

Summary
We developed an RNA sequencing-based pipeline to discover differentially expressed cell surface molecules in neuroblastoma that meet criteria for optimal immunotherapeutic target safety and efficacy. Here we show that GPC2 is a strong candidate immunotherapeutic target in this childhood cancer. We demonstrate high GPC2 expression in neuroblastoma due to MYCN transcriptional activation and/or somatic gain of the GPC2 locus. We confirm GPC2 to be highly expressed on most neuroblastomas, but not detectable at appreciable levels in normal childhood tissues. Additionally, we demonstrate that GPC2 is required for neuroblastoma proliferation. Finally, we develop a GPC2 directed antibody-drug conjugate that is potently cytotoxic to GPC2-expressing neuroblastoma cells. Collectively, these findings validate GPC2 as a non-mutated neuroblastoma oncoprotein and candidate immunotherapeutic target.
Keywords: glypican, neuroblastoma, MYCN, immunotherapy, antibody-drug conjugate, medulloblastoma
Graphical abstract
Bosse et al. show that GPC2 is expressed at high levels on most neuroblastomas (NB) but not at appreciable levels in normal childhood tissues and that GPC2 is critical for NB maintenance. They develop a GPC2 directed antibody-drug conjugate that is cytotoxic to GPC2-expressing NB cells in vitro and in vivo.
Introduction
Outcomes for children with high-risk neuroblastoma remain poor with long-term survival remaining less than 50% despite the dramatic intensification of cytotoxic therapy (Maris, 2010). Given the relative paucity of activating mutations in kinases or other druggable oncogenic molecules in primary neuroblastomas, alternative modalities must be sought to improve outcomes (Bosse and Maris, 2016). Neuroblastomas arise from neural crest progenitor cells of the developing sympathetic nervous system and thus continue to express biologically critical cell surface molecules not found on more mature tissues, potentially making them amenable to immune-based therapies (Maris, 2010). Recently the first neuroblastoma immunotherapeutic strategy utilizing a chimeric monoclonal antibody targeting the cell surface disialoganglioside GD2 showed improved outcomes in a randomized phase 3 clinical trial, credentialing immunotherapeutics for this malignancy (Yu et al., 2010). However, this therapy is quite toxic due to the presence of GD2 on nocireceptor containing peripheral nerves, and relapses are common on or after therapy (Yu et al., 2010). Thus, alternative immunotherapeutic strategies are urgently needed.
Despite decades of research on GD2 in neuroblastoma (Suzuki and Cheung, 2015), the remainder of the neuroblastoma cell surface proteome (surfaceome) remains unexplored. Furthermore, although this monoclonal antibody targeting GD2 has become the first FDA approved therapy for children with this disease, other types of immune-based therapies, such as antibody-drug conjugates (ADCs), have not been studied in this malignancy largely due to the lack of available differentially expressed cell surface molecules coupled with associated targeted therapeutics. Thus, a major hurdle to the development of effective immune-based therapies for pediatric cancers is in identifying tumor-specific cell surface molecules with limited expression on normal childhood tissues. Ideally these differentially expressed cell surface molecules will also be required for tumor sustenance, a second criterion that is becoming recognized as necessary to produce a long-lasting tumor cytotoxic effect without immune escape (Sotillo et al., 2015).
Glypicans are a family of six (GPC1-6) glycosylphosphatidylinositol (GPI)-anchored, extracellular proteoglycan signaling co-receptors that play diverse roles in growth factor signaling and cancer cell growth (Filmus et al., 2008; Matas-Rico et al., 2016). The glypican family member glypican-3 (GPC3) has been validated as a bona fide immunotherapeutic target in hepatocellular carcinoma (Gao et al., 2015). Here we use an RNA sequencing-based approach, coupled with protein level validation and tumor cell functional studies, to identify differentially expressed cell surface molecules in high-risk neuroblastoma that meet the criteria for an immunotherapeutic target.
