

Summary
RNAi factors and their catalytic activities are essential for heterochromatin assembly in S. pombe. This has led to the idea that siRNAs can promote H3K9 methylation by recruiting the Cryptic Loci Regulator Complex (CLRC), also known as Recombination In K Complex (RIKC), to the nucleation site. The conserved RNA-binding protein Rct1 (AtCyp59/SIG-7) interacts with splicing factors and RNA polymerase II. Here we show that Rct1 promotes processing of pericentromeric transcripts into siRNAs, via the RNA recognition motif. Surprisingly, loss of siRNA in rct1 mutants has no effect on H3K9 di- or tri-methylation, resembling other splicing mutants, and suggesting post-transcriptional gene silencing per se is not required to maintain heterochromatin. Splicing of the Argonaute gene is also defective in rct1 mutants, and contributes to loss of silencing but not to loss of siRNA. Our results suggest that Rct1 guides transcripts to the RNAi machinery by promoting splicing of elongating non-coding transcripts.
Keywords: RNA interference (RNAi), Heterochromatin silencing, H3K9 methylation, small interfering RNA (siRNA), Splicing, Rct1 (RRM-containing cyclophilin regulating transcription), non-coding transcripts, exosome
Graphical Abstract
Introduction
RNA interference (RNAi) is a mechanism for gene silencing conserved in most eukaryotes though with a few exceptions, including the budding yeast Saccharomyces cerevisiae. In plants, Drosophila and Caenorhabditis elegans, as well as in the fission yeast Schizosaccharomyces pombe, RNAi is involved in both post-transcriptional and transcriptional silencing by modifying chromatin into silent heterochromatin. Once assembled, chromatin modification is faithfully inherited, suggesting coordination between DNA replication and heterochromatin assembly (Castel and Martienssen, 2013).
The genome of S. pombe has blocks of heterochromatin at centromeres, subtelomeres, and at the mating-type locus. Heterochromatin is characterized by hypoacetylated histones H3 and H4, and H3 lysine 9 (H3K9) methylation in most eukaryotes including S. pombe. Methylated H3K9 serves as a binding site for heterochromatin protein HP1, encoded by Swi6 and Chp2 in S. pombe, leading to epigenetic repression. During the cell cycle, repressive histone marks are diluted by newly synthesized histones deposited on replicated DNA, and Swi6 is transiently evicted by phosphorylation of H3 serine 10, resulting in heterochromatic transcription (Kloc et al., 2008, Chen et al., 2008). Paradoxically, RNA polymerase II (Pol II) transcription is essential to re-establish repressive histone marks in G2: specific mutations in Pol II subunits Rpb2 and Rpb7 impair siRNA biogenesis and heterochromatin maintenance at the pericentromeric region (Djupedal et al., 2005, Kato et al., 2005).
Subsequently, pericentromeric transcripts are converted to double-stranded RNAs (dsRNAs) by the action of RNA-dependent RNA Polymerase 1 (Rdp1) (Motamedi et al., 2004). The RNase III family endonuclease Dicer (Dcr1) processes dsRNA precursors into 22-24 nt long siRNAs (Colmenares et al., 2007, Volpe et al., 2002). These siRNAs are loaded onto the RITS (RNA-Induced Transcriptional Silencing) complex, which contains Argonaute (Ago1), Chp1 and Tas3 (Verdel et al., 2004). The RITS complex targets homologous RNA through siRNA-mediated base pairing (Buker et al., 2007, Irvine et al., 2006), while the LIM domain protein Stc1 binds both the RITS complex and the CLRC (Bayne et al., 2010). The CLRC contains the H3K9 methyltransferase Clr4, which catalyzes H3K9 methylation (Hong et al., 2005, Horn et al., 2005, Jia et al., 2005), and recruits chromodomain proteins, including Swi6, Chp1, Chp2 and Clr4 itself (Bannister et al., 2001, Partridge et al., 2002, Sadaie et al., 2008, Zhang et al., 2008). Chp1 binding to methylated H3K9 mediates the association between RITS complex and heterochromatin (Petrie et al., 2005), while the binding of Clr4 to methylated H3K9 promotes H3K9 methylation at the neighboring histones, allowing heterochromatin spreading at the mating-type locus (Zhang et al., 2008). All components of the RNAi machinery and CLRC are required for robust siRNA biogenesis and efficient H3K9 methylation at pericentromeric heterochromatin in S. pombe, and especially for the spreading of H3K9 methylation into embedded reporter genes (Irvine et al., 2006).
Several histone deacetylases (HDACs) facilitate heterochromatin assembly by histone deacetylation and nucleosome repositioning, including H3K14 deacetylase Clr3 and the more general HDAC Clr6 and Sir2 (Bjerling et al., 2002, Jia et al., 2004, Shankaranarayana et al., 2003, Sugiyama et al., 2007). In the absence of HDACs, silencing is compromised without impairing siRNA biogenesis, and the combinatorial effect of intact RNAi and impaired heterochromatin silencing causes elevated siRNA levels in HDAC mutants (Sugiyama et al., 2007).
Rrp6 and Dis3 are exoribonucleases associated with the nuclear exosome, a crucial component of the RNA surveillance machinery (Houseley et al., 2006). Rrp6 mediates aberrant transcript degradation and retention at the transcription site (Eberle et al., 2010). Both Dis3 and Rrp6 contribute to H3K9 methylation and silencing at pericentromeric heterochromatin, but Rrp6 (which is non-essential in fission yeast) only does so in the absence of RNAi (Buhler et al., 2007, Yamanaka et al., 2013, Murakami et al., 2007). Rrp6 degrades centromeric transcripts following cleavage by Ago1 (Irvine et al., 2006), indicating that Rrp6 and the RNAi machinery target some of the same RNA substrates. In the absence of Rrp6, RNAi targets a handful of genes and retrotransposable elements, in addition to pericentromeric non-coding RNA, although the corresponding small RNA accumulate at a very low level (Yamanaka et al., 2013).
The enzymatic activities of RNAi components are required for pericentromeric H3K9 methylation, supporting a direct role of siRNAs in heterochromatin assembly (Colmenares et al., 2007, Irvine et al., 2006). However, artificial siRNAs introduced by a hairpin RNA fail to assemble H3K9 methylation in trans despite efficient siRNA production (Buhler et al., 2006). The ability of artificial siRNAs to act in trans depends on the context of their target sequences, and is promoted by inefficient poly-adenylation, convergent transcription and proximity to pre-existing H3K9 methylation (Yu et al., 2014). Furthermore, reporter transgenes integrated into pericentromeric repeats generate much less siRNAs than the repeats themselves, and yet are far more dependent on Ago1 and Dcr1 for H3K9 methylation (Buhler et al., 2006, Irvine et al., 2006, Volpe et al., 2002).
In this study, we describe a highly conserved RNA-binding protein that has co-evolved with RNAi components in S. pombe. This protein was named Rct1 (RRM-containing cyclophilin regulating transcription) because of the conserved RNA recognition motif (RRM), the Prolyl-Peptidyl Isomerase (PPIase) domain, and its role in regulating Pol II (Gullerova et al., 2007). It was first isolated as an essential SR domain protein (AtCyp59) in Arabidopsis, where it interacts with both Pol II and with other SR proteins that mediate alternative splicing (Gullerova et al., 2006, Thomas et al., 2012), and binds most cellular mRNA (Bannikova et al., 2013). We show that the RRM of Rct1 is required for siRNA biogenesis, heterochromatic silencing and for efficient intron splicing, but is dispensable for H3K9 methylation. The defect in siRNA accumulation can be partially suppressed by increased transcription in HDAC mutants, or by mutations in the exosome. Interestingly, the Rct1 homologue in C. elegans, SIG-7, has been recently shown to regulate transgene silencing in both somatic and germline cells (Ahn et al., 2016), suggesting that the role of Rct1 is likely to be conserved in higher eukaryotes.
Results
Rct1 Is Essential for siRNA Production and Heterochromatic Silencing
We performed a candidate gene knockout screen based on the observation that S. cerevisiae has lost all the key RNAi components found in S. pombe (Aravind et al., 2000, Nakayashiki et al., 2006). We hypothesized that any gene that is specific to S. pombe, with no apparent S. cerevisiae homologue and yet is conserved in higher eukaryotes, could potentially be involved in RNAi. This list contains 538 genes (Table S1), including rdp1, hrr1, cid12, dcr1, chp1 and ago1. One of the genes on this list was rct1, which overlaps with the presence of RNAi genes in diverse fungi (Figure S1).
