

Abstract
Mutations in the gene BEST1 are causally associated with as many as five clinically distinct retinal degenerative diseases, which are collectively referred to as the “bestrophinopathies”. These five associated diseases are: Best vitelliform macular dystrophy, autosomal recessive bestrophinopathy, adult-onset vitelliform macular dystrophy, autosomal dominant vitreoretinochoroidopathy, and retinitis pigmentosa. The most common of these is Best vitelliform macular dystrophy. Bestrophin 1 (Best1), the protein encoded by the gene BEST1, has been the subject of a great deal of research since it was first identified nearly two decades ago. Today we know that Best1 functions as both a pentameric anion channel and a regulator of intracellular Ca2+ signaling. Best1 is an integral membrane protein which, within the eye, is uniquely expressed in the retinal pigment epithelium where it predominantly localizes to the basolateral plasma membrane. Within the brain, Best1 expression has been documented in both glial cells and astrocytes where it functions in both tonic GABA release and glutamate transport. The crystal structure of Best1 has revealed critical information about how Best1 functions as an ion channel and how Ca2+ regulates that function. Studies using animal models have led to critical insights into the physiological roles of Best1 and advances in stem cell technology have allowed for the development of patient-derived, “disease in a dish” models. In this article we review our knowledge of Best1 and discuss prospects for near-term clinical trials to test therapies for the bestrophinopathies, a currently incurable and untreatable set of diseases.
Keywords: Bestrophin, Best1, retinal pigment epithelium, retinal disease, maculopathy, anion channel
Introduction
The bestrophins were first identified in the human genome as a result of the association of BEST1 mutations with Best vitelliform macular dystrophy (BVMD) (Marquardt et al., 1998; Petrukhin et al., 1998). To date, mutations in BEST1 have been found in association with at least five clinically distinct retinal degenerative diseases. Following the association of BEST1 (then known as VMD2) with BVMD, Kramer et al. identified three human homologues of BEST1 initially termed VMD2L1, VMD2L2, and VMD2L3 (Kramer et al., 2004). The HUGO nomenclature committee has since reassigned names of the genes as BEST1 (VMD2), BEST2 (VMD2L1), BEST3 (VMD2L2), and BEST4 (VMD2L3). None of these homologues are known to be associated with human disease, though functional deficiencies in sweating (Cui et al., 2012) and maintenance of intraocular pressure (Bakall et al., 2008; Zhang et al., 2009) in BEST2 knock-out mice suggest the possibility that BEST2 mutations may have as yet unrecognized effects on human health.
Bestrophins are an ancient family of proteins and they exhibit a remarkable level of evolutionary conservation. They are found throughout the animal kingdom and have been identified in virtually every organism studied (Hartzell et al., 2008; Milenkovic et al., 2008). These diverse bestrophin-containing organisms range in complexity from simple bacteria (Yang et al., 2014), to eye-regenerating planarian flatworms (Cross et al., 2015; Lapan and Reddien, 2012), and finally to complex mammals (Bakall et al., 2003; Marmorstein et al., 2000). Although each bestrophin possesses unique physiological functions, they are invariably ion channels (Hartzell et al., 2008; Xiao et al., 2010). Within the phylogenetic tree, bestrophin shows a diverse array of gene orthologs as well as gene paralogs (Hartzell et al., 2008). All mammals studied to date have at least four vestigial paralogues (Hartzell et al., 2008), though Bestrophin 4 is a pseudogene in mice (Kramer et al., 2004). Insects, such as the fruit fly Drosophila and the mosquito Anopheles, also have four paralogs (Hartzell et al., 2008; Petrukhin et al., 1998). In contrast, the nematode species Caenorhabditis elegans has 25 bestrophin paralogs and the primitive chordate Ciona savignyi has just one bestrophin gene (Hartzell et al., 2008; Petrukhin et al., 1998). Between bestrophin orthologs and paralogs, the first 350 amino acids show the most conservation (Hartzell et al., 2008).
All four human bestrophin paralogs function as calcium-activated anion channels (Qu and Hartzell, 2008; Xiao et al., 2010). Other than being reportedly expressed in absorptive cells in human colon and small intestine (Ito et al., 2013), very little is known about the Bestrophin 4 protein. Bestrophin 3 shows a very broad tissue distribution and emerging evidence suggests that this anion channels plays important cell protective roles (Svenningsen, 2015) against endoplasmic reticulum stress (Lee et al., 2012), oxidative stress (Jiang et al., 2013), and inflammation (Song et al., 2014). Bestrophin 2 has been shown to mediate bicarbonate transport in colonic goblet cells (Yu et al., 2010) and compelling data indicates that Bestrophin 2 also mediates bicarbonate transport in sweat glands (Cui et al., 2012) as well as nonpigmented epithelium (Bakall et al., 2008; Zhang et al., 2009). Knockout mice lacking Bestrophin 2 suffer from a complete inability to sweat (Cui et al., 2012). Best2 knockout mice also exhibit a significantly reduced intraocular pressure (Zhang et al., 2009; Zhang et al., 2010).
Best1 is predominantly expressed in the retinal pigment epithelium (RPE) (Marmorstein et al., 2000). Within the RPE, Best1 is an integral membrane protein localized to the basolateral plasma membrane (Marmorstein et al., 2000). The human protein is comprised of 585 amino acids and, evolutionarily, the first ~350 amino acids of Best1 are highly conserved between species. Best1 has intracellular N- and C-termini, the latter of which is a large cytosolic domain comprised of approximately 280 amino acids including the C-terminus (Kane Dickson et al., 2014; Yang et al., 2014). Within the RPE, Best1 appears to function as both an anion channel and a regulator of intracellular calcium signaling (Marmorstein et al., 2015; Milenkovic et al., 2015; Singh et al., 2013). Best1 itself is encoded by the gene BEST1, which is located on chromosome 11q13 (Marquardt et al., 1998; Petrukhin et al., 1998). Crystal structure data has revealed that Best1 forms homo-pentamers in both bacteria (Yang et al., 2014) and chicken (Kane Dickson et al., 2014), indicating that this pentameric configuration is very highly conserved throughout the animal kingdom.
Over 200 mutations throughout the entire BEST1 gene have been reported to cause at least five clinically distinct forms of retinal degeneration (http://www-huge.uni-regensburg.de/BEST1_database/home.php?select_db=BEST1). These retinal diseases are collectively referred to as the “bestrophinopathies” and consist of BVMD (Marquardt et al., 1998; Petrukhin et al., 1998), adult-onset vitelliform macular dystrophy (AVMD) (Allikmets et al., 1999; Kramer et al., 2000), autosomal recessive bestrophinopathy (ARB) (Burgess et al., 2008), autosomal dominant vitreoretinochoroidopathy (ADVIRC) (Yardley et al., 2004), and retinitis pigmentosa (RP) (Davidson et al., 2009). Despite being one of the most common retinal disorders caused by RPE mutations, the bestrophinopathies are currently an untreatable set of diseases. In addition to describing the function of Best1 and the clinical spectrum of BEST1 mutations, this review focuses on the pathogenesis of the bestrophinopathies as well as potential treatment options which could attenuate vision loss or fully restore vision in affected patients.
Clinical Spectrum of the Bestrophinopathies
Best vitelliform macular dystrophy
The most common of the bestrophinopathies is BVMD (Fig. 1), otherwise known as Best disease. BVMD is inherited in an autosomal dominant fashion but with variable expressivity (Marquardt et al., 1998; Petrukhin et al., 1998). The etymology of the disease comes from the physician Friedrich Best, who first described the disease in 1905. He described what he thought was a stationary disease in eight members of two familial generations (Best, 1905).
Figure 1. Clinical presentation of Best vitelliform macular dystrophy.

A classic vitelliform lesion is found in both the right (A) and left (D) eye of an 80 year old, female patient with Best disease. She presented with mild hyperopia, 20/40 vision in one eye, and 20/400 vision in the other eye. Both lesions were autofluorescent (B, E). OCT imaging of a horizontal section of the left (C) and right (F) maculas revealed retinal abnormalities. In particular, the left eye showed a raised retina and multiple fluid-filled, serous retinal detachments.
Studies on the prevalence of Best disease have been hampered by the fact that there is such variable expressivity within families as well as between families (Lacassagne et al., 2011; Walter et al., 1994). Unfortunately, all of the studies on incidence are in the white population. Even in this group, the data are skewed towards the incidence in a northern European population. In Sweden, the incidence was noted to be 2/10,000 (Nordstrom, 1974). In Denmark, the incidence was thought to be 1.5/100,000 (Bitner et al., 2012). Recently, we have shown that the incidence in a predominately white population in Olmsted County, Minnesota was between 1 in 16,500 and 1 in 21,000 (Dalvin et al., 2016b). A concern with the Swedish study is that it was performed prior to molecular genetic screening. Even in the more recent study performed in Minnesota, confirmatory genetic testing was rarely performed. AVMD, otherwise known as adult foveomacular dystrophy, was diagnosed three times more frequently than BVMD in our study due in most cases to absent electrooculogram (EOG) or genetic testing. Often family history was unknown. As a result it is possible that the incidence of BVMD was underestimated in our study (Dalvin et al., 2016b). More broadly, the incidence of rare diseases is likely underreported due to the fact that these diseases are frequently misdiagnosed.
Phenotypically, the main clinical findings in BVMD are in the posterior pole (Boon et al., 2009). Initially no lesions are seen and the posterior pole appears unremarkable (Stage 1). At this stage, the vision is normal and the only phenotypes are RPE window and granularity defects (Marmorstein et al., 2009). Stage 2 involves the development of a yellow, well-demarcated vitelliform lesion (Fig. 1). This is characterized by a 2–3 mm diameter central, yellowish, egg yolk elevation centered in the macula (Fig. 1A, D). At this stage, the vision might be slightly decreased. The yolk can layer down due to partial resorption of fluid (Marmorstein et al., 2009), causing a pseudohypopyon (Stage 3). Over time the egg yolk vitelliform lesion “scrambles” (vitelliruptive stage) and, when this occurs, vision can substantially worsen (Stage 4). Finally, there is an atrophic stage in which choroidal neovascularization can occur (Stage 5). This is called the atrophic/cicatricial stage and, usually, it is bilateral and relatively symmetric. While BVMD typically presents bilaterally, it sometimes presents unilaterally (Arora et al., 2016; Kaden et al., 2016). Macular holes can also occur as a severe complication in the end stage of BVMD (Liu et al., 2016).
As with many autosomal dominant diseases, however, there is variability in both expression and age of disease presentation (Boon et al., 2009; Marmorstein et al., 2009). As such, these prevalence studies are approximations based on clinical findings. The clinical presentation of BVMD in identified patients is not yet predictable, as approximately 7–9% of patients harboring disease-causing BEST1 mutations have normal vision and do not exhibit decreased visual acuity (Nordstrom and Thorburn, 1980). Other patients report experiencing vision loss that is episodic and, in general, visual acuity can range between 20/20 and 20/200 (Bard and Cross, 1975; Mohler and Fine, 1981). The visual fields are correlated with the fundus evaluation. There is a central scotoma that increases in density and correlates with the stage of the disease (Querques et al., 2011). Although it isn’t discussed much in the literature, the anterior segment is affected as well. Some patients will present with a shallow chamber, which causes hyperopia and can cause narrow angle glaucoma (Liu et al., 2016; Wittstrom et al., 2011).
The EOG is abnormal in all of these stages (Boon et al., 2009; Marmorstein et al., 2009). The Arden ratio, which is the ratio of the light peak/dark trough (LP/DT)], tends to be abnormally low in patients with BVMD. Per International Society for Clinical Electrophysiology of Vision standards (Brown et al., 2006), a normal LP/DT ratio is typically 2.0 or greater and, with Best disease, the LP/DT ratio falls below 1.55. Since it requires a certain level of expertise for a clinic to perform EOGs and interpret their results correctly, genetic testing is likely a more reliable assay for disease pathogenesis. In addition, there have been occasional reports of “normal” EOG responses in individuals with BVMD (Caldwell et al., 1999; Testa et al., 2008). Unlike the EOG, the full-field electroretinogram (Ganzfeld ERG) is normal. In late stages, however, the photopic ERG can sometimes be slightly decreased. Additionally, the multifocal ERG is affected in stages two through four and this is to be expected given the morphological changes (Palmowski et al., 2003). Multifocal ERG readings therefore do not add any further information than what can be gleaned from fundus findings. In our clinic we find electrophysiological testing to be unnecessary and instead we now routinely order genetic testing in its place. That some atypical BVMD patients can present with a normal EOG (Caldwell et al., 1999; Testa et al., 2008) further highlights the importance of performing genetic testing.
Short-wave length fundus autofluorescence in stage 2 of the disease usually shows hyperfluorescence of the vitelliform lesion (Fig. 1B, E). In most cases, the autofluorescence is either diffuse or patchy. As the yolk settles and scrambles, the smaller portions that are still yellow continue to have autofluorescence and this underlies the patchy pattern. Once the lesion is completely atrophic, the autofluorescence can disappear (Parodi et al., 2014). The near infrared autofluorescence presents analogously to the short-wavelength fundus autofluorescence, though it rarely is seen as diffuse and predominantly manifests as patchy. Fluorescein angiography reveals that, in early stages, there is blocked fluorescence from the vitelliform material. As the yolk layers scramble, there are windows of defects seen due to the underlying atrophy of the RPE and the choriocapillaris (Boon et al., 2009; Marmorstein et al., 2009). At least a portion of this fluorescence is likely due to the greater than normal accumulation of lipofuscin in the RPE (Weingeist et al., 1982) (Bakall et al., 2007). Optical coherence tomography (OCT) imaging has divulged that, in the vitelliform stage, there is an elevation of the retina as well as hyperreflective material between the photoreceptors and the RPE (Fig. 1C, F). Due to scrambling of the lesion and further atrophy of the RPE, over time the amount of hyperreflective material decreases and there is a concomitant loss of photoreceptors. As more atrophy occurs, the area of RPE atrophy and photoreceptor loss increases. Choroidal neovascular membranes can develop as well (Boon et al., 2009; Marmorstein et al., 2009).
BVMD can also manifest with multiple vitelliform lesions and, when it does, it is referred to as multi-focal Best disease (Fig. 2). The lesions can be large or small (Fig. 2A, B, C, E), though the extrafoveal lesions tend to be small and are more common in the superior macula than in the inferior macula. There can be a central lesion as well as extrafoveal lesions. Like in classical BVMD, the lesions are autofluorescent (Fig. 2D, F) There are cases where a patient presents with multifocal lesions but the patient’s family members show a classic, singular vitelliform lesion. The symptoms are otherwise similar to those of classical BVMD (Ciulla and Frederick, 1997; Lacassagne et al., 2011).
Figure 2. Clinical presentation of multifocal Best vitelliform macular dystrophy.

Fundus photographs revealed prominent multi-focal lesions in both the left (A, C) and right (B, E) eyes in a 33 year-old male patient with multifocal Best disease. These lesions were autofluorescent (D, F) and choroidal neovascularization was apparent in the fundus of the patient’s left eye (E).
Mutations in BEST1 associated with BVMD are mostly missense mutations, though single amino acid deletions have also been noted. How these mutations cause BVMD is not known. Hypotheses on this vary from loss of anion channel activity (Xiao et al., 2010) to functional activation of the protein (Zhang et al., 2010). Absolute loss of Best1 activity is unlikely the cause of BVMD as Best1−/− mice do not exhibit a BVMD phenotype (Marmorstein et al., 2006) while mice carrying the BVMD associated mutation W93C do exhibit classical BVMD symptoms (Zhang et al., 2010).
Adult-onset vitelliform macular dystrophy
There are certainly other diseases that cause vitelliform or pseudovitelliform lesions in the macula. The latter term pseudovitelliform has been used to describe patients who have AVMD (Fig. 3), otherwise known as adult-onset foveomacular vitelliform dystrophy. AVMD has been associated with mutations in BEST1 and PRPH2, though the majority of cases appear to be idiopathic. The overall incidence of AVMD, based on our study conducted in Olmsted county (Dalvin et al., 2016b), is 3x greater than the incidence of BVMD. However, a lack of genetic testing in these cases leaves us without the ability to determine whether any of these cases fall within the definition of BVMD (due to BEST1) or Pattern Dystrophy (due to PRPH2). It would be of interest to know if AVMD due to BEST1 mutations is clinically distinct from AVMD due to PRPH2 mutations. Although the presentation of AVMD was initially thought to be clinically distinct from BVMD, it is now unclear whether there are any notable differences between AVMD and BVMD and it is likely that AVMD is indistinguishable from milder cases of BVMD. Classically, AVMD has a vitelliform-like lesion that is about 500 to 700 microns in size (Fig. 3A, D) and is associated with only a minimal or mild amount of visual loss. These lesions are autofluorescent (Fig. 3B, E) and OCT imaging reveals the presence of hyperreflective material between the RPE and the photoreceptors as well as an elevation of the retina (Fig. 3C, F). AVMD is usually sporadic, though some families with multiple cases of AVMD have been reported. Although occasionally neovascularization can occur, this is a rare finding that is associated with more significant vision loss (Boon et al., 2009; Marmorstein et al., 2009). Since AVMD and BVMD are clinically very similar if not indistinguishable, we propose that individuals carrying mutations in BEST1 that are diagnosed with AVMD should be reclassified as BVMD.
Figure 3. Clinical presentation of adult-onset vitelliform macular dystrophy.

While initially thought to have age-related macular degeneration, further testing diagnosed this 88 year old female patient with adult-onset vitelliform macular dystrophy. The presentation is identical to Best vitelliform macular dystrophy, with a classical vitelliform lesion in the fundus of both eyes (A, D). These lesions are autofluorescent (B, E) and OCT imaging of a horizontal section of the macula shows retinal abnormalities (C, F). The OCT of the left eye, in particular, shows a fluid-filled retinal detachment (C).
Autosomal recessive bestrophinopathy
ARB (Fig. 4) was first recognized by Burgess et al in 2008 (Burgess et al., 2008). The parents of the proband did not have any abnormal fundus findings and their EOG was normal. Patients with ARB tend to be recognized because of decreased vision (typically around 20/40) in the first decade of life, though vision loss can occur in the second decade of life as well. As opposed to multifocal Best disease and classic Best disease which have autosomal dominant inheritance, the parents in many ARB cases do not have fundus findings and their EOG is normal (Boon et al., 2009; Marmorstein et al., 2009).
Figure 4. Clinical presentation of autosomal recessive bestrophinopathy.

