

Abstract
Pluripotent stem cells (PSCs), including induced PSCs, hold great potential for personalized disease modeling, drug testing, and cell-based therapeutics. However, cells differentiated from PSCs remain immature in a dish, and this has emerged as a major obstacle for their applications for adult-onset diseases such as cardiomyopathies and Alzheimer’s disease. By taking advantage of knowledge gained about mammalian development and from bioinformatics analyses, we recently developed a neonatal rat system that enables maturation of PSC-derived cardiomyocytes to cardiomyocytes analogous to those seen in adult animals. Here we describe a detailed protocol that describes how to initiate the in vitro differentiation of mouse and human PSCs into cardiac progenitor cells, followed by intramyocardial delivery of the progenitor cells into neonatal rat hearts, in vivo incubation, and analysis. The entire process takes about 6 weeks, and the resulting cardiomyocytes can be analyzed for morphology, function, and gene expression. The neonatal system provides a valuable tool to understand the maturation and pathogenesis of adult human heart muscle cells and this concept may be expanded to maturing other PSC-derived cell types, including those containing mutations that lead to development of diseases in the adult.
INTRODUCTION
Human induced pluripotent stem cells (hiPSCs) were first described in 2007 after Takahashi and colleagues reprogrammed somatic cells with certain transcription factors1. hiPSC can differentiate into any cell type of the body and thus hold great promise for disease modeling, drug discovery, repairing non-regenerative organs and studying human development2,3. Since their discovery numerous hiPSCs cell lines from patients with familial diseases have been developed3,4. Although iPSCs can differentiate into any type of body cell, they exhibit fetal-like characteristics, remain largely immature, and fail to fully integrate to the host organ upon transplantation5–8. This means they are not always suitable for studying diseases that manifest in the adult.
Characteristics of PSC-CMs
Heart disease supersedes all other causes of death worldwide9 and PSC-derived cardiomyocytes (PSC-CMs) offer tremendous opportunities for modeling genetic cardiomyopathies and treatment of heart failure with regenerative therapies4,10. However, nearly all cardiomyopathies develop in adult life, and many PSC-CMs do not truly recapitulate adult disease phenotypes, probably due to the immaturity of the cells.
Cardiac maturation initiates during early embryonic life and continues to early adulthood. During this process, CMs become rectangular, multinucleated, elongated and develop more organized sarcomeric structures5,16. Additionally, myosin heavy chain subtypes switch and T-tubule sarcolemma structures and intercalated discs to connect CMs are rapidly formed during the early postnatal period to enable functional maturation16,17. Analyzing numerous microarray datasets, we demonstrated that even after prolonged culture, PSC-CMs are comparable to late embryonic and neonatal stages7. In addition, their functional properties including Ca+2 transients and sarcomere shortening as well morphological characteristics such as size, shape, nucleation and presence of T-tubules are all consistent with immature fetal-like myocytes18,19. Finally, we have previously demonstrated that a number of transcription regulators are misregulated in long-term cultured PSC-CMs, which may explain the inability of the cells to mature beyond late embryonic/neonatal stages7.
Methods for PSC-CM maturation
Several groups have recently applied cellular engineering approaches to facilitate differentiation to more mature cardiomyocytes, including electrical stimulation, cell alignment techniques, culturing on different extracellular matrixes or mechanical stretching11–13. These approaches have resulted in CMs with more mature structural and functional properties, including increased conduction velocity, improved calcium handling properties etc. Additionally, treatment of PSC-CMs with either glucocorticoids or thyroid hormones promoted their maturation by increasing their size, sarcomere length, improving their contractility etc.14,15. Therefore, it appears that microenvironmental factors such as paracrine and endocrine signals, physical and electrical forces, and extracellular matrices might promote the maturation of PSC-CMs. Despite all these efforts, the resulting PSC-CMs partially mature and do not form T-tubules, acquire adult membrane potentials or shorten sarcomeres. Recently Kadota et al. used an in vivo approach by injecting hPSC-CMs in neonatal and adult rats, but the resulting CMs, determined by heart sections, did not exhibit the size and structure of adult CMs22. This might be due to the use of a different cell source, incubation time or analysis.
Experimental design
Islet 1 (Isl1) + CPCs are present in neonatal rodent and human hearts. Unlike PSC-CMs in culture, the vast majority of those neonatal CPCs give rise to fully mature CMs over the next few weeks in vivo5,23,24. Interestingly, recent comparative microarray data suggest that the maturation of CMs is regulated similarly in mice and humans25. Based on these findings, we explored the potential of the neonatal heart environment to facilitate maturation of CPCs by injecting mouse and human PSC-derived CPCs into neonatal (postnatal day 3–7) rat hearts26. To detect the injected cells in the rat hearts, we used fluorescent-labeled CPCs. We injected FACS-isolated mouse CPCs expressing the Isl1-RFP transgene or unsorted human CPCs expressing high percentages of Isl1. We elected to use rats instead of mice due to their larger heart size and the fact that their heart rate is closer to the human heart. To avoid cellular rejection we used immunosuppressed, athymic animals. We sacrificed the rats at various time points and noted that a relatively small percentage (~1%) of the transplanted cells engrafted26. CPCs had a much higher survival and engraftment rate and, within one month, in vivo-matured PSC-CMs developed the morphological and functional properties of adult CMs including T-tubule staining, Ca+2 transients and sarcomere shortening26. Moreover, their gene expression pattern, based on single cell RNA-sequencing analysis, was similar to adult CMs. It is worth noting that PSC-CMs do not mature properly when they are injected into older rat hearts (after postnatal day 14)26. This suggests the presence of a critical neonatal window that allows PSC-CM maturation.
Applications of the neonatal rat system for the generation of adult-like mature cardiomyocytes from pluripotent stem cells
Our system can be used to further understand the mechanisms of CM maturation as well as to study the pathogenesis of several adult cardiac diseases. Furthermore, rats with transplanted matured hiPSC-CMs can be used for the in vivo testing of drug therapies and for personalized medicine. Importantly, based on the same principal, our protocol may also be utilized for the maturation of other cell types like neurons, hepatocytes, skeletal muscle etc. derived from hiPSCs. In fact, a recent study used the neonatal brain to mature hiPSC-derived neurons in vivo for modeling Alzheimer’s disease27. Finally, considering the late onset of numerous diseases such as certain familial cardiomyopathies and Alzheimer’s disease, the neonatal system may be used to uncover the characteristics of cells at the point of disease onset, enabling earlier disease diagnosis, prevention, and better disease management.
Limitations
The main limitation of our protocol is the low cell engraftment, which is primarily due to the small size of the host heart and our current cell delivery method. However, it is very likely the use of larger animals such as pigs and a higher number of cell injections will markedly improve cellular engraftment and the yield of matured PSC-CMs, which will also potentially allow the in vivo modeling of human diseases in animal models. In addition, the need to generate a genetically modified reporter PSC line in order to locate the injected cells in the host’s heart may make our protocol less suitable for the production of mature cardiomyocytes for clinical applications.
MATERIALS
REAGENTS
Rats
-
We used athymic, T-cell deficient RNU (Charles River Laboratories) rats, between postnatal day 1 (P1) to postnatal day 7 (P7) as immunosuppressed hosts of injected cells.
! CAUTION Experiments using rodents must conform to all relevant institutional and governmental ethics regulations. This protocol was approved by the Johns Hopkins University animal care and use committee.