To test if Rct1 was involved in siRNA biogenesis, we analyzed siRNAs derived from dh/dg pericentromeric repeats in rct1Δ mutant cells and found they were barely detectable, similar to RNAi and CLRC mutants (Figure 1A). Small RNA-seq quantification revealed that dh/dg siRNA reads in rct1Δ were only about 1.3 % of the mapped reads in wild type cells, similar to ago1Δ and dcr1Δ, which have about 0.4% (Figure 1B). RNA-seq and semi-quantitative RT-PCR revealed that dh/dg transcripts accumulated in rct1Δ mutant cells, similar to RNAi or CLRC mutants (Figures 1C, 1D, S2A and S2B). In addition, the ura4 transgene inserted into the dg repeat (otr1R::ura4) was derepressed in rct1Δ mutant cells (Figure 1D), resemble other silencing mutants. Loss of pericentromeric silencing resulted in enhanced growth of strains carrying the ura4 gene inserted into dg and dh repeats on medium lacking uracil (Figures S2C and S2D). These results suggest a defect in the processing of repeat transcripts into siRNAs, rather than defects in transcription.
Figure 1. Rct1 is Required for siRNA Biogenesis and Pericentromeric Heterochromatin Silencing, See also Table S1, Figures S1 and S2.
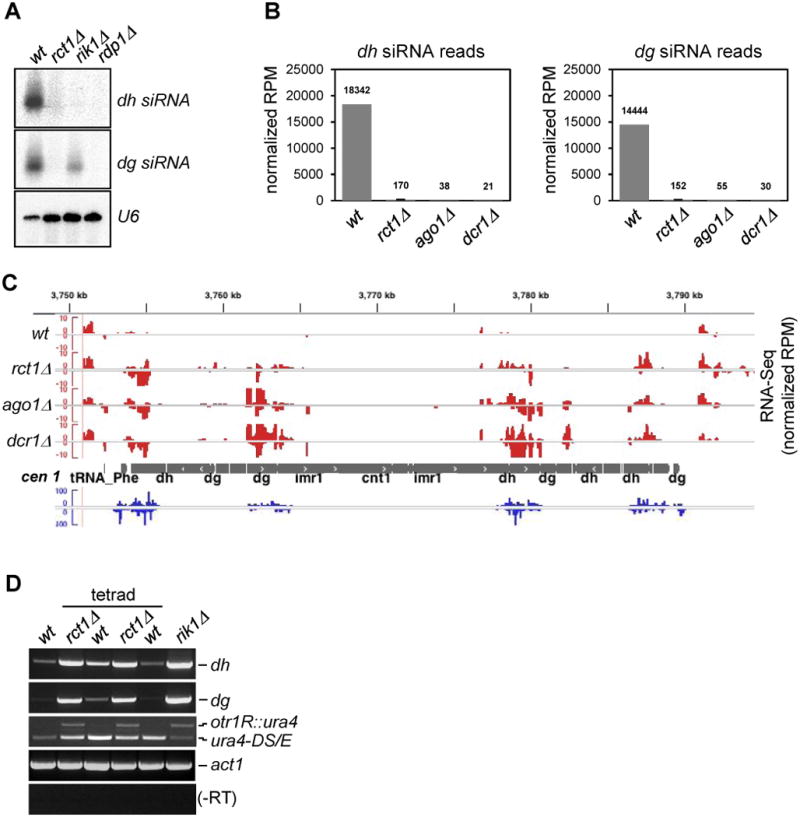
(A) Small RNA northern blots of pericentromeric dh/dg derived siRNAs. U6 serves as loading control.
(B) Quantification of cen siRNAs in indicated strains. Y-axis represents normalized reads in each library (read per million). Normalized reads mapped to dh/dg repeats are plotted separately. Data from two biological replicates of rct1Δ mutant cells were analyzed. Error bar indicates standard error from mean (SEM).
(C) RNA-seq reads at centromere (cen) 1 from indicated strains. RNA-seq tracks (red), small RNA-seq track (blue), cen 1 (grey). Y-axis represents normalized reads in each library (RPM). Only selected features are annotated on Integrated Genome Viewer screenshots in this and all subsequent figures.
(D) Semi-quantitative RT-PCR of dh/dg and otr1R::ura4 transcript levels in rct1Δ mutant cells. Truncated ura4-DS/E at the endogenous site and act1 serves as loading controls, -RT omits the reverse transcription step. A full tetrad was analyzed to show that the silencing defect phenotype segregates with rct1Δ alleles.
Rct1 Functions in the RNAi Pathway and requires the RRM domain
The HDAC Clr3 acts in parallel with the RNAi machinery (Sugiyama et al., 2007, Reyes-Turcu et al., 2011). To test if Rct1 also acts in parallel with Clr3, we generated rct1Δclr3Δ double mutant cells. Quantification of dh/dg expression levels revealed that pericentromeric silencing was further impaired relative to single mutants (Figure 2A). In contrast, rct1Δrdp1Δ, rct1Δdcr1Δ and rct1Δago1Δ double mutants did not further impair silencing (Figure 2B). Deletion of RNA exporting factor mlo3 rescues silencing in RNAi mutants but not in CLRC mutants (Reyes-Turcu et al., 2011). We generated rct1Δmlo3Δ double mutant cells and showed that pericentromeric silencing was efficiently restored in rct1Δmlo3Δ double mutants (Figure 2C). Taken together, we conclude that Rct1 is in the RNAi pathway.
Figure 2. Rct1 Functions in the RNAi Pathway, See also Figures S3A and S3B.
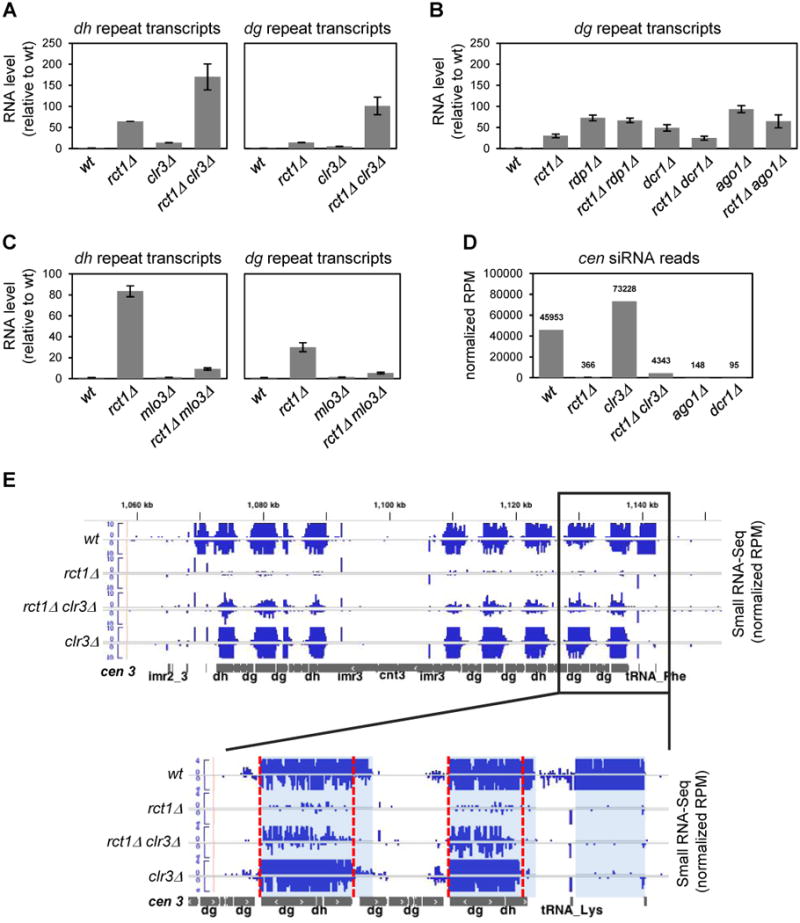
(A, B and C) RT-qPCR analysis of dh/dg transcript expression levels in mutant strains as indicated. actin transcript levels were used for normalization by ΔΔCT method. Y-axis represents RNA expression levels relative to wild type. At least two biological replicates were used for each genotype and qPCR reaction was performed at least twice for each strain. Error bars indicate SEM.
(D) Quantification of cen siRNAs in indicated strains. Y-axis represents normalized reads in each library (RPM). Data from two biological replicates of rct1Δ mutant cells were analyzed. Error bar indicates SEM.
(E) cen siRNA levels and distribution in indicated strains. Small RNA-seq tracks, (blue), cen 3 (grey). Y-axis represents normalized reads in each library (RPM). Lower panel is a blow up view of the pericentromeric boundary located at the right arm of cen 3, note the difference in scale. Blue shade indicates siRNA distribution from wild type cells, red dashed line marks confined distribution in the mutant cells.