Fundus photographs of a 17-year old girl diagnosed with autosomal recessive bestrophinopathy show classical findings, such as vitelliform lesions (A–F) and yellowish, subretinal deposits (G, H). These lesions are autofluorescent (I).
When a patient is first seen, there tends to be a central serous detachment with a fibrous subretinal central scar, which is likely caused by a choroidal neovascular membrane in at least one eye. There are small vitelliform lesions (Fig. 4A, B) proximal to the arcades (Boon et al., 2009; Johnson et al., 2015; Marmorstein et al., 2009). Yellowish, subretinal deposits are also common fundus findings (Fig. 4C–H). Like in BVMD, the EOG is decreased and the ERG tends to be normal. There is a large area of mild fundus autofluorescence that corresponds to the central area of serous detachment (Fig. 4I). Marked hyperautofluorescence is associated with the smaller vitelliform lesions. OCT imaging highlights both the serous detachment and the hyperreflectivity of the vitelliform lesions. Some patients also present with a cystoid macular edema and, like the vitelliform lesions, the central scar is hyperreflective. Fluorescence angiography shows staining of the vitelliform lesion as well as the serous detachment. Like in BVMD, some patients develop a choroidal neovascular membrane that can be seen by fluorescein and indocyanine angiography (Boon et al., 2009; Marmorstein et al., 2009). Vision decreases over time, but usually very slowly unless a choroidal neovascular membrane develops. If this occurs, then anti-VEGF agents can be employed (Hussain et al., 2015). ARB also has multiple peripheral vitelliform lesions that autofluoresce (Boon et al., 2013; Burgess et al., 2008; Marmorstein et al., 2009).
ARB has been hypothesized to represent the human “null” phenotype for Best1 (Burgess et al., 2008; Pomares et al., 2012). The mutation spectrum associated with ARB varies from missense to truncations to single base changes in introns (http://www-huge.uni-regensburg.de/BEST1_database/home.php?select_db=BEST1). Because ARB is a recessively inherited disease, both alleles of BEST1 must be mutated. In the majority of cases the patient will be a compound heterozygote. Evidence favoring the “null phenotype” hypothesis includes several ARB patients that are homozygous for truncating mutations such as BEST1R200X (Burgess et al., 2008). The prospect of ARB resulting from absence of BEST1 is also supported by a naturally occurring canine BEST1 knock-out model which presents with symptoms highly analogous to ARB (Guziewicz et al., 2007). In contrast, this hypothesis is disfavored by the findings that two independently derived Best1−/− mouse lines do not exhibit a phenotype similar to ARB (Marmorstein et al., 2006; Milenkovic et al., 2015).
Similarly presenting diseases with vitelliform lesions
Molecular studies have shown that some patients with vitelliform lesions have BEST1 mutations, characterizing their lesion as being induced by a bestrophinopathy. Other patients with vitelliform lesions may have peripherin/RDS mutations (Felbor et al., 1997) while others may have mutations in the IMPG1 or IMPG2 gene (Manes et al., 2013; Meunier et al., 2014). Vision loss in these patients is milder and could have been easily characterized as BVMD or AVMD in the past (Patrinely et al., 1985). Basal laminar drusen, also called cuticular drusen or early adult-onset group drusen, can develop a vitelliform-like localized serous detachment in the posterior pole. These drusen give a starry sky appearance when visualized via fluorescein angiography and are similar to the drusen seen in Malattia Leventinese. In the latter disease, however, the fine drusen can develop a radial distribution (Pilli et al., 2011). While vision is typically unaffected by these drusen, there have been cases where vision decreased to the 20/50 level. Typically the detachment resorbs and the vision improves at this point. Moreover, similar to BVMD, some patients can develop choroidal neovascularization (Sigford and Schaal, 2014). In contrast to Malattia Leventinese, where the genetic defect is caused by mutation of EFEMP1 (Stone et al., 1999), some patients with basal laminar drusen have a mutation in the complement factor H gene (van de Ven et al., 2012).
Pseudoxanthoma elasticum, which is associated with a mutation in the ABCA6 gene, is yet another disease associated with vitelliform macular lesions and is characterized by angiod streaks, a peau d’orange stippling of the RPE temporal to the fovea, and peripheral comets caused by breaks in Bruch’s membrane that stream out calcium deposits (Parodi et al., 2015). Choroidal neovascularization occurs frequently with this disease and the development of vitelliform lesions occurs less frequently (Parodi et al., 2015). Pattern dystrophy can have vitelliform lesions similar to those seen in AVMD, albeit these lesions are small. Central serous chorioretinopathy, especially in pregnancy and in someone that is pigmented, can present with whitish fibrin within the localized serous detachments that look like a vitelliform lesion. Paraneoplastic exudative vitelliform maculopathy and the non-cancer associated acute polymorphous exudative vitelliform maculopathy appear to have multiple small vitelliform lesions similar to those seen in multifocal Best disease. This disease is characterized by the presence of autoantibodies to the RPE. Although one report indicated that these autoantibodies recognize Best1 (Eksandh et al., 2008), we did not find anti-Best1 antibodies in a patient with paraneoplastic exudative vitelliform maculopathy due to multiple myeloma (Dalvin et al., 2015). Other diseases that should also be considered when a patient presents with vitelliform lesions include Waldenstrom’s macroglobulinemia, hypertensive choroidopathy, and Vogt-Koyanagi-Harada disease. Responses of the RPE to the use of MEK inhibitors or desferoxamine (Viola et al., 2014) should also be considered.
Because of all these analogous diseases or phenotypic mimickers, it is critically important to perform genetic testing in patients with vitelliform lesions to determine if the presented symptoms are indeed due to a bestrophinopathy.
Autosomal dominant vitreoretinochoroidopathy
Autosomal dominant vitreoretinochoroidopathy (Fig. 5) was first described by Kaufman et al in 1982 (Kaufman et al., 1982). The first proband was a 12 year old boy who had 20/50 and 20/100 vision in the right eye and left eye respectively. He had mild hyperopia, though axial length was smaller than average. There were pigmented cells in the vitreous and there was a cystoid macular edema in both eyes as well as preretinal neovascularization in one eye. In the midperiphery, there was a sharp demarcation between an area of normal retina and clumped hyperpigmentation at the level of the equator with tiny white dots within the areas of hyperpigmentation. His 18 year old sister had myopia and slightly enlarged axial lengths. There was peripheral avascularity and the same pigmented demarcation line. Two older brothers were affected as well as the father. Additionally, the scotopic ERG in the father had subnormal a-waves and b-waves. The father had undergone cataract surgery at an early age (Kaufman et al., 1982).
Figure 5. Clinical presentation of autosomal dominant vitreoretinochoroidopathy.

Fundus photographs of a patient diagnosed with autosomal dominant vitreoretinochoroidopathy reveal classical symptoms, including a sharp demarcation line between a region of normal retina and a region of clumped, hyperpigmentation. Whitish specs and yellowish deposits are distributed throughout the peripheral retina.
A second family showed similar findings and again vision was decreased from either cystoid macular edema or vitreous hemorrhage from the retinal neovascularization (Blair et al., 1984). The peripheral findings were similar to those seen in the first family and the commonalities between the families included an autosomal dominant inheritance, peripheral pigmentary changes, white dots in the area of pigmentary changes, cystoid macular edema, retinal neovascularization, early onset cataracts, and hypoplastic ciliary processes. The ERG was, for the most part, normal. A subsequent study by Han and Lewandowski showed that the EOG was subnormal but that the ERG was normal (Han and Lewandowski, 1992). Interestingly, the patients described in this study had narrow angles. Further studies have shown that there can be a progression of the fundus findings with downstream development of central cone dysfunction. Similar progression in the posterior pole has also been noted (Chen and Goldberg, 2016; Chen et al., 2016; Oh and Vallar, 2006). In 2004, Yardley et al described five families with ADVIRC and nanophthalmos that were genetically linked to mutations in BEST1 (Yardley et al., 2004). All had abnormal ERGs and pathologically low EOGs. Figure 5 shows the fundus of a patient with a classical presentation of ADVIRC, including the well-demarcated line between normal retina and abnormal retina.
MRCS comprising of microcornea, rod-cone dystrophy, cataract, and posterior staphyloma presents similarly to ADVIRC and has also been reported in association with mutation of BEST1. MRCS was first described in 2003 by Reddy et al (Reddy et al., 2003) in a three-generation English family with six affected members and three unaffected members. The affected members showed an autosomal dominant inheritance of the disease and presented with hyperopia, microcornea, and early onset cataracts. Narrow angles were reported in the younger patients. All affected patients exhibited a well-demarcated separation between normal retina posteriorly and clumped RPE anteriorly. There was also a posterior pole staphyloma in some patients but not others. The older patients had a decreased ERG while the younger patients had “subnormal” ERG results. The peripheral fundus findings were exactly similar to those seen in ADVIRC (Reddy et al., 2003). The same study by Yardley et al which identified BEST1 mutations in association with ADVIRC also linked BEST1 mutations to MRCS (Yardley et al., 2004). ADVIRC can, like MRCS, present with posterior pole changes, ERG changes, narrow angles, and early-onset cataracts. Given the near-identical presentation of MRCS described by Reddy et al (Reddy et al., 2003) and ADVIRC described by Yardley et al (Yardley et al., 2004), we find it likely that both MRCS and ADVIRC are the same disease.
An important diagnosis differential is autosomal dominant neovascular inflammatory vitreoretinopathy. This disease is associated with retinal and iris neovascularization, cystoid macular edema, and vitreitis. The ERG b wave is decreased in these patients and is thus different than what is typically seen in patients with ADVIRC (Bennett et al., 1990). This disease is associated with Calpain mutations (Mahajan et al., 2012).
Retinitis pigmentosa
RP is a peripheral retinal disease and its association with BEST1 mutations was first described by Davidson et al in 2009 (Davidson et al., 2009). The authors reported four missense mutations in BEST1 associated with patients diagnosed with RP in five unrelated families. Fundoscopy revealed symptoms of panretinal dystrophy associated with flecks localized to the midperiphery, retinal gliosis, and vascular attenuation. Although EOG testing was not performed, all tested individuals exhibited highly diminished ERGs. Other symptoms reported in these patients included dense pigmentary changes in all peripheral retinal quadrants, pale optic disks, yellow foveal deposits, macular edema, and reduced visual acuity. Three of the mutations appeared to be autosomal dominant while one of the mutations appeared to be autosomal recessive (Davidson et al., 2009). It has been suggested that RP associated with BEST1 mutation represents misdiagnosed ADVIRC (Traboulsi, 2012). However, we recently reported a case of RP associated with a heterozygous 10kbp deletion in the BEST1 gene (Dalvin et al., 2016a). It is not apparent how this mutation results in an RP phenotype and our patient had several heterozygous mutations in known RP genes. Thus, we suggest that RP due to BEST1 may be multi-genic and require other as yet unidentified mutations in other genes. Identification of additional families with RP associated with BEST1 mutations will be necessary to test this hypothesis.
Best1 Expression, Localization, and Function
Expression and localization in the retinal pigment epithelium
Based on Northern blot analysis, Petrukhin et al determined that BEST1 gene expression in man is highest in the retina followed by brain and spinal cord (Petrukhin et al., 1998). In situ hybridization identified the RPE as the sole site of BEST1 expression in the adult human eye. This was confirmed by Marquardt and co-workers using Northern blot analysis of human RPE (Marquardt et al., 1998). The first analysis of Best1 protein was reported in 2000 by Marmorstein et al (Marmorstein et al., 2000), who generated novel monoclonal and polyclonal antibodies to Best1. Using these tools, they discovered that Best1 protein is uniquely expressed in the RPE in human eyes (Marmorstein et al., 2000). No Best1 protein expression was found in the neurosensory retina, ciliary body, iris, cornea, or lens. The RPE-derived cell lines ARPE-19, D407, and RPE-J were found to express Best1 mRNA (detected by reverse transcription-PCR) but not Best1 protein (Marmorstein et al., 2000). Triton X-114 extraction and cell-surface biotinylation experiments demonstrated that Best1 is an integral membrane protein localized to the plasma membrane. Immunohistochemical staining of macaque and porcine eyes showed that this staining was unique to the basolateral plasma membrane and this was confirmed by confocal microscopy (Marmorstein et al., 2000). This RPE-specific ocular expression and/or basolateral plasma membrane localization of Best1 has been confirmed by many subsequent studies using several different native models, including fetal human RPE cells (Johnson et al., 2013; Marmorstein et al., 2015), induced pluripotent stem cell (iPSC)-derived RPE cells (Brandl et al., 2014; Johnson et al., 2015; Milenkovic et al., 2015; Singh et al., 2013), mice (Marmorstein et al., 2006; Zhang et al., 2010), rats (Marmorstein et al., 2004), dogs (Guziewicz et al., 2013), rhesus monkeys (Gouras et al., 2009), and humans (Dalvin et al., 2015; Mullins et al., 2007). Basolateral plasma membrane localization has also been confirmed in models where Best1 was heterologously expressed, namely Madin-darby canine kidney II cells (Davidson et al., 2011; Davidson et al., 2009; Johnson et al., 2014; Johnson et al., 2013; Milenkovic et al., 2011b) and ARPE-19 cells (Marmorstein et al., 2000; Rosenthal et al., 2006). Basolateral plasma membrane localization of Best1 in MDCK II cells, fhRPE, and iPSC-RPE is shown in Figure 6. This immunofluorescent localization in the X-Y and X-Z planes was visualized by confocal microscopy (Fig. 6).
Figure 6. Basolateral plasma membrane localization of Best1 in confluent MDCK II, fhRPE, and iPSC-RPE cells.

Best1 was expressed in MDCK II cells via adenovirus mediated gene transfer and stained for Best1 (green) and the apical plasma membrane marker Gp135 (red). Localization of endogenous Best1 (green) was assessed in both fhRPE and iPSC-RPE cells. For both fhRPE and iPSC-RPE cells, nuclei (blue) were stained as a positional marker. iPSC-RPE cells were additionally stained with the tight junction marker ZO-1 (red). Confocal X-Y and X-Z scans were generated to show the localization of Best1 in the X, Y, and Z planes for all three cell types. Scale bars: 20 μm.
Particularly powerful evidence that Best1 is localized to the basolateral plasma membrane comes from studies showing that endogenous Best1 can be cell-surface biotinylated (Brandl et al., 2014; Marmorstein et al., 2000) and that Best1-labeled gold particles are present in the basal membrane of RPE from rhesus monkeys (Gouras et al., 2009). Other evidence has come to suggest, however, that a second, sub-population of Best1 may be localized to an internal compartment very close to the basolateral plasma membrane (Strauss et al., 2014). Evidence for this comes predominantly from the laboratory of Olaf Strauss, which identified this sub-population using short-term cultured porcine RPE cells (Strauss et al., 2014). Support for this also comes from the lab of David Gamm, which performed subcellular fractionation and co-immunoprecipitation experiments in iPSC-RPE to identify a sub-population of Best1 not localized to the basolateral plasma membrane (Singh et al., 2013). The work however, was performed on cell lysates generated using Triton X-100, which solubilizes membranes and would result in a significant portion of Best1 remaining in the supernatant when centrifuged at >100,000 × g (Singh et al., 2013). This separate population has been proposed to assist in releasing and accumulating Ca2+ from intracellular stories by conducting Cl− as a counterion for Ca2+ (Gomez et al., 2013; Neussert et al., 2010; Strauss et al., 2014). Work from the Kunzelmann laboratory has also suggested that a population of Best1 localized to the endosplasmic reticulum functions as a counterion channel (Barro-Soria et al., 2010; Kunzelmann et al., 2011). Further studies are warranted to better understand the role of this sub-population of Best1 and how distinct its role is from that of the majority basolateral plasma membrane-bound population.
The expression profile of Best1 outside of the eye has been somewhat controversial. A systematic examination of multiple organs from pigs for Best1 expression using immunoprecipitation with well-characterized antibodies did not identify any Best1 outside of the eye (Marmorstein et al., 2009; Stanton et al., 2006). However, those experiments may have missed low levels of expression. Today it is becoming accepted that Best1 is likely expressed elsewhere in the central nervous system, a topic that will be covered later in this review.
Function of Best1 in the human retinal pigment epithelium
Until fairly recently, the only evidence for Best1 functioning as an anion channel came from transfected HEK293 cells (Hartzell et al., 2008). Since its anion channel properties in this heterologous system have been reviewed extensively in the past (Hartzell et al., 2008; Xiao et al., 2010), this review will focus on more recent data demonstrating anion channel activity in human RPE. At this time, there are only three studies which have provided strong evidence for Best1 anion channel function in RPE cells.
The first study was performed on fhRPE cells (Marmorstein et al., 2015). In this study, Marmorstein et al. expressed the BVMD mutant Best1W93C in confluent fhRPE monolayers using adenovirus-mediated gene transfer. The W93C mutation affects a critical amino acid present in the anion channel pore of Best1 and severely impairs channel function (Qu et al., 2003; Sun et al., 2002). Transepithelial electrical properties were then assessed in these fhRPE monolayers and compared to uninfected fhRPE (which express endogenous Best1) as well as fhRPE made to overexpress wild-type (WT) Best1 via adenovirus-mediated gene transfer. Fetal human RPE expressing Best1W93C exhibited a notable reduction in transepithelial potential. In contrast, overexpression of WT Best1 led to an increase in transepithelial potential. Substituting chloride in the bath media with gluconate reduced transepithelial potential in monolayers overexpressing Best1, but had no effect on monolayers overexpressing Best1W93C (Marmorstein et al., 2015). These data strongly demonstrate that Best1 contributes to the transepithelial potential of the RPE. Moreover, the data specifically show that Best1 can affect anion currents in a human model of RPE.
The second study demonstrating Best1 anion channel activity in human RPE was published shortly afterwards by Milenkovic et al (Milenkovic et al., 2015). Using iPSC-RPE derived from a healthy control as well as iPSC-RPE derived from patients with the BVMD mutations A243V or Q238R, the authors used whole-cell patch clamp to demonstrate that iPSC-RPE exhibit a volume-dependent chloride current. This current was revealed to have characteristic, functional properties of volume-regulated anion channels and to be outwardly rectifying. Compared to healthy iPSC-RPE, this volume-regulated current was significantly reduced in iPSC-RPE derived from the patients with BVMD (Milenkovic et al., 2015). The third study, from the laboratory of Dr. Stephen Tsang (Moshfegh et al., 2016) used anion sensitive fluorescent dyes to compare Ca2+ stimulated Cl− secretion in iPSC-RPE from unaffected and BVMD donors. The BVMD donors exhibited substantially lower levels of Cl− secretion than the control cells.
Taken together, these three studies support the hypothesis that Best1 functions as an anion channel in the RPE. However, all three studies suffer from some drawbacks. The changes in transepithelial potential observed by Marmorstein et al (Marmorstein et al., 2015) as well as the changes in Cl− secretion observed by Moshfegh et al (Moshfegh et al., 2016) are indirect observations of Best1 activity and could have been due to effects of Best1 on Ca2+ signaling rather than Best1-mediated anion transport. While Milenkovic et al used whole cell patch clamp recordings to more directly measure Best1 anion channel activity (Milenkovic et al., 2015), they used only one unaffected control cell line which may not be representative. Further studies on the potential role of Best1 as an anion channel in the RPE are necessary to fully understand the function of Best1 in the RPE.
In addition to mediating anion transport, Best1 has also been shown to regulate intracellular Ca2+ signaling in human RPE. The first evidence that Best1 functions to regulate Ca2+ signaling was reported in 2006 by Rosenthal et al., who demonstrated significant effects of Best1 on the kinetics of L-type voltage dependent Ca2+ channels in RPE-J cells (Rosenthal et al., 2006). Similar data were obtained by Yu et al using HEK293 cells (Yu et al., 2008). In fact, the β subunit of voltage-dependent calcium channels has been one of very few proteins that Best1 has been observed to physically interact with (Milenkovic et al., 2011a; Reichhart et al., 2010; Yu et al., 2008). This interaction appears to affect the targeting of Best1 to the plasma membrane (Reichhart et al., 2010). Burgess et al. also showed that Best1 affects the activity of calcium channels in transfected HEK293 cells (Burgess et al., 2008). From a functional perspective, Best1 not only alters the kinetics of voltage-dependent calcium channels but also appears to regulate the release of Ca2+ stores in response to ATP in RPE cells in mice (Marmorstein et al., 2006; Zhang et al., 2010). Additionally, several studies in native RPE (fhRPE, iPSC-RPE) strongly indicate that Best1 affects intracellular calcium signaling and that disease-causing mutations disrupt calcium homeostasis (Singh et al., 2013) (Marmorstein et al., 2015).
Taken together, these studies demonstrate that Best1 functions to both regulate calcium homeostasis and mediate anion transport in the RPE.
Brain distribution of Best1
In addition to showing robust expression in the RPE, Best1 is also reportedly expressed in the mouse brain (Lee et al., 2010; Woo et al., 2012) and dorsal root ganglion (Boudes et al., 2009). Recent studies have thoroughly elucidated the expression and function of Best1 in the murine brain and these works have significantly increased our understanding of Best1 function. Thus, these studies have also enhanced our comprehension of Best1’s role in the eye. The first strong evidence that Best1 functions as an anion channel in vivo, for example, comes from a study on Best1 function in the brain in Best1 knock-out mice (Woo et al., 2012).
Unlike Best1 in the eye, the expression profiles of the various bestrophins in the brain had not been clearly established until 2009. As early as 1998 (Petrukhin et al., 1998), it was observed using Northern blotting and PCR that Best1 mRNA was detected in brain and spinal cord. However, the level of expression was significantly lower than in RPE. Using in situ hybridization, BEST1 mRNA was identified in RPE. Despite the presence of mRNA for human Best1 in brain and spinal cord, protein expression for human (hBest1) and porcine Best1 has been reported only in RPE. Although the expression of hBest1 is highly restricted, in situ hybridization data revealed the widely distributed expression of mouse Best1 (mBest1) in the brain, with especially high levels in the olfactory bulb, hippocampus, and cerebellum. In particular, significant expression was identified in both neurons and astrocytes (Park et al., 2009). mBest1 expression both in cortical neurons and astrocytes was also detected by RT-PCR and confirmed by gene silencing for mBest1 with mBest1-shRNA (Lee et al., 2010; Oh et al., 2012; Park et al., 2009). Murine Best1 mRNA and protein have also been reportedly expressed in the dorsal spinal cord and dorsal root ganglion (Al-Jumaily et al., 2007; Andre et al., 2003; Pineda-Farias et al., 2015).
Although limited information is available about the relative Best1 protein levels in the brain, the expression of mBest1 has been most thoroughly analyzed in the hippocampus and cerebellum via both western blot and immunohistochemistry. The primary antibody used in most of the brain-related studies was a polyclonal antibody raised against the C-terminus of mBest1 (Barro Soria et al., 2009). Using this antibody, expression of mBest1 in both neurons and astrocytes in the hippocampal CA1 region was identified (Park et al., 2009). Following the identification of Best1 protein expression in the mouse brain, subsequent studies sought to elucidate the neuronal function of this anion channel. In cultured mouse cortical astrocytes, mBest1 expression was reliably detected by Western blot analysis (Lee et al., 2010). Immunohistochemical analysis showed that mBest1 was significantly expressed in astrocytes in both the hippocampal dentate gyrus (Jo et al., 2014) and CA1 regions (Park et al., 2015; Woo et al., 2012). Furthermore, immunogold electron microscopy of astrocytic mBest1 in the molecular layer of the dentate gyrus and the stratum radiatum revealed that mBest1 was localized to the microdomain near the synaptic region. Little to no localization was observed in the soma or the processes (Park et al., 2015; Woo et al., 2012). In the cerebellum, prominent expression of mBest1 was observed in Purkinje cells, Bergmann glia, and lamellar astrocytes in the molecular layers, but not in granule cells (Lee et al., 2010) Unlike hippocampal astrocytes, Bergmann glial cells in cerebellum were found to express mBest1 in the cell body as well as in main processes (Park et al., 2013). Neuronal expression demonstrated via Western blotting (Fig. 7A), immunofluorescent staining (Fig. 7B–D), and electron microscopy (Fig. 7E) of mouse Best1 is summarized in Figure 7.
Figure 7. Neuronal expression of localization of mBest1.