Results
Identification of glypican-2 (GPC2) as a candidate immunotherapeutic target in high-risk neuroblastoma
To identify proteins that might be most amenable to immunotherapeutic treatment approaches directed against high-risk neuroblastomas, we developed a multi-step discovery and prioritization algorithm (Figure 1A). First, to identify genes differentially expressed in neuroblastomas compared to normal tissues we compared RNA sequencing data from 126 high-risk primary neuroblastomas profiled via the Therapeutically Applicable Research to Generate Effective Treatments project (TARGET; https://ocg.cancer.gov/programs/target) to normal tissue RNA sequencing data (n = 7,859 samples across 31 unique normal tissues, range = 5 – 1,152 samples per tissue; GTEx; https://www.gtexportal.org) (Figure 1A). This approach identified 296 (0.48%) significantly differentially expressed genes (log-fold change (logFC) tumor vs. normal >1 for each tissue; adjusted p < 0.01) (Figures 1A and 1B). Notable among this list and validating this approach, ALK and L1CAM are cell surface molecules that have been previously explored for immune-based therapies in neuroblastoma (Carpenter et al., 2012; Kunkele et al., 2017). Next, to further identify those differentially expressed genes that have the qualities of an optimal candidate immunotherapeutic target, including potentially having extracellular epitopes susceptible to cell or protein based immunotherapeutics and being highly expressed in the majority of neuroblastomas cases, we further filtered this gene list by in silico plasma membrane prediction (11%, 33 of 296 genes) and by neuroblastoma tumor absolute RNA expression (9 of 33 genes; mean FPKM >50; Figures 1A and 1B). Based on these analyses, we prioritized the extracellular glycosylphosphatidylinositol (GPI) anchored signaling co-receptor glypican-2 gene (GPC2) for further validation based upon several factors (Figures 1A-C). First, GPC2 was found to have robust differential RNA expression (logFC tumor vs. normal tissue = 1.71 - 9.22; p = 1.99 × 10-9 - 1.88 × 10-300; Figure 1C), which we subsequently validated in a unique set of comprehensive normal tissue RNA sequencing studies (n = 32 unique normal tissues; http://www.proteinatlas.org/) (Uhlen et al., 2015). Next, we found most neuroblastomas to have high-level absolute GPC2 expression (median FPKM = 60; 87% of tumors with an FPKM > 20) and somatic DNA copy number gain of chromosome arm 7q, which includes the GPC2 locus encoded at chromosome 7q22.1, occurs in approximately 40% of primary neuroblastomas (Figure 1C) (Pugh et al., 2013). Further, GPC2 was similarly identified using mRNA microarray analysis as being differentially expressed between multiple pediatric tumors, including neuroblastomas, and normal tissues, further validating our findings here (orentas et al., 2012). Finally, the glypican family of proteins have been found to be involved in tumorigenesis and validated as safe and efficacious molecules to target with immune-based therapies (Filmus et al., 2008; Gao et al., 2015; Matas-Rico et al., 2016).
Figure 1. Identification of GPC2 as a differentially expressed cell surface molecule in high-risk neuroblastoma.
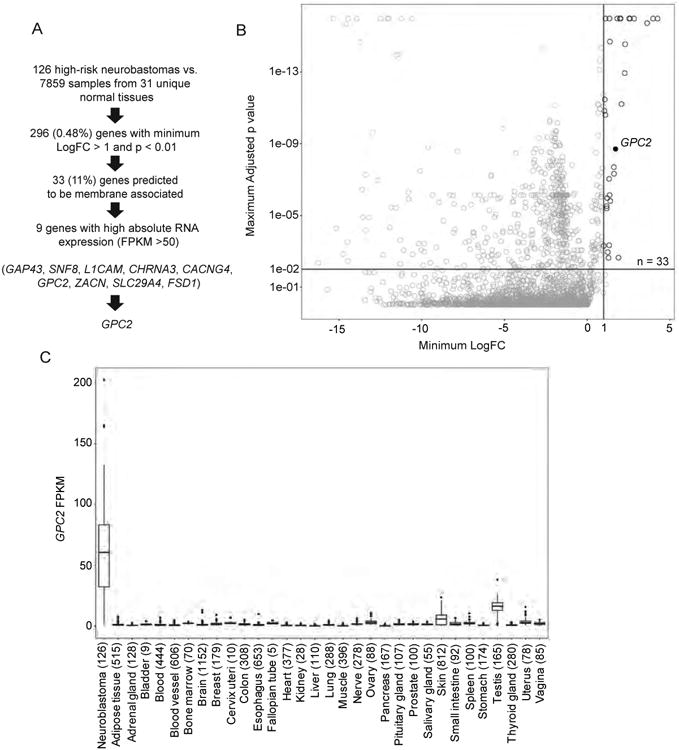
(A) Prioritization pipeline for identification of differentially expressed cell surface proteins in high-risk neuroblastoma.
(B) Plot displaying identification of 33 differentially expressed genes encoding cell surface proteins in high-risk neuroblastoma. GPC2 indicated with black circle.
(C) Plot displaying GPC2 expression in high-risk neuroblastoma (n = 126) compared to normal tissue RNA sequencing data profiled via the GTEx consortium (n = 7,859 samples across 31 unique normal tissues, n = 5 - 1,152 samples per tissue). Box plots extend from the first to the third-quartile, the horizontal line is the median, and the error bars represent the 1.5 interquartile range from the first-and third-quartile. n for each tissue indicated in parentheses.
