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature. 2017;545(7655):432–438. doi: 10.1038/nature22370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Liu X, et al. Maintenance of mouse hematopoietic stem cells ex vivo by reprogramming cellular metabolism. Blood. 2015;125(10):1562–5. doi: 10.1182/blood-2014-04-568949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vannini N, et al. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat Commun. 2016;7:13125. doi: 10.1038/ncomms13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ito K, et al. Self-renewal of a purified Tie2+ hematopoietic stem cell population relies on mitochondrial clearance. Science. 2016;354(6316):1156–1160. doi: 10.1126/science.aaf5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.van Galen P, et al. The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature. 2014;510(7504):268–72. doi: 10.1038/nature13228. [DOI] [PubMed] [Google Scholar]
- 154.Miharada K, et al. Dppa5 improves hematopoietic stem cell activity by reducing endoplasmic reticulum stress. Cell Rep. 2014;7(5):1381–92. doi: 10.1016/j.celrep.2014.04.056. [DOI] [PubMed] [Google Scholar]
- 155.Sigurdsson V, et al. Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell. 2016;18(4):522–32. doi: 10.1016/j.stem.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 156.Discher DE, et al. Growth factors, matrices, and forces combine and control stem cells. Science. 2009;324(5935):1673–7. doi: 10.1126/science.1171643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gattazzo F, et al. Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–19. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.LaIuppa JA, et al. Culture materials affect ex vivo expansion of hematopoietic progenitor cells. J Biomed Mater Res. 1997;36(3):347–59. doi: 10.1002/(sici)1097-4636(19970905)36:3<347::aid-jbm10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 159.Franke K, et al. Engineered matrix coatings to modulate the adhesion of CD133+ human hematopoietic progenitor cells. Biomaterials. 2007;28(5):836–43. doi: 10.1016/j.biomaterials.2006.09.031. [DOI] [PubMed] [Google Scholar]
- 160.Feng Q, et al. Expansion of engrafting human hematopoietic stem/progenitor cells in three-dimensional scaffolds with surface-immobilized fibronectin. J Biomed Mater Res A. 2006;78(4):781–91. doi: 10.1002/jbm.a.30829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dao MA, et al. Adhesion to fibronectin maintains regenerative capacity during ex vivo culture and transduction of human hematopoietic stem and progenitor cells. Blood. 1998;92(12):4612–21. [PubMed] [Google Scholar]
- 162.Chua KN, et al. Surface-aminated electrospun nanofibers enhance adhesion and expansion of human umbilical cord blood hematopoietic stem/progenitor cells. Biomaterials. 2006;27(36):6043–51. doi: 10.1016/j.biomaterials.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 163.Tan J, et al. Maintenance and expansion of hematopoietic stem/progenitor cells in biomimetic osteoblast niche. Cytotechnology. 2010;62(5):439–48. doi: 10.1007/s10616-010-9297-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Engler AJ, et al. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 165.Holst J, et al. Substrate elasticity provides mechanical signals for the expansion of hemopoietic stem and progenitor cells. Nat Biotechnol. 2010;28(10):1123–8. doi: 10.1038/nbt.1687. [DOI] [PubMed] [Google Scholar]
- 166.Miyoshi H, et al. Three-dimensional culture of mouse bone marrow cells within a porous polymer scaffold: effects of oxygen concentration and stromal layer on expansion of haematopoietic progenitor cells. J Tissue Eng Regen Med. 2011;5(2):112–8. doi: 10.1002/term.295. [DOI] [PubMed] [Google Scholar]
- 167.Sharma MB, et al. Mimicking the functional hematopoietic stem cell niche in vitro: recapitulation of marrow physiology by hydrogel-based three-dimensional cultures of mesenchymal stromal cells. Haematologica. 2012;97(5):651–60. doi: 10.3324/haematol.2011.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li Y, et al. Human cord cell hematopoiesis in three-dimensional nonwoven fibrous matrices: in vitro simulation of the marrow microenvironment. J Hematother Stem Cell Res. 2001;10(3):355–68. doi: 10.1089/152581601750288966. [DOI] [PubMed] [Google Scholar]
- 169.Ferreira MS, et al. Cord blood-hematopoietic stem cell expansion in 3D fibrin scaffolds with stromal support. Biomaterials. 2012;33(29):6987–97. doi: 10.1016/j.biomaterials.2012.06.029. [DOI] [PubMed] [Google Scholar]
- 170.Torisawa YS, et al. Bone marrow-on-a-chip replicates hematopoietic niche physiology in vitro. Nat Methods. 2014;11(6):663–9. doi: 10.1038/nmeth.2938. [DOI] [PubMed] [Google Scholar]
- 171.Florian MC, et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell. 2012;10(5):520–30. doi: 10.1016/j.stem.2012.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Morrison SJ, Weissman IL. The long-term repopulating subset of hematopoietic stem cells is deterministic and isolatable by phenotype. Immunity. 1994;1(8):661–73. doi: 10.1016/1074-7613(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 173.Takano H, et al. Asymmetric division and lineage commitment at the level of hematopoietic stem cells: inference from differentiation in daughter cell and granddaughter cell pairs. J Exp Med. 2004;199(3):295–302. doi: 10.1084/jem.20030929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Essers MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458(7240):904–8. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- 175.Majeti R, et al. Identification of a hierarchy of multipotent hematopoietic progenitors in human cord blood. Cell Stem Cell. 2007;1(6):635–45. doi: 10.1016/j.stem.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Choi JS, et al. Engineering the hematopoietic stem cell niche: Frontiers in biomaterial science. Biotechnol J. 2015;10(10):1529–45. doi: 10.1002/biot.201400758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Kobel SA, et al. Automated analysis of single stem cells in microfluidic traps. Lab Chip. 2012;12(16):2843–9. doi: 10.1039/c2lc40317j. [DOI] [PubMed] [Google Scholar]
- 178.de Lima M, et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N Engl J Med. 2012;367(24):2305–15. doi: 10.1056/NEJMoa1207285. [DOI] [PMC free article] [PubMed] [Google Scholar]