HDAC mutants have robust siRNA accumulation due to elevated transcription in the presence of RNAi. In rct1Δclr3Δ double mutant cells, we found that pericentromeric siRNA levels were increased more than 10-fold when compared to rct1Δ (Figure 2D). These siRNAs mapped to the same regions as in clr3Δ cells (Figure 2E), and retained their 5′ U bias and size distribution similar to siRNAs produced from RNAi pathway (data not shown), suggesting an additive effect. Surprisingly, siRNAs originating from IRC boundary elements (Noma et al., 2006, Keller et al., 2013) located just outside of the pericentromeric repeats, were completely absent in clr3Δ (Figures 2E, S3A and S3B), revealing a new role for Clr3 in siRNA accumulation
Rct1 has a PPIase domain at the N-terminus followed by a conserved RRM, with a less conserved C-terminus region (Gullerova et al., 2007). To address the role of each domain in siRNA biogenesis, we generated domain specific rct1 mutants and assessed dh/dg siRNA levels (Figure 3A). Deleting the PPIase and C-terminal domains had no effect, but mutation of the RRM domain resulted in complete loss of siRNAs (Figure 3B). Pericentromeric dh/dg transcripts accumulated in rct1-rrm, but not in rct1ΔIso or rct1ΔC mutant cells, consistent with the loss of siRNAs (Figure 3C). These results suggest that the RRM domain of Rct1 is required for siRNA biogenesis and pericentromeric silencing.
Figure 3. The RNA Recognition Motif of Rct1 is Essential for siRNA Biogenesis and Pericentromeric Heterochromatin Silencing.
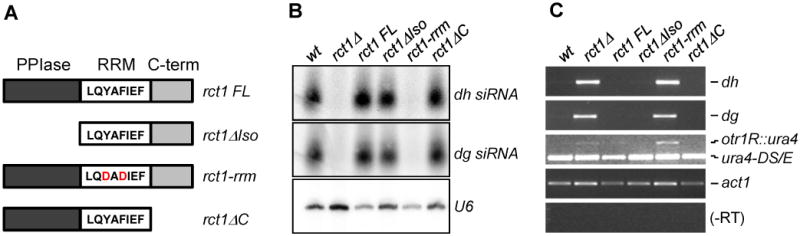
(A) Schematic representation of rct1 alleles. rct1 FL contains full-length Rct1 with no mutations. rct1ΔIso lacks the first 175 amino acids corresponding to PPIase domain. rct1-rrm includes two amino acid mutations (red) in the RRM,Y287D and F289D, both of which combined were predicted to abolish the RNA-binding ability of Rct1. rct1ΔC has amino acids 333-428 removed.
(B) Small RNA northern blots of pericentromeric dh/dg derived siRNAs. U6 serves as loading control.
(C) Semi-quantitative RT-PCR of dh/dg and otr1R::ura4 transcript levels in rct1 mutant cells. Truncated ura4-DS/E at the endogenous locus and act1 serves as loading controls, -RT omits the reverse transcription step.
Rct1 Mediates Splicing and Prevents Targeting by the Exosome
The Rrp6 exosome degrades pericentromeric transcripts, but does not process them into siRNAs (Irvine et al., 2006, Reyes-Turcu et al., 2011, Yamanaka et al., 2013). To test if pericentromeric transcripts in rct1 mutant cells were targeted by Rrp6, we sequenced siRNAs in rct1Δrrp6Δ and rct1-rrm rrp6Δ mutant cells. We found that pericentromeric siRNAs, including the boundary siRNAs, were partially restored in rct1Δrrp6Δ and rct1-rrm rrp6Δ double mutants as compared to rct1Δ and rct1-rrm single mutant cells (Figures 4A, S3C and S3D) by 20- to 40-fold (Figure 4B). siRNAs derived from otr1R::ura4 transgene were similarly restored in rct1Δrrp6Δ and rct1-rrm rrp6Δ mutant cells (Figure 4C), as were siRNAs from tf2 retrotransposons and a handful of genes (Figure S4A), as previously reported (Yamanaka et al., 2013). However, levels of these non-centromeric small RNA per kilobase of precursor transcript were less than 1% of pericentromeric siRNA, and extensive A-and U-tailing indicated these siRNA were, most likely, targeted by the TRAMP complex for degradation by the exosome (Figure S4A). This could account for their modest accumulation in rrp6 mutants, rather than being targets of a specialized RNA processing machinery (Yamanaka et al., 2013).
Figure 4. Rct1 Mediates Splicing and Prevents Targeting by the Exosome, See also Figures S3C, S3D and S4.

(A) cen siRNA levels and distribution in indicated strains. Small RNA-seq tracks (blue), cen 3 (grey). Y-axis represents normalized reads in each library (RPM).
(B) Quantification of cen siRNAs in indicated strains. Y-axis represents normalized reads in each library (RPM). Data from two biological replicates of each strain were analyzed with the exception of rct1-rrm mutant cells, where data from one strain was analyzed. Error bars indicate SEM.
(C) Quantitative analysis of ura4 siRNA levels in indicated strains as described in (B).
(D and E) RT-qPCR analysis of dh/dg transcript expression levels in mutant strains as indicated. actin transcript levels were used for normalization by ΔΔCT method. Y-axis represents RNA expression levels relative to wild type. Two biological replicates were used for each genotype and qPCR reactions were performed in triplicates for each strain. Error bars indicate SEM.
(F) Log2 fold changes are shown for all differentially expressed intron (purple) and exon (grey) observations in independent comparisons of mutant and wild type RNA-seq as determined by DEXSeq with false discovery rate < 0.05. Number indicates differentially expressed inton/exon number in each strain. Boxes represent the interquartile range bisected by the median. Whiskers extend to the lesser of IQR × 1.5 or the most extreme observation.
(G) RT-qPCR analysis of spliced dg transcript levels in mutant strains using primers complementary to exon splice junctions of the dg non-coding centromeric transcripts. Total dg transcript levels were used for normalization by ΔΔCT method. Y-axis represents spliced RNA expression levels relative to wild type. At least two biological replicates were used for each genotype and qPCR reaction was performed at least twice for each strain. Error bars indicate SEM. Note that overall dg transcript levels were much higher in mutant than in wild type cells (Figure 3).
The siRNAs detected in rct1Δrrp6Δ and rct1-rrm rrp6Δ mutant cells had strong 5′ U bias (Figure S4B) and were mostly 22-24 nt in length (Figure S4C), suggesting they were likely bound to Ago1. To test if these siRNAs restored silencing, we quantified dh/dg transcript levels, and found silencing was restored in rct1Δrrp6Δ and rct1-rrm rrp6Δ double mutant cells (Figures 4D and 4E). Our results indicate that pericentromeric transcripts, and/or their siRNA, were targeted by Rrp6 in rct1 mutant cells, resulting in loss of silencing.
Rrp6 mediates retention of unspliced transcripts at the transcription site (Eberle et al., 2010), and inefficient splicing in rct1 mutants could cause Rrp6 to target pericentromeric transcripts. We analyzed splicing efficiency in rct1 mutant cells by RNA-seq and revealed a striking upregulation in intron retention in rct1Δ and rct1-rrm mutant cells (Figure 4F). A role for Rct1 in splicing is supported by direct interaction of the Arabidopsis homolog AtCyp59 with SR splicing proteins, Pol II, and RNA (Baltz et al., 2012, Bannikova et al., 2013, Gullerova et al., 2007, Gullerova et al., 2006). Non-coding transcripts from dg repeats have introns (Chinen et al., 2010), and we tested for intron retention using RT-qPCR (Figure 4G). We found that dg introns were spliced in dcr1Δ and clr4Δ mutant cells, but that splicing was very inefficient in rct1Δ and rct1-rrm mutants, supporting the idea that defects in non-coding RNA splicing might direct them away from the siRNA biogenesis pathway (Bayne et al., 2008).
Splicing defects in three protein coding genes important for siRNA biogenesis (see Methods), namely ago1, ers1 and arb2, were validated by RT-PCR in rct1Δ and rct1-rrm mutant cells (Figure 5A). To test if splicing defects in these genes were responsible for loss of silencing, we deleted rct1 from strains in which ago1, esr1 and arb2 have been replaced by intron-less cDNA, in all combinations (Kallgren et al., 2014), and measured dg transcript levels (Figure 5B). We found that replacement of ago1 with intron-less ago1 at the endogenous locus resulted in restoration of silencing in rct1Δ mutant cells, while replacement of ers1 and arb2 with cDNA versions had little or no effect. Importantly, similar replacements had no effect on loss of silencing in dcr1Δ mutant cells or in RNAi wild type backgrounds (Figure S5A). Surprisingly, we found that levels of Ago1 protein (Figure 5C) and dh/dg siRNA (Figure 5D) were only partially restored, even when all three RNAi genes were replaced by cDNA. Ago1 protein levels were similar to those found in dcr1Δ mutant cells (Figure S5B), in which siRNA levels were also very low.
Figure 5. The Impaired RNAi Gene Splicing in rct1 Mutant Cells is Responsible for Loss of Silencing, but not Low Ago1 Protein or Pericentromeric siRNA Levels, See also Figure S5.
