

Abstract
Background
Flavaglines are a family of natural compounds shown to have anti-inflammatory and cytoprotective effects in neurons and cardiomyocytes. Flavaglines target prohibitins as ligands, which are scaffold proteins that regulate mitochondrial function, cell survival, and transcription. This study tested the therapeutic potential of flavaglines to promote intestinal epithelial cell homeostasis and to protect against a model of experimental colitis in which inflammation is driven by epithelial ulceration.
Methods
Survival and homeostasis of Caco2-BBE and IEC-6 intestinal epithelial cell lines were measured during treatment with the flavaglines FL3 or FL37 alone and in combination with the proinflammatory cytokines tumor necrosis factor (TNF) α and interferon γ. Wild-type mice were intraperitoneally injected with 0.1 mg/kg FL3 or vehicle once daily for 4 days during dextran sodium sulfate–induced colitis to test the in vivo anti-inflammatory effect of FL3.
Results
FL3 and FL37 increased basal Caco2-BBE and IEC-6 cell viability, decreased apoptosis, and decreased epithelial monolayer permeability. FL3 and FL37 inhibited TNFα- and interferon γ–induced nuclear factor kappa B and Cox2 expression, apoptosis, and increased permeability in Caco2-BBE cells. FL3 and FL37 protected against TNFα-induced mitochondrial superoxide generation by preserving respiratory chain complex I activity and prohibitin expression. p38-MAPK activation was essential for the protective effect of FL3 and FL37 on barrier permeability and mitochondrial-derived reactive oxygen species production during TNFα treatment. Mice administered FL3 during dextran sodium sulfate colitis exhibited increased colonic prohibitin expression and p38-MAPK activation, preserved barrier function, and less inflammation.
Conclusions
These results suggest that flavaglines exhibit therapeutic potential against colitis and preserve intestinal epithelial cell survival, mitochondrial function, and barrier integrity.
Keywords: barrier function, prohibitin, intestinal epithelium, TNFα, colitis
Inflammatory bowel diseases (IBD), the most common forms being Crohn’s disease and ulcerative colitis, are associated with disturbed intestinal epithelial cell (IEC) homeostasis. IECs structurally provide host defense by forming a single-cell barrier between luminal contents and the underlying intestinal tissue. Epithelial barrier dysfunction is an early event in the pathogenesis of IBD, resulting in increased exposure of intraluminal contents to the mucosal immune system, thereby aggravating the inflammatory condition.1 Disruption of the epithelial barrier can be manifested by increased epithelial cell apoptosis not equal to epithelial proliferation (ulceration) and/or by alteration of permeability at the tight junctions that establish a semipermeable barrier between epithelial cells, restricting passage of large molecules.2 Restoring and maintaining the epithelial barrier are critical to limit mucosal inflammation and promote healing.1 Identification of agents that act to promote epithelial cell homeostasis and barrier integrity in the context of tissue injury or IBD is critical for the development of therapeutic or preventative strategies to promote or maintain mucosal healing.
Flavaglines comprise a family of compounds found in medicinal plants of Southeast Asia that have shown anticancer, anti-inflammatory, and cytoprotective activities.3 Flavaglines have been reported to reduce neurotoxicity in a mouse model of Parkinson’s disease4 and protect cardiomyocytes from doxorubicin-induced cardiac toxicity.5 In addition, at nanomolar concentrations, flavaglines inhibit the production of interferon (IFN) γ, TNFα, interleukin 2, and interleukin 4 by T lymphocytes and proinflammatory mediators from microglia and endothelial cells.4,6 It is well established that flavaglines target prohibitins (PHBs) as their molecular ligands.3 PHB (B-cell receptor–associated protein 32) and PHB2 (repressor of estrogen receptor activity, B-cell receptor–associated protein 37) are highly conserved proteins with diverse functions, including regulation of cell cycle progression, apoptosis, and transcription, depending on their posttranslational modifications and subcellular localization.7 The best characterized function of PHB and PHB2 is their role in maintaining the structure and function of mitochondria, including respiration and protein metabolism, while residing in the inner mitochondrial membrane as heterodimers.8–10 In IECs, PHB is predominantly localized in the mitochondria, where it has been shown to be required for optimal activity of complexes I and IV of the electron transport chain (ETC).11–14 During IBD, expression of PHB is decreased in uninvolved and inflamed epithelium.12,14 Epithelial PHB is protective against intestinal inflammation as evidenced by less severe experimental colitis in transgenic mice with IEC-specific PHB overexpression.15,16 Gene silencing of PHB in cultured IECs induces mitochondrial membrane depolarization and cellular stress pathways, including intracellular reactive oxygen species (ROS) generation and apoptosis.17 Furthermore, cultured IECs overexpressing PHB exhibit less intracellular ROS and apoptosis,17 suggesting that relative levels of PHB modulate epithelial cell homeostasis.
In this study, we investigated the anti-inflammatory activity of the flavaglines FL3 and FL37, which are among the most potent of flavaglines tested in a wide variety of pharmacological assays,18 in cultured IECs and the dextran sodium sulfate (DSS) model of colitis. We also elucidated the mechanism of FL3 and FL37 protection in IECs.
MATERIALS AND METHODS
Cell Culture
Flavaglines FL3 and FL37 were synthesized in our laboratory as previously described.19,20 The Caco2-BBE human colonic adenocarcinoma epithelial cell line and the nontransformed IEC-6 rat small IEC line were used as in vitro models of polarized intestinal epithelium. Both cell lines were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Cells were grown in Dulbecco’s modified Eagle’s medium (Caco2-BBE) supplemented with penicillin (40 mg/L), streptomycin (90 mg/L), and 10% fetal bovine serum. Caco2-BBE cells and IEC-6 cells were plated on permeable supports (pore size, 0.4 μm; transwell-clear polyester membranes; Corning, Tewksbury, MA) and cultured for 8 days to allow the cells to polarize. All experiments performed on Caco2-BBE cells were between passages 32 and 45 and IEC-6 cells were between passages 10 and 18. FL3 and FL37 were administered at 1, 10, and 50 nM because previous studies showed that flavaglines exhibit cardioprotective and neuroprotective effects at 1 to 10 nM.18 Caco2-BBE or IEC-6 cells were treated with 10 ng/mL recombinant human or rat TNFα or 50 ng/mL recombinant human or rat IFNγ, respectively (R&D Systems, Minneapolis, MN).
PHB Knockdown
To achieve PHB knockdown, Caco2-BBE cells were transiently transfected with Stealth RNAi against PHB1 (5′-CAGAAUGUCAACAUCACACUGCGCA-3′) or Stealth RNAi Negative Control Med GC (Life Technologies, Carlsbad, CA) at 20 μm concentration using Amaxa electroporation with Nucleofector kit T (Lonza, Basel, Switzerland).
Induction of Colitis in Mice
Eight-week-old wild-type (C57BL/6) male and female mice were administered orally DSS (molecular weight, 50,000; MP Biomedicals, Solon, OH) at 2.5% (wt/vol) in tap water ad libitum for 6 days. Controls were administered normal tap water throughout the treatment period. Mice were intraperitoneally injected with 0.1 mg/kg FL3 or vehicle (veh) once daily on days 0 to 4. We chose this dose because it proved to be adequate in our previous assays of cardioprotection in mice.5 Mean DSS water consumption, body weight, and clinical signs of inflammation were assessed daily during the treatment period. All mice were group housed in standard cages under a controlled temperature (25°C) and photoperiod (12-hour light/dark cycle) and were allowed standard chow and tap water ad libitum. All experiments were approved by the Baylor Research Institute Institutional Animal Care and Use Committee.
Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis and Western Immunoblot Analysis
Total protein was isolated from cultured cells or distal colon mucosa from wild-type mice. The samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis using Laemmli’s 2× sodium dodecyl sulfate sample buffer and AnyKD gradient polyacrylamide gels (Bio-Rad, Hercules, CA) followed by electrotransfer to nitrocellulose membranes (Bio-Rad). Membranes were incubated with primary antibodies at 4°C overnight and subsequently incubated with corresponding peroxidase-conjugated secondary antibodies. Membranes were washed and immunoreactive proteins were detected using Amersham ECL Plus reagent (GE Healthcare, Piscataway, NJ). PHB antibody was purchased from Thermo Fisher (Waltham, MA); PHB2, Stat3, and ERK1/2 from Santa Cruz Biotechnology (Santa Cruz, CA); phospho-p38-MAPK, p38-MAPK, pERK1/2, pS727-Stat3, pY705-Stat3, pSAPK/JNK, SAPK/JNK, pAKT, AKT, and cleaved caspase 3 from Cell Signaling Technology (Danvers, MA); proliferating cell nuclear antigen (PCNA) from Abcam (Cambridge, MA); p65 from BD Biosciences (San Jose, CA); and Cox2 from Cayman Chemicals (Ann Arbor, MI). Blots were reprobed with β-tubulin or β-actin (Sigma-Aldrich Corp., St. Louis, MO) antibody as a loading control.
Measuring Mitochondrial ROS
Cells were incubated with Hank’s balanced salt solution with 5 μM MitoSOX Red Mitochondrial Superioxide Indicator dye (Life Technologies) for 10 minutes at 37°C. Cells were washed twice with warm Hank’s balanced salt solution, and fluorescent intensity was measured at 510 nm excitation/580 nm emission. For experiments using complex I or III inhibitors, 15 minutes before collection, cells were treated with 5 μM rotenone (Sigma-Aldrich), a complex I inhibitor, or 1 μM antimycin A (Sigma-Aldrich), a complex III inhibitor. For experiments using p38-MAPK inhibitor, cells were incubated with 20 μM p38-MAPK inhibitor SB203580 (Sigma-Aldrich) for 1 hour before treatment with FL3, FL37, and TNFα.
Measuring ATP Concentration
The concentration of ATP was determined using the Enlighten ATP Assay Bioluminescence Detection kit (Promega, Madison, WI) according to the manufacturer’s protocol.
Detection of Mitochondrial Complex I Activity
The activity of complex I was measured using the Complex I Dipstick Assay kit (Abcam) according to the manufacturer’s protocol using 20 μg of protein.
Cytotoxicity Test
Lactate dehydrogenase cytotoxicity detection kit (Clontech, Mountain View, CA) was used to measure cell viability. An aliquot of 100 μL of culture media was added to 100 μL of lactate dehydrogenase reagent, and percent cytotoxicity and percent viable cells were measured according to the manufacturer’s protocol.
Measuring Cell Apoptosis
Percentage of apoptotic cells was measured using the Cell Death Detection ELISA Plus kit (Roche, Indianapolis, IN) as described by the manufacturer. As a second measure of apoptosis, cells or colon sections were stained for terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick-end labeling (TUNEL) as described by the manufacturer’s protocol (Roche). The nuclei of cells were stained with 4′,6-diamidino-2-phenylindole (Life Technologies). The number of TUNEL-positive cells were quantitated using a fluorescent microscope across 20 fields per treatment for in vitro experiments or across 20 well-oriented crypts per animal for in vivo experiments.
Colonic epithelial cells were isolated from mice as previously described.21 Total protein was extracted and analyzed by Western blotting for cleaved caspase 3.
Measurement of Transepithelial Electrical Resistance and Macromolecular Permeability In Vitro
Transepithelial electrical resistance was measured with an epithelial voltohmmeter (Millicell-ers; Millipore, Billerica, MA). For permeability assays, cells were incubated in Hank’s balanced salt solution. Fluorescein isothiocyanate (FITC)-dextran (10 mg/mL) (molecular weight 4 kDa; Sigma-Aldrich) was added to the apical chamber. The apical and basolateral chambers were sampled at 30 minutes, 1 hour, and 2 hours after the addition of FITC-dextran to the apical chamber. FITC-dextran concentration was quantified through spectrofluorimetry (excitation, 492 nm, emission, 510 nm). Values are shown as rate (nanograms per milliliter per minute) of FITC-dextran translocation to the basolateral reservoir.
Clinical Score Assessment
A clinical activity score was generated using body weight loss, stool consistency, and the presence of occult blood by a guaiac test (Hemoccult Sense; Beckman Coulter, Fullerton, CA) as described previously.16 The scores for each parameter were added to get a clinical activity score with 12 being the maximal score.
Histological Damage Score
Distal colon was fixed in formalin and stained with hemotoxylin and eosin for histology. Sections were coded for blind microscopic assessment of inflammation. Histological scoring was performed on the basis of 3 parameters: the severity of inflammation, crypt damage, and ulceration as described previously.16 These values were added to give a maximal histological score of 11.
Myeloperoxidase Activity
Neutrophil infiltration into the distal colon was quantified by measuring myeloperoxidase activity. Briefly, a portion of colon or cecum was homogenized in 1:20 (wt/vol) 50 mmol/L phosphate buffer (pH 6.0) containing 0.5% hexadecyltrimethyl ammonium bromide on ice by using a Polytron homogenizer. The homogenate was sonicated for 10 seconds, freeze thawed 3 times, and centrifuged at 14,000 rpm for 15 minutes. Supernatant was added to 1 mg/mL of o-dianisidine hydrochloride and 5 × 10−4% hydrogen peroxide, and the change in absorbance at 460 nm was measured. One unit of myeloperoxidase activity was defined as the amount that degraded 1 μmol of peroxidase per minute at 25°C.
In Vivo Permeability Assay
Barrier function was assessed using an FITC-labeled dextran method. Briefly, on day 6 of DSS administration, mice were gavaged with permeability tracer (60 mg/100 g body weight of FITC-labeled dextran, FD-4, Mr 4000; Sigma-Aldrich). Serum was collected retro-orbitally 4 hours after FD-4 gavage, and fluorescence intensity of each sample was measured (excitation, 492 nm; emission, 525 nm). FITC-dextran concentrations were determined from standard curves generated by serial dilution of FITC-dextran and normalized to total protein.
Measuring In Vivo Oxidative Damage
Five-micrometer paraffin-embedded sections of colon were analyzed for 4-hydroxynoneal (4-HNE) staining as a marker of lipid peroxidation. Sections were deparaffinized in xylene, rehydrated in ethyl alcohol gradient, incubated in 0.3% H2O2 for 30 minutes, and treated with 10 mM sodium citrate buffer (pH 6.0) at 110°C for 20 minutes in a pressure cooker. Sections were blocked with 5% normal goat serum and incubated with 4-HNE antibody (Abcam) overnight at 4°C. After washing with phosphate-buffered saline, sections were incubated with biotinylated secondary antibodies for 30 minutes at room temperature, and color development was performed using the Vectastain ABC kit (Vector Laboratories) and 3,3′-Diaminobenzidine (Dako, Carpinteria, CA). Sections were counterstained with hematoxylin.
The oxidative damage to proteins was assessed using a protein carbonyl assay kit (Cayman Chemicals) according to the manufacturer’s protocol.
Statistical Analyses
Values are expressed as mean ± SEM. Comparisons between FL3 or FL37 treatment versus vehicle control were analyzed by unpaired Student’s t test. Comparisons between FL3 or FL37 combined with TNFα or IFNγ treatment were analyzed by 2-way analysis of variance, and subsequent pairwise comparisons used Bonferroni post hoc tests to test for significant differences between 2 particular groups. P value <0.05 was considered statistically significant in all analyses.