A) Western blot analysis of mBest1 in cultured astrocytes and gene silencing for mBest1 by infection with lentivirus carrying mB1-shRNA or control-shRNA. B) Immunostaining of DAPI (blue), GFAP (green), and mBest1 (red) of hippocampal CA1 region in wild-type mouse (left) and Best1 KO mouse (right). Best1 is strongly co-localized with GFAP, an astrocytic marker, in wild-type mice, while mBest1 is not expressed in mBest1 KO mice. C) Immunostaining for mBest1 (red) and GFP (green) in cerebellum from GFAP-GFP transgenic mice. mBest1 is expressed in Purkinje cells (asterisk), interneurons (white arrowheads), Bergmann glia (arrows), and lamellar astrocytes (pale blue arrowheads), but not in granule cells. All GFP-positive astrocytes robustly expressed mBest1. D) Immunostaining and quantification of Best1 in the molecular layer of DG in mouse hippocampus. Top, representative confocal images for mBest1 (red) and GFP (green) in astrocytes. Bottom, percentage of Best1-positive areas in the cell body and process or in the microdomain over total area. **P < 0.01 (Student’s t-test). E) Immunogold electron microscopy of Best1 in the molecular layer of DG in mouse hippocampus. Top, representative images of mBest1 labeling (black dots indicated by arrowheads) in DAB-stained astrocytes (outlined with dashed lines). Pre, presynaptic terminal; Post, postsynapse. Bottom left, density of gold particles for Best1 in cell body, process and microdomain. Bottom right, percentage of gold particles for Best1 located on the plasma membrane of the cell body, process, and microdomain. ***P < 0.001 (Student’s t-test). Number on each bar refers to the number of cells (d) or images (e) analyzed. Data are presented as mean ± s.e.m.
Recently, markedly different patterns of mBest1 expression were observed in reactive astrocytes of hippocampal dentate gyrus in APP/PS1 mice (Jo et al., 2014), a mouse model of Alzheimer’s disease. In APP/PS1 mice, mBest1 staining exhibited reduced fluorescent intensity and fewer puncta in the microdomains of astrocytes were identified by immunohistochemical analysis and immunogold electron microscopic labeling (Jo et al., 2014). mBest1 seems to be redistributed within reactive astrocytes that are commonly observed not only in Alzheimer’s disease but also in other brain diseases, such as Parkinson’s disease, stroke, epilepsy, and traumatic brain injury. This suggests that the dynamic redistribution of Best1 might play an important role for the function of astrocytes both physiologically and pathologically. Thus, the regulators underlying this dynamic neuronal localization and distribution of Best1 should be explored in future studies.
Physiological roles of Best1 in the mouse brain
One of the most striking features of Best1 is its permeability to large anions and osmolytes in addition to chloride. In its initial characterization in HEK293 cells, hBest1 was shown to be highly permeable to both chloride (Sun et al., 2002) and bicarbonate (Qu and Hartzell, 2008). In cultured astrocytes, the Ca2+-activated anion channel was found to be encoded by mouse Best1 and was permeable to both glutamate and isethionate (Park et al., 2009). Permeability to these compounds as well as gluconate was also documented in astrocytes from hippocampal CA1 slices (Park et al., 2013). The precise permeability ratio between glutamate and chloride ion was estimated to be around 0.67 for heterologously expressed mBest1 (Woo et al., 2012), 0.53 for mBest1 expressed by hippocampal CA1 astrocytes, and 0.47 for cultured astrocytes. These ratios are fairly high considering the molecular size difference between chloride and glutamate. Glutamate-permeable Best1 was separately demonstrated to be permeable to GABA, which is normally present in zwitterionic form (Lee et al., 2010). The measured GABA permeability ratio of Best1 (0.19) was lower than that of glutamate, probably because most of the permeating GABA is in zwitterionic form and only a small portion is in an ionic form that carries the current (Lee et al., 2010).
The ability of Best1 to permeate large anions and osmolytes renders a unique function of releasing important transmitters, such as GABA and glutamate. Using the sniffer-patch technique, Best1 was shown to Ca2+-dependently and pore-dependently release GABA and glutamate from astrocytes upon activation of GPCR (Jo et al., 2014; Lee et al., 2010; Woo et al., 2012). This release also arose from any other stimulation that caused a rise in intracellular Ca2+. Even at resting intracellular Ca2+ concentrations, Best1 was capable of tonically releasing GABA. This is due to Best1’s half maximal effective concentration (EC50) for Ca2+ of 150 nM, which is slightly higher than resting Ca2+ concentration levels (Lee et al., 2010). In fact, it was demonstrated that cell-type specific gene silencing of Best1 by shRNA-carrying lentivirus significantly reduced the tonic inhibition current in cerebellar granule cells. This indicates that astrocytic Best1 is responsible for the tonic release of GABA that results in tonic inhibition of granule cell excitability in the cerebellum (Lee et al., 2010; Yoon et al., 2011). Unlike the cerebellum, hippocampal astrocytes do not contain GABA and do not release GABA under normal conditions (Jo et al., 2014; Yoon et al., 2011). Instead of GABA, hippocampal astrocytes release glutamate at the microdomains near the synaptic junctions where mBest1 is localized (Woo et al., 2012). The released glutamate targets post-synaptically localized NMDA receptors, enhances NMDA receptor-mediated current (Han et al., 2013; Woo et al., 2012), and contributes to enhanced synaptic plasticity by lowering the threshold for NMDA receptor-dependent long-term potentiation (Park et al., 2015).
In addition to GABA and glutamate, Best1 might be permeable to other transmitters such as D-serine. D-serine is a well known co-agonist of NMDA receptors that binds to the glycine binding site of the GluN1 subunit (Mothet et al., 2000). D-serine is converted from L-serine by serine racemase and is reportedly expressed mostly in astrocytes (Wolosker et al., 1999). It is possible that D-serine is synthesized in astrocytes and released through Best1. If this were the case, the two important endogenous agonists of NMDA receptors, both glutamate and D-serine, would be provided by astrocytes. Future work is needed to explore this exciting possibility.
Pathological roles of Best1 in the mouse brain
In a mouse model of Alzheimer’s disease, Best1 was shown to dynamically change its hippocampal distribution pattern from the synaptic distal microdomain to the soma and processes of reactive astrocytes (Jo et al., 2014). Reactive astrocytes accompany morphological changes as well as molecular changes in the hippocampus of Alzheimer’s disease mice or in brain samples of Alzheimer’s disease patients. Among these various changes, the most prominent molecular change was the phenotypical switch from GABA-lacking to GABA-containing astrocytes in the diseased dentate gyrus of hippocampus (Jo et al., 2014; Wu and Sun, 2015). These GABA-containing reactive astrocytes, acompanied by redistrution of Best1 to the soma and processes, began to release GABA tonically through Best1 and contributed to impaired synaptic transmission, synaptic plasticity, and spatial memory in Alzheimer’s disease by tonically inhibiting dentate granule celll excitability (Jo et al., 2014). Because reactive astrocytes appear in many neurodegenerative diseases and psychiatric disorders, it is possible that this proposed model (Fig. 8) is a general mechanism that occurs in various neurological diseases, such as Parkinson’s disease. These exciting possibilities call for future investigations.
Figure 8. Model diagram of memory impairment in Alzheimer’s disease.
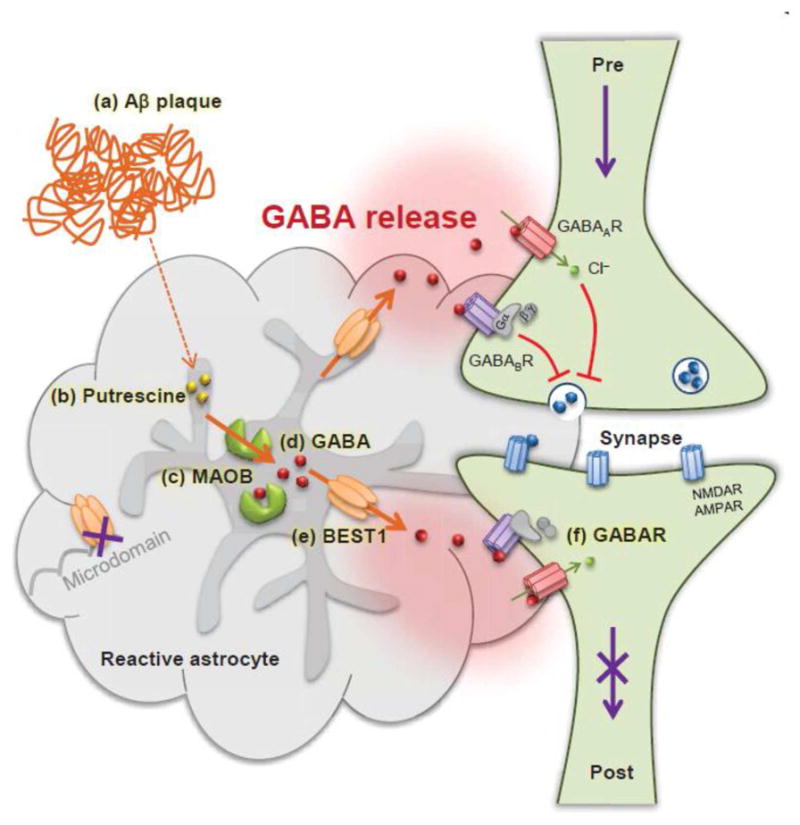
In Alzheimer’s disease, astrocytes near Amyloid β plaques (a) have more putrescine (b). Putrescine is degraded by MAOB (c) to produce the inhibitory neurotransmitter GABA (d). GABA is then abnormally released via BEST1 channels (e) which is redistributed away from microdomains. The released GABA binds to extrasynaptic GABAA and GABAB receptors (f) and strongly inhibits presynaptic release and spike probability. Consequently, granule cells of the dentate gyrus receive less glutamatergic inputs at perforant path synapses and show reduced synaptic plasticity. This finally leads to memory impairment in Alzheimer disease. Pre: presynaptic terminal, Post: postsynapse, NMDAR: N-methyl-D-aspartate receptor, AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazole propionate receptor.
Work from a few different studies has also demonstrated that Bestrophin plays a role in the dorsal root ganglia and spinal cord. Upregulation of Best1 expression and enhanced Ca2+-activated Cl− current in dorsal root ganglion neurons after peripheral nerve axotomy (Boudes et al., 2009) or spinal nerve ligation (Pineda-Farias et al., 2015) have suggested a function for Best1 in nociceptive processing. Another study reported that Best1 had a positive, supportive role in the regenerative process of mechanosensitive afferent fibers after peripheral nerve injury (Boudes and Scamps, 2012), suggesting a function for Bestrophin in the regeneration of injured sensory neurons as well as the maintenance of neuropathic pain. These studies indicate that this anion channel may be a useful target for treating neuropathic pain in a clinical setting.
BEST1 was first identified as the gene causing BVMD (Marquardt et al., 1998; Petrukhin et al., 1998). Subsequently, a great many disease-causing mutations have been reported (http://www-huge.uni-regensburg.de/BEST1_database/home.php?select_db=BEST1). However, there seems to be no systemic defects associated with BEST1 mutations outside of the eye. This could be, in part, due to the fact that the role of Best1 in the aberrant tonic GABA release, for example, would be observed only in pathological conditions. Based on these findings, one would expect to see rather a resistance to both tonic GABA release and memory impairment in people carrying mutations that cause eye diseases. In contrast, the physiological role of Best1 in tonic GABA release in cerebellum is expected to control motor coordination and learning. Due to its inhibitory nature, the tonic GABA would contribute negatively to motor coordination and learning. Therefore, it is possible that people with mutations in BEST1 would show improved motor coordination and motor learning. Future studies are needed to test these interesting possibilities in the cerebellum. If these theories are true, it would not be surprising that these predicted positive outcomes (e.g., resistance to tonic inhibition in potential Alzheimer’s disease patients and improved motor coordination and learning in the cerebellum) have gone unnoticed in people carrying BEST1 mutations.
In conclusion, the brain functions of bestrophin are beginning to unravel as the unique properties of the channel become further elucidated. Its ability to permeate large anions and osmolytes gives rise to its surprising role as a mediator of tonic release of various important transmitters, such as glutamate and GABA. These novel functions that are found in the brain may be applicable to the RPE and may help us understand how BEST1 mutations lead to retinal disease. The relevance is quite palpable, as receptors for both GABA (Cheng et al., 2015; Peterson and Miller, 1995) and glutamate (Miyamoto and Del Monte, 1994) have been reported in the RPE. Moreover, RPE have been reported to secrete glutamate (Harned et al., 2014) and the drug vigabatrin, which inhibits GABA transaminase, reversibly alters the EOG Arden ratio without affecting the ERG a-wave (Arndt et al., 1999).
Best1 Protein Structure and Topology
General architecture of a Best1 ion channel
Recently, the crystal structure for both eukaryotic chicken Best1 (Kane Dickson et al., 2014) and prokaryotic Klebsiella Best1 (Yang et al., 2014) was identified. Both the structures are homo-pentameric comprising a continuous central pore (Fig. 9). Each protomer has four transmembrane helices with both the N- and C-termini residing on the cytoplasmic side. The five protomers are symmetrically arranged around a central axis (Fig. 9A), forming a funnel-shaped transmembrane ion conduction pore (Fig. 9B). While chicken Best1 is an anion channel that robustly conducts Cl−, bacterial Best1 is a cation channels that conducts Na+. As such, the surface of the pore is negatively charged in Klebsiella Best1.
Figure 9. Best1 forms homo-pentameric anion channels.

A) Cartoon representation showing the pentameric arrangement of the subunits of the Best1 channel. B) A detailed surface representation of the Best1 channel from the top, showing the opening of the pore formed from five identical subunits.
Topologically, each protomer (Fig. 10A) of Best1 comprises of 4 transmembrane (TM) helices. On the extracellular side, TM1-TM2 helices are connected by a 12-residue loop and TM3-TM4 helices are connected by a short 3-residue loop. Towards the intracellular side, TM2 and TM3 are connected by a 5 alpha-helices comprising 105 residues. Towards the end, TM4 is connected to a C-terminal helix through a conserved carboxylate-rich loop (residues: EDDDDFE). One of the defining feature of the mammalian bestrophin channel family is the RFP (Arg-Phe-Pro) signal, which is conserved in all mammalian bestrophin channels (Hartzell et al., 2008). Bacterial Best1 counterparts do not have this signal and, instead, Klebsiella Best1 harbors the RIL (ArgIle-Leu) residues (Yang et al., 2014). While the purpose of this motif in the mammalian channel is not known, these residues are located on the outer perimeter of the intracellular domains.
Figure 10. The Best1 protomer and neck region of the pore.