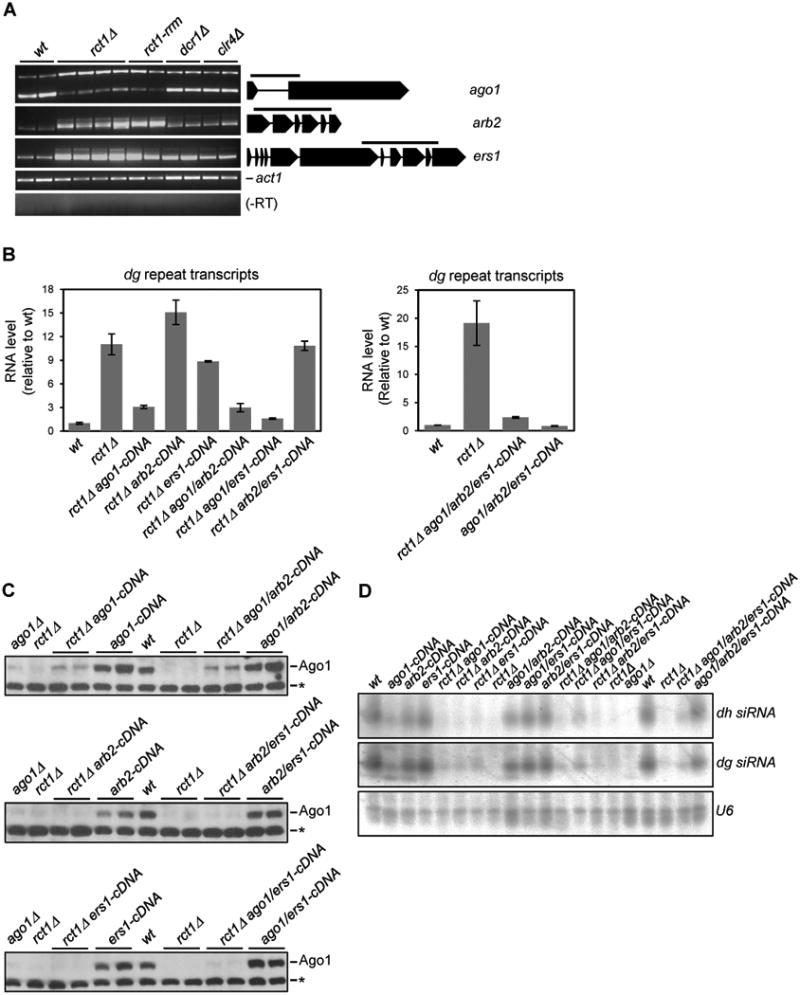
(A) Semi-quantitative RT-PCR analysis of ago1, arb2, and ers1 splicing in indicated strains. Amplicons spanning one or more introns are indicated as solid line above each gene cartoon. act1 serves as loading controls, -RT omits the reverse transcription step. At least two biological replicates were used for each genotype. Upper bands represent unspliced transcripts, while lower bands are spliced.
(B) RT-qPCR analysis of dg transcript levels in mutant strains as indicated. actin transcript levels were used for normalization by ΔΔCT method. Y-axis represents RNA expression levels relative to wild type. At least two biological replicates were used for each genotype and qPCR reaction was performed at least three times for each strain. Error bars indicate SEM.
(C) Ago1 protein levels were analyzed by western blot in indicated strains using anti-Ago1 antibody (Abcam, ab18190). Two biological replicates were used for each genotype. Asterisk indicates a prominent non-specific band which shows equal loading between samples.
(D) Small RNA northern blots of pericentromeric dh/dg derived siRNAs. U6 serves as loading control.
H3K9 Methylation and RNA polymerase II accumulation in rct1 Mutants
At least five genes required for spliceosome assembly are needed for the biogenesis of siRNAs, prp5, prp8, prp10, prp12 and cwf10. Curiously, however, these mutants had little or no effect on the maintenance of heterochromatic H3K9me2 (Bayne et al., 2008). We performed H3K9me2 and H3K9me3 ChIP-seq, and found that rct1Δ and rct1-rrm mutant cells also retained normal levels of H3K9 methylation (Figures 6 and S6), even at the otr1R::ura4 transgene (Figures 6D and 6H) which is more sensitive to the loss of RNAi components (Irvine et al., 2006). This observation distinguishes Rct1 from RNAi components such as Ago1, Dcr1 and Rdp1, in which histone H3K9 methylation is substantially reduced at centromeric repeats, and completely abolished at the otr1R::ura4 transgene.
Figure 6. Rct1 Is Not Required for H3K9 Methylation at Pericentromeric Heterochromatin, See also Figure S6.
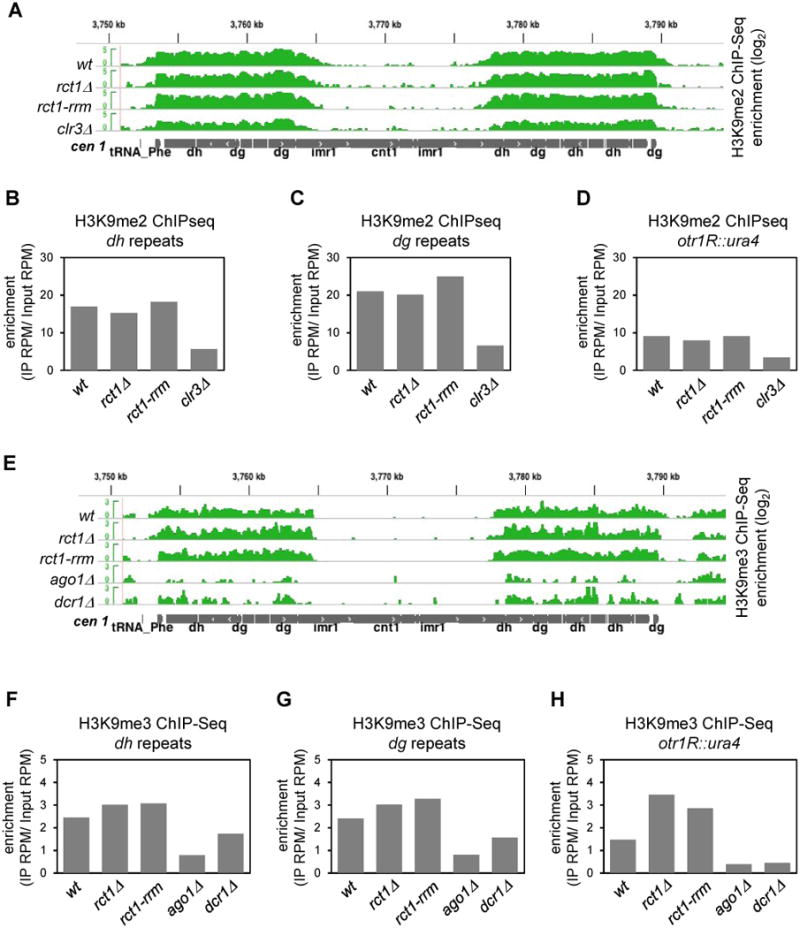
(A) H3K9me2 enrichment and distribution in indicated strains. ChIP-seq tracks (green), cen 1 (grey). Y-axis represents the log scale of enrichment. Positive value indicates enrichment after IP as compared to input controls, only positive values were shown.
(B, C and D) Quantification of H3K9me2 enrichment in indicated strains at endogenous dh/dg repeats (B) (C) and otr1R::ura4 transgene region (D).
(E) H3K9me3 enrichment and distribution in indicated strains. ChIP-seq tracks (green). cen 1 (grey). Y-axis represents the log scale of enrichment. Positive value indicates enrichment after IP as compared to input controls, only positive values were shown.
(F, G and H) Quantification of H3K9me3 enrichment was performed as in (B, C and D). Data from two biological replicates of rct1Δ mutant cells were analyzed.
Spreading of H3K9 methylation from heterochromatic repeats into embedded reporter genes requires the coupling of Clr4 with the leading strand DNA polymerase Pol ε (Li et al., 2011, Zaratiegui et al., 2011). During S phase, when the replication machinery encounters Pol II, the failure to remove Pol II from pericentromeric repeats in dcr1Δ mutant cells interferes with fork progression. This results in the loss of H3K9 methylation due to fork restart by homologous recombination (Zaratiegui et al., 2011). We performed Pol II ChIP-seq in rct1-rrm mutant cells and found that Pol II failed to accumulate at the pericentromeric repeats (Figures 7A and S7A), consistent with the H3K9me2 retention. In contrast, Pol II accumulated at siRNA clusters in RNAi mutants (Zaratiegui et al., 2011). In rct1Δ mutant cells, Pol II was also found to accumulate but extended into the intervening regions between neighboring repeats (Figures 7A and S7A). Pol II enrichment within siRNA clusters was confirmed in rct1Δ mutant cells but not in rct1-rrm mutant cells (Figure 7B), and suggested that Rct1, but not the RRM domain, was required for Pol II release from the pericentromeric repeats. ChIP-seq with antibodies specific for initiating and elongating Pol II (phosphorylated on CTD Serine-5 and Serine-2, respectively) revealed that the accumulated Pol II was largely trapped in the initiating form, even in intergenic regions between non-coding repeats (Figures S7B and S7C). Our results are in agreement with previous observations that Rct1 interacts with Pol II and is thought to be required for CTD phosphorylation (Gullerova et al., 2007, Gullerova et al., 2006).