RESULTS
FL3 and FL37 Decrease IEC Apoptosis
An intact intestinal epithelial barrier that prevents the translocation of intraluminal contents and subsequent immune cell activation is an initial event in suppressing inflammation deeper in the bowel wall.1 Disruption of the epithelial barrier can be manifested by increased epithelial cell apoptosis not balanced with epithelial proliferation. To determine whether very low (nanomolar) doses of FL3 or FL37 affect IEC viability, polarized Caco2-BBE or IEC-6 cell monolayers were treated with increasing concentrations of FL3 or FL37 for 16 hours and markers of cell proliferation or apoptosis were measured. Ten and 50 nanomolar of FL3 or FL37 enhanced cell viability in Caco2-BBE cells as measured by lactate dehydrogenase release (Fig. 1A). IEC-6 nontransformed cells were used as a second in vitro model of IECs. FL3 and FL37 increased viability of IEC-6 cells at the higher doses tested (see Fig. A, Supplemental Digital Content 1, http://links.lww.com/IBD/B116). To determine whether increased cell viability was associated with changes in cell proliferation, PCNA protein expression was measured. FL3 and FL37 did not affect PCNA protein expression in Caco2-BBE cells, suggesting that FL3 and FL37 do not increase cell proliferation (Fig. 1B). FL3 and FL37 decreased Caco2-BBE cell apoptosis measured by enzyme-linked immunosorbent assay (Fig. 1C) and TUNEL staining (Fig. 1D). The number of apoptotic cells was decreased to approximately 5% during treatment with FL3 or FL37 compared with 10% in vehicle-treated cells (Fig. 1D).
FIGURE 1.
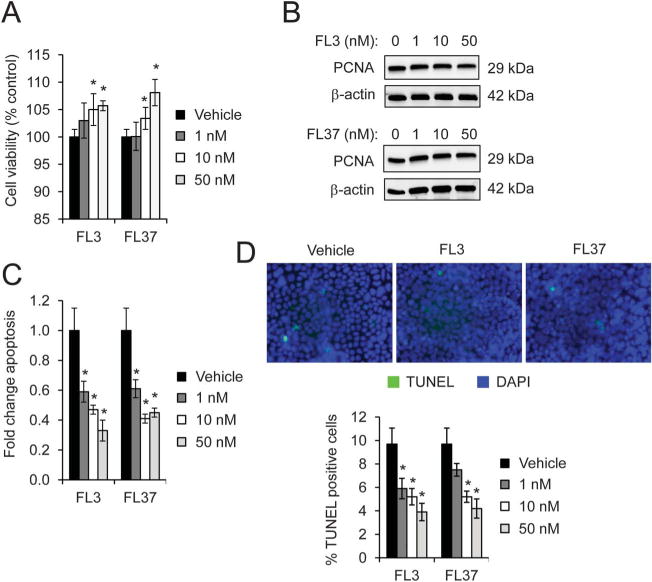
FL3 and FL37 decrease apoptosis in Caco2-BBE cells. Cells were treated with increasing concentrations of FL3 or FL37 for 16 hours A, Cell viability using lactate dehydrogenase assay; *P < 0.05 versus vehicle; n = 8 per treatment across 2 separate experiments. B, Representative Western blots of PCNA protein expression, a marker of cell proliferation. C, Apoptosis measured by enzyme-linked immunosorbent assay; *P < 0.05 versus vehicle; n = 4 per treatment. D, TUNEL-positive cells were quantified across 20 fields; *P < 0.05 versus vehicle.
FL3 and FL37 Increase Protein Expression of Known Flavagline Targets PHB and PHB2 in IECs
To determine whether FL3 or FL37 alters IEC expression of PHB or PHB2, which are established flavagline targets,3 Caco2-BBE cells were treated with increasing doses of FL3 or FL37. Treatment with FL3 or FL37 at very low (nanomolar) concentrations increased PHB and PHB2 protein levels in Caco2-BBE (Fig. 2A). FL3 and FL37 increased PHB protein expression rapidly, as early as 15 minutes, with peak induction for FL3 between 1 and 2 hours and for FL37 between 15 minutes and 1 hour (Fig. 2B). The induction of PHB2 by FL3 and FL37 showed an identical pattern as PHB. FL3- and FL37-induced PHB and PHB2 protein expression was associated with p38-MAPK activation (Fig. 2B), but not activation of Stat3, SAPK/JNK, ERK, or AKT (see Fig., Supplemental Digital Content 2, http://links.lww.com/IBD/B117). FL3 and FL37 induced PHB and PHB2 protein expression and activation of p38-MAPK in IEC-6 cells in a similar pattern as induction in Caco2-BBE cells (see Fig. B, Supplemental Digital Content 1, http://links.lww.com/IBD/B116).
FIGURE 2.
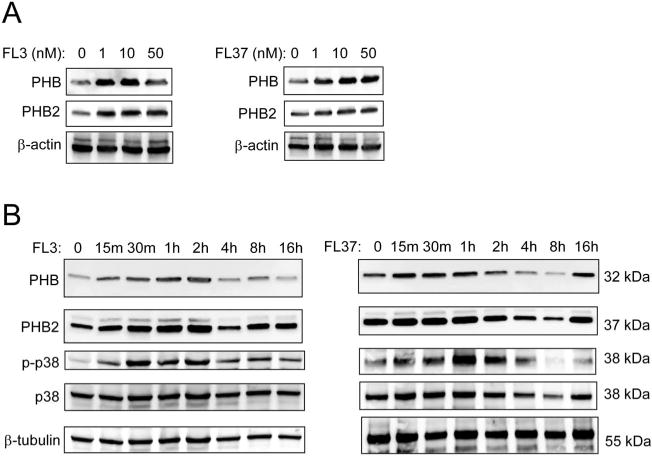
FL3 and FL37 increase protein expression of known flavagline targets PHB and PHB2 in IECs. A, Polarized Caco2-BBE cells were treated with increasing concentrations of FL3 or FL37 for 2 hours. Representative Western blots are shown for PHB, PHB2, and β-actin (loading control). B, Caco2-BBE cells were treated with 10 nM FL3 or FL37 for increasing time. Representative Western blots are shown for PHB, PHB2, phospho-p38-MAPK, total p38-MAPK, and β-tubulin (loading control).
Pretreatment with FL3 or FL37 Decreases TNFα- or IFNγ-induced Expression of Nuclear Factor Kappa B p65 and Cox2 in IECs
To determine whether flavaglines exhibit anti-inflammatory action in IECs, Caco2-BBE cells were pretreated with increasing concentrations of FL3 or FL37 followed by treatment with tumor necrosis factor (TNF) α or IFNγ, 2 proinflammatory cytokines upregulated during intestinal inflammation. Western blotting revealed that FL3 and FL37 prevented TNFα- or IFNγ-induced nuclear factor kappa B p65 and Cox2 protein expression, which are 2 key proinflammatory pathways in the intestine. This effect of FL3 and FL37 was associated with sustained PHB expression, which is decreased by TNFα or IFNγ (Fig. 3A, B). Similar results were evident in IEC-6 cells, in which 1 and 10 nM FL3 or FL37 pretreatment prevented TNFα-induced nuclear factor kappa B p65 and Cox2 induction (see Fig. A, Supplemental Digital Content 3, http://links.lww.com/IBD/B118). All further experiments were performed with FL3 or FL37 at a concentration of 10 nM because this was the lowest most effective dose at preventing TNFα- or IFNγ-induced loss of PHB protein expression.
FIGURE 3.
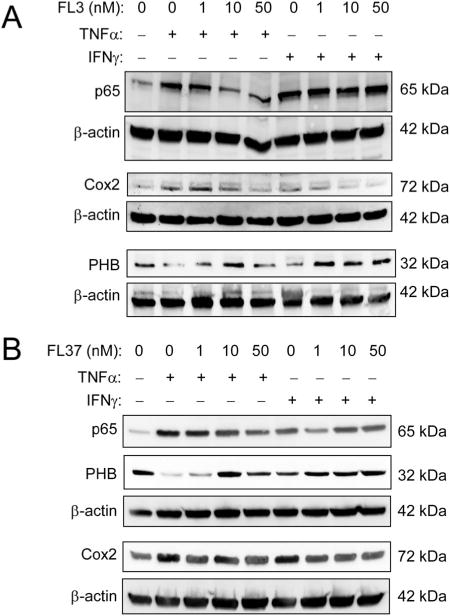
Pretreatment with FL3 or FL37 decreases TNFα- or IFNγ-induced expression of nuclear factor kappa B (NFκB) p65 and Cox2 in Caco2-BBE cells. Cells were pretreated with increasing concentrations of FL3 (A) or FL37 (B) for 1 hour, followed by treatment with 10 ng/mL TNFα or 50 ng/mL IFNγ for 16 hours. Total protein was isolated for Western blotting for expression of NFκB p65, Cox2, PHB, and β-actin (loading control).