A) Ribbon diagram of a Best1 protomer. B) Cartoon representation of the pentameric channel. View from top, showing the amino acids which form the first restriction site in the neck region of the pore. Ile 76 in black, Phe 80 in red, and Phe 84 in blue.
Ion pore
The pore is a continuous funnel-shaped vestibule penetrating midway into the membrane with no sidewise openings and a total length of ~95 Å (Fig. 10B and Fig. 11) (Kane Dickson et al., 2014; Yang et al., 2014). Entryway on the extracellular side is electronegative and hence repels most of the anions (especially divalent anions). The opening of the pore is ~20 Å across and ~12 Å into the pore it begins to narrow, forming the neck region (Fig. 11). The opening here is restricted to ~6 Å across and the first restriction is due to the hydrophobic amino acids Ile 76, Phe 80 and Phe 84 lining the neck in chicken Best1 (Fig. 11B). These residues are highly conserved in human Best1 and the equivalent residues in Klebsiella Best1 are Ile 62, Ile 66 and Phe 70. These hydrophobic amino acids lining the neck exclude both anions and cations and the selectivity for the passage of small anions but not cations comes from the phenylalanine residues lining the narrowest part of the neck. After this restriction, the pore opens into a larger inner cavity (Fig. 11) which is 45 Å long and 20 Å across. This inner cavity constitutes the bulk of the cytosolic portion of the channel and is highly positively charged. The purpose of the positively charged inner cavity is to attract anions from inside the cell. Towards the end, the pore again narrows down one more time due to restriction from Val 205 (chicken Best1), Ile 180 (Klebsiella Best1), or Ile 205 (human Best1). The purpose of this restriction lying below the inner cavity is to prevent the entry of bulkier anions which otherwise would block the pore. The hydrophobic gates, which cause restriction and selectivity, are conserved and are important for the function of many ion channels. The pores in these pentameric states are neither too small nor too large, allowing for easy access to both the closed and the open states, a hallmark of ion channels. The amino acid residues Phe 80 and Ile 205 are responsible for the two restrictions (one below the neck and the other below the inner cavity) in the Best1 channel. The importance of these is exemplified by the fact that the I205T BEST1 mutation has been reported to cause retinitis pigmentosa (Davidson et al., 2009). Recent electrophysiological studies performed by Vaisey et al have revealed specific regions that control both ion selectivity and calcium activation (Vaisey et al., 2016). Their experiments strongly demonstrate that the neck does not significantly affect ion selectivity but instead primarily serves to act as a gate to calcium-dependently control chloride permeation. Moreover, they find that the cytosolic aperture of the pore controls the relative permeability of the channel to different anions (Vaisey et al., 2016).
Figure 11. A cut-away view of Best1 showing the pore.

The outer entryway, neck, and inner cavity are the three major compartments that comprise the pore.
Although Best1 functions as a calcium-activated chloride channels, other studies have also shown that Best1 channels have a unique permeability to large anions and osmolytes such as GABA and glutamate (Han et al., 2013; Lee et al., 2010; Woo et al., 2012). This is a bit paradoxical based on the extant crystal structures for bacterial (Yang et al., 2014) and chicken (Kane Dickson et al., 2014) Best1. It is also paradoxical because ions in solution are hydrated and dehydration of ions costs energy. A dehydration step is the fundamental basis for ion selectivity and it is very well established in the case of K+ channels. Based on the crystal structure of chicken Best1 (Kane Dickson et al., 2014), the channel is quite permeable to other monovalent anions like Cl−, Br−, I−, SCN−, HCO3− and NO3−. NO3− is more permeable than Cl− and the Br−, I, and Cl− ions l fit very well into the gate of the ion pore. Although the anions SCN− and HCO3− are permeable, the fit is tight. Based on the existing crystal structure for a pentameric Best1 channel, there is no room for GABA or glutamate to fit or pass through the ion pore. The stoke radius of these osmolytes is substantially higher than that of Cl−.
While it is difficult to envision permeability to these anions with our current structural data, it is important to note that crystal structures generally provide a static image of channels and that Best1 was crystallized in the absence of membrane lipids, making the resultant crystal structure not truly physiological. Gating is the process by which the channels open and close and the selectivity filter would determine which ions and water molecules could pass. In the case of smaller molecules (like F−) and other than intended ions (like Cl−), the filter would collapse and the smaller ion would get trapped within the selectivity filter. In the case of larger ions or osmolytes (like GABA or glutamate), the filter would have to stretch quite a bit to allow the larger ions. It is not clear if this stretching is energetically feasible. In the case of Best1’s crystal structure, it is also not clear in which state the crystal structure was captured and it is possible that the visualized protein complex is one of an inactivated state. Hence, this may be why we see no room for passage of GABA or glutamate in the crystal structure data. Typically, anionic channels are less specific than cation channels. Therefore, with the existing structural data, it is difficult to shed light on the permeability of glutamate and GABA. More experimental data is required to further understand this apparent paradox. Cryo-EM structures of Best1 in the presence and absence of GABA or glutamate would help to probe the issue of large osmolyte permeability.
Calcium clasp
Each Best1 protomer (Fig. 10A) has a calcium-binding site, called the Ca2+ clasp (Fig. 12A) (Kane Dickson et al., 2014). The individual protomers (Fig. 10A) come together to form a pentameric channel (Fig. 9) and all five symmetrical Ca2+ clasps resemble a belt around the central section of the channel. This clasp is located within the intracellular part of the channel, close to the neck region (Fig. 12A). This neck region is hydrophobic in nature and hosts the first restriction into the pore. The proximity of the Ca2+ clasp to the restriction site of the neck region controls the closing and opening of the neck upon binding of calcium ions due to calcium induced conformational changes in the protein. Thus far, the crystal structure available has only captured the calcium-bound state and this calcium-binding site is completely buried by protein (Kane Dickson et al., 2014). Until we obtain the crystal structure of Best1 in a calcium-free state, it would be difficult to speculate on the calcium binding induced conformational changes that control the opening of the neck region. Work by Mladenova et al using Fourier transform infrared analyses suggests that binding of Ca2+ results in substantial conformational changes in Best1’s secondary structure, including changes in the molecular and macro-organization of Best1 in RPE monolayers (Mladenova et al., 2017).
Figure 12. Best1 pentameric channels contain a calcium clasp region.

A) The view of the calcium clasp site. Both calcium (red sphere) and the amino acid side chains (blue) involved in the coordination of Ca2+ are highlighted. The amino acids are Pro297, Glu 300, Asp 301–304. B) Left. The view of location of Trp 93. Trp is not part of the restriction of the pore. Right. Trp 93 is located closer to the Ca2+ clasp region.
Mutations
There is no clear information on the non-pore mutations associated with the bestrophinopathies. In the case of cardiac action potential disorders, there is a strong correlation between pore and non-pore mutations and different levels of risk for cardiac arrhythmias. Exemplar of this is the KCNH2 subunit, in which the non-pore mutation is in the transmembrane region which results in impaired trafficking (Liu et al., 2013). A corresponding BEST1 mutation may be W93C, where the tryptophan at amino acid 93 is not part of the pore restriction site and does not interact directly with Ca2+. This residue is close to the Ca2+ clasp region (Fig. 12B). It is therefore possible that the mutation of Trp 93 would have an effect on the local maintenance of [Ca2+]i and hence affect activation of the channel. This is supported by a variety of studies indicating that the W93C mutation abrogates channel activity (Marmorstein et al., 2015) and affects calcium homeostasis (Marmorstein et al., 2015; Rosenthal et al., 2006; Zhang et al., 2010). We currently don’t have information on the calcium binding induced conformational states. It would be of interest to learn if these calcium changes are local and if there are any long-range structural changes. It would also be of interest to learn if any of these changes affect the geometry of the pore and in turn affect anion conduction.
It is evident from the crystal structure that there are at least three important regions that have functional implications: 1) First restriction site in the neck region (amino acids Ile 76, Phe 80, Phe 84); 2) Calcium clasp site (amino acids Pro 297, Glu 300, Asp 301, Asp 302, Asp 303, Asp 304, Ala 10 and Gln 293); 3) Second restriction site towards the bottom of the pore (Val 205 in chicken; Ile 205 in human). Although disease-causing BEST1 mutations exist throughout the entire Best1 protomer (http://www-huge.uni-regensburg.de/BEST1_database/home.php?select_db=BEST1), many mutations associated with the bestrophinopathies are prevalent in or around the first restriction site and the Ca2+ clasp site. A fourth critical region may be the cytosolic aperture of the pore, which significantly affects relative permeabilities among anions (Vaisey et al., 2016).
Pathogenesis of the Bestrophinopathies
Protein mistrafficking
Despite the discovery of the gene BEST1 in 1998 (Marquardt et al., 1998; Petrukhin et al., 1998), exactly how BEST1 mutations lead to retinal degeneration remains unknown. More enigmatic is the question of why different BEST1 mutations are capable of causing clinically distinct retinopathies.
As evinced by numerous studies in native systems from various laboratories (Brandl et al., 2014; Gouras et al., 2009; Marmorstein et al., 2000), Best1 is localized to the basolateral plasma membrane of the RPE (Fig. 6). Given that defects in protein trafficking are known to underlie other channelopathies (Jentsch et al., 2005; Pedemonte and Galietta, 2012), one viable hypothesis is that some mutations disrupt Best1’s trafficking to the plasma membrane. Work from our laboratory as well as others support this hypothesis, demonstrating that numerous mutations associated with BVMD, AVMD, and ARB cause Best1 to accumulate in intracellular compartments (Davidson et al., 2011; Davidson et al., 2009; Johnson et al., 2014; Johnson et al., 2013; Milenkovic et al., 2011b). In MDCK cells, the majority of BVMD and ARB mutants tested were found to be localized in intracellular compartments, though a significant number still exhibited basolateral plasma membrane localization (Davidson et al., 2011; Davidson et al., 2009; Johnson et al., 2014; Johnson et al., 2013; Milenkovic et al., 2011b). In our hands, tested RP and ADVIRC mutants were properly localized when expressed in confluent MDCK cells via adenovirus-mediated gene transfer (Johnson et al., 2014). Work from a separate laboratory has reported that two tested RP mutants failed to integrate into the plasma membrane when transfected into MDCK cells (Davidson et al., 2009). The latter portion of the cytoplasmic C-terminal domain is unnecessary for proper trafficking, as the truncated ARB mutants L472PfsX10 and H490QfsX24 are properly localized in MDCK cells (Johnson et al., 2014). Mutations scattered throughout the entire Best1 protein are capable of inducing mislocalization in MDCK cells, including mutations in the short intracellular N-terminus (e.g., T6P, V9M), the cytoplasmic loop between transmembrane domains two and three (e.g., R92S, P101T, P152A, L174Qfs*57, R200X, L224M), transmembrane domain three (e.g., T237R), and the early portion of the cytoplasmic C-terminal domain (e.g., F305S, V311G, D312N, V317M, M325T).
Mutant mistrafficking of Best1 has thus far been demonstrated predominantly through heterologous expression in MDCK II cells (Davidson et al., 2011; Davidson et al., 2009; Johnson et al., 2014; Johnson et al., 2013; Milenkovic et al., 2011b). Although numerous mutants have been found to exhibit mistrafficking compared to WT Best1 in MDCK cells, some discrepancies in Best1 localization have been observed between MDCK and RPE cells. For example, the ARB mutant Best1R141H is mislocalized when expressed in confluent MDCK cells via adenovirus-mediated gene transfer (Johnson et al., 2014). When co-expressed with Best1 in MDCK cells, both WT and mutant Best1 co-localize together predominantly in intracellular compartments (Johnson et al., 2014). In contrast, Best1R141H is properly localized to the plasma membrane when expressed via adenovirus-mediated gene transfer in confluent, iPSC-RPE cells (Johnson et al., 2015). That Best1R141H is properly localized in the presence of endogenous Best1 in iPSC-RPE cells yet mislocalized in the presence of Best1 in heterologous MDCK cells would suggest that trafficking results in MDCK cells should be validated in a native model (i.e., iPSC-RPE, fhRPE). One possibility for these discrepancies is that mislocalization due to overexpression in heterologous systems is a common problem known to befuddle trafficking studies (Lisenbee et al., 2003).
A further discrepancy comes from the BVMD mutant Best1W93C, which is mislocalized in MDCK cells (Johnson et al., 2013) yet properly localized in fhRPE cells (Johnson et al., 2013; Marmorstein et al., 2015) as well as in RPE in a rat eye (Marmorstein et al., 2004). While MDCK cells have been a useful model for illustrating that Best1 mutants traffic differently compared to each other and WT Best1, trafficking studies should be additionally performed or validated in a native RPE system. To date, only a few laboratories have assessed Best1 mutant trafficking in RPE. We previously showed that the BVMD associated mutant V9M is mislocalized in fhRPE cells as well as in RPE in the mouse eye (Johnson et al., 2013). Best1Q238R, another mutant associated with BVMD, was similarly reported to be mislocalized in iPSC-RPE (Milenkovic et al., 2015). A recent study by Carter et al analyzed Best1 localization in iPSC-RPE derived from a patient with ADVIRC and the associated BEST1 mutation V235A (Carter et al., 2016).
While Best1V235A was found to be properly localized in MDCK cells (Johnson et al., 2014), Best1 was found to be mislocalized in these ADVIRC iPSC-RPE (Carter et al., 2016). This is further evidence that future trafficking studies should strive to analyze Best1 localization in a human RPE model (e.g., iPSC-RPE, fhRPE, or RPE in situ). Work by Mullins et al has also shown that postmortem, BVMD eyes harboring the mutations Y227N or T6R show an anomalous immunofluorescent localization in the RPE (Mullins et al., 2007; Mullins et al., 2005).
With the exception of Best1T6R, Best1V9M, Best1Y227N, Best1V235A, and Best1Q238R, all other Best1 mutants tested have been found to be properly localized in native system like iPSC-RPE (Johnson et al., 2015; Singh et al., 2013), fhRPE (Johnson et al., 2013; Marmorstein et al., 2015), or RPE in the rat eye (Marmorstein et al., 2004). These include the BVMD mutants Best1A146K, Best1N296H (Singh et al., 2013), Best1A243V (Milenkovic et al., 2015), Best1W93C, Best1R218C (Johnson et al., 2013; Marmorstein et al., 2004) as well as the ARB mutants Best1R141H and Best1I366fsX18 (Johnson et al., 2015). To date, only mutants associated with BMVD, ADVIRC, and ARB have been assessed for localization in an RPE system. Further studies are warranted to assess trafficking of mutants associated with RP.
Presently, there is no obvious correlation between trafficking and disease phenotype. While numerous BVMD, AVMD, and ARB mutants have been found to be mislocalized in MDCK II cells, several others traffic properly to the basolateral plasma membrane (Davidson et al., 2011; Johnson et al., 2014; Johnson et al., 2013; Milenkovic et al., 2011b). Furthermore, the BVMD mutant W93C is mislocalized on its own in MDCK II cells but is properly localized in the presence of WT Best1 in both fhRPE and RPE in the rat eye (Johnson et al., 2013; Marmorstein et al., 2004). Despite this, patients homozygous or heterozygous for W93C exhibit an identical disease phenotype (Bakall et al., 2007). Prior mapping of the trafficking status of tested Best1 mutants on a topology model of Best1 reveals no coherent correlation between localization status, associated disease, and location in a Best1 monomer (Johnson et al., 2014). Since most trafficking data to date has been obtained in heterologous MDCK cells, however, additional evidence in native RPE cells is required to better determine the contribution of mislocalization to each bestrophinopathy.
Defects in Best1 Oligomerization
As indicated above, the crystal structure for Best1 has demonstrated that the channel is formed as a homo-pentamer with an ion conductance pathway at its center (Kane Dickson et al., 2014; Yang et al., 2014). It is therefore possible that some BEST1 mutations cause disease by disrupting oligomerization and preventing proper channel formation. We previously theorized that, because ARB mutations are recessive, ARB mutants would be unable to interact with WT Best1 or with each other. This would explain why ARB mutations are benign in heterozygous patients but cause a retinopathy in patients with homozygous or compound heterozygous BEST1 mutations. To test this theory, we hitherto studied co-immunoprecipitation, live-cell FRET acceptor photobleaching, and/or confocal co-localization of 32 disease-causing mutants associated with ADVIRC, AVMD, ARB, RP, and BVMD (Johnson et al., 2015; Johnson et al., 2014; Johnson et al., 2013). We found that every single mutant tested was capable of physically interacting with WT Best1, suggesting that an inability to oligomerize is not a pathogenic component of any of these diseases. Earlier work by Sun et al. corroborates these findings by demonstrating that several disease-causing mutants are capable of co-immunoprecipitation with WT Best1 (Sun et al., 2002). Even severely truncated mutants– such as Best1174Qfs*57 and Best1R200X – are capable of interacting with WT Best1, indicating that the latter portion of Best1 is dispensable with regard to physical interaction (Johnson et al., 2014). While the bulk of this work was done in MDCK II cells (Johnson et al., 2014; Johnson et al., 2013), we recently demonstrated that the ARB mutants Best1R141H and Best1I366fsx18 are both capable of interacting with endogenous Best1 in iPSC-RPE cells. In addition to co-immunoprecipitating with both endogenous and overexpressed WT Best1, both Best1R141H and Best1I366fsX18 are capable of physically interacting with each other (Johnson et al., 2015). These data stemming from a patient with ARB (Johnson et al., 2015) suggest that ARB mutants are capable of physical interaction physiologically. One unexplored possibility is that specific mutations result in wrongly numbered oligomers besides the correct pentameric structure formed by WT Best1. Crystal structrues of Best1 disease-causing mutants would be extremely valuable for helping to understand how mutations disrupt pentameric formation in disease.
In 2002 we reported that Best1 can oligomerize and functionally interact with protein phosphatase 2A (PP2A) (Marmorstein et al., 2002). Interaction with PP2A was demonstrated in both porcine and human RPE via reciprocal co-immunoprecipitation in these native systems. This interaction was mediated through the C-terminal cytoplasmic domain of Best1. In the same manuscript, we showed that Best1 was phosphorylated when expressed in RPE-J cells and that purified PP2A was capable of deposphorylating Best1 in vitro (Marmorstein et al., 2002). Work from the Hartzell laboratory has also provided evidence of Best1 phosphorylation, finding that Best1 possesses a protein kinase C phosphorylation site at serine 358 and that Best1 channel activity is maintained by both protein kinase C activators and protein phosphatase inhibitors (Xiao et al., 2009). One interesting possibility is that some disease-causing mutations uniquely affect interaction of Best1 and PP2A, resulting in dysregulation of Best1’s channel function. Whether or not mutations associated with one bestrophinopathy or another differentially affect Best1’s phosphorylation-induced regulation remains unknown.
Anion channel activity
A number of studies have shown that, in transfected HEK293 cells, anion currents of Best1 mutants associated with BVMD, AVMD, ARB, and RP are severely attenuated (Hartzell et al., 2008; Xiao et al., 2010). To date, no published manuscripts have examined the effects of ADVIRC mutations on Best1 currents. Recent efforts from various laboratories have expanded upon the in vitro data and have shown that the removal or mutation of BEST1 affects anion currents both in vivo and in native systems. Within the mouse brain, selective silencing of Best1 in glial cells blocks GABA release and eliminates tonic inhibition (Lee et al., 2010). Similarly, the slow mode of glutamate release was reportedly eliminated in cultured astrocytes from Best1−/− mice (Woo et al., 2012). Unlike the brain, no group has documented any defects on channel activity in mouse RPE resulting from the absence or mutation of Best1. Chloride currents were previously found to be normal in RPE from Best1−/− mice (Marmorstein et al., 2006) as well as in mice homozygous or heterozygous for the BVMD mutation W93C (Zhang et al., 2010).
Work in human RPE has been more successful in this regard. We previously showed that overexpression of Best1W93C via adenovirus-mediated gene transfer in fhRPE notably reduces transepithelial potential and short-circuit current (Marmorstein et al., 2015). Substituting Cl− in the bath media with gluconate significantly reduced short-circuit current for monolayers overexpressing Best1, but had no effect on monolayers expressing Best1W93C (Marmorstein et al., 2015). These data strongly indicate that, in a human RPE model expressing endogenous Best1, a disease-causing mutant can severely disrupt anion currents and transepithelial electrical properties. Recent work by Milenkovic et al. has found that volume-regulated chloride currents are severely reduced in iPSC-RPE derived from patients with the BVMD mutations A243V or Q238R (Milenkovic et al., 2015). This work substantiates a significant body of literature demonstrating that Drosophila Bestrophin-1 is regulated by volume and functions as a swell-activated anion channel (Chien and Hartzell, 2007, 2008; Duran et al., 2013; Fischmeister and Hartzell, 2005; Stotz and Clapham, 2012). It is also supported by our study in fhRPE, where overexpression of WT Best1 appeared to cause cell shrinkage in response to ATP stimulation (Marmorstein et al., 2015). iPSC-RPE derived from patients with BVMD were similarly found to have abrogated calcium-induced chloride export, as measured by a chloride biosensor (Moshfegh et al., 2016).
While it is clear that BEST1 mutations perturb the channel activity of Best1 in the RPE, anomalous anion currents have only been reported in association with BVMD mutations. Further studies are warranted to elucidate how mutations associated with the remaining bestrophinopathies disrupt Best1’s function as an anion channel. For example, Best1 is known to be permeable to a wide array of anions beyond chloride, such as bicarbonate (Qu and Hartzell, 2008) and glutamate (Woo et al., 2012). Whether or not different disease-causing BEST1 mutations more obviously affect the transport of other anions besides chloride in RPE cells would be of value to know. Relevant to this, work by Vaisey et al indicates that mutations in the cytosolic aperture of the pore significantly affect Best1’s relative permeabilities among anions.
Intracellular calcium signaling
In addition to functioning as an anion channel, Best1 also serves as a regulator of intracellular Ca2+ signaling and Ca2+ homeostasis. Interestingly, the only recorded RPE phenotype in Best1−/− mice is anomalous intracellular Ca2+ levels following stimulation with ATP (Marmorstein et al., 2006; Zhang et al., 2010). More specifically, [Ca2+]i in RPE from Best1−/− mice was significantly greater than [Ca2+]i in RPE from their Best1+/+ littermates following stimulation with extracellular ATP (Marmorstein et al., 2006).
Conversely, RPE from mice homozygous or heterozygous for W93C exhibited suppressed [Ca2+]i levels compared to their Best1+/+ littermates (Zhang et al., 2010). These data demonstrate that, in murine RPE, Best1 serves to suppress intracellular Ca2+ levels post-ATP stimulation. Compared to control monolayers, fhRPE overexpressing Best1W93C displayed notably reduced [Ca2+]i following application with ATP (Marmorstein et al., 2015). In contrast, iPSC-RPE harboring the BVMD mutations A146K or N296H showed a significant increase in ATP-induced Ca2+ release (Singh et al., 2013). The differential responses observed between Best1W93C and the mutants Best1A146K and Best1N296H reveal that each mutant likely affects Ca2+ signaling differently. Work in RPE models from the laboratory of Olaf Strauss suggests that, independent of ATP stimulation, Best1 functions to conduct counterions for Ca2+, thereby helping to accumulate and release Ca2+ from stores (Gomez et al., 2013; Neussert et al., 2010; Strauss et al., 2014). An interesting, unanswered question is whether or not Best1’s effects on calcium are downstream of its anion channel activity or if they are a wholly independent function.
In vitro data strongly suggest that Best1 can alter the kinetics of voltage dependent calcium channels (Milenkovic et al., 2011a; Reichhart et al., 2010; Rosenthal et al., 2006; Yu et al., 2008). Furthermore, Best1 physically and functionally interacts with voltage-dependent calcium channels and this interaction is mediated by Best1’s large, intracellular C-terminal domain (Milenkovic et al., 2011a; Reichhart et al., 2010; Rosenthal et al., 2006; Yu et al., 2008). In particular, Best1 has been shown to physically interact with the β3 and β4 subunits of L-type voltage-dependent calcium channels (Milenkovic et al., 2011a). Work from the laboratory of Olaf Strauss suggests that the trafficking of Best1 to the plasma membrane is facilitated by its interaction with β4 subunits (Milenkovic et al., 2011a; Reichhart et al., 2010). We have shown previously that mice lacking the β4 subunit have a DC-ERG light peak that is diminished and similar to what is seen in Best1W93C knock-in mice (Marmorstein et al., 2006; Zhang et al., 2010). Studies seeking to expand on these data in native RPE systems are warranted. Unique changes in calcium homeostasis may selectively underlie and help to distinguish between the bestrophinopathies. Another possibility is that some mutations uniquely affect Best1’s ability to physically and functionally interact with voltage-dependent calcium channel subunits.
Animal Models of the Bestrophinopathies
Canine model of BEST1-associated maculopathies
The natural canine model of BEST1-associated maculopathies, canine multifocal retinopathy (cmr) a.k.a. canine bestrophinopathy, was found to recapitulate a full spectrum of clinical, molecular, and histological features characteristic to its human disease counterpart (Figs. 13–15). This retinal disorder in the dog is caused by one of three distinct mutations in the canine BEST1 ortholog (R25X, G161D, or P463Pfs) spontaneously occurring in 11 dog breeds worldwide, all inherited in an autosomal recessive fashion (Guziewicz et al., 2007; Hoffmann et al., 2012; Zangerl et al., 2010).
Figure 13. Clinical manifestation of canine bestrophinopathy.