Embryonic Stem Cells
-
We used the mouse ESC reporter line (mESCIsl1-Cre; Rosa-RFP; aMHC-GFP) which we have previously generated28 and the human iPSC line 2016 which was provided by Takahashi et al.1
! CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
Glasgow’s MEM (GMEM) (Gibco, Cat no: 11710035)
Dulbecco’s Modified Eagle’s Medium high glucose (DMEM) (Gibco, Cat no: 11965-092)
Characterized Fetal Bovine Serum (FBS) 500mL (Invitrogen, Cat no: SH30071.03)
Sodium Pyruvate 100mM (Gibco, Cat no: 11360)
β-mercaptoethanol (Sigma, Cat no: M6250)
ESGRO® (LIF) (Millipore, Cat no: ESG1106)
PD0325901 (Selleckchem, Cat no: S1036)
CHIR99021 (Selleck chemicals, Cat no: S2924)
0.1% (w/v) Gelatin (EMD Millipore, Cat no: ES-006-B)
1X DPBS w/Calcium and Magnesium (Thermo Fisher Scientific, Cat no: 21-031-CV)
1X PBS w/o Calcium and Magnesium (Thermo Fisher Scientific, Cat no: 21-040-CV)
TrypLE (Gibco, Cat no: 12604)
IMDM (Gibco, Cat no: 12440053)
Ham’s F12 (Gibco, Cat no: 10-080-CV)
N2-SUPPLEMENT (Gibco, Cat no: 17502-048)
B27® minus vitamin A (50×) (Thermo Fisher Scientific, Cat no: 12587010)
B27® minus insulin (50×) (Thermo Fisher Scientific, Cat no: A1895601)
Bovine Serum Albumin (BSA) (Sigma, Cat no: A2153)
100X Pen/Strep (Gibco, Cat no: 15070-063)
Monothioglycerol (Sigma, Cat no: M-6145)
Ascorbic Acid (Sigma, Cat no: A-4544)
BMP4 (R & D Systems, Cat no: 314-BP)
Geltrex™ LDEV-Free Reduced Growth Factor Basement Membrane Matrix (ThermoFisher Scientific, Cat no: A1413202)
Essential 8 Medium (Gibco, Cat no: A1517001)
RPMI 1640 Medium (Gibco, Cat no: 11875119)
Y-27632 (ROCK inhibitor) (Stem cell technologies, Cat no: 72304)
XAV939 (Sigma, Cat no: X3004)
Isoflurane (Forane) (Baxter)
Penicillin-streptomycin (Gibco, Cat no: 15070)
Non-essential amino acid solution (NEAA; Invitrogen, Cat no: 11140-050)
-
Pierce™ 16% (w/v) Formaldehyde (Thermo Fisher Scientific, Cat no: 28906)
! CAUTION It is a hazardous solution and a cross-linking agent. Wear gloves and a lab coat when handle it. Dispose it appropriately after use.
GlutaMAX (100 x) (Gibco, Cat no: 35050-061)
Saponin (Sigma, Cat no: S4521)
Mouse anti-Islet1 (Developmental Studies Hybridoma Bank, Iowa City, IA)
Donkey anti-mouse IgG secondary antibody, Alexa Fluor® 647 conjugate (Thermo Fisher Scientific, Cat no: A-31571, Lot #1757130)
Sodium Chloride (Sigma, Cat no: S9888)
Potassium Chloride (Sigma, Cat no: P9333)
Magnesium Sulfate (Sigma, Cat no: M7506)
Sodium Phosphate Monobasic (Sigma, Cat no: S3139)
Sodium Bicarbonate (Sigma, Cat no: S5761)
Glucose (Sigma, Cat no: D9434)
HEPES (Sigma, Cat no: H3375)
Magnesium Chloride (Sigma, Cat no: M8266)
Calcium Chloride (Sigma, Cat no: C1016)
Collagenase type II (Worthington Biochemical Corporation, Cat no: LS004176)
Protease type XIV (Sigma, Cat no: P5147)
Fura-2AM (Thermo Fisher Scientific, Cat no: F1221)
Fluor 594-conjugated WGA antibody (Thermo Fisher Scientific, Cat no: W11262)
ProLong® Diamond Antifade mount. (Thermo Fisher Scientific, Cat no: P36961)
RNase inhibitor (Thermo Fisher Scientific, Cat no: N8080119)
DNase I (RNase free) (New England Biolabs, Cat no: M0303S)
RNase free water (Qiagen, Cat no: 129112)
SMARTscribe reverse transcriptase (Clontech, Cat no: 639536)
dNTP Mix (10mM each) (Thermo Fisher Scientific, Cat no: R0191)
DTT, 1M (Thermo Fisher Scientific, Cat no: P2325)
Advantage® 2 Polymerase mix (Clontech, Cat no: 639201)
Custom-designed PCR primers (Integrated DNA Technologies)
AMPure XP PCR purification kit (Beckman Coulter, Cat no: A63880)
RPMI 1640 without Glucose (Thermo Fisher Scientific, Cat no 11879020)
Sodium L-Lactate (Sigma, Cat no: 71718)
-
Mouse anti-Isl1 antibody, clone 39.4D5-c (Developmental Studies Hybridoma Bank)
! CRITICAL It is crucial that anti-islet1 antibody from this supplier is used rather than an alternative. We found other anti-islet1 antibodies were not as specific.
EQUIPMENT
Cell culture plate, six wells (Corning, Cat no. 3506)
T25 flasks (Corning, Cat no: 353109)
Falcon™ 15mL Conical Centrifuge Tubes (Corning, Cat no: 100150)
Falcon™ 50mL Conical Centrifuge Tubes (Corning, Cat no: 100151)
Cell culture petri dish, 100 mm × 20 mm (Corning, Cat no: 430293)
Suspension culture dish 150 mm × 25mm (Corning, Cat no: 430597)
Scepter™ 2.0 Handheld Automated Cell Counter with pack of 60μm Sensors (Millipore, Cat no: PHCC20060)
Corning Ultra Low Attachment T75 flask (Fisher Scientific, Cat no: 07-200-875)
Corning BioCoat™ Laminin 60mm TC-Treated Culture Dishes (Corning, Cat no: 354405)
Cell strainer 70μm (Fisher Scientific, Cat no: 08-771-2)
Cell strainer 100μm (Fisher Scientific, Cat no: 08-771-19)
5 mL Polystyrene round-bottom tube with a 40μm cell strainer (BD Falcon, Cat no: 352235)
Eppendorf® FemtoJet® Microinjector (Eppendorf, Cat no: 10910)
CO2 Incubator (Thermo Fisher, Cat no: 51030285)
Cell sorter (Sony SH800 or any other fluorescence-activated cell sorter)
BD Accuri C6 plus flow cytometer (BD Biosciences)
EVOS®FL microscope (Thermo Fisher scientific, Cat no: AMF4300)
Eclipse TE2000 inverted microscope (Nikon)
Centrifuge Sorvall Legend XT (ThermoFisher, Cat no: 75004508)
Fine Scissors 2.5 mm cutting edge (Fine Science Tools, Cat no: 15000-08)
Ultra-Fine Forceps (Fine Science Tools, Cat no: 11370-40)
Student Fine Scissors (Fine Science Tools, Cat no: 91460-11)
Tissue adhesive glue 3M VETBOND (Fisher Scientific, Cat no: NC9259532)
Mini Rocker (Bio-Rad, Cat no: 166-0710EDU)
Dissecting stereoscopic microscope Discovery V8 (Zeiss)
Sure-Seal Large Mouse/Rat Induction Chamber (WPI, cat. no. EZ-1785)
Stericup 500mL/1000mL Millipore Express® (Millipore, Cat no: SCGPU10RE)
Stericup 500mL/500mL Millipore Express® (Millipore, Cat no: SCGPU05RE)
Steriflip® 50mL Disposable Vacuum Filtration System (Millipore, Cat no: SCGP00525)
IonOptix software (Myocam-S)
pClamp 10 software (Molecular Devices)
Image J software Version 1.50e (NIH, http://imagej.nih.gov/ij)
Confocal microscope (Leica DM2500)
5mm round cover glass (Warner instruments, Cat no: W4 64-0700)
FlowJo software ver. 10.0.8r1
REAGENT SETUP
2i medium
1L of the medium contains 870 mL GMEM, 100 mL FBS, 10 mL GlutaMAX™, 10 mL NEM NEAA, 10 mL Sodium Pyruvate, 3 μL beta-mercaptoethanol, 20 μL of Lif (200 U/mL), 0.3 μM CHIR99021 and 0.1 μM PD0325901. Filter to sterilize it using a 1L filter and store at 4° C for up to one month.