Pretreatment with FL3 or FL37 Prevented IEC Apoptosis Induced by TNFα or IFNγ
We next determined whether FL3 or FL37 affected cytokine-induced proliferation and/or apoptosis in IECs. TNFα or IFNγ significantly decreased Caco2-BBE cell viability in vehicle-treated cells, with TNFα reducing cell viability by 24% and IFNγ by 11% (Fig. 4A). Pretreatment with 10 nM FL3 or FL37 prevented the reduction in cell viability by TNFα or IFNγ (Fig. 4A). Similar results were evident in IEC-6 cells, in which pretreatment with 10 nM FL3 or FL37 increased cell viability during TNFα or IFNγ treatment (see Fig. B, Supplemental Digital Content 3, http://links.lww.com/IBD/B118). Pretreatment with 10 nM FL3 or FL37 did not affect PCNA protein expression when normalized to β-actin expression during TNFα or IFNγ treatment (Fig. 4B). These results suggest that the protection of cell viability by FL3 and FL37 during TNFα or IFNγ treatment is not associated with increased cell proliferation. TUNEL staining revealed that pretreatment with FL3 or FL37 prevented TNFα- or IFNγ-induced Caco2-BBE apoptosis (Fig. 4C).
FIGURE 4.
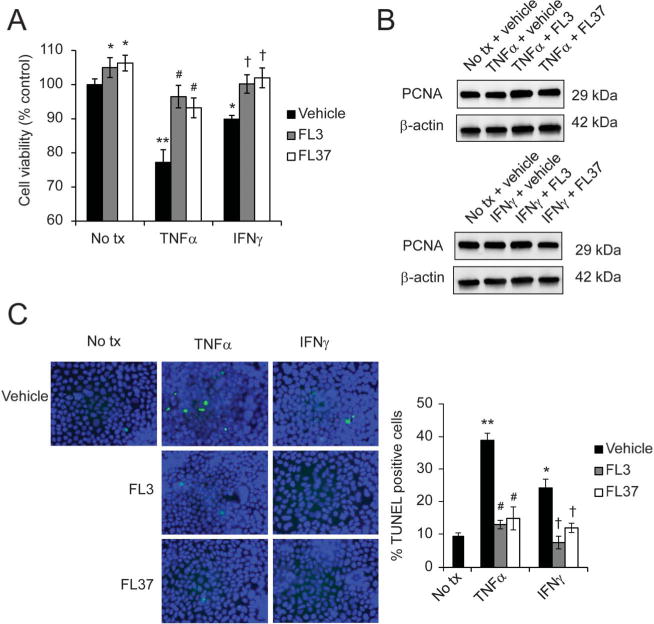
Pretreatment with FL3 or FL37 prevented Caco2-BBE apoptosis induced by TNFα or IFNγ. Cells were pretreated with 10 nM FL3 or FL37 for 1 hour, followed by treatment with 10 ng/mL TNFα or 50 ng/mL IFNγ for 16 hours. A, Cell viability using lactate dehydrogenase assay; n = 8 per treatment across 2 separate experiments. B, Representative Western blots of PCNA protein expression, a marker of cell proliferation. C, TUNEL-positive cells were quantified across 20 fields; *P < 0.05, **P < 0.01 versus no tx + vehicle; #P < 0.01 versus TNFα + vehicle; †P < 0.05 versus IFNγ + vehicle. No tx, no treatment.
Pretreatment with FL3 or FL37 Protects Against Cytokine-induced Barrier Dysfunction in Caco2-BBE Cell Monolayers
Ten nanomolar of FL3 or FL37 increased transepithelial electrical resistance (Fig. 5A) and decreased translocation of FITC-dextran between Caco2-BBE monolayers from the apical chamber to the basolateral chamber (Fig. 5B). Pretreatment with 10 nM FL3 or FL37 prevented TNFα- or IFNγ-induced reduction in transepithelial electrical resistance (Fig. 5A) and increase in FITC-dextran across Caco2-BBE cells (Fig. 5B). These results suggest that FL3 and FL37 enhance intestinal epithelial barrier function basally and protect from barrier dysfunction induced by TNFα and IFNγ.
FIGURE 5.
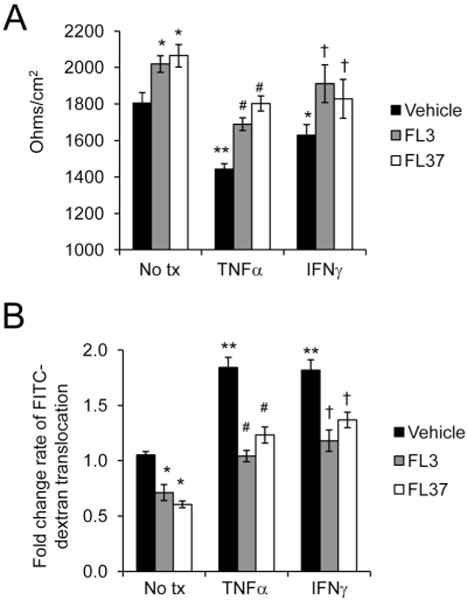
Pretreatment with FL3 or FL37 protects against cytokine-induced permeability changes in IECs. Monolayers of polarized Caco2-BBE cells were pretreated with 10 nM FL3 or FL37 for 1 hour, followed by treatment with 10 ng/mL TNFα or 50 ng/mL IFNγ for 16 hours. A, Transepithelial electrical resistance. B, Macromolecular permeability as measured by rate of 4 kDa FITC-dextran translocation from apical to basolateral chamber (nanograms per milliliter per minute); *P < 0.05, **P < 0.01 versus no tx + vehicle; #P <0 0.01 versus TNFα + vehicle; †P < 0.05 versus IFNγ + vehicle.
FL3 and FL37 Preserve Mitochondrial Function During TNFα Treatment
PHB and PHB2, known targets of flavaglines, are predominantly localized to the inner mitochondrial membrane of IECs.14 Because treatment with FL3 or FL37 decreased basal and TNFα- or IFNγ-induced apoptosis, we next assessed mitochondrial function, which is known to be dysregulated during IBD and in animal models of colitis22–28 and plays a central role in cell fate decisions, especially apoptosis.29 Blockade of forward electron flow through inhibition of ETC complexes leads to electrons accumulating at upstream complexes, ROS generation, and reduced ATP production.30 Ten nanomolar of FL3 or FL37 increased basal ATP levels, but this did not reach statistical significance (Fig. 6A). TNFα decreased ATP levels in vehicle control cells as shown previously31–33; pretreatment with 10 nM FL3 or FL37 prevented TNFα-induced decrease in ATP (Fig. 5A). IFNγ did not affect ATP production and pretreatment with FL3 or FL37 before IFNγ treatment affected ATP levels similar to treatment with the flavaglines alone (Fig. 5A). For this reason, remaining experiments assessed the effect of FL3 or FL37 on changes to mitochondrial function induced by TNFα and not IFNγ.
FIGURE 6.