(A) Fundus photograph of a 23-week-old cBest1-R25X-affected dog with focal canine macular lesion in vitelliform stage (arrowhead). (B) A 36-week-old cBest1-R25X/P463fs-affected dog exhibiting early stage lesions associated with both canine fovea-like region (arrowhead) and aligned along visual streak. (C) Multifocal presentation of canine bestrophinopathy in 40-week-old dog harboring cBest1-R25X/P463fs compound heterozygous mutation. Arrowhead indicates lesion in fovea-like region. OD: right eye; OS: left eye; cBest1: canine bestrophin 1.
Figure 15. Immunohistochemical evaluation of an early lesion in the canine model of Best1-associated maculopathies.

A vitelliform lesion of a 112-week-old cBest1-R25X-affected dog immunolabeled with anti-cone arrestin (hCAR, red). Note the massive autofluorescent deposits within RPE cell monolayer and in the subretinal space (green and yellowish-green: native autofluorescence). RPE: retinal pigment epithelium; PRs: photoreceptors. ONL: outer nuclear layer; INL: inner nuclear layer. Scale bar 40 μm.
Although parallel to human BEST1-related disorders, canine bestrophinopathy presents considerable heterogeneity in its clinical manifestation. Its phenotypic spectrum always involves a canine macular component (a.k.a. area centralis) (Figs. 13 and 14) (Beltran et al., 2014). Canine bestrophinopathy arises as a focal detachment between the RPE and the neural retina in the area centralis (Beltran et al., 2014) and can stay limited to the canine fovea-like region (Fig. 13A and Fig. 14) or can develop extramacular satellite lesions (Fig. 13B, C), similar to a subset of BVMD and ARB patients (Boon et al., 2013; Lacassagne et al., 2011; Pineiro-Gallego et al., 2011). The factors underlying this variability in the clinical presentation of canine bestrophinopathy remain unclear and are under active investigation.
Figure 14. Progression of unifocal disease in canine bestrophinopathy.

(A) cSLO/SD-OCT images of central lesion in previtelliform stage (OD) in a 15-week-old cBest1-R25X-affected dog. Note the subtle dissociation of the neural retina from the RPE on the OCT scan (arrowhead). (B) En face infrared view of representative cBEST1-C73T/R25X mutant dog. (*) indicates canine fovea-like area in OS; black arrows signify locations of cross-sectional OCT scans shown in the below panel. Outer photoreceptor nuclear layer (ONL) and retinal pigment epithelium (RPE) are highlighted for visibility on OCT scans. (C) Topographic localization of the sites (*) of the early macular lesions in cBest1-mutant dogs (ages: 10–62 weeks; n = 7, left) correspond to the localization of the fovea-like area in wildtype dogs (ages: 7 weeks – 8 years; n = 13, right). (D) A 17-week-old cBest1-R25X-affected dog with a classic circular vitelliform lesion (OD) resembling Stage II of Best Vitelliform Macular Dystrophy (BVMD). OD: right eye; OS: left eye; NIR - near infrared reflectance. Images B & C taken from Beltran et al, 2014 (doi:10.1371/journal.pone.0090390.g002).
The typical canine bestrophinopathy presents bilaterally, has an early onset (usually between 12–25 weeks of age), and progresses slowly following the well-defined clinical stages described in BVMD (Fig. 14) (Mohler and Fine, 1981). While Stage 0 is characterized by a normal fundus appearance in both BVMD patients and canine bestrophinopathy-affected dogs, Stage I, pre-vitelliform shows a discreet disruption between the RPE and the neural retina in the canine macular region (Fig. 14A). Although this is not noticeable on a routine eye exam, this can be detected by in vivo imaging. Stage II is the vitelliform lesion stage and is characterized by a circular, well-demarcated, yolk-like macular lesion that is highly comparable between affected human patients and dogs (Fig. 14B). The subsequent disease stages (Stage III - known as pseudohypopyon phase, Stage IV- marked by a scrambled egg appearance, and Stage V - atrophic) also occur, yet tend to develop slowly and are currently being characterized. Immunohistochemical evaluation of vitelliform lesions from the R25X-mutant dog revealed extensive accumulation of autofluorescent material that occupied RPE cells as well as the subretinal space (Fig. 15). The RPE monolayer was otherwise intact and there was no indication of photoreceptor cell loss in this early clinical stage. These findings are consistent with the histopathological reports on BVMD donor eyes (Bakall et al., 2007; Mullins et al., 2007).
In summary, canine bestrophinopathy recapitulates all fundamental aspects of human bestrophinopathies, including involvement of the canine macula in the disease phenotype, disease onset and course, and molecular consequences of BEST1 mutations (Guziewicz et al., 2012; Guziewicz et al., 2011). These factors as well as the salient predilection of lesions to the canine macular region make canine bestrophinopathy an extremely attractive model system for the bestrophinopathies. The canine model is very suitable for studies aiming to understand the pathogenesis of the bestrophinopathies. Because BEST1-associated maculopathies are autosomal recessive in dog, this model is also well suited for the development of novel therapeutics such as gene replacement therapy (Guziewicz et al., 2013). Proof-of concept studies using AAV-mediated BEST1 gene therapy are currently in progress.
Mouse models
Both Best1 knock-out and knock-in mice have been produced. The first Best1−/− knockout mouse was generated by our laboratory and published in 2006 (Marmorstein et al., 2006). A second knock-out mouse was recently published by Milenkovic et al in 2015 (Milenkovic et al., 2015). Although there are significant differences between the knock-out mice, neither exhibits a phenotype reminiscent of a bestrophinopathy.
Given that ARB has been theorized to be associated with a null phenotype for Best1 (Burgess et al., 2008; Pomares et al., 2012), the Best1 knockout model could be viewed as a theoretical model for some types of ARB. However, the lack of a disease phenotype in the Best1−/− mouse (Marmorstein et al., 2006; Milenkovic et al., 2015) has caused some question regarding both the utility of mice as bestrophinopathy models as well as whether ARB is a true “null” phenotype in man. Retinal health was completely normal in the original Best1 knockout mouse and, electrophysiologically, the only anomalous phenotype was an enhancement of light peak luminance responsiveness (Marmorstein et al., 2006). Interestingly, no differences in chloride currents were found between Best1 knockout and WT mice. The only phenotype observed relevant to ion transport was specific to calcium. When stimulated with ATP, RPE from Best1−/− mice showed a fivefold greater increase in [Ca2+]i compared to their Best1+/+ littermates (Marmorstein et al., 2006).
In contrast to Best1−/− mice, the knock-in mouse model does exhibit a phenotype similar to BVMD. This phenotype includes a dominant inheritance and an incomplete penetrance (Zhang et al., 2010). As shown in Figure 16, the Best1 knock-in mouse develops a serous retinal detachment that is visible in the fundus. Compared to a healthy WT mouse (Fig. 16A), the fundus of the BVMD knock-in mouse is notably anomalous (Fig. 16B). Histopathology also confirms and highlights this serous retinal detachment (Fig. 16C) as well as an increased accumulation of lipofuscin granules (Fig. 16D) and unphagocytosed photoreceptor outer segments (Fig. 16D). While a- and b-waves were normal in the ERG, the light peak luminance response was diminished. Moreover, RPE from Best1W93C/W93C and Best1+/W93C mice showed suppressed calcium levels post-ATP stimulation compared to their Best1+/+ littermates (Zhang et al., 2010). This is the opposite of the response observed in Best1−/− mice. Like the Best1 knockout mice, chloride currents were strangely found to be normal in RPE from knock-in mice harboring the W93C mutation. It remains to be explored whether Best1’s effects on calcium levels in these mouse models are due to changes in calcium stores and/or voltage-dependent calcium channels.
Figure 16. Comparison of Best1+/+ and Best1 knock-in mice.