MEF medium
500 mL of this medium contain 435 mL DMEM, 50 mL fetal bovine serum (FBS), 5 mL non-essential amino acids, 5 mL Sodium Pyruvate and 5 mL of GlutaMAX™. Filter to sterilize using a 500 mL filter and store at 4° C for up to one month.
SFD medium
1L of this medium contains 715 mL IMDM, 250 mL Ham’s F12, 5 mL N2-Supplement (0.5% v/v), 10 mL B27 minus Vitamin A, 5 mL of 10% (w/v) BSA (in PBS), 7.5 mL GlutaMAX™ and 7.5 mL Pen-Strep. Filter to sterilize using a 500 mL filter and store at 4° C for up to one month. ▲ CRITICAL To differentiate mESCs, add Ascorbic Acid (50μg/mL) and 3.9 × 10−3% (v/v) of Monothioglycerol (MTG) prior to using.
FACS sorting solution 10X
This solution contains 1% (v/v) FBS, 200 mM HEPES and 10 mM of EDTA in PBS. Filter to sterilize using a 50 mL filter and store at 4° C for up to two months.
RPMI plus B27 minus insulin medium
Mix 500mL of RPMI 1640 Medium with 10 mL of B27 minus insulin and store at 4° C for up to two months.
RPMI plus B27 minus Vitamin A medium
Mix 500mL of RPMI 1640 Medium with 10 mL of B27 minus Vitamin A and store at 4° C for up to two months.
FACS immunostaining solution
This solution contains 5% (v/v) FBS and 0.75% (w/v) of Saponin in PBS. Filter to sterilize using a 50 mL filter and store at 4° C for up to 3 months.
Tyrode’s solution 10X
This solution contains 1.37 M NaCl, 49 mM KCl, 12 mM MgSO4, 12 mM NaH2PO4, 150 mM Glucose and 200 mM HEPES in distilled water. Store this solution at room temperature (23–27° C) up to 3 months.
Perfusion buffer 10X
This solution contains 1.2 M NaCl, 54 mM KCl, 12 mM MgSO4, 10 mM NaH2PO4, 200 mM NaHCO3 and 56 mM Glucose in distilled water. Filter to sterilize using a 500 mL filter and keep at room temperature for up to 3 months.
Collagenase solution
Add 50 mg collagenase and 1 mg protease to 50 mL of 1× perfusion buffer prior to using and mix well. This solution cannot be stored and needs to be made fresh every time.
PROCEDURE
Generation of ESC-derived cardiac progenitor cells in vitro
Generate mouse (option A) or human (option B) progenitor cells.
A Generation of mouse ESC-derived cardiac progenitor cells in vitro ● TIMING 5 d
Grow mESCs (mESCsIsl1-Cre; Rosa-RFP; aMHC-GFP)28 on 0.1% (w/v) gelatin coated T25 flasks in 2i medium. When cells reach 70–80% confluence, proceed to the next step to start cell differentiation.
Rinse the cells once with Ca2+, Mg2+ free DPBS and then dissociate into single cells by adding 1 mL of TrypLE and incubating at 37° C for 3 min.
-
Inactivate TrypLE by adding 4 mL of MEF medium. Prepare the cells for counting by diluting the resuspended cells 1:10 in MEF medium and mixing well with a pipette. Attach the 60 μm Scepter sensor to the Scepter 2.0 handheld automated cell counter and then aspirate the cells. The total cell number equals the number provided by the automated counter multiplied by 10 times the number of the total mLs.
▲ CRITICAL STEP 10 × 106 cells are needed for cell differentiation. Use the remaining mESCs for cell maintenance.
Centrifuge 10 × 106 cells for 3 min at 270 g, at room temperature. Then aspirate the supernatant and resuspend the cells in 100 mL of SFD.
Split the resuspended cells into four 150 mm × 25 mm sterile plates and incubate in the 5% CO2 incubator for 48 hours. The mESCs should form embryoid bodies (EBs) during this 48 hour incubation.
-
Collect all the EBs in two 50 mL tubes and centrifuge for 3 min at 145 g, at room temperature.
▲ CRITICAL STEP Using a low speed (145 g) enables isolation of almost exclusively EBs and avoids the precipitation of single cells.
-
Aspirate the supernatant and resuspend the EBs in 100 mL of SFD medium, 6 μM of CHIR99021 and 0.4 ng mL−1 of BMP4 for differentiation induction. Split the resuspended EBs into four 150 mm × 25mm sterile plates and incubate them in the 5% CO2 incubator for 24 hours. Collect all the EBs in two 50 mL tubes and centrifuge at 145 g for 3 min, room temperature. Aspirate the supernatant and resuspend the EBs in 25 mL of SFD medium. Transfer the resuspended EBs in an ultra-low attachment 75 cm2 flask and incubate the EBs in 5% CO2 incubator for 48 hours.
CRITICAL STEP If you wish to also or alternatively mature EBs to mESC-CMs in vitro, centrifuge the EBs at 145 g for 3 min, room temperature and resuspend them in SFD medium. Culture cells; beating and aMHC-GFP expression should appear 2 days later. Continue to culture the CMs as EBs by changing the medium every other day or dissociating the EBs as in step A(ii), counting the cells and replating 5–6 × 105 cells/well in 0.1% (w/v) gelatin coated 24-well plates. The cells will attach to the bottom and should be incubated in the 5% CO2 incubator for one month, changing the SFD medium every other day. Cells can then be analyzed as described in Box 3
-
To dissociate the EBs and collect single CPCs, first centrifuge them at 145 g for 3min, room temperature and aspirate the supernatant. Add 1 mL of TrypLE and incubate at 37° C for 3 min. Mix well by pipetting to dissociate the cells.
? TROUBLESHOOTING
CRITICAL STEP At this time point almost all the EBs should express Isl1-RFP, this can be checked under the microscope (Fig. 2A).
Add 4 mL of MEF medium to inactivate TrypLE and mix well by pipetting. To remove the non-dissociated EBs, filter the mix using a 70 μm strainer and centrifuge the filtrated cells for 3 min at 270 g, room temperature. Aspirate the supernatant and add 1 mL of FACS sorting solution to resuspend.
To remove all cell clusters prior to sorting, filter the cells once more using a 5 mL polystyrene round-bottom tube with a 40 μm cell strainer. Keep the cells on ice until sorting.
-
Sort Isl1-RFP positive cells by FACS (Sony SH800) (Fig. 2B, Supplemental Figure 1) and collect the sorted cells in 1 mL of FBS.
▲ CRITICAL STEP Avoid long sorting times and keep the FACS sorting solution, the cell sample and sorted cells at low temperatures (~4–5° C) to decrease the amount of cell death. Injecting unsorted cells can lead to teratoma formation (Supplemental Figure 2)
? TROUBLESHOOTING
-
Centrifuge sorted Isl1-RFP positive CPCs for 3 min at 270 g, room temperature. Aspirate the supernatant and resuspend the cells in IMDM with Geltrex at 60:1 ratio to a final concentration of 2 × 103 cells mL−1. Keep CPCs on ice before transplantation.
▲ CRITICAL STEP To avoid the polymerization of Geltrex which will increase the viscosity of the cell sample and will clog the microneedle during cell injection, keep the sample on ice at all times for no more than 4 hours.
PAUSEPOINT
Figure 2.