Figure 7. Rct1 Releases Pol II from siRNA Cluster Regions Independently of the RRM, See also Figure S7.
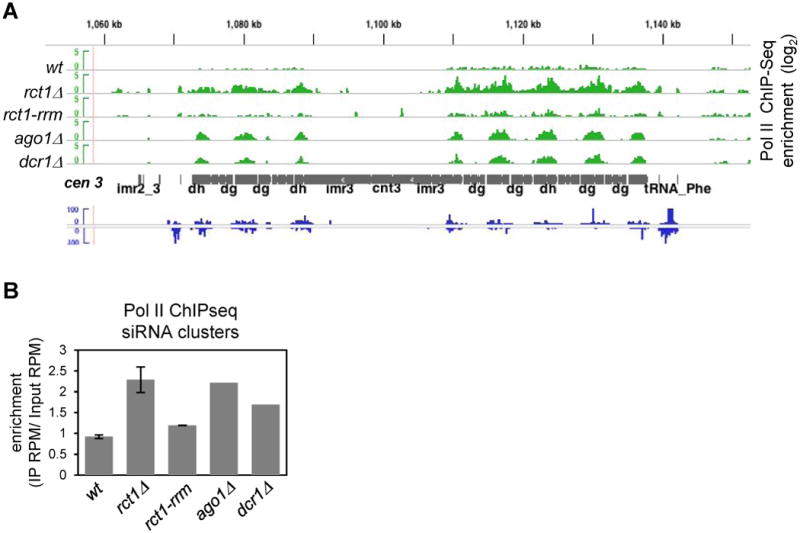
(A) Pol II enrichment and distribution in indicated strains. ChIP-seq tracks (green), cen 3 (grey), wild type cell small RNA-seq tracks, (blue). Y-axis represents the log scale of enrichment. Positive value indicates enrichment after IP as compared to input controls, only positive values were shown.
(B) Quantification of Pol II enrichment within siRNA clusters in indicated strains. siRNA cluster regions were defined by > 100 siRNA counts on genome browser tracks. Data from two biological replicates of rct1Δ mutant cells were analyzed. Two independent ChIP experiments were performed and libraries were constructed independently, with the exception of ago1Δ and dcr1Δ mutants, where one ChIP experiment was done. Error bar indicates SEM.
Discussion
Rct1 Is Required for siRNA Accumulation and Post-Transcriptional Silencing
Genetic interactions and compromised siRNA biogenesis indicate Rct1 is required for RNAi. However, we were not able to detect interactions between Rct1 and siRNAs or Rct1 and Ago1 (data not shown). Additionally, siRNA biogenesis was partially restored in rct1Δclr3Δ and rct1Δrrp6Δ mutant cells, further indicating that unlike other RNAi components, Rct1 does not directly participate in siRNA biogenesis, so that mutants that increase siRNA levels indirectly, such as rrp6 and clr3, can bypass the requirement of Rct1 to some extent. Instead, Rct1 promotes splicing of both non-coding and coding transcripts, as well as promoting the release of elongating Pol II. Each of these functions contributes indirectly to siRNA production, and to silencing, independently of changes in histone H3K9 methylation.
Rct1 is engaged with transcription by interacting with the C-terminal domain of Pol II (Gullerova et al., 2007) and its homolog AtCyp59 interacts with mRNA (Bannikova et al., 2013), most likely through its RRM. The splicing machinery is further recruited to this transcript, and properly spliced transcripts are exported to the cytosol for translation. We found that protein-coding genes required for RNAi were not properly spliced in rct1 mutants, which has been proposed as an explanation for loss of silencing in splicing mutants. Furthermore, replacement of ago1, arb2, and ers1 genes with cDNA restored silencing to rct1Δ mutant cells resembling some other splicing mutants in this respect (Kallgren et al., 2014). However, levels of Ago1 protein were very low in rct1Δ mutant cells, much lower than expected given the substantial levels of correctly spliced mRNA, and were only marginally increased by removing the introns from ago1 and arb2, only rising to levels similar to those observed in dcr1Δ mutant cells which lack siRNA. We conclude that the splicing defect in RNAi genes was responsible for the loss of silencing, but was most likely not responsible for the loss of siRNA. Though formally, we cannot exclude the possibility that unknown intron-containing RNAi genes are mis-spliced in rct1 mutant cells, or that multiple minor defects are responsible.
Non-coding transcripts from centromeric repeats stay in the nucleus to be processed into double stranded RNA, stimulated by the presence of spliceosomes that interact with the RDRC (Bayne et al., 2008). Weak splicing signals are found in non-coding transcripts from dg repeats, and the introns are partially spliced (Chinen et al., 2010). Stalled spliceosomes at weak splice site are thought to stimulate siRNA production in Cryptococcus neoformans (Dumesic et al., 2013). In cells lacking Rct1 or a functional RRM, nascent pericentromeric transcripts are not processed into siRNA, and accumulate as unspliced RNAs. It is possible, that failure to recruit the splicing machinery is responsible for loss of siRNA. Rrp6 could potentially mediate unspliced transcript retention at the transcription site, preventing RNAi from targeting these transcripts.
Alternatively, Rrp6 could simply degrade the siRNA that still accumulate in rct1 mutants, following 3′ end modification by tailing. Our analysis revealed that A and U tailed small RNA were prevalent in rrp6Δ small RNA samples compared to wild type. This was especially prominent in ectopic small RNA from the myb2, and SPCC1442.04c genes as well as tf2 elements, which only occur in rrp6 mutant cells (Yamanaka et al., 2013), suggesting they might be degraded by the exosome in wild type cells. A similar function has been proposed for the TRAMP complex, which is likely responsible for polyadenylation of small RNA (Buhler et al., 2008). However, these ectopic small RNAs are less than 1% of those derived from centromeric dg/dh repeats, when normalized for genomic content, so that their function, if any, is obscure.
Rct1 in H3K9 Methylation and Pol II Accumulation
Unlike other RNAi mutants, but resembling other splicing mutants (Bayne et al., 2008), cells lacking Rct1 uncouple siRNA biogenesis and H3K9 methylation. RNAi factors resolve the collision between the replication and transcription machinery during S phase, by promoting transcriptional termination and allowing replication to proceed, along with H3K9 methylation (Zaratiegui et al., 2011). Thus most siRNAs generated from centromeric repeats could be considered as degradation byproducts of co-transcriptional silencing by the RNAi pathway, rather than functional components of heterochromatic modification per se (Halic and Moazed, 2010). In rct1-rrm mutant cells, Pol II is released normally from the pericentromeric repeats and H3K9 methylation is retained, despite the loss of 98-99% of siRNAs. In rct1Δ mutant cells, however, Pol II accumulates. One possibility is that Rct1 directly promotes Pol II release or degradation via protein interactions with Pol II outside of the RRM. The ubiquitin ligase Cul3 triggers Pol II degradation in response to DNA damage in S. cerevisiae (Ribar et al., 2007), and the Rct1 homolog in Drosophila melanogaster, CG5808, exists in a protein complex with Cul3 (Fujiyama-Nakamura et al., 2009). Several protein interactions in this complex are conserved in S. pombe (Geyer et al., 2003, Pintard et al., 2004).
Surprisingly, both Pol II and H3K9 methylation accumulate in rct1Δ mutant cells, though transcript accumulation is much lower than in RNAi mutants. The trapped Pol II is predominantly phosphorylated on Serine-5 of the CTD, rather than Serine-2, consistent with a defect in elongation (Gullerova et al., 2007). Thus domains of Rct1 other than the RRM seem to have additional functions distinct from defects in splicing, that appear to bypass the requirement of RNAi for H3K9 methylation (Reddy et al., 2011). A role in transcriptional elongation has recently been proposed for the Rct1 homolog in C. elegans, CYN-14 (Ahn et al., 2016). Furthermore, mutants in the Pol II associated factor complex, Paf1C, also have an elongation defect, and have been shown to lose pericentromeric siRNA while retaining H3K9me2, highly reminiscent of rct1 (Sadeghi et al., 2015). RNAi mediated silencing driven by hairpin siRNA is actually enhanced in Paf1c deficient backgrounds (Kowalik et al., 2015). These observations are consistent with the model proposed here.