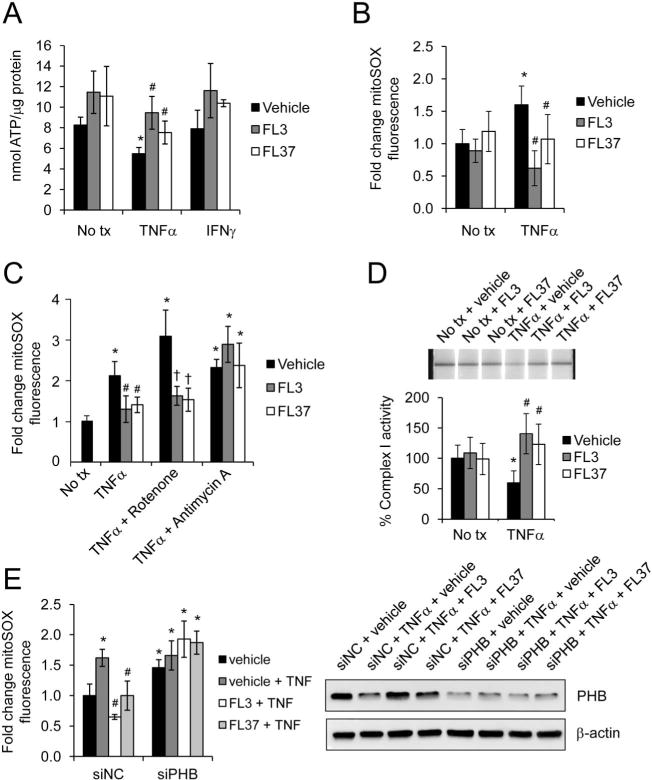
FL3 and FL37 preserve mitochondrial function during TNFα treatment. Caco2-BBE cells were pretreated with 10 nM FL3 or FL37 for 1 hour, followed by treatment with 10 ng/mL TNFα or 50 ng/mL IFNγ for 16 hours. A, ATP concentration. B, Mitochondrial ROS levels were measured using mitoSOX dye; n = 8 per treatment across 2 separate experiments. C, 15 minutes before collection, cells were treated with 5 μM rotenone, a complex I inhibitor, or 1 μM antimycin A, a complex III inhibitor. Mitochondrial ROS levels were measured using mitoSOX dye; n = 8 per treatment. D, Mitochondrial complex I activity was determined using the Mitochondrial Dipstick Assay kit. Activity of no tx + vehicle control cells was set to 100%. E, Before pretreatment with FL3 or FL37, cells were transfected with siPHB or siNegative control (siNC) for 72 hours. Mitochondrial ROS levels were measured using mitoSOX dye. Total protein was analyzed by Western blotting to ensure efficiency of PHB knockdown by siPHB; *P < 0.05 versus no tx + vehicle; #P < 0.01 versus TNFα + vehicle; †P < 0.05 versus TNFα + vehicle + rotenone.
To determine mitochondrial ROS generation, we used the mitochondrial superoxide detection dye MitoSox Red. Ten nanomolar of FL3 or FL37 did not significantly affect basal mitochondrial ROS production (Fig. 6B). TNFα significantly increased mitoSOX fluorescence in Caco2-BBE cells, as shown previously.32,33 Pretreatment with 10 nM FL3 or FL37 prevented TNFα-induced mitochondrial ROS production.
Previous studies have shown that TNFα promotes cellular injury predominantly through mitochondrial ROS production resulting from decreased activity of ETC complex I.34 To determine whether FL3 or FL37 suppresses ROS production induced by complex I, mitoSOX fluorescence was measured in Caco2-BBE cells pretreated with 10 nM FL3 or FL37 followed by TNFα treatment and rotenone (5 μM; complex I inhibitor). The protective effect of FL3 or FL37 on TNFα-induced mitochondrial ROS production was sustained during the addition of rotenone, suggesting that complex I activity is resistant to rotenone inhibition in FL3- or FL37-treated cells (Fig. 6C). FL3 or FL37 did not protect against ROS production induced by the complex III inhibitor antimycin A (1 μM) during TNFα treatment. These results suggest that FL3 and FL37 preserve complex I activity, but not complex III activity, during TNFα treatment. Complex I activity was then measured by the Mitochondrial Dipstick Assay kit. FL3 or FL37 did not alter basal complex I activity in Caco2-BBE cells compared with vehicle control cells (Fig. 6D). TNFα treatment decreased complex I activity in vehicle-treated cells, whereas pretreatment with FL3 or FL37 prevented TNFα inhibition of complex I (Fig. 6D).
Because FL3 or FL37 treatment increases expression of PHB in Caco2-BBE cells (Fig. 2) and because PHB is crucial for complex I assembly and function,11 we next determined whether PHB was necessary for FL3 or FL37 protection against TNFα-induced mitochondrial ROS production (Fig. 6E). During loss of PHB expression by small interfering RNA transfection, FL3 or FL37 did not protect against mitochondrial ROS production induced by TNFα, suggesting that PHB is necessary for FL3 and FL37 protection against TNFα-induced mitochondrial dysfunction.
p38-MAPK Activation Is Necessary for Protective Effect of FL3 and FL37 on TNFα-induced Mitochondrial-derived ROS and Increased Permeability in Caco2-BBE Cells
Because FL3- and FL37-induced PHB and PHB2 protein expression was associated with p38-MAPK activation (Fig. 2B), but not activation of Stat3, SAPK/JNK, ERK, or AKT (see Fig., Supplemental Digital Content 2, http://links.lww.com/IBD/B117), we next determined whether p38-MAPK activation was essential for FL3 and FL37 protection against TNFα-induced epithelial mitochondrial and barrier dysfunction. SB203580, a p38-MAPK inhibitor, abolished the protective effect of FL3 or FL37 to inhibit TNFα-induced mitochondrial-derived ROS (Fig. 7A) and increased permeability in Caco2-BBE cells (Fig. 7B). SB203580 alone had no effect on mitochondrial ROS production or barrier function.
FIGURE 7.
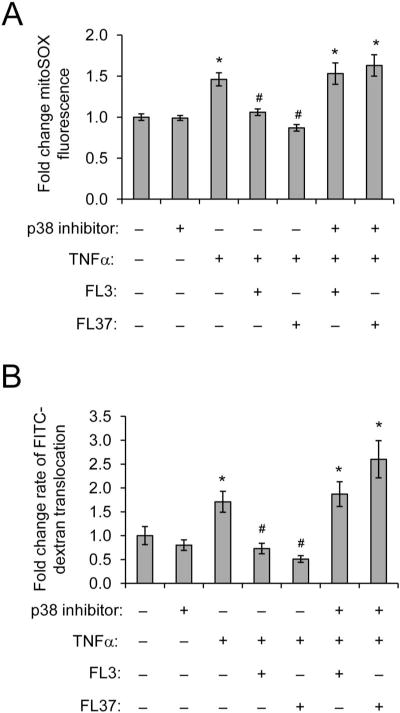
p38-MAPK activation is necessary for protective effect of FL3 and FL37 on TNFα-induced mitochondrial-derived ROS and increased permeability. Caco2-BBE monolayers were pretreated with 20 mM p38-MAPK inhibitor SB203580 for 1 hour, then treated with 10 nM FL3 or FL37 for 1 hour, and finally treated with 10 ng/mL TNFα for 16 hours A, Mitochondrial ROS levels were measured using mitoSOX dye; n = 8 per treatment. B, Rate of 4 kDa FITC-dextran translocation from apical to basolateral chamber (nanograms per milliliter per minute); n ≥ 6 per treatment; *P < 0.05 versus no tx; #P < 0.05 versus TNFα.
FL3 Protects Against DSS-induced Colitis
To determine the therapeutic potential of FL3 to reduce acute colitis, wild-type mice were intraperitoneally injected with 0.1 mg/kg FL3 or vehicle once daily on days 0 to 4 and administered DSS on days 0 to 6, which is a well-characterized model of colonic epithelial ulceration.35 Mice given vehicle during DSS administration showed significant weight loss starting on day 5 of DSS treatment (Fig. 8A). In contrast, FL3-treated mice maintained their body weight during the course of DSS treatment and exhibited body weights similar to control mice given water. On day 6, the mice treated with DSS were assigned a clinical score consisting of severity of body weight loss, stool consistency, and the presence of gross bleeding or blood in the stool. FL3-treated mice given DSS exhibited a significantly lower clinical score compared with vehicle-treated mice given DSS (Fig. 8B).
FIGURE 8.

FL3 protects against DSS-induced colitis. Mice were given 2.5% DSS for 6 days and intraperitoneally injected with 0.1 mg/kg FL3 or vehicle (veh) once daily on days 0 to 4. Control mice were given regular drinking water throughout the protocol; n = 6 per treatment group across 2 separate experiments with similar results. A, Percent change in body weight; *P < 0.05 versus DSS + FL3. B, Clinical and histological damage score; *P < 0.05 versus DSS + vehicle. C, Representative photomicrographs of paraffin-embedded hemotoxylin and eosin–stained sections of distal colon. Original magnification, ×20. D, Colon length measured on day 6 of DSS treatment. Photos of representative colons from DSS-treated mice; *P < 0.05. E, Neutrophil infiltration into the colon, quantified by measuring myeloperoxidase activity; *P < 0.05. F, On day 3 of DSS treatment, mice were gavaged with 4 kDa FITC-dextran. Translocation of fluorescent FITC-dextran across the intestinal epithelium was measured in serum collected 4 hours after gavage; *P < 0.05.