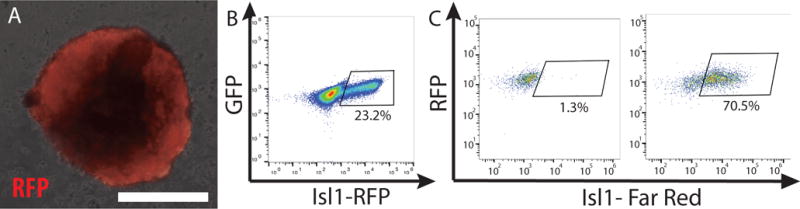
Isl1 positive cardiac progenitor cells. A. Isl1-Cre driven expression of Red Fluorescent Protein (RFP) in a mouse embryoid body. B. RFP+ CPCs are purified by FACS. (62,000 live cells were sorted 14,410 of those were Isl1-positive (23.3%)). C. Immunostaining for Isl1 and flow cytometry analysis of fixed human CPCs. Left plot: negative control with only secondary antibody. (2384 cells were analyzed and of those 31 were only Isl1-positive (1.3%)). Right plot: immunostaining with Isl1 primary antibody. (2750 cells were analyzed and of those 1940 were Isl1-positive (70.3%)). Scale bar 50 μm.
B Generation of human iPSC-derived cardiac progenitor cells in vitro ● TIMING 5–6 d
-
Generate a constitutively expressing GFP/RFP, hiPSC line (Box 1). Grow hiPSCs on Geltrex coated T25 flasks using Essential 8 medium until they reach 70–80% confluence (this usually occurs after 3–4 days), at which point you should proceed to the next step to start cell differentiation.
▲ CRITICAL STEP Coat each T25 flask with 3 mL of 1:60 Geltrex in DMEM, for 30–120 min, room temperature. Determine how many T25 flasks are needed based on the desired cell number. Each T25 flask when 70–80% confluent contains approximately 3–4 × 106 cells.
Coat at least 2 wells of a 6-well plate with Geltrex in DMEM (1:60) using 2 mL per well
Rinse the cells from step B(i) once with Ca2+, Mg2+ free DPBS and then dissociate into single cells by adding 1 mL of TrypLE and incubating at 37° C for 3 min.
-
Inactivate TrypLE by adding 4 mL of MEF medium. Prepare the cells for counting by diluting the resuspended cells 1:10 in MEF medium and mixing well with a pipette. Attach the 60 μm Scepter sensor to the Scepter 2.0 handheld automated cell counter and then aspirate the cells. The total cell number equals the number provided by the automated counter multiplied by 10 times the number of the total mLs.
▲ CRITICAL STEP 3 × 106 cells are needed for cell differentiation.
-
Centrifuge 3 × 106 hiPSCs for 3 min at 270 g, room temperature.
▲ CRITICAL STEP For cell maintenance keep 3–6 × 105 hiPSCs and replate them in a T25 flask.
-
Aspirate the supernatant and resuspend the cells you are differentiating in Essential 8 medium with 10 μM of Rock inhibitor (Y27632) at a concentration of 6 × 105 hiPSCs/mL and plate 2.5 mL per well of a 6-well plate.
▲ CRITICAL STEP 100% confluence is critical for efficient CM differentiation. Therefore, after adding the cells, tap the plate from all sides and leave it inside the hood at room temperature for approximately 15min. This will enable more homogenous plating of the cells.
-
To initiate cell differentiation the next day, change the medium to RPMI plus B27 minus insulin and 6 μM of CHIR99021 and incubate in the 5% CO2 incubator for 48 hours.
▲ CRITICAL STEP To maintain consistency and increase differentiation efficiency, all incubation times stated in this procedure should remain unchanged. In addition, the media should always be added slowly to avoid mechanical stress, which can affect cell differentiation. Use 2.5mL of medium per well.
Change the medium to RPMI plus B27 minus insulin and incubate in the 5% CO2 incubator for 24 hours.
-
Change the medium to RPMI plus B27 minus insulin and 10 μM of XAV939 and incubate in the 5% CO2 incubator for 48 hours.
CRITICAL STEP If you wish to continue to grow cells in culture rather than proceed to transplantation, change the medium of the cells to RPMI plus B27 minus insulin and incubate in the 5% CO2 incubator for a further 48 hours. Then change the medium to RPMI plus B27 without Vitamin A and incubate in the 5% CO2 incubator for 48 hours. At this stage, most cells will be contracting CMs. Cells can be cultured for one month, during which time continue changing the medium to RPMI plus B27 without Vitamin A every other day. Cells can then be analyzed as described in Box 3.
? TROUBLESHOOTING
-
The majority of the cells at this stage should express Isl1. To test the percentage of Isl1+ CPCs, perform immunofluorescent staining and flow cytometry of CPCs Box2 (Fig. 2C).
▲ CRITICAL STEP To improve the yield of mature CMs, proceed to CPC injection only if approximately more than 60–70% of CPCs are Isl1+. Alternatively, fresh cardiomyocytes appearing at days 8–9 may be used with the caveat that their engraftment and survival would be significantly lower in comparison to cardiac progenitor cells.
? TROUBLESHOOTING
Add 500 μL of TrypLE per well to dissociate the cells and incubate at 37° C for 3 min. Pipet and mix the cells gently for approximately 30 sec, to mechanically dissociate the CPCs and generate single cells. Then add 2 mL of MEF medium per well, mix and transfer all the cells to a 15 mL tube. Count the cells as above and centrifuge the CPCs for 3 min at 270 g, room temperature.
-
Aspirate the supernatant and resuspend the cells in cold (4° C) IMDM with Geltrex at 60:1 ratio and 10 μM of Rock inhibitor (Y27632). The cell concentration should be 2 × 103 cells mL−1.
PAUSEPOINT Keep CPCs on ice prior to transplantation for a maximum of 4 hours.
Box 2 Immunofluorescence staining of fixed cells for flow cytometry from step B(x)● TIMING 2–2.5 h
Centrifuge the cell mix for 3 min at 270 g, room temperature. Aspirate the supernatant and resuspend the cells in 4% (w/v) paraformaldehyde in PBS. Incubate for 30 min, room temperature to fix the cells.
Centrifuge the cell mix for 3 min at 895 g, room temperature. Aspirate the supernatant and resuspend the cells in PBS to wash the PFA. Repeat this step once more.
Aspirate the supernatant and resuspend the cells in FACS immunostaining solution. Split the sample in two and add mouse anti-Isl1 antibody (1:500 dilution) in one sample and use the other one as negative control. Incubate for 30 min, room temperature.
Wash the cells twice as described in step 2 using FACS immunostaining solution instead of PBS. Aspirate the supernatant and resuspend both cell samples in FACS immunostaining solution with 1:500 donkey anti-mouse IgG (H+L) secondary antibody, Alexa Fluor 647 conjugate. Incubate for 30 min, room temperature.
-
Wash twice with FACS immunostaining solution as in step E(ii). Aspirate the supernatant and resuspend the cells in PBS. Use the BD Accuri C6 plus flow cytometer to analyze the cells (Fig. 2c, Supplemental Figure 1).
▲ CRITICAL STEP Use an anti-mouse secondary antibody with high excitation and emission wavelengths to avoid non-specific separation of the GFP-tagged CPCs.
Surgery ● TIMING 7–15 min per rat pup
▲ CRITICAL Prior to rat surgery, autoclave all surgical tools.
-
2 To anesthetize the rat pups wrap in foil and place them on an ice bed (Fig. 3A). Pups should be anesthetized on the ice bed within 5–10 min.
▲ CRITICAL STEP Take one rat pup at a time and use only postnatal day 1–7 rats. Direct contact of pups with ice is associated with higher post-procedure mortality, therefore keep pups covered with aluminum foil.
▲ CRITICAL STEP Hypothermia minimizes blood loss during heart surgery and reduces the pup’s heart rate allowing for better cell engraftment.