Fundus from a wild type mouse (A) and a Best1 knock-in mouse harboring the disease-causing mutation W93C (B). The dotted line highlights the anomalous portion of the fundus in the Best1 knock-in mouse (B). Panel C shows an electron micrograph of a chronic serous detachment of the retina (*) in a Best1 knock-in mouse harboring the disease-causing mutation W93C. Note the RPE microvilli lying horizontally (mv). Photoreceptor outer segments show damage and internal debris (arrows). Panel D shows that, in some regions, unphagocytosed photoreceptor outer segments (arrowheads) are observed “sitting” atop RPE microvilli. Note the extensive accumulation of lipofuscin granules in both C and D.
Taken all together, these data demonstrate that Best1 knock-in mice are a good model of BVMD and support the hypothesis that Best1 plays an important role in regulating calcium levels in the RPE.
Rat model of Best vitelliform macular dystrophy
In 2004, our laboratory created a transient model of BVMD in rats by overexpressing the common BVMD mutants Best1W93C or Best1R218C in RPE via injection of replication-defective adenoviruses (Marmorstein et al., 2004). We found that overexpressed WT, W93C, and R218C Best1 all localized to the basolateral plasma membrane of rat RPE. ERG recordings demonstrated that neither mutant nor WT Best1 impacted the a- or b-waves in live, post-injected rats. While overexpression of WT Best1 increased both fast oscillation and the c-wave, overexpression of mutant Best1 reduced the amplitude of the light peak. Best1W93C significantly altered the light peak response function while Best1R218C had no effect on this parameter. Overexpressed WT Best1 led to an overall desensitization of the luminance response function (Marmorstein et al., 2004). In addition to recapitulating some disease characteristics in humans and validating itself as a transient model, these data suggest that localization is a non-issue for these two BVMD mutants. They also indicate that, in an otherwise healthy animal with endogenous WT bestrophin, overexpression of mutant Best1 is sufficient to disrupt the ERG and therefore impact retinal health.
Prospective Therapies
Drug treatment
At the moment, no concrete therapies or treatments exist for a patient suffering from any bestrophinopathy. Contemporary discoveries and fresh avenues of research, however, have created some excitingly tangible clinical possibilities. A recent study from the Forbes Manson laboratory reported that four ARB mutants (L41P, R141H, R202W, M325T) were mislocalized to the cytoplasm, underwent proteasomal degradation, and failed to conduct Cl− anions in transfected HEK293 cells (Uggenti et al., 2016). The authors found that treatment with proteasome inhibitor4-phenylbutyrate restored the chloride conductances of these mutant proteins and that a combination treatment with 4-phenylbutyrate and a second proteasome inhibitor, bortezomib, rescued their localization back to the basolateral plasma membrane. Both bortezomib and 4-phenylbutyrate are clinically approved for long-term use. Thus, this may be a viable therapy for ARB patients. Since these studies were performed in MDCK II cells however, subsequent confirmation in iPSC-RPE or fhRPE, is warranted to determine if this rescue effect also occurs in a human model.
The laboratory of David Gamm has also explored small molecule drugs in an iPSC-RPE model of a bestrophinopathy. Using iPSC-RPE derived from patients with BVMD, Singh et al demonstrated that RPE lines harboring BVMD mutations displayed reduced rates of photoreceptor outer segment breakdown (Singh et al., 2013). In a follow-up study, Singh et al discovered that these BVMD cultures concomitantly exhibited anomalous rates of exosome secretion, higher protein oxidation, and decreased levels of free-ubiquitin (Singh et al., 2015). They discovered that treatment with valproic acid alone or in combination with rapamycin was able to increase the rate of photoreceptor outer segment degradation in BVMD iPSC-RPE (Singh et al., 2015). Treatment with the drug bafilomycin-A1 decreased photoreceptor outer segment degradation rates and this decrease was completely reversible by valproic acid (Singh et al., 2015). Since anomalies in photoreceptor outer segments are implicated in the pathogenesis of the bestrophinopathies (Abramoff et al., 2013), these data suggest that valproic acid may be or similar compounds may be of use in the treatment of BVMD. A large array of other drug candidates remain to be explored (Edelhauser et al., 2010; Johnson and Riehle, 2015).
Gene therapy
Given the recessive nature of ARB and that one WT copy of BEST1 is sufficient to prevent an ARB mutation from causing disease, ARB is an excellent prospect for gene therapy. Several gene therapy trials in the retina are currently underway and, for some diseases like RPE65-Leber congenital amaurosis, gene therapy has been shown to be highly efficacious (Boye et al., 2013; Cideciyan et al., 2008; Cideciyan et al., 2009a; Cideciyan et al., 2009b). Administration of gene therapy to provide functional RPE65 to patients has been consistently shown to be clinically effective, devoid of serious complications, and clinically safe (Boye et al., 2013). Bolstering support for the idea that gene therapy could be successful for ARB like it has been for Leber congenital amaurosis, work by Guziewicz et al. has shown that recombinant adeno-associated virus-mediated gene transfer of BEST1 is safe and feasible in a canine model (Guziewicz et al., 2013).
Stem cell-based RPE transplants
The advent of iPSC technology (Takahashi and Yamanaka, 2006) brings the exciting prospect that diseased or damaged tissue could be replaced with healthy, autologous tissue free of immune rejection issues. While initially there were concerns that iPSCs may be different than embryonic stem cells and less suitable for transplantation (Rohani et al., 2014), more recent work has demonstrated that iPSCs and embryonic stem cells are highly analogous if not completely identical (Shutova et al., 2016). Autologous stem cell transplantation is commonly performed with mesenchymal stem cells and numerous strategies have been devised to enhance their therapeutic efficacy. For example, pre-incubating stem cells with certain compounds can increase their genetic stability (Johnson et al., 2016) while rearing stem cells under specific conditions, such as hypoxia (Naaldijk et al., 2015), can enhance their transplantation efficiency.
With regards to RPE-specific transplants, iPSC-RPE cell sheets have already been optimized to meet strict clinical use requirements. When generated as a monolayer without any artificial scaffolds, autologous transplantation of iPSC-RPE sheets into cynomolgus monkeys was performed safely without any issues of tumor formation or immune rejection (Kamao et al., 2014). Transplantation of iPSC-RPE into Royal College of Surgeons dystrophic rats, a rat model of inherited retinal degeneration, was sufficient to restore their ERG responses and increase the outer nuclear thickness of their retinas (Kamao et al., 2014). In MHC-matched monkey models, transplantation of iPSC-RPE allografts has also been performed safely with no signs of rejection (Sugita et al., 2016). A clinical trial involving an autologous transplantation of iPSC-RPE has begun in Japan and is being performed by the RIKEN Institute (Kimbrel and Lanza, 2015). In this trial, a 70-year old female patient with exudative age-related macular degeneration became the historic first human recipient of iPSC-derived cells. After one year the patient has exhibited no negative effects.
Given that iPSC-RPE transplantation is likely to be deemed safe, this therapeutic approach is an appealing one for the bestrophinopathies. Replacing damaged or dysfunctional RPE with healthy RPE could alleviate or entirely cure BVMD, AVMD, ARB, ADVIRC, and RP. For autologous transplants, the primary concern becomes that the generated iPSC-RPE would harbor the same disease-causing mutation as the patient they were derived from. To rectify this, the BEST1 mutation or BEST1 mutations in iPSC-RPE cells could be fixed with gene editing technology such as CRISPR/Cas9 (Xue et al., 2016) or TALENs (Joung and Sander, 2013). Following genetic modification, healthy, patient-derived iPSC-RPE could then be transplanted and used to replace or supplement unhealthy RPE. We feel that this therapeutic path holds significant promise and should be strongly pursued.
Concluding Remarks
The bestrophinopathies are a diverse spectrum of retinal diseases caused by mutations in the gene BEST1. Since the discovery of the BEST1 gene in 1998, research from many laboratories has generated an impressive amount of information about the protein Best1 and its associated retinopathies. Although we still do not understand how different mutations lead to clinically distinct diseases, we now know that Best1 is a pentameric channel protein with defined roles in mediating anion transport and regulating calcium signaling in human RPE. While the anion permeability of retinal Best1 as well as the mechanistic details of how Best1 impacts calcium signaling both require further elucidation, we are optimistic that such details will be unveiled given the current research trajectory. More excitingly, the use of “disease in a dish” models developed around patient specific iPSC-RPE cells should permit rapid advances in our understanding of disease pathogenesis and provide screening tools for novel pharmaceuticals, gene therapy, and regenerative medicine based therapies. All are being actively pursued as viable therapeutics for patients suffering from the bestrophinopathies. Future studies are strongly warranted to develop these therapies and bring them to the patients who continue to await them in the clinic.
Highlights.
Mutations in the gene BEST1 are associated with five clinically distinct diseases
We suggest that BVMD and AVMD are the same disease and should be both grouped as BVMD
iPSC technology shows great potential for the treatment of the bestrophinopathies
Bestrophin 1 has been unambiguously shown to be a Ca2+-activated, pentameric anion channel
Bestrophin 1 shows robust expression and activity in both human RPE and mouse brain
Acknowledgments
This work is supported by the National Institutes of Health (NIH) grants EY13160 (ADM) and EY014465 (LYM), by Research to Prevent Blindness (unrestricted grant to the Department of Ophthalmology at the Mayo Clinic in Rochester, Minnesota, United States of America), and by the Creative Research Initiative Program, Korean National Research Foundation (2015R1A3A2066619; CJL). The authors thank Gustavo D. Aguirre (University of Pennsylvania) for his input and expert advice on the development and testing of ocular therapies in canine models.
Abbreviations
- AVMD
Adult-onset vitelliform macular dystrophy
- ADVIRC
autosomal dominant vitreoretinochoroidopathy
- ARB
autosomal recessive bestrophinopathy
- BVMD
Best vitelliform macular dystrophy
- Best1
Bestrophin 1
- cBest1
Canine bestrophin-1
- EOG
electrooculogram
- ERG
electroretinogram
- fhRPE
fetal human retinal pigment epithelial
- hBest1
human Bestrophin 1
- iPSC-RPE
induced pluripotent stem cell derived retinal pigment epithelium
- mBest1
mouse Bestrophin 1
- RPE
retinal pigment epithelium
- RP
retinitis pigmentosa
- TM
transmembrane
- WT
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abramoff MD, Mullins RF, Lee K, Hoffmann JM, Sonka M, Critser DB, Stasheff SF, Stone EM. Human photoreceptor outer segments shorten during light adaptation. Investigative ophthalmology & visual science. 2013;54:3721–3728. doi: 10.1167/iovs.13-11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jumaily M, Kozlenkov A, Mechaly I, Fichard A, Matha V, Scamps F, Valmier J, Carroll P. Expression of three distinct families of calcium-activated chloride channel genes in the mouse dorsal root ganglion. Neuroscience bulletin. 2007;23:293–299. doi: 10.1007/s12264-007-0044-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets R, Seddon JM, Bernstein PS, Hutchinson A, Atkinson A, Sharma S, Gerrard B, Li W, Metzker ML, Wadelius C, Caskey CT, Dean M, Petrukhin K. Evaluation of the Best disease gene in patients with age-related macular degeneration and other maculopathies. Human genetics. 1999;104:449–453. doi: 10.1007/s004390050986. [DOI] [PubMed] [Google Scholar]
- Andre S, Boukhaddaoui H, Campo B, Al-Jumaily M, Mayeux V, Greuet D, Valmier J, Scamps F. Axotomy-induced expression of calcium-activated chloride current in subpopulations of mouse dorsal root ganglion neurons. Journal of neurophysiology. 2003;90:3764–3773. doi: 10.1152/jn.00449.2003. [DOI] [PubMed] [Google Scholar]
- Arndt CF, Derambure P, Defoort-Dhellemmes S, Hache JC. Outer retinal dysfunction in patients treated with vigabatrin. Neurology. 1999;52:1201–1205. doi: 10.1212/wnl.52.6.1201. [DOI] [PubMed] [Google Scholar]
- Arora R, Khan K, Kasilian ML, Strauss RW, Holder GE, Robson AG, Thompson DA, Moore AT, Michaelides M. Unilateral BEST1-Associated Retinopathy. American journal of ophthalmology. 2016;169:24–32. doi: 10.1016/j.ajo.2016.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakall B, Marmorstein LY, Hoppe G, Peachey NS, Wadelius C, Marmorstein AD. Expression and localization of bestrophin during normal mouse development. Investigative ophthalmology & visual science. 2003;44:3622–3628. doi: 10.1167/iovs.03-0030. [DOI] [PubMed] [Google Scholar]
- Bakall B, McLaughlin P, Stanton JB, Zhang Y, Hartzell HC, Marmorstein LY, Marmorstein AD. Bestrophin-2 is involved in the generation of intraocular pressure. Investigative ophthalmology & visual science. 2008;49:1563–1570. doi: 10.1167/iovs.07-1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakall B, Radu RA, Stanton JB, Burke JM, McKay BS, Wadelius C, Mullins RF, Stone EM, Travis GH, Marmorstein AD. Enhanced accumulation of A2E in individuals homozygous or heterozygous for mutations in BEST1 (VMD2) Experimental eye research. 2007;85:34–43. doi: 10.1016/j.exer.2007.02.018. [DOI] [PubMed] [Google Scholar]
- Bard LA, Cross HE. Genetic counseling of families with Best macular dystrophy. Transactions. Section on Ophthalmology. American Academy of Ophthalmology and Otolaryngology. 1975;79:OP865–873. [PubMed] [Google Scholar]
- Barro-Soria R, Aldehni F, Almaca J, Witzgall R, Schreiber R, Kunzelmann K. ER-localized bestrophin 1 activates Ca2+-dependent ion channels TMEM16A and SK4 possibly by acting as a counterion channel. Pflugers Arch. 2010;459:485–497. doi: 10.1007/s00424-009-0745-0. [DOI] [PubMed] [Google Scholar]
- Barro Soria R, Spitzner M, Schreiber R, Kunzelmann K. Bestrophin-1 enables Ca2+-activated Cl− conductance in epithelia. The Journal of biological chemistry. 2009;284:29405–29412. doi: 10.1074/jbc.M605716200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Guziewicz KE, Iwabe S, Swider M, Scott EM, Savina SV, Ruthel G, Stefano F, Zhang L, Zorger R, Sumaroka A, Jacobson SG, Aguirre GD. Canine retina has a primate fovea-like bouquet of cone photoreceptors which is affected by inherited macular degenerations. PloS one. 2014;9:e90390. doi: 10.1371/journal.pone.0090390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett SR, Folk JC, Kimura AE, Russell SR, Stone EM, Raphtis EM. Autosomal dominant neovascular inflammatory vitreoretinopathy. Ophthalmology. 1990;97:1125–1135. doi: 10.1016/s0161-6420(90)32447-8. discussion 1135–1126. [DOI] [PubMed] [Google Scholar]
- Best F. Uber eine hereditare Maculaaffektion Beitrag zur Vererbungslehre. Z Augenheilk. Z Augenheilk. 1905;13:199–212. [Google Scholar]
- Bitner H, Schatz P, Mizrahi-Meissonnier L, Sharon D, Rosenberg T. Frequency, genotype, and clinical spectrum of best vitelliform macular dystrophy: data from a national center in Denmark. American journal of ophthalmology. 2012;154:403–412. e404. doi: 10.1016/j.ajo.2012.02.036. [DOI] [PubMed] [Google Scholar]
- Blair NP, Goldberg MF, Fishman GA, Salzano T. Autosomal dominant vitreoretinochoroidopathy (ADVIRC) The British journal of ophthalmology. 1984;68:2–9. doi: 10.1136/bjo.68.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Progress in retinal and eye research. 2009;28:187–205. doi: 10.1016/j.preteyeres.2009.04.002. [DOI] [PubMed] [Google Scholar]
- Boon CJ, van den Born LI, Visser L, Keunen JE, Bergen AA, Booij JC, Riemslag FC, Florijn RJ, van Schooneveld MJ. Autosomal recessive bestrophinopathy: differential diagnosis and treatment options. Ophthalmology. 2013;120:809–820. doi: 10.1016/j.ophtha.2012.09.057. [DOI] [PubMed] [Google Scholar]
- Boudes M, Sar C, Menigoz A, Hilaire C, Pequignot MO, Kozlenkov A, Marmorstein A, Carroll P, Valmier J, Scamps F. Best1 is a gene regulated by nerve injury and required for Ca2+-activated Cl− current expression in axotomized sensory neurons. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:10063–10071. doi: 10.1523/JNEUROSCI.1312-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudes M, Scamps F. Calcium-activated chloride current expression in axotomized sensory neurons: what for? Frontiers in molecular neuroscience. 2012;5:35. doi: 10.3389/fnmol.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Lewin AS, Hauswirth WW. A comprehensive review of retinal gene therapy. Molecular therapy: the journal of the American Society of Gene Therapy. 2013;21:509–519. doi: 10.1038/mt.2012.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandl C, Zimmermann SJ, Milenkovic VM, Rosendahl SM, Grassmann F, Milenkovic A, Hehr U, Federlin M, Wetzel CH, Helbig H, Weber BH. In-depth characterisation of Retinal Pigment Epithelium (RPE) cells derived from human induced pluripotent stem cells (hiPSC) Neuromolecular medicine. 2014;16:551–564. doi: 10.1007/s12017-014-8308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M, Marmor M, Vaegan Zrenner E, Brigell M, Bach M. ISCEV Standard for Clinical Electro-oculography (EOG) 2006. Documenta ophthalmologica. Advances in ophthalmology. 2006;113:205–212. doi: 10.1007/s10633-006-9030-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess R, Millar ID, Leroy BP, Urquhart JE, Fearon IM, De Baere E, Brown PD, Robson AG, Wright GA, Kestelyn P, Holder GE, Webster AR, Manson FD, Black GC. Biallelic mutation of BEST1 causes a distinct retinopathy in humans. American journal of human genetics. 2008;82:19–31. doi: 10.1016/j.ajhg.2007.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell GM, Kakuk LE, Griesinger IB, Simpson SA, Nowak NJ, Small KW, Maumenee IH, Rosenfeld PJ, Sieving PA, Shows TB, Ayyagari R. Bestrophin gene mutations in patients with Best vitelliform macular dystrophy. Genomics. 1999;58:98–101. doi: 10.1006/geno.1999.5808. [DOI] [PubMed] [Google Scholar]
- Carter DA, Smart MJ, Letton WV, Ramsden CM, Nommiste B, Chen LL, Fynes K, Muthiah MN, Goh P, Lane A, Powner MB, Webster AR, da Cruz L, Moore AT, Coffey PJ, Carr AF. Mislocalisation of BEST1 in iPSC-derived retinal pigment epithelial cells from a family with autosomal dominant vitreoretinochoroidopathy (ADVIRC) Scientific reports. 2016;6:33792. doi: 10.1038/srep33792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CJ, Goldberg MF. Progressive Cone Dysfunction and Geographic Atrophy of the Macula in Late Stage Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC) Ophthalmic genetics. 2016;37:81–85. doi: 10.3109/13816810.2014.889171. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Kaufman S, Packo K, Stohr H, Weber BH, Goldberg MF. Long-Term Macular Changes in the First Proband of Autosomal Dominant Vitreoretinochoroidopathy (ADVIRC) Due to a Newly Identified Mutation in BEST1. Ophthalmic genetics. 2016;37:102–108. doi: 10.3109/13816810.2015.1039893. [DOI] [PubMed] [Google Scholar]