DSS-induced colitis is histopathologically characterized by infiltration of inflammatory cells into the mucosa and submucosa, epithelial ulceration, and crypt damage, with the distal colon the most severely affected. Hemotoxylin and eosin–stained sections of distal colon of water control mice treated with FL3 showed similar histology to mice given water and vehicle (Fig. 8C). Distal colon sections of DSS-treated mice given vehicle showed severe inflammatory infiltration, complete crypt loss in focal areas, and increased ulceration. In contrast, mice treated with FL3 during DSS showed moderate inflammatory infiltration, less crypt loss, and less ulceration compared with mice given vehicle (Fig. 8C). Histological scoring of inflammation revealed that FL3-treated mice given DSS exhibited a significantly less damage (severity of inflammatory infiltration, ulceration, and crypt damage) compared with vehicle-treated mice given DSS (Fig. 8B).
A reduction in colon length is a gross indicator of disease severity in the DSS model of colitis. All animals treated with DSS showed reduced colon length compared with water controls; however, shrinkage was less severe in FL3-treated mice compared with vehicle controls (Fig. 8D). In addition, FL3 treatment significantly reduced myeloperoxidase activity, a marker of neutrophil infiltration, in the distal colon during DSS colitis (Fig. 8E). Intestinal permeability was measured using translocation of 4 kDa FITC-dextran into serum in DSS-treated mice. Barrier dysfunction is one of the earliest events in DSS-induced colitis that precedes evident inflammation or mucosal damage. Therefore, we measured intestinal permeability after 3 days of DSS treatment. FL3-treated mice showed decreased FITC-dextran translocation compared with vehicle-treated mice during DSS colitis (Fig. 8F).
To determine whether in vivo effects of FL3 corroborate our in vitro results in cultured IECs, oxidative damage and colonic epithelial apoptosis were measured. FL3 decreased 4-HNE staining (Fig. 9A) and protein carbonyl content (Fig. 9B), markers of lipid peroxidation and oxidative damage to protein, respectively, in the colon of DSS-treated mice compared with vehicle. FL3 did not affect 4-HNE staining or generation of protein carbonyls in water-treated control mice. FL3 treatment abolished cleaved caspase 3 protein expression during DSS colitis in isolated colonic epithelial cells (Fig. 9C), suggesting that FL3 protects against DSS-induced epithelial apoptosis. This was further supported by TUNEL staining of distal colon, which demonstrated that FL3 treatment significantly decreased the number of TUNEL-positive epithelial cells per crypt during DSS colitis (Fig. 9D, E). FL3 did not significantly alter epithelial cleaved caspase 3 expression or the number of TUNEL-positive cells in water-treated control mice.
FIGURE 9.
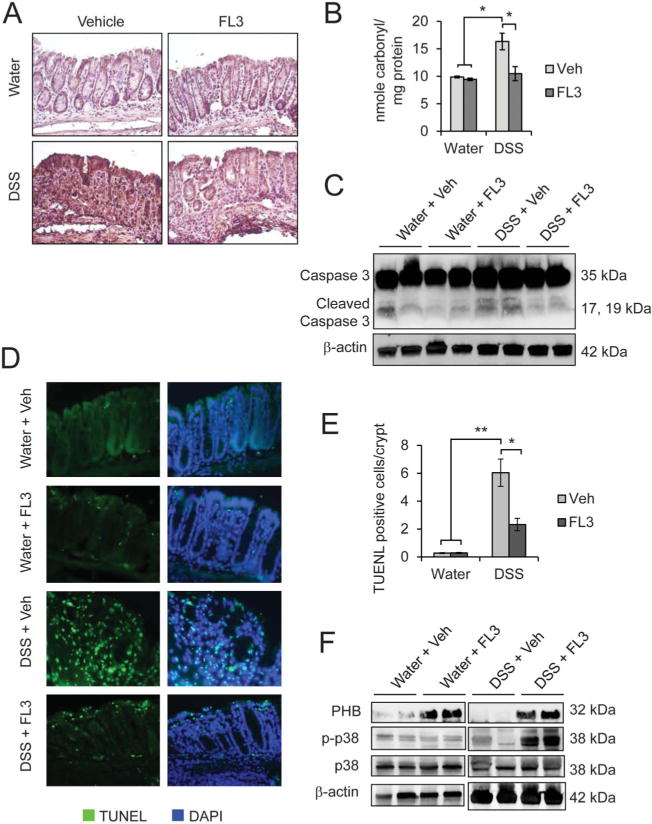
FL3 decreased colonic oxidative damage and epithelial apoptosis induced by DSS colitis, and these protective effects were associated with increased PHB and phospho-p38 MAPK expression. A, Immunohistochemistry staining of 4-HNE. B, Protein carbonyl content in the distal colon; *P < 0.05, n = 6 per treatment. C, Representative Western blots showing total and cleaved caspase 3 levels in isolated colonic epithelial cells. β-actin is included as a loading control. D, TUNEL staining (green) of colonic sections from mice treated with FL3 or vehicle (veh) during DSS colitis or control (water). Sections were stained with 4′,6-diamidino-2-phenylindole to visualize nuclei (blue). E, Number of TUNEL-positive cells per crypt in well-oriented crypts; *P < 0.05, **P < 0.01; n = 5 per treatment with a minimum of 20 crypts counted per animal. F, Representative Western blots showing PHB, phospho-p38-MAPK, p38-MAPK, and β-actin in total protein isolated from whole colon.
To determine whether the protective effects of FL3 treatment on DSS-induced colitis were associated with increased expression of PHB or p38-MAPK activation, total protein from distal colon was isolated and assessed by Western blotting. DSS-treated mice given FL3 exhibited increased PHB expression and p38-MAPK activation (Fig. 9F), but not activation of Stat3, SAPK/JNK, ERK, or AKT (see Fig., Supplemental Digital Content 4, http://links.lww.com/IBD/B119).
DISCUSSION
Our study is the first to characterize the therapeutic potential of flavaglines in the intestinal epithelium during inflammation. We show that the flavaglines FL3 and FL37 protect against apoptosis and barrier dysfunction in cultured IECs basally and during proinflammatory cytokine treatment. Protective effects of FL3 were corroborated in vivo, with FL3 administration reducing the severity of DSS-induced colitis and barrier permeability.
Since the first flavagline, rocaglamide, was first isolated in 1992, more than 60 natural flavaglines have been identified.20,36 Crude extracts from leaves and flowers of different Aglaia (family Meliaceae) plants, from which flavaglines are extracted, are used in several countries of southeast Asia as traditional medicine for the treatment of inflammatory skin diseases and allergic inflammatory disorders, such as asthma.6 However, these plants contain many classes of pharmacologically active agents, and the above-cited activities may not involve flavaglines but other classes of drugs. Interest in flavaglines as therapeutic compounds stems from their anticancer, anti-inflammatory, and cytoprotective properties. At very low nanomolar concentrations, flavaglines enhance survival of neurons and cardiomyocytes when challenged with numerous stressors.3 In this study, we assessed the effect of FL3 and FL37 in IECs because both compounds were shown to display in vivo anticancer and cardioprotective effects. As an in vitro model of inflammation, cultured IECs were challenged with TNFα and IFNγ, 2 proinflammatory cytokines involved in IBD pathogenesis.37,38 Similar to results in neurons and cardiomyocytes, FL3 and FL37 exhibited prosurvival and anti-inflammatory effects in IECs at nanomolar concentrations during TNFα and IFNγ treatment. FL3 and FL37 inhibited TNFα and IFNγ downstream proinflammatory signaling as evidenced by reduced nuclear factor kappa B and Cox2 expression, maintained cell viability, reduced TNFα- and IFNγ-induced apoptosis, and prevented TNFα- and IFNγ-increased epithelial permeability. We went on to demonstrate that mitochondrial dysfunction, as characterized by increased mitochondrial-derived ROS, reduced activity of ETC complexes and ultimately decreased ATP production, was induced by TNFα33 but not IFNγ, and was prevented by FL3 or FL37. Importantly, multiple studies have reported mitochondrial dysfunction in the epithelium during IBD and experimental models of colitis.12,25,27,28,39,40 Recent studies indicate that mitochondria integrate cellular homeostasis signaling and that mitochondrial stress participates in the pathology of IBD.22,24,41 Our results suggest that FL3 and FL37 protection of mitochondrial function in IECs is associated with decreased apoptosis, enhanced cell viability, and sustained epithelial barrier function during TNFα-induced damage. FL3 and FL37 also protected against IFNγ-induced apoptosis and epithelial barrier dysfunction, but unlike TNFα, these effects of IFNγ were not associated with altered mitochondrial function, suggesting that FL3 and FL37 cytoprotective effects against IFNγ involve signaling pathways beyond those regulating mitochondrial function.