▲ CRITICAL STEP The time to reach anesthesia varies and depends greatly on the age and weight of the pups. Usually 5 min is enough for P1 (5–7 g) rat pups, however for P7 (11–13 g) pups longer times up to 10 min are usually needed.
? TROUBLESHOOTING
-
3 Transfer the pup from the ice bed to the surgical bed. Put the rat pup in the supine position and tape all 4 limbs for immobilization. To sterilize the skin and prevent infections, wipe the chest with a Betadine swap.
▲ CRITICAL STEP For longer anesthesia during surgery, consider cooling the surgical bed using ice or ice packs.
4 Use the fine scissors (2.5 mm cutting edge) to make an incision of approximately 5–6 mm in the skin of the left upper chest wall, between the third and fourth ribs (Fig. 3B). Gently separate the skin from the underlying muscles using fine forceps. Dissect the muscles of the intercostal space and create an opening of approximately 4–5 mm using the forceps (Fig. 3B).
-
5 Visualize the left ventricular apex and proceed immediately to cell transplantation (Fig. 3C).
▲ CRITICAL STEP At this stage the skin and muscles of the chest wall are thin and the heart can usually be located. Entering the anterior mediastinum will require very gentle manipulations and practice to avoid injuring any of the major blood vessels.
▲ CRITICAL STEP Although a wider skin incision can help visualize the heart better, it will decrease the survival of the pup.
? TROUBLESHOOTING
Figure 3.

Surgical transplantation of cardiac progenitor cells for in vivo maturation. A. Neonatal rats 1–7 days old are placed on ice to induce anesthesia. B. Use the fine scissors to make a ~5 mm incision in the skin between the third and fourth ribs and gently separate the skin from the underlying muscles using fine forceps. Dissect the muscles of the intercostal space to create an opening of ~4 mm using the forceps C. Inject CPCs to the left ventricular apex using a microneedle connected to the Eppendorf FemtoJet Microinjector. D. Pinch the edges of the incision with the forceps and no more than 5 μL of the tissue adhesive glue to seal it the incision.
Cell transplantation and recovery ● TIMING 10–20 min per rat pup
-
6 Load immediately 10 μL of the CPC mix into the glass needle and inject all 10 μL into the left ventricular apex using the Eppendorf FemtoJet Microinjector.
▲ CRITICAL STEP Use the coarse mode of Eppendorf FemtoJet Microinjector for faster manipulations and overall quicker injections.
▲ CRITICAL STEP To avoid penetrating the ventricular wall and improve the pup survival and cell engraftment, do not advance the needle deep in the ventricular wall. Stop advancing after feeling resistance.
-
7 After the cell injection, pinch the edges of the chest wall incision with the forceps for 10 sec and then use no more than 5 μL of the tissue adhesive glue to seal it (Figure 3D).
▲ CRITICAL STEP Higher amounts of tissue glue can be toxic to the pup or can increase the probability of cannibalization by the mother not recognizing her own pups.
-
8 Transfer the pup immediately to a heating pad for 10–15 min and then return to the mother.
▲ CRITICAL STEP Ensure the pup is fully rewarmed and active prior to return to its mother’s cage. Additionally, rub the pup with cage bedding prior to return to its mother to improve survival.
▲ CRITICAL STEP Since prolonged hypothermia increases pup mortality, surgery should be completed within 5–10 min.
? TROUBLESHOOTING
Isolation of transplanted cardiomyocytes from injected rat heart ● TIMING ~1–1.5 h per heart, 30 d after cell injection
9 Transfer the 1 month-old injected rat to the anesthesia induction chamber and euthanize it by adding isoflurane for 3 min.
-
10 Quickly remove the heart and place it in perfusion buffer29. Cannulate the heart via the aorta and use a Langendorff system to perfuse the heart with perfusion buffer for 3 min at 35–37° C at a flow of 10 mL min−1. Then perfuse for 15 min using the collagenase solution at a flow of ~3mL min−1. Within 2 min into this perfusion add 50 μM of CaCl2.
▲ CRITICAL STEP Decrease the perfusion flow to 2–3 mL min−1 during collagenase treatment and then increase to 10mL min−1 immediately after.
11 Localize the area of transplanted cells by the presence of an obvious myocardial scar or by visualization of the heart under a fluorescence stereoscopic microscope (Fig. 4A). Cut the transplanted area using scissors (Student Fine Scissors) and transfer it into a small plastic container, which contains 5 mL of the collagenase solution and cut it to smaller pieces (approximately 20 times).
12 Add 45 mL of collagenase solution and mix gently up to 10 times using a plastic pipette. Transfer 35 mL of supernatant into a 50 mL tube.
13 Centrifuge the supernatant at 46 g for 1min, room temperature. Transfer 30 mL of the supernatant into the small plastic container with the rest of the heart pieces from step (iv). Keep the pellet.
14 Place the container on the Rocker at low speed for 1 min, room temperature.
-
15 Transfer the mix to a new 50 mL tube and centrifuge at 46 g for 1 min, room temperature.
▲ CRITICAL STEP To remove the non-dissociated pieces of heart tissue, filter the mix using a 100μm cell strainer.
16 Remove the supernatant and add 10 mL of Tyrode’s solution buffer with 1% (w/v) BSA (in PBS) and 250 μM of CaCl2 and gently invert to resuspend the CM pellet.
17 Add 10 mL of Tyrode’s solution with 0.5% (w/v) BSA (in PBS) to the pellet in the 50 mL tube from step v. Invert gently to resuspend the CMs.
18 Combine the two mixes from step (vi) and (vii), gently invert to mix and centrifuge for 1 min at 46 g, room temperature. Cells cannot be stored and will need to be analyzed immediately after this step. (Fig. 4B)
Figure 4.
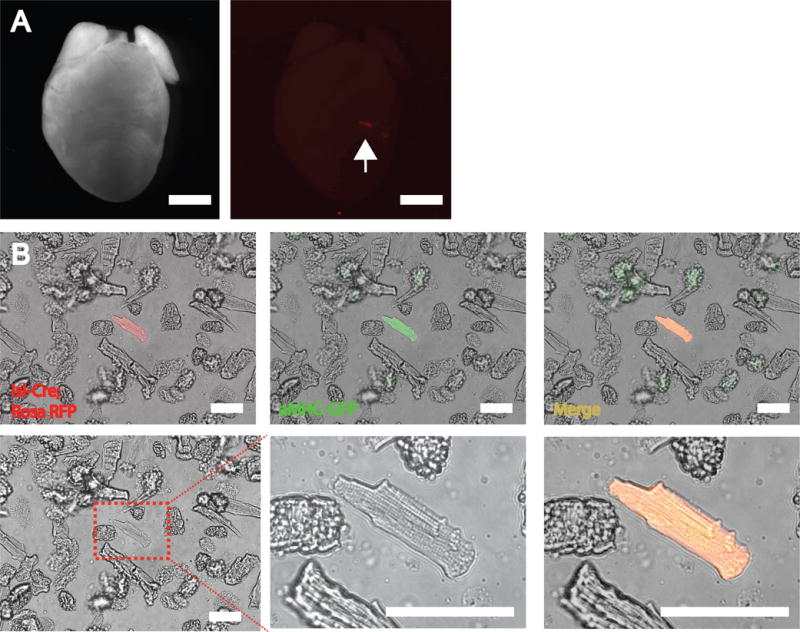
Isolation of mESC-CMs from injected rat hearts. A. Isolated rat hearts with injected RFP+ mouse CMs (arrow). Scale bars 500mm. B. Isolated adult-like mESC-CMs after processing of injected rat heart. The injected mESC-CMs are marked with GFP. Scale bars 100 μm.