- Cheng ZY, Wang XP, Schmid KL, Han XG, Song H, Tang X. GABAAalpha1 and GABAArho1 subunits are expressed in cultured human RPE cells and GABAA receptor agents modify the intracellular calcium concentration. Mol Vis. 2015;21:939–947. [PMC free article] [PubMed] [Google Scholar]
- Chien LT, Hartzell HC. Drosophila bestrophin-1 chloride current is dually regulated by calcium and cell volume. The Journal of general physiology. 2007;130:513–524. doi: 10.1085/jgp.200709795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien LT, Hartzell HC. Rescue of volume-regulated anion current by bestrophin mutants with altered charge selectivity. The Journal of general physiology. 2008;132:537–546. doi: 10.1085/jgp.200810065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, Pang JJ, Sumaroka A, Windsor EA, Wilson JM, Flotte TR, Fishman GA, Heon E, Stone EM, Byrne BJ, Jacobson SG, Hauswirth WW. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Pang JJ, Roman AJ, Byrne BJ, Jacobson SG. Human RPE65 gene therapy for Leber congenital amaurosis: persistence of early visual improvements and safety at 1 year. Human gene therapy. 2009a;20:999–1004. doi: 10.1089/hum.2009.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cideciyan AV, Hauswirth WW, Aleman TS, Kaushal S, Schwartz SB, Boye SL, Windsor EA, Conlon TJ, Sumaroka A, Roman AJ, Byrne BJ, Jacobson SG. Vision 1 year after gene therapy for Leber’s congenital amaurosis. The New England journal of medicine. 2009b;361:725–727. doi: 10.1056/NEJMc0903652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciulla TA, Frederick AR., Jr Acute progressive multifocal Best’s disease in a 61-year-old man. American journal of ophthalmology. 1997;123:129–131. doi: 10.1016/s0002-9394(14)71007-4. [DOI] [PubMed] [Google Scholar]
- Cross SD, Johnson AA, Gilles BJ, Bachman LA, Inoue T, Agata K, Marmorstein LY, Marmorstein AD. Control of Maintenance and Regeneration of Planarian Eyes by ovo. Investigative ophthalmology & visual science. 2015;56:7604–7610. doi: 10.1167/iovs.15-17458. [DOI] [PubMed] [Google Scholar]
- Cui CY, Childress V, Piao Y, Michel M, Johnson AA, Kunisada M, Ko MS, Kaestner KH, Marmorstein AD, Schlessinger D. Forkhead transcription factor FoxA1 regulates sweat secretion through Bestrophin 2 anion channel and Na-K-Cl cotransporter 1. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:1199–1203. doi: 10.1073/pnas.1117213109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvin LA, Abou Chehade JE, Chiang J, Fuchs J, Iezzi R, Marmorstein AD. Retinitis pigmentosa associated with a mutation in BEST1. American Journal of Ophthalmology Case Reports. 2016a;2:11–17. doi: 10.1016/j.ajoc.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvin LA, Johnson AA, Pulido JS, Dhaliwal R, Marmorstein AD. Nonantibestrophin Anti-RPE Antibodies in Paraneoplastic Exudative Polymorphous Vitelliform Maculopathy. Translational vision science & technology. 2015;4:2. doi: 10.1167/tvst.4.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalvin LA, Pulido JS, Marmorstein AD. Vitelliform dystrophies: Prevalence in Olmsted County, Minnesota, United States. Ophthalmic genetics. 2016b:1–5. doi: 10.1080/13816810.2016.1175645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AE, Millar ID, Burgess-Mullan R, Maher GJ, Urquhart JE, Brown PD, Black GC, Manson FD. Functional characterization of bestrophin-1 missense mutations associated with autosomal recessive bestrophinopathy. Investigative ophthalmology & visual science. 2011;52:3730–3736. doi: 10.1167/iovs.10-6707. [DOI] [PubMed] [Google Scholar]
- Davidson AE, Millar ID, Urquhart JE, Burgess-Mullan R, Shweikh Y, Parry N, O’Sullivan J, Maher GJ, McKibbin M, Downes SM, Lotery AJ, Jacobson SG, Brown PD, Black GC, Manson FD. Missense mutations in a retinal pigment epithelium protein, bestrophin-1, cause retinitis pigmentosa. American journal of human genetics. 2009;85:581–592. doi: 10.1016/j.ajhg.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran C, Chien LT, Hartzell HC. Drosophila bestrophin-1 currents are regulated by phosphorylation via a CaMKII dependent mechanism. PloS one. 2013;8:e58875. doi: 10.1371/journal.pone.0058875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelhauser HF, Rowe-Rendleman CL, Robinson MR, Dawson DG, Chader GJ, Grossniklaus HE, Rittenhouse KD, Wilson CG, Weber DA, Kuppermann BD, Csaky KG, Olsen TW, Kompella UB, Holers VM, Hageman GS, Gilger BC, Campochiaro PA, Whitcup SM, Wong WT. Ophthalmic drug delivery systems for the treatment of retinal diseases: basic research to clinical applications. Investigative ophthalmology & visual science. 2010;51:5403–5420. doi: 10.1167/iovs.10-5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eksandh L, Adamus G, Mosgrove L, Andreasson S. Autoantibodies against bestrophin in a patient with vitelliform paraneoplastic retinopathy and a metastatic choroidal malignant melanoma. Archives of ophthalmology. 2008;126:432–435. doi: 10.1001/archopht.126.3.432. [DOI] [PubMed] [Google Scholar]
- Felbor U, Schilling H, Weber BH. Adult vitelliform macular dystrophy is frequently associated with mutations in the peripherin/RDS gene. Human mutation. 1997;10:301–309. doi: 10.1002/(SICI)1098-1004(1997)10:4<301::AID-HUMU6>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Fischmeister R, Hartzell HC. Volume sensitivity of the bestrophin family of chloride channels. The Journal of physiology. 2005;562:477–491. doi: 10.1113/jphysiol.2004.075622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez NM, Tamm ER, Straubeta O. Role of bestrophin-1 in store-operated calcium entry in retinal pigment epithelium. Pflugers Archiv: European journal of physiology. 2013;465:481–495. doi: 10.1007/s00424-012-1181-0. [DOI] [PubMed] [Google Scholar]
- Gouras P, Braun K, Ivert L, Neuringer M, Mattison JA. Bestrophin detected in the basal membrane of the retinal epithelium and drusen of monkeys with drusenoid maculopathy. Graefe’s archive for clinical and experimental ophthalmology = Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie. 2009;247:1051–1056. doi: 10.1007/s00417-009-1091-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Aguirre GD, Zangerl B. Modeling the structural consequences of BEST1 missense mutations. Advances in experimental medicine and biology. 2012;723:611–618. doi: 10.1007/978-1-4614-0631-0_78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Slavik J, Lindauer SJ, Aguirre GD, Zangerl B. Molecular consequences of BEST1 gene mutations in canine multifocal retinopathy predict functional implications for human bestrophinopathies. Investigative ophthalmology & visual science. 2011;52:4497–4505. doi: 10.1167/iovs.10-6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Komaromy AM, Iwabe S, Chiodo VA, Boye SL, Hauswirth WW, Beltran WA, Aguirre GD. Recombinant AAV-mediated BEST1 transfer to the retinal pigment epithelium: analysis of serotype-dependent retinal effects. PloS one. 2013;8:e75666. doi: 10.1371/journal.pone.0075666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD. Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Investigative ophthalmology & visual science. 2007;48:1959–1967. doi: 10.1167/iovs.06-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han DP, Lewandowski MF. Electro-oculography in autosomal dominant vitreoretinochoroidopathy. Archives of ophthalmology. 1992;110:1563–1567. doi: 10.1001/archopht.1992.01080230063021. [DOI] [PubMed] [Google Scholar]
- Han KS, Woo J, Park H, Yoon BJ, Choi S, Lee CJ. Channel-mediated astrocytic glutamate release via Bestrophin-1 targets synaptic NMDARs. Molecular brain. 2013;6:4. doi: 10.1186/1756-6606-6-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harned J, Nagar S, McGahan MC. Hypoxia controls iron metabolism and glutamate secretion in retinal pigmented epithelial cells. Biochim Biophys Acta. 2014;1840:3138–3144. doi: 10.1016/j.bbagen.2014.06.012. [DOI] [PubMed] [Google Scholar]
- Hartzell HC, Qu Z, Yu K, Xiao Q, Chien LT. Molecular physiology of bestrophins: multifunctional membrane proteins linked to best disease and other retinopathies. Physiological reviews. 2008;88:639–672. doi: 10.1152/physrev.00022.2007. [DOI] [PubMed] [Google Scholar]
- Hoffmann I, Guziewicz KE, Zangerl B, Aguirre GD, Mardin CY. Canine multifocal retinopathy in the Australian Shepherd: a case report. Veterinary ophthalmology. 2012;15(Suppl 2):134–138. doi: 10.1111/j.1463-5224.2012.01005.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain RN, Shahid FL, Empeslidis T, Ch’ng SW. Use of Intravitreal Bevacizumab in a 9-Year-Old Child with Choroidal Neovascularization Associated with Autosomal Recessive Bestrophinopathy. Ophthalmic genetics. 2015;36:265–269. doi: 10.3109/13816810.2014.962706. [DOI] [PubMed] [Google Scholar]
- Ito G, Okamoto R, Murano T, Shimizu H, Fujii S, Nakata T, Mizutani T, Yui S, Akiyama-Morio J, Nemoto Y, Okada E, Araki A, Ohtsuka K, Tsuchiya K, Nakamura T, Watanabe M. Lineage-specific expression of bestrophin-2 and bestrophin-4 in human intestinal epithelial cells. PloS one. 2013;8:e79693. doi: 10.1371/journal.pone.0079693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ, Poet M, Fuhrmann JC, Zdebik AA. Physiological functions of CLC Cl- channels gleaned from human genetic disease and mouse models. Annual review of physiology. 2005;67:779–807. doi: 10.1146/annurev.physiol.67.032003.153245. [DOI] [PubMed] [Google Scholar]
- Jiang L, Liu Y, Ma MM, Tang YB, Zhou JG, Guan YY. Mitochondria dependent pathway is involved in the protective effect of bestrophin-3 on hydrogen peroxide-induced apoptosis in basilar artery smooth muscle cells. Apoptosis: an international journal on programmed cell death. 2013;18:556–565. doi: 10.1007/s10495-013-0828-4. [DOI] [PubMed] [Google Scholar]
- Jo S, Yarishkin O, Hwang YJ, Chun YE, Park M, Woo DH, Bae JY, Kim T, Lee J, Chun H, Park HJ, Lee da Y, Hong J, Kim HY, Oh SJ, Park SJ, Lee H, Yoon BE, Kim Y, Jeong Y, Shim I, Bae YC, Cho J, Kowall NW, Ryu H, Hwang E, Kim D, Lee CJ. GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med. 2014;20:886–896. doi: 10.1038/nm.3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Bachman LA, Gilles BJ, Cross SD, Stelzig KE, Resch ZT, Marmorstein LY, Pulido JS, Marmorstein AD. Autosomal Recessive Bestrophinopathy Is Not Associated With the Loss of Bestrophin-1 Anion Channel Function in a Patient With a Novel BEST1 Mutation. Investigative ophthalmology & visual science. 2015;56:4619–4630. doi: 10.1167/iovs.15-16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Lee YS, Chadburn AJ, Tammaro P, Manson FD, Marmorstein LY, Marmorstein AD. Disease-causing mutations associated with four bestrophinopathies exhibit disparate effects on the localization, but not the oligomerization, of Bestrophin-1. Experimental eye research. 2014;121:74–85. doi: 10.1016/j.exer.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Lee YS, Stanton JB, Yu K, Hartzell CH, Marmorstein LY, Marmorstein AD. Differential effects of Best disease causing missense mutations on bestrophin-1 trafficking. Human molecular genetics. 2013;22:4688–4697. doi: 10.1093/hmg/ddt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Naaldijk Y, Hohaus C, Meisel HJ, Stolzing A. Protective effects of alpha phenyl-tert-butyl nitrone and ascorbic acid in human adipose derived mesenchymal stem cells from differently aged donors. Aging. 2016 doi: 10.18632/aging.101035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AA, Riehle MA. Resveratrol Fails to Extend Life Span in the Mosquito Anopheles stephensi. Rejuvenation research. 2015;18:473–478. doi: 10.1089/rej.2015.1670. [DOI] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nature reviews Molecular cell biology. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaden TR, Tan AC, Feiner L, Freund KB. Unilateral Best Disease: A Case Report. Retinal cases & brief reports. 2016 doi: 10.1097/ICB.0000000000000431. [DOI] [PubMed] [Google Scholar]
- Kamao H, Mandai M, Okamoto S, Sakai N, Suga A, Sugita S, Kiryu J, Takahashi M. Characterization of human induced pluripotent stem cell-derived retinal pigment epithelium cell sheets aiming for clinical application. Stem cell reports. 2014;2:205–218. doi: 10.1016/j.stemcr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane Dickson V, Pedi L, Long SB. Structure and insights into the function of a Ca(2+)-activated Cl(−) channel. Nature. 2014;516:213–218. doi: 10.1038/nature13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SJ, Goldberg MF, Orth DH, Fishman GA, Tessler H, Mizuno K. Autosomal dominant vitreoretinochoroidopathy. Archives of ophthalmology. 1982;100:272–278. doi: 10.1001/archopht.1982.01030030274008. [DOI] [PubMed] [Google Scholar]
- Kimbrel EA, Lanza R. Current status of pluripotent stem cells: moving the first therapies to the clinic. Nature reviews Drug discovery. 2015;14:681–692. doi: 10.1038/nrd4738. [DOI] [PubMed] [Google Scholar]
- Kramer F, Stohr H, Weber BH. Cloning and characterization of the murine Vmd2 RFP-TM gene family. Cytogenetic and genome research. 2004;105:107–114. doi: 10.1159/000078016. [DOI] [PubMed] [Google Scholar]
- Kramer F, White K, Pauleikhoff D, Gehrig A, Passmore L, Rivera A, Rudolph G, Kellner U, Andrassi M, Lorenz B, Rohrschneider K, Blankenagel A, Jurklies B, Schilling H, Schutt F, Holz FG, Weber BH. Mutations in the VMD2 gene are associated with juvenile-onset vitelliform macular dystrophy (Best disease) and adult vitelliform macular dystrophy but not age-related macular degeneration. European journal of human genetics: EJHG. 2000;8:286–292. doi: 10.1038/sj.ejhg.5200447. [DOI] [PubMed] [Google Scholar]
- Kunzelmann K, Kongsuphol P, Chootip K, Toledo C, Martins JR, Almaca J, Tian Y, Witzgall R, Ousingsawat J, Schreiber R. Role of the Ca2+-activated Cl- channels bestrophin and anoctamin in epithelial cells. Biol Chem. 2011;392:125–134. doi: 10.1515/BC.2011.010. [DOI] [PubMed] [Google Scholar]
- Lacassagne E, Dhuez A, Rigaudiere F, Dansault A, Vetu C, Bigot K, Vieira V, Puech B, Defoort-Dhellemmes S, Abitbol M. Phenotypic variability in a French family with a novel mutation in the BEST1 gene causing multifocal best vitelliform macular dystrophy. Molecular vision. 2011;17:309–322. [PMC free article] [PubMed] [Google Scholar]
- Lapan SW, Reddien PW. Transcriptome analysis of the planarian eye identifies ovo as a specific regulator of eye regeneration. Cell reports. 2012;2:294–307. doi: 10.1016/j.celrep.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Yoon BE, Berglund K, Oh SJ, Park H, Shin HS, Augustine GJ, Lee CJ. Channel-mediated tonic GABA release from glia. Science. 2010;330:790–796. doi: 10.1126/science.1184334. [DOI] [PubMed] [Google Scholar]
- Lee WK, Chakraborty PK, Roussa E, Wolff NA, Thevenod F. ERK1/2-dependent bestrophin-3 expression prevents ER-stress-induced cell death in renal epithelial cells by reducing CHOP. Biochimica et biophysica acta. 2012;1823:1864–1876. doi: 10.1016/j.bbamcr.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Lisenbee CS, Karnik SK, Trelease RN. Overexpression and mislocalization of a tail-anchored GFP redefines the identity of peroxisomal ER. Traffic. 2003;4:491–501. doi: 10.1034/j.1600-0854.2003.00107.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Xuan Y, Zhang Y, Liu W, Xu G. Bilateral macular holes and a new onset vitelliform lesion in Best disease. Ophthalmic genetics. 2016:1–4. doi: 10.3109/13816810.2015.1126614. [DOI] [PubMed] [Google Scholar]
- Liu L, Hayashi K, Kaneda T, Ino H, Fujino N, Uchiyama K, Konno T, Tsuda T, Kawashiri MA, Ueda K, Higashikata T, Shuai W, Kupershmidt S, Higashida H, Yamagishi M. A novel mutation in the transmembrane nonpore region of the KCNH2 gene causes severe clinical manifestations of long QT syndrome. Heart rhythm: the official journal of the Heart Rhythm Society. 2013;10:61–67. doi: 10.1016/j.hrthm.2012.09.053. [DOI] [PubMed] [Google Scholar]
- Mahajan VB, Skeie JM, Bassuk AG, Fingert JH, Braun TA, Daggett HT, Folk JC, Sheffield VC, Stone EM. Calpain-5 mutations cause autoimmune uveitis, retinal neovascularization, and photoreceptor degeneration. PLoS genetics. 2012;8:e1003001. doi: 10.1371/journal.pgen.1003001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manes G, Meunier I, Avila-Fernandez A, Banfi S, Le Meur G, Zanlonghi X, Corton M, Simonelli F, Brabet P, Labesse G, Audo I, Mohand-Said S, Zeitz C, Sahel JA, Weber M, Dollfus H, Dhaenens CM, Allorge D, De Baere E, Koenekoop RK, Kohl S, Cremers FP, Hollyfield JG, Senechal A, Hebrard M, Bocquet B, Ayuso Garcia C, Hamel CP. Mutations in IMPG1 cause vitelliform macular dystrophies. American journal of human genetics. 2013;93:571–578. doi: 10.1016/j.ajhg.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Cross HE, Peachey NS. Functional roles of bestrophins in ocular epithelia. Progress in retinal and eye research. 2009;28:206–226. doi: 10.1016/j.preteyeres.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Kinnick TR, Stanton JB, Johnson AA, Lynch RM, Marmorstein LY. Bestrophin-1 influences transepithelial electrical properties and Ca2+ signaling in human retinal pigment epithelium. Molecular vision. 2015;21:347–359. [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Marmorstein LY, Rayborn M, Wang X, Hollyfield JG, Petrukhin K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:12758–12763. doi: 10.1073/pnas.220402097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein AD, Stanton JB, Yocom J, Bakall B, Schiavone MT, Wadelius C, Marmorstein LY, Peachey NS. A model of best vitelliform macular dystrophy in rats. Investigative ophthalmology & visual science. 2004;45:3733–3739. doi: 10.1167/iovs.04-0307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmorstein LY, McLaughlin PJ, Stanton JB, Yan L, Crabb JW, Marmorstein AD. Bestrophin interacts physically and functionally with protein phosphatase 2A. The Journal of biological chemistry. 2002;277:30591–30597. doi: 10.1074/jbc.M204269200. [DOI] [PubMed] [Google Scholar]
- Marmorstein LY, Wu J, McLaughlin P, Yocom J, Karl MO, Neussert R, Wimmers S, Stanton JB, Gregg RG, Strauss O, Peachey NS, Marmorstein AD. The light peak of the electroretinogram is dependent on voltage-gated calcium channels and antagonized by bestrophin (best-1) The Journal of general physiology. 2006;127:577–589. doi: 10.1085/jgp.200509473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquardt A, Stohr H, Passmore LA, Kramer F, Rivera A, Weber BH. Mutations in a novel gene, VMD2, encoding a protein of unknown properties cause juvenile-onset vitelliform macular dystrophy (Best’s disease) Human molecular genetics. 1998;7:1517–1525. doi: 10.1093/hmg/7.9.1517. [DOI] [PubMed] [Google Scholar]
- Meunier I, Manes G, Bocquet B, Marquette V, Baudoin C, Puech B, Defoort-Dhellemmes S, Audo I, Verdet R, Arndt C, Zanlonghi X, Le Meur G, Dhaenens CM, Hamel CP. Frequency and clinical pattern of vitelliform macular dystrophy caused by mutations of interphotoreceptor matrix IMPG1 and IMPG2 genes. Ophthalmology. 2014;121:2406–2414. doi: 10.1016/j.ophtha.2014.06.028. [DOI] [PubMed] [Google Scholar]