Small RNA and Heterochromatin at Pericentromeric Boundaries
Clr3 contributes to silent heterochromatin in part by the elimination of the nucleosome free region (NFR) found within the repeats, inhibiting Pol II engagement. However, unlike the dh/dg repeat NFRs, the NFRs at the pericentromeric boundaries are resistant to Clr3 mediated elimination (Garcia et al., 2010). Nonetheless, we have found that some pericentromeric small RNAs, especially those from pericentromeric boundaries, are Clr3 dependent (Figure 2E). Boundary siRNAs on chromosome 1 are thought to load only rarely onto Ago1 and are not associated with H3K9me2 (Keller et al., 2013). However, IRC3-R boundary siRNA on chromosome 3 are efficiently loaded onto Ago1 (Halic and Moazed, 2010), and yet are still not associated with high levels of H3K9me2 in wild type cells (Figure S6B), nor does Pol II accumulate there in the absence of RNAi (Figure 7A). Instead, these boundaries appear to be transcribed by Pol III (Noma et al., 2006). Interestingly, deletion of Ago1 has a pronounced effect on H3K9me3 accumulation, and Clr3-dependent boundary siRNA appear to be associated with H3K9me3 (Figures S6C, S6D and 2E). The regulation of H3K9 di- and tri-methylation by boundary siRNAs is largely unknown and we revealed an unexpected link between Clr3, Ago1 and boundary siRNA biogenesis.
Experimental Procedures
Fission Yeast Strains and Standard Manipulation, See also Tables S2 and S3
S. pombe strains and primers used in this study are described in Tables S2 and S3, respectively. Deletion mutants were generated by standard PCR or plasmid-based methods (Gregan et al., 2006). All yeast strains were cultured in YES (yeast exact with supplements) media at 30 °C. rct1 mutant strains were generated by transforming freshly prepared diploid cells to avoid accumulation of suppressors. Tetrad dissection was done following meiosis and PCR genotyping performed to confirm allele segregation.
Small RNA Northern
Cells were grown to a concentration of ∼1 × 107 cells/ml. Total RNA was extracted by the hot phenol method (Leeds et al., 1991). mirVana miRNA isolation kit (Ambion) was used to enrich the small RNA fraction. 10 to 15 ug of enriched small RNAs were used, and RNAs were chemically cross-linked to membranes (Pall and Hamilton, 2008). Radiolabeled riboprobes were generated by T3/T7 in vitro transcription kit (Ambion) using dh or dg DNA as templates and αP32-UTP for radiolabeling. Riboprobes were hydrolyzed before hybridization. U6 radiolabeled oligoprobe was prepared by P32-ATP end labeling with T4 PNK (Polynucleotide Kinase). Radioactive signals were detected by Fuji phosphoimager.
Small RNA Sequencing Library Construction and Data Analysis
Small RNA libraries were constructed by NEBNext multiplex Small RNA library prep kit (NEB E7300) following manufacturer's protocol. Libraries were size selected (125-160 bp) by Blue Pippin machine (Sage Science). Barcoded libraries were pooled and sequenced on Illumina MiSeq platform. Obtained reads were quality filtered using Trimmomatic and aligned to the S. pombe genome assembly ASM294v2.21 using Bowtie v2.1.0 and local alignment, with multi-mappers randomly distributed. Only reads between 15 and 36 nucleotides were used for the analysis. Read counts were normalized to reads per million (RPM) using total library size. Reads mapping to the sense strands of tRNA and rRNA were discarded before producing genome browser pileups.
RNA Sequencing Library Construction
ScriptSeq V2 kit (Epicentre) was used to prepare barcoded RNA-seq libraries. 50 ng ribosomal RNA (rRNA)-depleted RNA samples were used as starting material following manufacturer's protocol. Ribo-Zero Gold kit (Epicentre) was used to remove rRNA from total RNA (DNA free) samples. Barcoded libraries were pooled and sequenced on Illumina HiSeq platform.
RNA-seq Preprocessing, Alignment and Coverage Visualization
Sequencing adapters were trimmed from reads using Trimmomatic 0.30 (Bolger et al., 2014), and surviving read pairs with both mates longer than 25 bp were retained. Reads were then mapped to isolated rDNA annotations with Bowtie 2 2.1.0 with default options (Langmead and Salzberg, 2012). Only read pairs that failed to map concordantly to rDNA were retained. Subsequently, reads were aligned to the Ensembl 21 S. pombe genome release with STAR 2.3.1z (Dobin et al., 2013). Genome index construction was performed with the option --sjdbOverhang 100 and the Ensembl 21 annotations supplied to --sjdbGTFfile. Alignment was performed with the following options: --outFilterMultimapNmax 100 --outFilterMismatchNmax 5. Non-primary and non-concordant alignments were removed with samtools 0.1.19 (Li et al., 2009). One random placement was chosen for multi-mapping reads. Coverage tracks were prepared from STAR alignments with Bedtools 2.19.0-7 and UCSC BigWig utilities (Quinlan and Hall, 2010). BAM alignments were converted to BED format, and the strand of the second read in each aligned pair inverted so base coverage for both mates would be counted on the origin strand. Base coverage was tallied with Bedtools genomecov for each strand and normalized by millions of reads mapped. Figures were produced in IGV (Robinson et al., 2011, Thorvaldsdottir et al., 2013).
Differential Intron and Exon Usage
Independent pairwise comparisons of RNA-seq reads from rct1Δ, rct1-rrm, rct1Δ-clr3Δ and clr3Δ with wild type were performed with DEXSeq 1.8.0 (Anders et al., 2012). Two replicates were used in all comparisons. A non-overlapping set of exon counting bins in gff format was generated with the dexseq_prepare_annotation.py script. The resulting gff was modified by adding intron counting bins between all exons, which were distinguished by appending an “i” to the preceding exon ID. dexseq_count.py was run with parameters “-p yes -s yes” to generate raw counts of reads overlapping the bins. DEXSeq routines were called with default arguments to test for differential expression and estimate log2 fold changes for the counting bins. Differential exon/intron usage events with a Benjamini-Hochberg adjusted p-value less than 0.05 were considered significant. Boxplots of log2 fold change estimates for these events were generated with ggplot2 (Wickham, 2009). RNA-seq analysis showed statistical evidence of differential exon and/or intron usage in the following genes required for RNAi: ago1, air1, arb1, arb2, chp2, clr6, ers1, hip1, hob1, hrr1, hst2, mst2, pir2, pmt3, pst1, rpb2, sir2. However, only ago1, ers1, arb1 and arb2 had alterations in exon and/or intron usage >5% compared to wild type levels. Three of these genes were validated by RT-PCR but only one (ago1) had phenotypic consequences when the introns were removed.
Semi-Quantitative RT-PCR
DNA-free total RNA was isolated by hot phenol extraction followed by Turbo DNase (Ambion) treatment. 20 to 30 ng of total RNA were used in one-step RT-PCR reactions (Qiagen) following manufacturer's protocol. Primers used are listed in Table S3. RT-omitted the reverse transcription step and proceeded directly to enzyme inactivation at 95 °C.
RT-qPCR
Super Script III First-Strand Synthesis System (Life technologies) was used to reverse transcribe total RNA into cDNA. cDNA was amplified by IQ SYBR Green Super Mix with CFX96 real time PCR detection system (Bio-Rad). Primers used are listed in Table S3. Expression levels relative to wild type were calculated by ΔΔCT method using act1 levels for normalization.
ChIP, ChIP Sequencing Library Construction and Data Analysis
Cells were grown to a concentration of ∼1 × 107 cells/ml, then fixed in 1% formaldehyde at 25°C for 20 min. Fixation was stopped by adding glycine to a final concentration of 0.125 M, and cells were washed twice in 1XPBS then stored in -80°C. Cells were spheroplasted by zymolyase at 37°C and then sonicated using a bioruptor for 8 cycles (30s ON 60s OFF). For each IP, 500-750 ug of chromatin were used with 3 to 5 ul antibody. Antibodies used in ChIP experiments were H3K9 dimethylation (Upstate 07-441), H3K9 trimethylation (Abcam ab8898), Pol II 8WG16 (Abcam ab817), Pol II phospho-serine 5 (Abcam ab5131) and Pol II phospho-serine 2 antibody (Abcam ab5095).
DNA (1ng) purified from ChIP experiments was made into libraries by using NEB enzymes. In brief, DNA was end-repaired by T4 DNA polymerase, Klenow fragment and T4 DNA PNK. “A” bases were added to the 3′ end of end-repaired DNA fragment with Klenow 3′ to 5′ exo minus and dATP. Barcoded Truseq adaptors (Illumina) were ligated to DNA fragments using quick ligase at 25°C. Five PCR cycles were performed prior to size selection. After size selection, purified DNA was PCR amplified with 6 to 12 cycles (Kapa HiFi HotStart ready mix). Barcoded libraries were pooled and sequenced on Illumina HiSeq platform. Obtained reads were quality filtered using Trimmomatic and aligned to the S. pombe genome assembly ASM294v2.21 using Bowtie v2.1.0 and local alignment, with multi-mappers randomly distributed. All read counts were normalized to reads per million (RPM) using total library size. ChIP enrichment was calculated as the log2 of the ratio of normalized IP reads to normalized input (whole cell extract) reads. Quantification at individual features was performed by intersecting reads with the feature of interest.
Supplementary Material
Table S1. Conserved Eukaryotic Genes Present in S. pombe with No Apparent S. cerevisiae Ortholog, Related to Figure 1.
Table S2. Strains Used in This Study, Related to Experimental Procedures.