Analysis of transplanted cardiomyocytes from injected rat heart ● TIMING 1.5–5 h per heart
19 Transplanted cardiomyocytes can be analyzed in various ways. To assess sarcomere shortening and calcium transients, follow option A. To fix cells for T-tubule or other analysis, follow option B. For cDNA generation for single cell gene expression analysis, follow option C.
(A) Sarcomere shortening and Calcium transients
Aspirate the supernatant and add 10 mL of Tyrode’s solution with 250 μM of CaCl2 and invert gently to resuspend the CM pellet. Leave the tube upright for 10 min at room temperature to let the CMs sink to the bottom. Remove the supernatant and add 10 mL of Tyrode’s solution with 500 μM of CaCl2.
Repeat the previous step, but add 10 mL of Tyrode’s solution with 1 mM of CaCl2.
Aspirate the supernatant and add 15 mL of Tyrode’s solution with 1 mM of CaCl2 containing 1 μM of the ratiometric Ca2+ indicator dye Fura-2AM and incubate for 10 min, room temperature.
Transfer the cells to a heated (at 37° C) perfusion chamber placed on the stage of an inverted microscope. Perfuse the CMs with Tyrode’s solution with 1mM of CaCl2 at a flow-through rate of 2 mL min−1. Stimulate the cells at 0.5 Hz with electrical pulses from two electrodes placed in the perfusion chamber.
Record CM shortening by video-edge detection using the IonOptix software. Use the same software for data analysis.
Record intracellular Fluo-3 fluorescence for Ca+2 transients and analyze the recorded data using pClamp 10 software29,30.
(B) Cell fixation for T-tubule staining and analysis
Plate isolated CMs in Tyrode’s solution with 1 mM of CaCl2 on a Laminin coated 60 mm dish and incubate in 5% CO2 incubator at 37° C for 30 min to let them attach to the dish.
-
Aspirate Tyrode’s solution and add 3 mL of 4% (w/v) paraformaldehyde in PBS and incubate for 20 min at room temperature for cell fixation. Wash paraformaldehyde twice with PBS. The cells can now be used for immunostaining and nucleation assessment with DAPI staining.
▲ CRITICAL STEP For confocal microscopy analysis plate the cells on Laminin coated coverslips.
PAUSEPOINT The cells can be stained immediately or after overnight storage at 4° C.
-
Block the fixed cells with 1% (w/v) BSA in PBS by incubating for 1 hour at room temperature. For T-tubule staining, add Fluor 594-conjugated WGA antibody at 1:1000 dilution and incubate at 4° C overnight. Wash the cells twice for 5 min with PBS.
▲ CRITICAL STEP Avoid shorter incubation times to prevent low intensity of the staining.
▲ CRITICAL STEP Alternative antibody combinations can be substituted, and a standard immunofluorescence staining performed.
Add DAPI to stain the nuclei and incubate for 10 min at room temperature.
-
Wash the cells twice for 5 minutes with PBS and mount the cells with ProLong Diamond Antifade mount.
PAUSEPOINT The cells can either be imaged immediately or stored overnight at 4° C and imaged the next day.
Image the T-tubules using a confocal microscope with a 403 (1.15 NA) oil-immersion lens.
Analyze WGA staining using ImageJ. Apply a median filter, create a binary mask of the WGA signal using the plugin Auto Local Threshold and erode the mask once.
Create a region of interest in each cell. Exclude the boundary membrane to isolate the T-tubule network for segmentation analysis.
-
Use the Analyze Particles plugin to measure the T-tubule area (mm2), which is normalized to the total cell area to calculate the fractional area.
▲ CRITICAL STEP Compare two groups using two-tailed t-test. Use the same optical field for statistical comparisons between host CMs and in vivo matured CMs to eliminate staining errors.
(C) cDNA generation for single cell gene expression analysis
Use a 20 μL pipette to manually pick the isolated GFP/RFP positive CMs (in Tyrode’s solution) under a fluorescent microscope (EVOS). Transfer each cell into a single well of a 96 well plate containing 2.4 μL RNase-free water, 0.2 μL RNase-free DNase I and 0.25 μL RNase inhibitor.
Incubate at 72° C for 3 min to inactivate DNase I and then quench on ice. Add 1 μL of custom-designed primer (with 30dT anchor) and transfer to 72° C for 2 min to anneal to polyadenylated RNA and then quench on ice.
Add a mixture of 1 μL SMARTscribe reverse transcriptase, 1 μL of custom-designed TSO oligo (12 mM), 0.3 μL MgCl2 (200 mM), 0.5 μL RNase inhibitor, 1 μL dNTP mix, 0.25 μL DTT (100 mM) and incubate at 42° C for 90 min, followed by enzyme inactivation at 70° C for 10 min.
Add a mixture of 29 μL water, 5 μL Advantage2 taq polymerase buffer, 2μL dNTPs, 2 μL custom-designed amplifying PCR primer (12 mM) and 2 μL of Advantage2 taq polymerase to the reverse transcription product and amplify for 20 cycles.
Purify the amplification product using Ampure XP beads. Use the cDNA to test gene expression by qPCR or perform RNA sequencing based on previously published protocols26,31.
Box 3 Analysis of in vitro matured PSC-derived cardiomyocytes ● TIMING 1.5–24 h per heart, 30 d after cell culture
1. Various methods can be used to analyse the cells. Follow option A for immunostaining and nucleation analysis, option B for a further in vitro cell culture and option C for sorting.
(A) Immunostaining and nucleation analysis
Fix in vitro plated PSC-CMs directly with 4% w/v paraformaldehyde in PBS for 30 min and use for immunostaining and nucleation analysis (DAPI staining).
Wash the fixed cells twice for 5 min with PBS at room temperature.
Stain for 5 min with DAPI (1:2000) in PBS followed by two washings for 5 min with PBS at room temperature. Keep the cells in PBS and either store overnight at 4° C or image immediately.
Save images and manually count and analyze the number of nuclei per cell.
(B) Further culture of cells
Replate the cells by dissociating with TrypLE (as described in step1 A(ii) and (iii)) and replating on gelatin coated 5mm round cover glass in either SFD medium for mouse CMs or RPMI plus B27 minus Vitamin A with 10% FBS and 10 μM of Rock inhibitor for human CMs to increase cell survival.
Transfer the cover glass with plated CMs into the perfusion chamber just before measuring Ca+2 transients and sarcomere shortening.
Measure whole-cell Ca+2 transients and sarcomere shortening as described in step 19A.
(C) Sorting of cells
Sort cells as single cells using a cell sorter or pick single cells directly under microscope (EVOS) and transfer them to 96-well plate for cDNA preparation as above.
Box 1 - Generation of GFP/RFP-tagged human iPSCs ● TIMING ~ 3 w
ADDITIONAL MATERIALS
REAGENTS
Lentivirus
-
We used a CAG-GFP lentivirus (Cellomics Technology, Cat no: PLV-10057-50). Alternatively, a CAR-RFP lentivirus can also be used (Cellomics Technology, Cat no: PLV-10071-50)
! CAUTION The handling of lentiviral vectors should be carried out using the proper biosafety containment. See Guidance on Biosafety Considerations for Research with Lentiviral Vectors (http://osp.od.nih.gov/sites/default/files/resources/Lenti_Containment_Guidance.pdf)
DMSO (Corning Cellgro, Cat no: 25-950-CQC)
EQUIPMENT
• Cryogenic vials (Corning, Cat no: CLS431421)
-
1 Coat with Geltrex 3 wells of a 6-well plate as described in step 1B(i). Dissociate plated hiPSCs as described in steps 1A(ii) and 1A(iii). Resuspend 6 × 105 cells in 6 mL of Essential 8 medium with 10 μM of Rock inhibitor and split them in three 15 mL tubes. Quickly thaw one vial of the CAG-GFP/RFP lentivirus and test three different volumes by adding 2, 5 and 10 μL to determine the optimal transduction efficiency of the virus. Mix well and plate the infected hiPSCs in the wells. Incubate in the 5% CO2 incubator overnight.