- Milenkovic A, Brandl C, Milenkovic VM, Jendryke T, Sirianant L, Wanitchakool P, Zimmermann S, Reiff CM, Horling F, Schrewe H, Schreiber R, Kunzelmann K, Wetzel CH, Weber BH. Bestrophin 1 is indispensable for volume regulation in human retinal pigment epithelium cells. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:E2630–2639. doi: 10.1073/pnas.1418840112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic VM, Krejcova S, Reichhart N, Wagner A, Strauss O. Interaction of bestrophin-1 and Ca2+ channel beta-subunits: identification of new binding domains on the bestrophin-1 C-terminus. PloS one. 2011a;6:e19364. doi: 10.1371/journal.pone.0019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic VM, Langmann T, Schreiber R, Kunzelmann K, Weber BH. Molecular evolution and functional divergence of the bestrophin protein family. BMC evolutionary biology. 2008;8:72. doi: 10.1186/1471-2148-8-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic VM, Rohrl E, Weber BH, Strauss O. Disease-associated missense mutations in bestrophin-1 affect cellular trafficking and anion conductance. Journal of cell science. 2011b;124:2988–2996. doi: 10.1242/jcs.085878. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Del Monte MA. Na(+)-dependent glutamate transporter in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1994;35:3589–3598. [PubMed] [Google Scholar]
- Mladenova K, Petrova SD, Andreeva TD, Moskova-Doumanova V, Topouzova-Hristova T, Kalvachev Y, Balashev K, Bhattacharya SS, Chakarova C, Lalchev Z, Doumanov JA. Effects of Ca2+ ions on bestrophin-1 surface films. Colloids and surfaces B, Biointerfaces. 2017;149:226–232. doi: 10.1016/j.colsurfb.2016.10.023. [DOI] [PubMed] [Google Scholar]
- Mohler CW, Fine SL. Long-term evaluation of patients with Best’s vitelliform dystrophy. Ophthalmology. 1981;88:688–692. doi: 10.1016/s0161-6420(81)34965-3. [DOI] [PubMed] [Google Scholar]
- Moshfegh Y, Velez G, Li Y, Bassuk AG, Mahajan VB, Tsang SH. BESTROPHIN1 mutations cause defective chloride conductance in patient stem cell-derived RPE. Human molecular genetics. 2016 doi: 10.1093/hmg/ddw126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothet JP, Parent AT, Wolosker H, Brady RO, Jr, Linden DJ, Ferris CD, Rogawski MA, Snyder SH. D-serine is an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:4926–4931. doi: 10.1073/pnas.97.9.4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RF, Kuehn MH, Faidley EA, Syed NA, Stone EM. Differential macular and peripheral expression of bestrophin in human eyes and its implication for best disease. Investigative ophthalmology & visual science. 2007;48:3372–3380. doi: 10.1167/iovs.06-0868. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Oh KT, Heffron E, Hageman GS, Stone EM. Late development of vitelliform lesions and flecks in a patient with best disease: clinicopathologic correlation. Arch Ophthalmol. 2005;123:1588–1594. doi: 10.1001/archopht.123.11.1588. [DOI] [PubMed] [Google Scholar]
- Naaldijk Y, Johnson AA, Ishak S, Meisel HJ, Hohaus C, Stolzing A. Migrational changes of mesenchymal stem cells in response to cytokines, growth factors, hypoxia, and aging. Experimental cell research. 2015;338:97–104. doi: 10.1016/j.yexcr.2015.08.019. [DOI] [PubMed] [Google Scholar]
- Neussert R, Muller C, Milenkovic VM, Strauss O. The presence of bestrophin-1 modulates the Ca2+ recruitment from Ca2+ stores in the ER. Pflugers Archiv: European journal of physiology. 2010;460:163–175. doi: 10.1007/s00424-010-0840-2. [DOI] [PubMed] [Google Scholar]
- Nordstrom S. Hereditary macular degeneration--a population survey in the country of Vsterbotten, Sweden. Hereditas. 1974;78:41–62. doi: 10.1111/j.1601-5223.1974.tb01427.x. [DOI] [PubMed] [Google Scholar]
- Nordstrom S, Thorburn W. Dominantly inherited macular degeneration (Best’s disease) in a homozygous father with 11 children. Clinical genetics. 1980;18:211–216. doi: 10.1111/j.1399-0004.1980.tb00874.x. [DOI] [PubMed] [Google Scholar]
- Oh KT, Vallar C. Central cone dysfunction in autosomal dominant vitreoretino choroidopathy (ADVIRC) American journal of ophthalmology. 2006;141:940–943. doi: 10.1016/j.ajo.2005.11.041. [DOI] [PubMed] [Google Scholar]
- Oh SJ, Han KS, Park H, Woo DH, Kim HY, Traynelis SF, Lee CJ. Protease activated receptor 1-induced glutamate release in cultured astrocytes is mediated by Bestrophin-1 channel but not by vesicular exocytosis. Molecular brain. 2012;5:38. doi: 10.1186/1756-6606-5-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmowski AM, Allgayer R, Heinemann-Vernaleken B, Scherer V, Ruprecht KW. Detection of retinal dysfunction in vitelliform macular dystrophy using the multifocal ERG (MF-ERG). Documenta ophthalmologica. Advances in ophthalmology. 2003;106:145–152. doi: 10.1023/a:1022550008074. [DOI] [PubMed] [Google Scholar]
- Park H, Han KS, Oh SJ, Jo S, Woo J, Yoon BE, Lee CJ. High glutamate permeability and distal localization of Best1 channel in CA1 hippocampal astrocyte. Molecular brain. 2013;6:54. doi: 10.1186/1756-6606-6-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Han KS, Seo J, Lee J, Dravid SM, Woo J, Chun H, Cho S, Bae JY, An H, Koh W, Yoon BE, Berlinguer-Palmini R, Mannaioni G, Traynelis SF, Bae YC, Choi SY, Lee CJ. Channel-mediated astrocytic glutamate modulates hippocampal synaptic plasticity by activating postsynaptic NMDA receptors. Molecular brain. 2015;8:7. doi: 10.1186/s13041-015-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Oh SJ, Han KS, Woo DH, Mannaioni G, Traynelis SF, Lee CJ. Bestrophin-1 encodes for the Ca2+-activated anion channel in hippocampal astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:13063–13073. doi: 10.1523/JNEUROSCI.3193-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parodi MB, Iacono P, Bandello F. Pseudoxanthoma elasticum associated with vitelliform macular lesion. Ophthalmic surgery, lasers & imaging retina. 2015;46:287–288. doi: 10.3928/23258160-20150213-11. [DOI] [PubMed] [Google Scholar]
- Parodi MB, Iacono P, Campa C, Del Turco C, Bandello F. Fundus autofluorescence patterns in Best vitelliform macular dystrophy. American journal of ophthalmology. 2014;158:1086–1092. doi: 10.1016/j.ajo.2014.07.026. [DOI] [PubMed] [Google Scholar]
- Patrinely JR, Lewis RA, Font RL. Foveomacular vitelliform dystrophy, adult type. A clinicopathologic study including electron microscopic observations. Ophthalmology. 1985;92:1712–1718. doi: 10.1016/s0161-6420(85)34097-6. [DOI] [PubMed] [Google Scholar]
- Pedemonte N, Galietta LJ. Pharmacological Correctors of Mutant CFTR Mistrafficking. Frontiers in pharmacology. 2012;3:175. doi: 10.3389/fphar.2012.00175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson WM, Miller SS. Identification and functional characterization of a dual GABA/taurine transporter in the bullfrog retinal pigment epithelium. J Gen Physiol. 1995;106:1089–1122. doi: 10.1085/jgp.106.6.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrukhin K, Koisti MJ, Bakall B, Li W, Xie G, Marknell T, Sandgren O, Forsman K, Holmgren G, Andreasson S, Vujic M, Bergen AA, McGarty-Dugan V, Figueroa D, Austin CP, Metzker ML, Caskey CT, Wadelius C. Identification of the gene responsible for Best macular dystrophy. Nature genetics. 1998;19:241–247. doi: 10.1038/915. [DOI] [PubMed] [Google Scholar]
- Pilli S, Zawadzki RJ, Werner JS, Park SS. High-resolution Fourier-domain optical coherence tomography findings in vitelliform detachment associated with basal laminar drusen. Retina. 2011;31:812–814. doi: 10.1097/IAE.0b013e318207d112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineda-Farias JB, Barragan-Iglesias P, Loeza-Alcocer E, Torres-Lopez JE, Rocha-Gonzalez HI, Perez-Severiano F, Delgado-Lezama R, Granados-Soto V. Role of anoctamin-1 and bestrophin-1 in spinal nerve ligation-induced neuropathic pain in rats. Molecular pain. 2015;11:41. doi: 10.1186/s12990-015-0042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineiro-Gallego T, Alvarez M, Pereiro I, Campos S, Sharon D, Schatz P, Valverde D. Clinical evaluation of two consanguineous families with homozygous mutations in BEST1. Molecular vision. 2011;17:1607–1617. [PMC free article] [PubMed] [Google Scholar]
- Pomares E, Bures-Jelstrup A, Ruiz-Nogales S, Corcostegui B, Gonzalez-Duarte R, Navarro R. Nonsense-mediated decay as the molecular cause for autosomal recessive bestrophinopathy in two unrelated families. Investigative ophthalmology & visual science. 2012;53:532–537. doi: 10.1167/iovs.11-7964. [DOI] [PubMed] [Google Scholar]
- Qu Z, Hartzell HC. Bestrophin Cl- channels are highly permeable to HCO3. American journal of physiology. Cell physiology. 2008;294:C1371–1377. doi: 10.1152/ajpcell.00398.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Wei RW, Mann W, Hartzell HC. Two bestrophins cloned from Xenopus laevis oocytes express Ca(2+)-activated Cl(−) currents. The Journal of biological chemistry. 2003;278:49563–49572. doi: 10.1074/jbc.M308414200. [DOI] [PubMed] [Google Scholar]
- Querques G, Atmani K, Bouzitou-Mfoumou R, Leveziel N, Massamba N, Souied EH. Preferential hyperacuity perimeter in best vitelliform macular dystrophy. Retina. 2011;31:959–966. doi: 10.1097/IAE.0b013e3181f441c1. [DOI] [PubMed] [Google Scholar]
- Reddy MA, Francis PJ, Berry V, Bradshaw K, Patel RJ, Maher ER, Kumar R, Bhattacharya SS, Moore AT. A clinical and molecular genetic study of a rare dominantly inherited syndrome (MRCS) comprising of microcornea, rod-cone dystrophy, cataract, and posterior staphyloma. The British journal of ophthalmology. 2003;87:197–202. doi: 10.1136/bjo.87.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichhart N, Milenkovic VM, Halsband CA, Cordeiro S, Strauss O. Effect of bestrophin-1 on L-type Ca2+ channel activity depends on the Ca2+ channel beta-subunit. Experimental eye research. 2010;91:630–639. doi: 10.1016/j.exer.2010.08.001. [DOI] [PubMed] [Google Scholar]
- Rohani L, Johnson AA, Arnold A, Stolzing A. The aging signature: a hallmark of induced pluripotent stem cells? Aging cell. 2014;13:2–7. doi: 10.1111/acel.12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal R, Bakall B, Kinnick T, Peachey N, Wimmers S, Wadelius C, Marmorstein A, Strauss O. Expression of bestrophin-1, the product of the VMD2 gene, modulates voltage-dependent Ca2+ channels in retinal pigment epithelial cells. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2006;20:178–180. doi: 10.1096/fj.05-4495fje. [DOI] [PubMed] [Google Scholar]
- Shutova MV, Surdina AV, Ischenko DS, Naumov VA, Bogomazova AN, Vassina EM, Alekseev DG, Lagarkova MA, Kiselev SL. An integrative analysis of reprogramming in human isogenic system identified a clone selection criterion. Cell cycle. 2016;15:986–997. doi: 10.1080/15384101.2016.1152425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigford DK, Schaal S. Bilateral choroidal neovascularization associated with basal laminar drusen in a 31-year-old. Canadian journal of ophthalmology. Journal canadien d’ophtalmologie. 2014;49:e80–82. doi: 10.1016/j.jcjo.2014.03.015. [DOI] [PubMed] [Google Scholar]
- Singh R, Kuai D, Guziewicz KE, Meyer J, Wilson M, Lu J, Smith M, Clark E, Verhoeven A, Aguirre GD, Gamm DM. Pharmacological Modulation of Photoreceptor Outer Segment Degradation in a Human iPS Cell Model of Inherited Macular Degeneration. Molecular therapy: the journal of the American Society of Gene Therapy. 2015;23:1700–1711. doi: 10.1038/mt.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R, Shen W, Kuai D, Martin JM, Guo X, Smith MA, Perez ET, Phillips MJ, Simonett JM, Wallace KA, Verhoeven AD, Capowski EE, Zhang X, Yin Y, Halbach PJ, Fishman GA, Wright LS, Pattnaik BR, Gamm DM. iPS cell modeling of Best disease: insights into the pathophysiology of an inherited macular degeneration. Human molecular genetics. 2013;22:593–607. doi: 10.1093/hmg/dds469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Yang Z, He B. Bestrophin 3 ameliorates TNFalpha-induced inflammation by inhibiting NF-kappaB activation in endothelial cells. PloS one. 2014;9:e111093. doi: 10.1371/journal.pone.0111093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton JB, Goldberg AF, Hoppe G, Marmorstein LY, Marmorstein AD. Hydrodynamic properties of porcine bestrophin-1 in Triton X-100. Biochimica et biophysica acta. 2006;1758:241–247. doi: 10.1016/j.bbamem.2006.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone EM, Lotery AJ, Munier FL, Heon E, Piguet B, Guymer RH, Vandenburgh K, Cousin P, Nishimura D, Swiderski RE, Silvestri G, Mackey DA, Hageman GS, Bird AC, Sheffield VC, Schorderet DF. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nature genetics. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- Stotz SC, Clapham DE. Anion-sensitive fluorophore identifies the Drosophila swell-activated chloride channel in a genome-wide RNA interference screen. PloS one. 2012;7:e46865. doi: 10.1371/journal.pone.0046865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss O, Muller C, Reichhart N, Tamm ER, Gomez NM. The role of bestrophin-1 in intracellular Ca(2+) signaling. Advances in experimental medicine and biology. 2014;801:113–119. doi: 10.1007/978-1-4614-3209-8_15. [DOI] [PubMed] [Google Scholar]
- Sugita S, Iwasaki Y, Makabe K, Kamao H, Mandai M, Shiina T, Ogasawara K, Hirami Y, Kurimoto Y, Takahashi M. Successful Transplantation of Retinal Pigment Epithelial Cells from MHC Homozygote iPSCs in MHC-Matched Models. Stem cell reports. 2016 doi: 10.1016/j.stemcr.2016.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Tsunenari T, Yau KW, Nathans J. The vitelliform macular dystrophy protein defines a new family of chloride channels. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:4008–4013. doi: 10.1073/pnas.052692999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsen P. Stressed podocytes - Bestrophin-3 is not just Bestrophin-3. Acta physiologica. 2015;214:430–431. doi: 10.1111/apha.12539. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Testa F, Rossi S, Passerini I, Sodi A, Di Iorio V, Interlandi E, Della Corte M, Menchini U, Rinaldi E, Torricelli F, Simonelli F. A normal electro-oculography in a family affected by best disease with a novel spontaneous mutation of the BEST1 gene. Br J Ophthalmol. 2008;92:1467–1470. doi: 10.1136/bjo.2008.143776. [DOI] [PubMed] [Google Scholar]
- Traboulsi EI. Genetic diseases of the eye. 2. Oxford University Press; New York: 2012. [Google Scholar]
- Uggenti C, Briant K, Streit AK, Thomson S, Koay YH, Baines RA, Swanton E, Manson FD. Disease models & mechanisms. 2016. Restoration of mutant bestrophin-1 expression, localisation and function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaisey G, Miller AN, Long SB. Distinct regions that control ion selectivity and calcium-dependent activation in the bestrophin ion channel. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E7399–E7408. doi: 10.1073/pnas.1614688113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ven JP, Boon CJ, Fauser S, Hoefsloot LH, Smailhodzic D, Schoenmaker-Koller F, Klevering J, Klaver CC, den Hollander AI, Hoyng CB. Clinical evaluation of 3 families with basal laminar drusen caused by novel mutations in the complement factor H gene. Archives of ophthalmology. 2012;130:1038–1047. doi: 10.1001/archophthalmol.2012.265. [DOI] [PubMed] [Google Scholar]
- Viola F, Barteselli G, Dell’Arti L, Vezzola D, Mapelli C, Villani E, Ratiglia R. Multimodal imaging in deferoxamine retinopathy. Retina. 2014;34:1428–1438. doi: 10.1097/IAE.0000000000000073. [DOI] [PubMed] [Google Scholar]
- Walter P, Brunner R, Heimann K. Atypical presentations of Best’s vitelliform macular degeneration: clinical findings in seven cases. German journal of ophthalmology. 1994;3:440–444. [PubMed] [Google Scholar]
- Weingeist TA, Kobrin JL, Watzke RC. Histopathology of Best’s macular dystrophy. Archives of ophthalmology. 1982;100:1108–1114. doi: 10.1001/archopht.1982.01030040086016. [DOI] [PubMed] [Google Scholar]
- Wittstrom E, Ponjavic V, Bondeson ML, Andreasson S. Anterior segment abnormalities and angle-closure glaucoma in a family with a mutation in the BEST1 gene and Best vitelliform macular dystrophy. Ophthalmic genetics. 2011;32:217–227. doi: 10.3109/13816810.2011.567884. [DOI] [PubMed] [Google Scholar]
- Wolosker H, Blackshaw S, Snyder SH. Serine racemase: a glial enzyme synthesizing D-serine to regulate glutamate-N-methyl-D-aspartate neurotransmission. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:13409–13414. doi: 10.1073/pnas.96.23.13409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo DH, Han KS, Shim JW, Yoon BE, Kim E, Bae JY, Oh SJ, Hwang EM, Marmorstein AD, Bae YC, Park JY, Lee CJ. TREK-1 and Best1 channels mediate fast and slow glutamate release in astrocytes upon GPCR activation. Cell. 2012;151:25–40. doi: 10.1016/j.cell.2012.09.005. [DOI] [PubMed] [Google Scholar]
- Wu C, Sun D. GABA receptors in brain development, function, and injury. Metabolic brain disease. 2015;30:367–379. doi: 10.1007/s11011-014-9560-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Hartzell HC, Yu K. Bestrophins and retinopathies. Pflugers Archiv: European journal of physiology. 2010;460:559–569. doi: 10.1007/s00424-010-0821-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Yu K, Cui YY, Hartzell HC. Dysregulation of human bestrophin-1 by ceramide-induced dephosphorylation. The Journal of physiology. 2009;587:4379–4391. doi: 10.1113/jphysiol.2009.176800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Wu J, Li S, Rao MS, Liu Y. Genetic Modification in Human Pluripotent Stem Cells by Homologous Recombination and CRISPR/Cas9 System. Methods in molecular biology. 2016;1307:173–190. doi: 10.1007/7651_2014_73. [DOI] [PubMed] [Google Scholar]
- Yang T, Liu Q, Kloss B, Bruni R, Kalathur RC, Guo Y, Kloppmann E, Rost B, Colecraft HM, Hendrickson WA. Structure and selectivity in bestrophin ion channels. Science. 2014;346:355–359. doi: 10.1126/science.1259723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yardley J, Leroy BP, Hart-Holden N, Lafaut BA, Loeys B, Messiaen LM, Perveen R, Reddy MA, Bhattacharya SS, Traboulsi E, Baralle D, De Laey JJ, Puech B, Kestelyn P, Moore AT, Manson FD, Black GC. Mutations of VMD2 splicing regulators cause nanophthalmos and autosomal dominant vitreoretinochoroidopathy (ADVIRC) Investigative ophthalmology & visual science. 2004;45:3683–3689. doi: 10.1167/iovs.04-0550. [DOI] [PubMed] [Google Scholar]
- Yoon BE, Jo S, Woo J, Lee JH, Kim T, Kim D, Lee CJ. The amount of astrocytic GABA positively correlates with the degree of tonic inhibition in hippocampal CA1 and cerebellum. Molecular brain. 2011;4:42. doi: 10.1186/1756-6606-4-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Lujan R, Marmorstein A, Gabriel S, Hartzell HC. Bestrophin-2 mediates bicarbonate transport by goblet cells in mouse colon. The Journal of clinical investigation. 2010;120:1722–1735. doi: 10.1172/JCI41129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xiao Q, Cui G, Lee A, Hartzell HC. The best disease-linked Cl- channel hBest1 regulates Ca V 1 (L-type) Ca2+ channels via src-homology-binding domains. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:5660–5670. doi: 10.1523/JNEUROSCI.0065-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zangerl B, Wickstrom K, Slavik J, Lindauer SJ, Ahonen S, Schelling C, Lohi H, Guziewicz KE, Aguirre GD. Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3) Molecular vision. 2010;16:2791–2804. [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Davidson BR, Stamer WD, Barton JK, Marmorstein LY, Marmorstein AD. Enhanced inflow and outflow rates despite lower IOP in bestrophin-2-deficient mice. Investigative ophthalmology & visual science. 2009;50:765–770. doi: 10.1167/iovs.08-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Stanton JB, Wu J, Yu K, Hartzell HC, Peachey NS, Marmorstein LY, Marmorstein AD. Suppression of Ca2+ signaling in a mouse model of Best disease. Human molecular genetics. 2010;19:1108–1118. doi: 10.1093/hmg/ddp583. [DOI] [PMC free article] [PubMed] [Google Scholar]