FL3 and FL37 rapidly induced expression of PHB and PHB2 in IECs, which are established flavagline targets.3 Our study demonstrated that PHB expression was essential for FL3 or FL37 inhibition of TNFα-induced mitochondrial-derived ROS production in IECs. Furthermore, we show that FL3 and FL37 preserve ETC complex I activity during TNFα treatment. Complex I of the ETC is a predominant site of PHB binding, resulting in optimal activity of complex I and the respiratory chain.11,13 Previous studies have shown that TNFα reduces PHB expression in IECs,32,42 which we also confirm in the current study (Fig. 3). Therefore, our results suggest that FL3 and FL37 promote mitochondrial function in IECs during stress induced by TNFα by sustaining PHB expression and ETC complex I activity, which are targets of TNFα-induced damage.33,42
FL3 and FL37 rapidly induced activation of p38-MAPK in IECs, but not activation of Stat3, SAPK/JNK, ERK, or AKT. The pattern of p38-MAPK activation during a time course of FL3 or FL37 treatment was similar to that of PHB and PHB2 induction. Activation of p38-MAPK by flavaglines has been demonstrated in lymphocytes, which causes immunosuppression through selective inhibition of the transcription factor nuclear factor of activated T cells (NFAT).6 NFAT is expressed in the intestinal epithelium where it regulates differentiation, cell cycle, and apoptosis.43 It is not known whether PHBs mediate activation of p38-MAPK or NFAT by flavaglines.7 We show that p38-MAPK activation is essential for the protective effects of FL3 and FL37 on IEC barrier permeability and mitochondrial-derived ROS production during TNFα treatment. Future studies will elucidate whether p38-MAPK activation by FL3 or FL37 is downstream of PHBs and whether p38-MAPK activation involves inhibition of NFAT in IECs.
The in vivo therapeutic effect of FL3 was demonstrated using the DSS model of acute colitis in mice. This experimental model of colitis has similarities to human ulcerative colitis, including epithelial cell ulceration and loss of integrity of the mucosal barrier triggering inflammation.35 Given our results showing that FL3 and FL37 enhance cultured IEC survival and barrier function, the DSS model was optimal to test whether FL3 elicited similar protective effects in vivo. FL3 alone did not alter body weight or induce signs of toxicity in mice, as shown in previous in vivo studies.5,44 Additionally, colon histology of mice administered FL3 alone was similar to vehicle control mice. Once-daily injection of FL3 concurrent with DSS administration through day 4 prevented weight loss, colon shrinkage, neutrophil activation, histological damage including crypt loss and ulceration, epithelial apoptosis, ROS-induced damage, and epithelial barrier dysfunction. Reduced severity of colitis by FL3 was associated with increased colonic expression of PHB and activation of p38-MAPK, similar to our findings in cultured IECs. Flavaglines are chemical compounds that do not act as antioxidants through a chemical mechanism (as a reductant or radical scavenger) but through their action on PHB signaling. Not surprisingly, there has been considerable interest in developing antioxidant-based therapeutic strategies for the treatment of IBD. Commonly used drugs, in particular sulfasalazine and its active moiety 5-aminosalicylic acid, are potent ROS scavengers. However, targeted antioxidant therapies have not reached clinical efficacy perhaps because of limited cell permeability, short circulating half-life, and/or immunogenicity and the need to be used in large excess compared with the quantity of ROS. In contrast, compounds that promote the resistance and the destruction of ROS by activation of specific signaling pathways may be effective at low concentrations. Our study suggests that flavaglines may provide therapeutic potential against IBD by protecting the intestinal epithelium and reducing oxidative stress. Future studies will determine the bioavailability of flavaglines and their side effects.
Collectively, our in vitro and in vivo data demonstrate that flavaglines exhibit anti-inflammatory effects during colitis and promote IEC survival, mitochondrial function, and barrier integrity. Further preclinical investigations elucidating flavagline mechanism underlying protection against intestinal inflammation and promotion of epithelial cell homeostasis are warranted.
Supplementary Material
Acknowledgments
The authors thank the late Dr. Shanthi V. Sitaraman from Emory University, Atlanta, GA, for her scientific guidance. The authors are also grateful to AAREC Filia Research and ANRT for fellowships to C. Basmadjian and Q. Zhao.
Supported by National Institutes of Health grants R03-DK098229 (A. L. Theiss) and funds from the Baylor Research Institute. L. Désaubry was supported by the “Association pour la Recherche sur le Cancer.” C. Basmadjian and Q. Zhao received fellowships from AAREC Filia Research and ANRT.
Footnotes
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal’s Web site (www.ibdjournal.org).
The authors have no conflicts of interest to disclose.
Author contributions: Study concept and design, L. Désaubry and A. L. Theiss; acquisition of data, J. Han, Q. Zhao, C. Basmadjian, and A. L. Theiss; analysis and interpretation of data, J. Han and A. L. Theiss; drafting the manuscript, J. Han, L. Désaubry, and A. L. Theiss.