Table S3. Oligonucleotides Used in This Study, Related to Experimental Procedures.
Acknowledgments
We thank W. Kelly for discussion and for communicating results prior to publication, R. Sternglanz, D. Spector, J. Hicks and E. Bernstein for advice and discussion, S. Jia for strains, and J. Simorowski for carefully reading the manuscript. This work is in partial fulfillment of the requirements for a Doctor of Philosophy (A.-Y.C.). S.C. was supported by a fellowship from the Natural Sciences and Engineering Research Council of Canada (PGSD) and a Cashin Scholarhip from the Watson School of Biological Sciences. This work was supported by the Howard Hughes Medical Institute-Gordon and Betty Moore Foundation (GMBF3033) and a grant from the National Institutes of Health (GM076396) to R.A.M. R.A.M acknowledges assistance from the Cold Spring Harbor Laboratory Shared Resources which are funded in part by the Cancer Center Support Grant (5PP30CA045508), and a Chaire Blaise Pascal (Region Ile-de-France) at Institut Biologie École Normale Superieure, Paris. The authors declare no competing financial interests.
Footnotes
Author Contributions: A.-Y.C. and H.S.K. performed experiments, and S.C. and E.E. analyzed the data. A.-Y.C. and R.A.M. designed experiments and wrote the manuscript.
Accession Numbers: The accession numbers for the ChIP-seq, RNA-seq, and small RNA-seq data presented in this paper are GEO: GSE97749.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JH, Rechsteiner A, Strome S, Kelly WG. A Conserved Nuclear Cyclophilin Is Required for Both RNA Polymerase II Elongation and Co-transcriptional Splicing in Caenorhabditis elegans. PLoS Genet. 2016;12:e1006227. doi: 10.1371/journal.pgen.1006227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Reyes A, Huber W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012;22:2008–17. doi: 10.1101/gr.133744.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci U S A. 2000;97:11319–24. doi: 10.1073/pnas.200346997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, Wyler E, Bonneau R, Selbach M, Dieterich C, Landthaler M. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012;46:674–90. doi: 10.1016/j.molcel.2012.05.021. [DOI] [PubMed] [Google Scholar]
- Bannikova O, Zywicki M, Marquez Y, Skrahina T, Kalyna M, Barta A. Identification of RNA targets for the nuclear multidomain cyclophilin atCyp59 and their effect on PPIase activity. Nucleic Acids Res. 2013;41:1783–96. doi: 10.1093/nar/gks1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC, Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–4. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- Bayne EH, Portoso M, Kagansky A, Kos-Braun IC, Urano T, Ekwall K, Alves F, Rappsilber J, Allshire RC. Splicing factors facilitate RNAi-directed silencing in fission yeast. Science. 2008;322:602–6. doi: 10.1126/science.1164029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayne EH, White SA, Kagansky A, Bijos DA, Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J, Allshire RC. Stc1: a critical link between RNAi and chromatin modification required for heterochromatin integrity. Cell. 2010;140:666–77. doi: 10.1016/j.cell.2010.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjerling P, Silverstein RA, Thon G, Caudy A, Grewal S, Ekwall K. Functional divergence between histone deacetylases in fission yeast by distinct cellular localization and in vivo specificity. Mol Cell Biol. 2002;22:2170–81. doi: 10.1128/MCB.22.7.2170-2181.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Haas W, Gygi SP, Moazed D. RNAi-dependent and -independent RNA turnover mechanisms contribute to heterochromatic gene silencing. Cell. 2007;129:707–21. doi: 10.1016/j.cell.2007.03.038. [DOI] [PubMed] [Google Scholar]
- Buhler M, Spies N, Bartel DP, Moazed D. TRAMP-mediated RNA surveillance prevents spurious entry of RNAs into the Schizosaccharomyces pombe siRNA pathway. Nat Struct Mol Biol. 2008;15:1015–23. doi: 10.1038/nsmb.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler M, Verdel A, Moazed D. Tethering RITS to a nascent transcript initiates RNAi- and heterochromatin-dependent gene silencing. Cell. 2006;125:873–86. doi: 10.1016/j.cell.2006.04.025. [DOI] [PubMed] [Google Scholar]
- Buker SM, Iida T, Buhler M, Villen J, Gygi SP, Nakayama J, Moazed D. Two different Argonaute complexes are required for siRNA generation and heterochromatin assembly in fission yeast. Nat Struct Mol Biol. 2007;14:200–7. doi: 10.1038/nsmb1211. [DOI] [PubMed] [Google Scholar]
- Castel SE, Martienssen RA. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat Rev Genet. 2013;14:100–12. doi: 10.1038/nrg3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Zhang K, Nicolas E, Cam HP, Zofall M, Grewal SI. Cell cycle control of centromeric repeat transcription and heterochromatin assembly. Nature. 2008;451:734–7. doi: 10.1038/nature06561. [DOI] [PubMed] [Google Scholar]
- Chinen M, Morita M, Fukumura K, Tani T. Involvement of the spliceosomal U4 small nuclear RNA in heterochromatic gene silencing at fission yeast centromeres. J Biol Chem. 2010;285:5630–8. doi: 10.1074/jbc.M109.074393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colmenares SU, Buker SM, Buhler M, Dlakic M, Moazed D. Coupling of double-stranded RNA synthesis and siRNA generation in fission yeast RNAi. Mol Cell. 2007;27:449–61. doi: 10.1016/j.molcel.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Djupedal I, Portoso M, Spahr H, Bonilla C, Gustafsson CM, Allshire RC, Ekwall K. RNA Pol II subunit Rpb7 promotes centromeric transcription and RNAi-directed chromatin silencing. Genes Dev. 2005;19:2301–6. doi: 10.1101/gad.344205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic PA, Natarajan P, Chen C, Drinnenberg IA, Schiller BJ, Thompson J, Moresco JJ, Yates JR, 3RD, Bartel DP, Madhani HD. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell. 2013;152:957–68. doi: 10.1016/j.cell.2013.01.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle AB, Hessle V, Helbig R, Dantoft W, Gimber N, Visa N. Splice-site mutations cause Rrp6-mediated nuclear retention of the unspliced RNAs and transcriptional down-regulation of the splicing-defective genes. PLoS One. 2010;5:e11540. doi: 10.1371/journal.pone.0011540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiyama-Nakamura S, Ito S, Sawatsubashi S, Yamauchi Y, Suzuki E, Tanabe M, Kimura S, Murata T, Isobe T, Takeyama K, Kato S. BTB protein, dKLHL18/CG3571, serves as an adaptor subunit for a dCul3 ubiquitin ligase complex. Genes Cells. 2009;14:965–73. doi: 10.1111/j.1365-2443.2009.01323.x. [DOI] [PubMed] [Google Scholar]
- Garcia JF, Dumesic PA, Hartley PD, El-Samad H, Madhani HD. Combinatorial, site-specific requirement for heterochromatic silencing factors in the elimination of nucleosome-free regions. Genes Dev. 2010;24:1758–71. doi: 10.1101/gad.1946410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer R, Wee S, Anderson S, Yates J, Wolf DA. BTB/POZ domain proteins are putative substrate adaptors for cullin 3 ubiquitin ligases. Mol Cell. 2003;12:783–90. doi: 10.1016/s1097-2765(03)00341-1. [DOI] [PubMed] [Google Scholar]
- Gregan J, Rabitsch PK, Rumpf C, Novatchkova M, Schleiffer A, Nasmyth K. High-throughput knockout screen in fission yeast. Nat Protoc. 2006;1:2457–64. doi: 10.1038/nprot.2006.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullerova M, Barta A, Lorkovic ZJ. AtCyp59 is a multidomain cyclophilin from Arabidopsis thaliana that interacts with SR proteins and the C-terminal domain of the RNA polymerase II. Rna. 2006;12:631–43. doi: 10.1261/rna.2226106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullerova M, Barta A, Lorkovic ZJ. Rct1, a nuclear RNA recognition motifcontaining cyclophilin, regulates phosphorylation of the RNA polymerase II C-terminal domain. Mol Cell Biol. 2007;27:3601–11. doi: 10.1128/MCB.02187-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halic M, Moazed D. Dicer-independent primal RNAs trigger RNAi and heterochromatin formation. Cell. 2010;140:504–16. doi: 10.1016/j.cell.2010.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, Villen J, Gerace EL, Gygi SP, Moazed D. A cullin E3 ubiquitin ligase complex associates with Rik1 and the Clr4 histone H3-K9 methyltransferase and is required for RNAi-mediated heterochromatin formation. RNA Biol. 2005;2:106–11. doi: 10.4161/rna.2.3.2131. [DOI] [PubMed] [Google Scholar]