▲ CRITICAL STEP Split the rest of the virus in 2 μL aliquots and store in −80° C to avoid thaw and refreeze which will decrease the transduction efficiency of the virus. Use bleach to clean all equipment that is in contact with the virus.
2 The next morning change the medium to Essential 8 to avoid cell toxicity. Then over the next days, change the medium to Essential 8 daily and determine the transduction efficiency using a fluorescent microscope to check GFP/RFP expression. When cells reach 60–70% confluence, dissociate the cells in the well with the highest GFP/RFP expression and replate ~5 × 104 cells in two Geltrex coated sterile 10 cm petri dishes (in Essential 8 with 10 μM of Rock inhibitor).
3 Continue to change Essential 8 medium daily and then 2–3 days later single cells will form colonies. Pick single GFP/RFP-only colonies using a 20 μL pipette under a fluorescent microscope in a cell culture hood. Transfer colonies in Geltrex coated wells of a 24-well plate containing Essential 8, 10 μM of Rock inhibitor and Penicillin-streptomycin.
4 Grow the cells for 3–4 days and then replate only the colonies where all cells have GFP/RFP expression. Replate each colony to a new well of 6-well plate and then subsequently expand the cells by growing them in a T25 flask.
-
5 When GFP-tagged hiPSCs reach 70–80% confluence split them in 4 cryovials with 10% DMSO in MEF medium and freeze them in −80° C for two days and then store them in liquid nitrogen.
▲ CRITICAL STEP To improve the survival of the colonies picked in step 3, keep the cells in the same Essential 8 medium with Rock inhibitor for 48 hours.
PAUSEPOINT Cells can be stored in liquid nitrogen for at least 10 years.
6 To thaw the cells, prepare 4mL of prewarmed at 37° C MEF medium in a 15 mL tube. Quickly thaw the frozen cells by moving the cryovial from liquid nitrogen directly to a 37° C water bath and before the cells are completely thaw add 1mL of the prewarmed MEF medium directly to the cells in the cryovial. Mix well with the rest of the MEF medium in the 15 mL tube. Centrifuge for 3 min at 270 g, room temperature. Aspirate the supernatant and resuspend the cells in Essential 8 medium with 10 μM of Rock inhibitor (Y27632) and replate them in a T25 flask coated with matrigel as in step 1(B)i.
-
? TROUBLESHOOTING
Step Problem Solution
1A(viii) Usually 25–35% of the cells will be positive for Isl1 but occassionaly the percentage of Isl1-RFP cells is less than 10% Incubate the EBs for 12–24 hours longer to increase the percentage of isl1+ cells
1A(xi) High percentsge of sorted dead cells Add DAPI to FACS sorting solution (dilution 1:2000) to stain and exclude dead cells. In addition, to improve cell survival, consider adding 10mM Rock inhibitor to the FACS sorting solution
B(ix) Low percentage of hiPSC-CMs Incubate the cells at day 9, in RPMI without glucose and 5 mM of sodium lactate. Change the medium every other day for a maximum of 4 days. Then continue culturing the cells in RPMI-B27 without Vitamin A.
B(x) Low percentage of isl1+ human CPCs optimize the cell differentiation protocol by testing different concentrations of CHIR99021 (up to 10mM depending on the hiPSC line) and XAV939. Additionally, try analyzing CPCs 12–24 hours later.
2 Increased post-procedure mortality due to prolonged hypothermia Monitor closely the pups while they are on ice.
5 Pups develop bleeding during surgery Use hypothermia during surgery and fine forceps with blunt ends.
8 CPCs do not engraft or the final number of cells is low, Use a previously published prosurvival cocktail32 to increase the engraftment and survival of the transplanted cells
• TIMING
Steps A(i)-(xii), Generation of mouse ESC-derived cardiac progenitor cells in vitro: 5 d
Steps B(i)-(xii), Generation of mouse ESC-derived cardiac progenitor cells in vitro: 5–6 d
Steps 2–5, Surgery: 7–15 min per rat pup
Steps 6–8, Cell transplantation and recovery: 10–20 min per rat pup
Steps 9–18, Isolation of transplanted cardiomyocytes from injected rat heart: ~1–1.5 h per heart, one month after cell injection
Step 19 Analysis of transplanted cardiomyocytes from injected rat heart: 1.5–5 h per heart
Box 1, Generation of GFP/RFP-tagged human iPSCs: ~ 3 w
Box 2 Immunofluorescence staining of fixed cells for flow cytometry: 2–2.5 h
Box 3, Analysis of in vitro matured PSC-derived cardiomyocytes: 1.5–24 h per heart, 30 d after cell culture
ANTICIPATED RESULTS
This protocol describes a method to generate mature, adult-like mouse or human PSC-CMs, using neonatal immunosuppressed rats as a live bioincubator. It takes advantage of the environmental cues of a neonatal heart to advance CM development, which is otherwise blocked, during their in vitro growth, at late embryonic stages.
After one month of CPC transplantation, isolated mouse or human PSC-CMs should acquire the morphologic and functional characteristics of an adult CM (Fig. 5). Those cells can be compared with in vitro matured CMs. We have not observed any teratomas after the injection of highly enriched CPCs. Importantly, as we have demonstrated, in vivo matured hiPSCs can recapitulate adult cardiomyopathies and can expedite the modeling of diseases that are otherwise challenging to study in vitro26. The resulting human-rat chimeras can potentially be used for in vivo drug testing, bringing us a step closer to personalized medicine.
Figure 5.
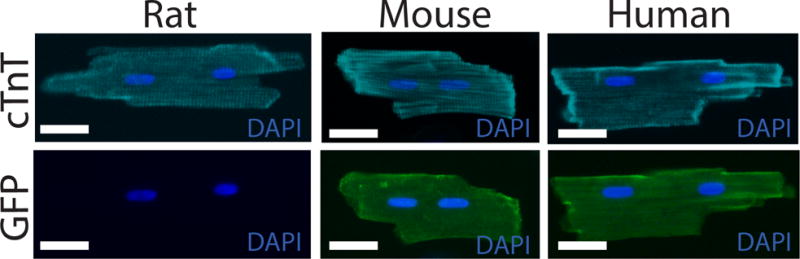
In vivo matured mouse and human PSC-cardiomyocytes. Injected mouse and human cardiomyocytes were identified by GFP expression. In vivo matured CMs had sarcomere structure, size and binucleation very similar to adult CMs. cTnT: cardiac Troponin T. For all animal experiments appropriate institutional regulatory board permission was obtained. Scale bars 25 μm.
More examples of typical morphologic results can be found in Figure 1 of the ref. 23. Examples of the functional properties of in vivo matured PSC-CMs can be found in Figures 2 and 4 of the ref. 23.
Supplementary Material
Supplemental Figure 1. FACS gating strategy. A. FACS gating strategy for the sorting of live Isl-1+ mouse CPCs. First plot gating for live cells, then second plot for single and then only for Isl1-1+ RFP cells. B. FACS gating strategy for the analysis of human CPCs. The cells were fixed and stained with a primary mouse anti-Islet-1 antibody and a secondary far red (647 nm emission wavelength) conjugated antibody. First plot gating for fixed cells, then single cells and then Isl-1+ Far Red CPCs.
Supplemental Figure 2. Teratoma formation (asterisk) after injection of unsorted mouse ESC-derived cells. Arrow points to the dissected part of the heart.
Figure 1.

Experimental protocol for in vivo and in vitro cardiomyocyte maturation. Left: Schematic illustration showing in vitro differentiation of mouse Isl1+ CPCs in red and human Isl1+ CPCs in green. Right: in vitro vs. in vivo maturation of cardiomyocytes derived from mouse and human-PSCs.