References
- 1.Neurath MF, Travis SP. Mucosal healing in inflammatory bowel diseases: a systematic review. Gut. 2012;61:1619–1635. doi: 10.1136/gutjnl-2012-302830. [DOI] [PubMed] [Google Scholar]
- 2.Fries W, Belvedere A, Vetrano S. Sealing the broken barrier in IBD: intestinal permeability, epithelial cells and junctions. Curr Drug Targets. 2013;14:1460–1470. doi: 10.2174/1389450111314120011. [DOI] [PubMed] [Google Scholar]
- 3.Thuaud F, Ribeiro N, Nebigil CG, et al. Prohibitin ligands in cell death and survival: mode of action and therapeutic potential. Chem Biol. 2013;20:316–331. doi: 10.1016/j.chembiol.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fahrig T, Gerlach I, Horvath E. A synthetic derivative of the natural product rocaglaol is a potent inhibitor of cytokine-mediated signaling and shows neuroprotective activity in vitro and in animal models of Parkinson’s disease and traumatic brain injury. Mol Pharmacol. 2005;67:1544–1555. doi: 10.1124/mol.104.008177. [DOI] [PubMed] [Google Scholar]
- 5.Bernard Y, Ribeiro N, Thuaud F, et al. Flavaglines alleviate doxorubicin cardiotoxicity: implication of Hsp27. PLoS One. 2011;6:e25302. doi: 10.1371/journal.pone.0025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Proksch P, Giaisi M, Treiber MK, et al. Rocaglamide derivatives are immunosuppressive phytochemicals that target NFAT activity in T cells. J Immunol. 2005;174:7075–7084. doi: 10.4049/jimmunol.174.11.7075. [DOI] [PubMed] [Google Scholar]
- 7.Basmadjian C, Thuaud F, Ribeiro N, et al. Flavaglines: potent anticancer drugs that target prohibitins and the helicase eIF4A. Future Med Chem. 2013;5:2185–2197. doi: 10.4155/fmc.13.177. [DOI] [PubMed] [Google Scholar]
- 8.Nijtmans LG, de Jong L, Artal Sanz M, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steglich G, Neupert W, Langer T. Prohibitins regulate membrane protein degradation by the m-AAA protease in mitochondria. Mol Cell Biol. 1999;19:3435–3442. doi: 10.1128/mcb.19.5.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatsuta T, Model K, Langer T. Formation of membrane-bound ring complexes by prohibitins in mitochondria. Mol Biol Cell. 2005;16:248–259. doi: 10.1091/mbc.E04-09-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bourges I, Ramus C, Mousson de Camaret B, et al. Structural organization of mitochondrial human complex I: role of the ND4 and ND5 mitochondria-encoded subunits and interaction with prohibitin. Biochem J. 2004;383:491–499. doi: 10.1042/BJ20040256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hsieh SY, Shih TC, Yeh CY, et al. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. 2006;6:5322–5331. doi: 10.1002/pmic.200500541. [DOI] [PubMed] [Google Scholar]
- 13.Tsutsumi T, Matsuda M, Aizaki H, et al. Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology. 2009;50:378–386. doi: 10.1002/hep.22998. [DOI] [PubMed] [Google Scholar]
- 14.Theiss AL, Idell RD, Srinivasan S, et al. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 15.Kathiria AS, Butcher MA, Hansen JM, et al. Nrf2 is not required for epithelial prohibitin-dependent attenuation of experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2013;304:G885–G896. doi: 10.1152/ajpgi.00327.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theiss AL, Vijay-Kumar M, Obertone TS, et al. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology. 2009;137:199–208. 208 e1–6. doi: 10.1053/j.gastro.2009.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kathiria AS, Butcher LD, Feagins LA, et al. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS One. 2012;7:e31231. doi: 10.1371/journal.pone.0031231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribeiro N, Thuaud F, Bernard Y, et al. Flavaglines as potent anticancer and cytoprotective agents. J Med Chem. 2012;55:10064–10073. doi: 10.1021/jm301201z. [DOI] [PubMed] [Google Scholar]
- 19.Ebada SS, Lajkiewicz N, Porco JA, Jr, et al. Chemistry and biology of rocaglamides (= flavaglines) and related derivatives from aglaia species (meliaceae) Prog Chem Org Nat Prod. 2011;94:1–58. doi: 10.1007/978-3-7091-0748-5_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ribeiro N, Thuaud F, Nebigil C, et al. Recent advances in the biology and chemistry of the flavaglines. Bioorg Med Chem. 2012;20:1857–1864. doi: 10.1016/j.bmc.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 21.Nenci A, Becker C, Wullaert A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446:557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 22.Bar F, Bochmann W, Widok A, et al. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology. 2013;145:1055–1063.e3. doi: 10.1053/j.gastro.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 23.O’Morain C, Smethurst P, Levi J, et al. Subcellular fractionation of rectal biopsy homogenates from patients with inflammatory bowel disease. Scand J Gastroenterol. 1985;20:209–214. doi: 10.3109/00365528509089659. [DOI] [PubMed] [Google Scholar]
- 24.Rath E, Berger E, Messlik A, et al. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61:1269–1278. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Restivo NL, Srivastava MD, Schafer IA, et al. Mitochondrial dysfunction in a patient with crohn disease: possible role in pathogenesis. J Pediatr Gastroenterol Nutr. 2004;38:534–538. doi: 10.1097/00005176-200405000-00014. [DOI] [PubMed] [Google Scholar]
- 26.Santhanam S, Rajamanickam S, Motamarry A, et al. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm Bowel Dis. 2012;18:2158–2168. doi: 10.1002/ibd.22926. [DOI] [PubMed] [Google Scholar]
- 27.Sifroni KG, Damiani CR, Stoffel C, et al. Mitochondrial respiratory chain in the colonic mucosal of patients with ulcerative colitis. Mol Cell Bio-chem. 2010;342:111–115. doi: 10.1007/s11010-010-0474-x. [DOI] [PubMed] [Google Scholar]
- 28.Tirosh O, Levy E, Reifen R. High selenium diet protects against TNBS-induced acute inflammation, mitochondrial dysfunction, and secondary necrosis in rat colon. Nutrition. 2007;23:878–886. doi: 10.1016/j.nut.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Dorn GW, II, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular Ensemble. Circ Res. 2015;116:167–182. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szczepanek K, Chen Q, Larner AC, et al. Cytoprotection by the modulation of mitochondrial electron transport chain: the emerging role of mitochondrial STAT3. Mitochondrion. 2012;12:180–189. doi: 10.1016/j.mito.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goossens V, Grooten J, De Vos K, et al. Direct evidence for tumor necrosis factor-induced mitochondrial reactive oxygen intermediates and their involvement in cytotoxicity. Proc Natl Acad Sci U S A. 1995;92:8115–8119. doi: 10.1073/pnas.92.18.8115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han J, Yu C, Souza RF, et al. Prohibitin 1 modulates mitochondrial function of Stat3. Cell Signal. 2014;26:2086–2095. doi: 10.1016/j.cellsig.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hansen JM, Zhang H, Jones DP. Mitochondrial thioredoxin-2 has a key role in determining tumor necrosis factor-alpha-induced reactive oxygen species generation, NF-kappaB activation, and apoptosis. Toxicol Sci. 2006;91:643–650. doi: 10.1093/toxsci/kfj175. [DOI] [PubMed] [Google Scholar]
- 34.Lopez-Armada MJ, Riveiro-Naveira RR, Vaamonde-Garcia C, et al. Mitochondrial dysfunction and the inflammatory response. Mitochondrion. 2013;13:106–118. doi: 10.1016/j.mito.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 35.Chassaing B, Aitken JD, Malleshappa M, et al. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr Protoc Immunol. 2014;104 doi: 10.1002/0471142735.im1525s104. Unit 15.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ng SC, Lam YT, Tsoi KK, et al. Systematic review: the efficacy of herbal therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2013;38:854–863. doi: 10.1111/apt.12464. [DOI] [PubMed] [Google Scholar]
- 37.Gyires K, Toth EV, Zadori SZ. Gut inflammation: current update on pathophysiology, molecular mechanism and pharmacological treatment modalities. Curr Pharm Des. 2014;20:1063–1081. doi: 10.2174/13816128113199990417. [DOI] [PubMed] [Google Scholar]
- 38.Slebioda TJ, Kmiec Z. Tumour necrosis factor superfamily members in the pathogenesis of inflammatory bowel disease. Mediators Inflamm. 2014;2014:325129. doi: 10.1155/2014/325129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delpre G, Avidor I, Steinherz R, et al. Ultrastructural abnormalities in endoscopically and histologically normal and involved colon in ulcerative colitis. Am J Gastroenterol. 1989;84:1038–1046. [PubMed] [Google Scholar]
- 40.Vanderborght M, Nassogne MC, Hermans D, et al. Intractable ulcerative colitis of infancy in a child with mitochondrial respiratory chain disorder. J Pediatr Gastroenterol Nutr. 2004;38:355–357. doi: 10.1097/00005176-200403000-00023. [DOI] [PubMed] [Google Scholar]
- 41.Rath E, Haller D. Mitochondria at the interface between danger signaling and metabolism: role of unfolded protein responses in chronic inflammation. Inflamm Bowel Dis. 2012;18:1364–1377. doi: 10.1002/ibd.21944. [DOI] [PubMed] [Google Scholar]
- 42.Theiss AL, Jenkins AK, Okoro NI, et al. Prohibitin inhibits tumor necrosis factor alpha-induced nuclear factor-kappa B nuclear translocation via the novel mechanism of decreasing importin alpha3 expression. Mol Biol Cell. 2009;20:4412–4423. doi: 10.1091/mbc.E09-05-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Q, Zhou Y, Jackson LN, et al. Nuclear factor of activated T cells (NFAT) signaling regulates PTEN expression and intestinal cell differentiation. Mol Biol Cell. 2011;22:412–420. doi: 10.1091/mbc.E10-07-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thuaud F, Bernard Y, Turkeri G, et al. Synthetic analogue of rocaglaol displays a potent and selective cytotoxicity in cancer cells: involvement of apoptosis inducing factor and caspase-12. J Med Chem. 2009;52:5176–5187. doi: 10.1021/jm900365v. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.