- Horn PJ, Bastie JN, Peterson CL. A Rik1-associated, cullin-dependent E3 ubiquitin ligase is essential for heterochromatin formation. Genes Dev. 2005;19:1705–14. doi: 10.1101/gad.1328005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J, Lacava J, Tollervey D. RNA-quality control by the exosome. Nat Rev Mol Cell Biol. 2006;7:529–39. doi: 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- Irvine DV, Zaratiegui M, Tolia NH, Goto DB, Chitwood DH, Vaughn MW, Joshua-Tor L, Martienssen RA. Argonaute slicing is required for heterochromatic silencing and spreading. Science. 2006;313:1134–7. doi: 10.1126/science.1128813. [DOI] [PubMed] [Google Scholar]
- Jia S, Kobayashi R, Grewal SI. Ubiquitin ligase component Cul4 associates with Clr4 histone methyltransferase to assemble heterochromatin. Nat Cell Biol. 2005;7:1007–13. doi: 10.1038/ncb1300. [DOI] [PubMed] [Google Scholar]
- Jia S, Noma K, Grewal SI. RNAi-independent heterochromatin nucleation by the stress-activated ATF/CREB family proteins. Science. 2004;304:1971–6. doi: 10.1126/science.1099035. [DOI] [PubMed] [Google Scholar]
- Kallgren SP, Andrews S, Tadeo X, Hou H, Moresco JJ, Tu PG, Yates JR, 3RD, Nagy PL, Jia S. The proper splicing of RNAi factors is critical for pericentric heterochromatin assembly in fission yeast. PLoS Genet. 2014;10:e1004334. doi: 10.1371/journal.pgen.1004334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Goto DB, Martienssen RA, Urano T, Furukawa K, Murakami Y. RNA polymerase II is required for RNAi-dependent heterochromatin assembly. Science. 2005;309:467–9. doi: 10.1126/science.1114955. [DOI] [PubMed] [Google Scholar]
- Keller C, Kulasegaran-Shylini R, Shimada Y, Hotz HR, Buhler M. Noncoding RNAs prevent spreading of a repressive histone mark. Nat Struct Mol Biol. 2013;20:994–1000. doi: 10.1038/nsmb.2619. [DOI] [PubMed] [Google Scholar]
- Kloc A, Zaratiegui M, Nora E, Martienssen R. RNA interference guides histone modification during the S phase of chromosomal replication. Curr Biol. 2008;18:490–5. doi: 10.1016/j.cub.2008.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalik KM, Shimada Y, Flury V, Stadler MB, Batki J, Buhler M. The Paf1 complex represses small-RNA-mediated epigenetic gene silencing. Nature. 2015;520:248–52. doi: 10.1038/nature14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeds P, Peltz SW, Jacobson A, Culbertson MR. The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev. 1991;5:2303–14. doi: 10.1101/gad.5.12a.2303. [DOI] [PubMed] [Google Scholar]
- Li F, Martienssen R, Cande WZ. Coordination of DNA replication and histone modification by the Rik1-Dos2 complex. Nature. 2011;475:244–8. doi: 10.1038/nature10161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motamedi MR, Verdel A, Colmenares SU, Gerber SA, Gygi SP, Moazed D. Two RNAi complexes, RITS and RDRC, physically interact and localize to noncoding centromeric RNAs. Cell. 2004;119:789–802. doi: 10.1016/j.cell.2004.11.034. [DOI] [PubMed] [Google Scholar]
- Murakami H, Goto DB, Toda T, Chen ES, Grewal SI, Martienssen RA, Yanagida M. Ribonuclease activity of Dis3 is required for mitotic progression and provides a possible link between heterochromatin and kinetochore function. PLoS One. 2007;2:e317. doi: 10.1371/journal.pone.0000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayashiki H, Kadotani N, Mayama S. Evolution and diversification of RNA silencing proteins in fungi. J Mol Evol. 2006;63:127–35. doi: 10.1007/s00239-005-0257-2. [DOI] [PubMed] [Google Scholar]
- Noma K, Cam HP, Maraia RJ, Grewal SI. A role for TFIIIC transcription factor complex in genome organization. Cell. 2006;125:859–72. doi: 10.1016/j.cell.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Pall GS, Hamilton AJ. Improved northern blot method for enhanced detection of small RNA. Nat Protoc. 2008;3:1077–84. doi: 10.1038/nprot.2008.67. [DOI] [PubMed] [Google Scholar]
- Partridge JF, Scott KS, Bannister AJ, Kouzarides T, Allshire RC. cis-acting DNA from fission yeast centromeres mediates histone H3 methylation and recruitment of silencing factors and cohesin to an ectopic site. Curr Biol. 2002;12:1652–60. doi: 10.1016/s0960-9822(02)01177-6. [DOI] [PubMed] [Google Scholar]
- Petrie VJ, Wuitschick JD, Givens CD, Kosinski AM, Partridge JF. RNA interference (RNAi)-dependent and RNAi-independent association of the Chp1 chromodomain protein with distinct heterochromatic loci in fission yeast. Mol Cell Biol. 2005;25:2331–46. doi: 10.1128/MCB.25.6.2331-2346.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintard L, Willems A, Peter M. Cullin-based ubiquitin ligases: Cul3-BTB complexes join the family. Embo j. 2004;23:1681–7. doi: 10.1038/sj.emboj.7600186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy BD, Wang Y, Niu L, Higuchi EC, Marguerat SB, Bahler J, Smith GR, Jia S. Elimination of a specific histone H3K14 acetyltransferase complex bypasses the RNAi pathway to regulate pericentric heterochromatin functions. Genes Dev. 2011;25:214–9. doi: 10.1101/gad.1993611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Zhang K, Zofall M, Chen E, Grewal SI. Defects in RNA quality control factors reveal RNAi-independent nucleation of heterochromatin. Nat Struct Mol Biol. 2011;18:1132–8. doi: 10.1038/nsmb.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribar B, Prakash L, Prakash S. ELA1 and CUL3 are required along with ELC1 for RNA polymerase II polyubiquitylation and degradation in DNA-damaged yeast cells. Mol Cell Biol. 2007;27:3211–6. doi: 10.1128/MCB.00091-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–6. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadaie M, Kawaguchi R, Ohtani Y, Arisaka F, Tanaka K, Shirahige K, Nakayama J. Balance between distinct HP1 family proteins controls heterochromatin assembly in fission yeast. Mol Cell Biol. 2008;28:6973–88. doi: 10.1128/MCB.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadeghi L, Prasad P, Ekwall K, Cohen A, Svensson JP. The Paf1 complex factors Leo1 and Paf1 promote local histone turnover to modulate chromatin states in fission yeast. EMBO Rep. 2015;16:1673–87. doi: 10.15252/embr.201541214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankaranarayana GD, Motamedi MR, Moazed D, Grewal SI. Sir2 regulates histone H3 lysine 9 methylation and heterochromatin assembly in fission yeast. Curr Biol. 2003;13:1240–6. doi: 10.1016/s0960-9822(03)00489-5. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Cam HP, Sugiyama R, Noma K, Zofall M, Kobayashi R, Grewal SI. SHREC, an effector complex for heterochromatic transcriptional silencing. Cell. 2007;128:491–504. doi: 10.1016/j.cell.2006.12.035. [DOI] [PubMed] [Google Scholar]
- Thomas J, Palusa SG, Prasad KV, Ali GS, Surabhi GK, Ben-Hur A, Abdel-Ghany SE, Reddy AS. Identification of an intronic splicing regulatory element involved in auto-regulation of alternative splicing of SCL33 pre-mRNA. Plant J. 2012;72:935–46. doi: 10.1111/tpj.12004. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D. RNAi-mediated targeting of heterochromatin by the RITS complex. Science. 2004;303:672–6. doi: 10.1126/science.1093686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA. Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science. 2002;297:1833–7. doi: 10.1126/science.1074973. [DOI] [PubMed] [Google Scholar]
- Wickham H. Ggplot2 : elegant graphics for data analysis. New York: Springer; 2009. [Google Scholar]
- Yamanaka S, Mehta S, Reyes-Turcu FE, Zhuang F, Fuchs RT, Rong Y, Robb GB, Grewal SI. RNAi triggered by specialized machinery silences developmental genes and retrotransposons. Nature. 2013;493:557–60. doi: 10.1038/nature11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Jih G, Iglesias N, Moazed D. Determinants of heterochromatic siRNA biogenesis and function. Mol Cell. 2014;53:262–76. doi: 10.1016/j.molcel.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, De Castro E, Marin L, Chang AY, Goto D, Cande WZ, Antequera F, Arcangioli B, Martienssen RA. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature. 2011;479:135–8. doi: 10.1038/nature10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Mosch K, Fischle W, Grewal SI. Roles of the Clr4 methyltransferase complex in nucleation, spreading and maintenance of heterochromatin. Nat Struct Mol Biol. 2008;15:381–8. doi: 10.1038/nsmb.1406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Conserved Eukaryotic Genes Present in S. pombe with No Apparent S. cerevisiae Ortholog, Related to Figure 1.
Table S2. Strains Used in This Study, Related to Experimental Procedures.
Table S3. Oligonucleotides Used in This Study, Related to Experimental Procedures.