Editorial summary.
This protocol describes how to generate mature adult-like cardiomyocytes by culturing mouse or human PSCs in vitro initially and then transferring to neonatal rats for further cell maturation.
Acknowledgments
The authors thank Kwon laboratory members for critical reading and discussions. E.T. was supported by Johns Hopkins School of Medicine Clinician Scientist Award. This work was supported by the Magic that Matters Fund, MSCRF (2015-MSCRFI-1622), NHLBI/NIH (R01HL111198), and NICHD/NIH (R01HD086026) to C.K.
Footnotes
AUTHOR CONTRIBUTIONS
G.C. and C.K. designed the experiments. G.C., E.T., and P.A. performed the experiments. G.C., E.T., and P.A. analyzed the data. E.T. and P.A. made the figures. E.T., G.C., P.A., and C.K wrote the manuscript. All authors approved the manuscript.
COMPETING FINANCIAL INTERESTS
Authors declare no competing financial interests.
References
- 1.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 2.Burridge PW, Keller G, Gold JD, Wu JC. Production of de novo cardiomyocytes: human pluripotent stem cell differentiation and direct reprogramming. Cell stem cell. 2012;10:16–28. doi: 10.1016/j.stem.2011.12.013. S1934-5909(11)00594-7 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bellin M, Marchetto MC, Gage FH, Mummery CL. Induced pluripotent stem cells: the new patient? Nat Rev Mol Cell Biol. 2012;13:713–726. doi: 10.1038/nrm3448. nrm3448 [pii] [DOI] [PubMed] [Google Scholar]
- 4.Moretti A, Laugwitz KL, Dorn T, Sinnecker D, Mummery C. Pluripotent stem cell models of human heart disease. Cold Spring Harbor perspectives in medicine. 2013;3 doi: 10.1101/cshperspect.a014027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circulation research. 2014;114:511–523. doi: 10.1161/CIRCRESAHA.114.300558. CIRCRESAHA.114.300558 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabar V, Studer L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature reviews Genetics. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uosaki H, et al. Transcriptional Landscape of Cardiomyocyte Maturation. Cell reports. 2015;13:1705–1716. doi: 10.1016/j.celrep.2015.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cho GS, Fernandez L, Kwon C. Regenerative medicine for the heart: perspectives on stem-cell therapy. Antioxidants & redox signaling. 2014;21:2018–2031. doi: 10.1089/ars.2014.6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. International journal of cardiology. 2013;168:934–945. doi: 10.1016/j.ijcard.2012.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshida Y, Yamanaka S. iPS cells: a source of cardiac regeneration. Journal of molecular and cellular cardiology. 2011;50:327–332. doi: 10.1016/j.yjmcc.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 11.Nunes SS, et al. Biowire: a platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nature methods. 2013;10:781–787. doi: 10.1038/nmeth.2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chun YW, et al. Combinatorial polymer matrices enhance in vitro maturation of human induced pluripotent stem cell-derived cardiomyocytes. Biomaterials. 2015;67:52–64. doi: 10.1016/j.biomaterials.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruan JL, et al. Mechanical Stress Promotes Maturation of Human Myocardium From Pluripotent Stem Cell-Derived Progenitors. Stem cells. 2015;33:2148–2157. doi: 10.1002/stem.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rog-Zielinska EA, et al. Glucocorticoids promote structural and functional maturation of foetal cardiomyocytes: a role for PGC-1alpha. Cell death and differentiation. 2015;22:1106–1116. doi: 10.1038/cdd.2014.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang X, et al. Tri-iodo-l-thyronine promotes the maturation of human cardiomyocytes-derived from induced pluripotent stem cells. Journal of molecular and cellular cardiology. 2014;72:296–304. doi: 10.1016/j.yjmcc.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robertson C, Tran DD, George SC. Concise review: maturation phases of human pluripotent stem cell-derived cardiomyocytes. Stem cells. 2013;31:829–837. doi: 10.1002/stem.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smolich JJ. Ultrastructural and functional features of the developing mammalian heart: a brief overview. Reproduction, fertility, and development. 1995;7:451–461. doi: 10.1071/rd9950451. [DOI] [PubMed] [Google Scholar]
- 18.Kane C, Couch L, Terracciano CM. Excitation-contraction coupling of human induced pluripotent stem cell-derived cardiomyocytes. Frontiers in cell and developmental biology. 2015;3:59. doi: 10.3389/fcell.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knollmann BC. Induced pluripotent stem cell-derived cardiomyocytes: boutique science or valuable arrhythmia model? Circulation research. 2013;112:969–976. doi: 10.1161/CIRCRESAHA.112.300567. discussion 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lundy SD, Zhu WZ, Regnier M, Laflamme MA. Structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Stem Cells Dev. 2013;22:1991–2002. doi: 10.1089/scd.2012.0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mihic A, et al. The effect of cyclic stretch on maturation and 3D tissue formation of human embryonic stem cell-derived cardiomyocytes. Biomaterials. 2014;35:2798–2808. doi: 10.1016/j.biomaterials.2013.12.052. [DOI] [PubMed] [Google Scholar]
- 22.Kadota S, Pabon L, Reinecke H, Murry CE. In Vivo Maturation of Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes in Neonatal and Adult Rat Hearts. Stem cell reports. 2017;8:278–289. doi: 10.1016/j.stemcr.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Laugwitz KL, et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature. 2005;433:647–653. doi: 10.1038/nature03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Genead R, et al. Islet-1 cells are cardiac progenitors present during the entire lifespan: from the embryonic stage to adulthood. Stem Cells Dev. 2010;19:1601–1615. doi: 10.1089/scd.2009.0483. [DOI] [PubMed] [Google Scholar]
- 25.Uosaki H, Taguchi YH. Comparative Gene Expression Analysis of Mouse and Human Cardiac Maturation. Genomics, proteomics & bioinformatics. 2016;14:207–215. doi: 10.1016/j.gpb.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho GS, et al. Neonatal Transplantation Confers Maturation of PSC-Derived Cardiomyocytes Conducive to Modeling Cardiomyopathy. Cell reports. 2017;18:571–582. doi: 10.1016/j.celrep.2016.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espuny-Camacho I, et al. Hallmarks of Alzheimer’s Disease in Stem-Cell-Derived Human Neurons Transplanted into Mouse Brain. Neuron. 2017;93:1066–1081 e1068. doi: 10.1016/j.neuron.2017.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Shenje LT, et al. Precardiac deletion of Numb and Numblike reveals renewal of cardiac progenitors. eLife. 2014;3:e02164. doi: 10.7554/eLife.02164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee DI, et al. PDE5A suppression of acute beta-adrenergic activation requires modulation of myocyte beta-3 signaling coupled to PKG-mediated troponin I phosphorylation. Basic research in cardiology. 2010;105:337–347. doi: 10.1007/s00395-010-0084-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tandon N, et al. Electrical stimulation systems for cardiac tissue engineering. Nature protocols. 2009;4:155–173. doi: 10.1038/nprot.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shin J, et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell stem cell. 2015;17:360–372. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laflamme MA, et al. Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nature biotechnology. 2007;25:1015–1024. doi: 10.1038/nbt1327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. FACS gating strategy. A. FACS gating strategy for the sorting of live Isl-1+ mouse CPCs. First plot gating for live cells, then second plot for single and then only for Isl1-1+ RFP cells. B. FACS gating strategy for the analysis of human CPCs. The cells were fixed and stained with a primary mouse anti-Islet-1 antibody and a secondary far red (647 nm emission wavelength) conjugated antibody. First plot gating for fixed cells, then single cells and then Isl-1+ Far Red CPCs.
Supplemental Figure 2. Teratoma formation (asterisk) after injection of unsorted mouse ESC-derived cells. Arrow points to the dissected part of the heart.