LogFC, log fold-change.
See also Figures S1 and S2.
Given the significant differential GPC2 expression revealed by our discovery analysis (Figure 1C), we next queried the expression of the other glypican family members (GPC1, GPC3-6). Analysis of both our neuroblastoma RNA sequencing data and an additional large RNA sequencing data set (n = 498 tumors; SEQC) (Zhang et al., 2015) showed that GPC2 is the predominantly expressed glypican in neuroblastoma, and further that GPC2 is the only differentially expressed glypican between high-risk neuroblastomas and normal tissues (Figures S1 and S2A). However, the small subset of primary neuroblastomas with low GPC2 expression consistently showed high GPC3 expression (Figure S2B).
Next, to investigate if high GPC2 expression may be a clinically relevant finding in neuroblastoma, we queried for any association between GPC2 expression and overall neuroblastoma patient survival using three large neuroblastoma tumor data sets with available survival endpoints via the Genomics Analysis and Visualization Platform (R2; http://r2.amc.nl; Kocak; n = 649, SEQC; n = 498, and Versteeg; n = 88; Figures S2C and S2D) (Kocak et al., 2013; Valentijn et al., 2012; Zhang et al., 2015). These analyses revealed that high GPC2 expression is associated with worse overall survival consistently across these three data sets (Figure S2C), especially in neuroblastoma patients with high-GPC2 expressing, MYCN non-amplified tumors (Figure S2D). We next utilized the SEQC neuroblastoma RNA sequencing dataset to examine for any significant correlation between GPC2 expression and neuroblastoma clinical risk group (low-, intermediate- or high-risk) and found significantly higher GPC2 expression in the tumors of patients with high-risk neuroblastoma (p < 0.0001). Finally, considering clinically important relapsed neuroblastomas, we additionally examined RNA sequencing data from primary tumor vs. relapsed paired samples (n = 7 paired intermediate and high-risk neuroblastomas; TARGET) and found no overall difference in GPC2 expression between these tumor subsets (p > 0.05). However, similar to the above analysis, the one paired primary-relapsed tumor set that had a significant decrease in GPC2 expression from the primary to the relapsed tumor (21-fold decreased GPC2 expression; FPKM of 43.29 to 2.06), had a concurrent significant increase in GPC3 expression in the relapsed tumor (20-fold increased GPC3 expression, FPKM of 8.66 to 180.66).
Somatic genomic gain and MYCN are drivers of aberrant GPC2 expression in neuroblastoma
Considering that GPC2 is the major cell surface glypican that is differentially over-expressed in neuroblastoma (Figures S1 and S2A), we next sought to further define the neuroblastoma specific drivers of high GPC2 expression. GPC2 is localized on chromosome 7q22.1, which is somatically gained in approximately 40% of high-risk primary neuroblastoma tumors, typically either as entire chromosome 7 or chromosome arm 7q gain (Pugh et al., 2013). Although not as well defined as a prognostic marker as other acquired segmental chromosomal aberrations in primary neuroblastomas, somatic gain of chromosome 7 is a frequent and potentially biologically relevant event in neuroblastoma tumorigenesis (Stallings et al., 2003). First, to determine any potential correlation between somatic gain of chromosome 7q/GPC2 with GPC2 expression, we stratified and compared tumors across 7q/GPC2 copy numbers in two high-risk neuroblastoma tumor data sets with paired GPC2 expression and genomic copy number data (TARGET; n = 64 and n = 118 tumors, respectively) and found that genomic gain of chromosome 7q/GPC2 was consistently associated with significantly higher GPC2 expression (Figure 2A). Additionally, this stratification revealed that MYCN amplification was also associated with significantly higher GPC2 expression, most notably in those tumors that were diploid at chromosome 7q (Figure 2A). These findings suggested the possibility of a biologic intersection between GPC2 and MYCN. Thus, we next confirmed that GPC2's promoter contains a canonical MYCN binding E-box motif (CACGTG) (Figure 2B). To determine if MYCN directly mediates GPC2 expression we performed MYCN chromatin immunoprecipitation (ChIP) sequencing studies in MYCN-amplified Kelly, NGP, and NB-1643 neuroblastoma cells. These experiments revealed that MYCN binds the endogenous GPC2 promoter at the site of the E-box motif (Figure 2B). MYCN was not found to significantly bind to the other glypicans, except for GPC1, despite the multiple E-box motifs present in these genes. Next, utilizing a GPC2 luciferase reporter assay in MYCN tolerant SHEP neuroblastoma cells and HEK293T cells, we additionally confirmed MYCN as a significant driver of GPC2 expression (Figure 2C). Further, utilizing SKNAS-NmycER cells we selectively activated the NmycER protein with addition of 4-hydroxytamoxifen (4-OHT) and observed concurrent and significant upregulation of GPC2 (Figure 2D) (Valentijn et al., 2005). Finally, we performed MYCN depletion studies in MYCN-amplified Kelly and NGP cells and found that MYCN depletion correlates with significant downregulation of GPC2 expression at both the RNA and protein level (Figures 2E and 2F). Collectively, these data suggest that MYCN mediates neuroblastoma GPC2 differential expression and further that gain of chromosome 7q also correlates with higher GPC2 expression in neuroblastoma tumors.
Figure 2. Mechanisms of increased GPC2 expression in neuroblastoma.
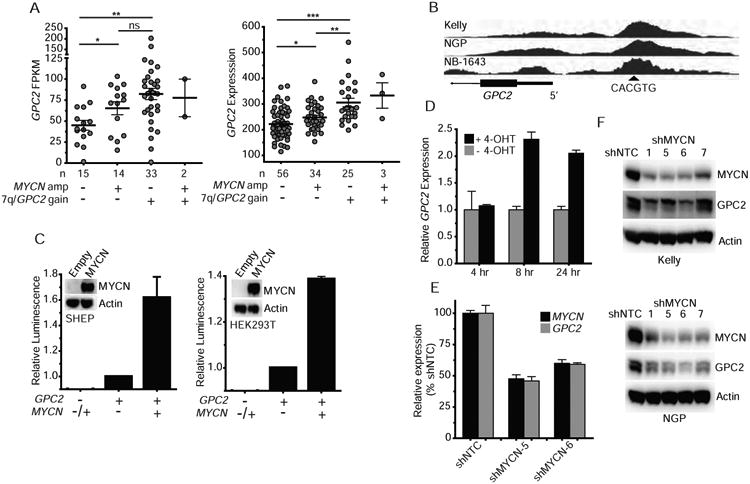
(A) GPC2 expression in two neuroblastoma tumor data sets stratified by chromosome 7q/GPC2 locus gain and MYCN amplification status (left, RNA sequencing, n = 64; right, mRNA microarray, n = 118; TARGET).
(B) MYCN ChIP plot showing MYCN binds an Ebox motif upstream of the GPC2 promoter in the MYCN amplified neuroblastoma cell lines Kelly, NGP, and NB-1643. Arrow head indicates location of Ebox motif (CACGTG).
(C) GPC2 reporter assay with and without forced overexpression of MYCN in SHEPneuroblastoma and HEK293T cells. Inset with Western blot displaying MYCN overexpression incorresponding cells.
(D) GPC2 quantitative PCR in 4-oHT MYCN inducible SKNAS neuroblastoma cells (SKNAS-NmycER) ± 4-OHT.
(E) MYCN/GPC2 quantitative PCR in the MYCN amplified neuroblastoma cell line Kelly after MYCN depletion with 2 unique shRNAs, shMYCN-5 and 6.
(F) Western blot of MYCN and GPC2 in the MYCN amplified neuroblastoma cell lines Kelly and NGP after MYCN depletion with an expanded set of 4 unique shRNAs, shMYCN-1, 5, 6 and 7.
Horizontal lines in A indicate mean ± SEM. Data in C are represented as mean ± SEM of at least 3 biological replicates. Data in D and E are represented as mean ± SEM of one experiment. Each experiment was repeated 2-4 times with similar results.
*p < 0.05; **p < 0.01; ***p < 0.0001; ns, not significant were derived via unpaired t tests.
Amp, amplification; Empty, pLenti CMV Puro empty vector; MYCN, MYCN pLenti CMV Puro vector; hr, hour. shNTC, non-targeting control shRNA.
Plasma membrane localized GPC2 is expressed in a majority of high-risk neuroblastomas
We next sought to confirm high-level cell surface localized GPC2 protein expression across high-risk neuroblastomas. First, using Western blot analyses with a highly specific monoclonal antibody targeting GPC2 C-terminus epitopes we confirmed GPC2 expression in all high- and intermediate-risk neuroblastoma primary tumors (n = 11), patient-derived xenografts (PDXs) derived from high-risk human primary tumors (n = 12) and a majority of neuroblastoma cell lines (n = 24; Figures 3A-C and S3A; Table S1). Similar to other members of the glypican family, GPC2 is expressed on neuroblastoma cells as both a core 62 kDa protein, and also as a higher molecular weight glycanated GPC2 species (80-100 kDa) with glycosaminoglycan post-translation modifications (Figures 3A-C and S3A; Table S1). GPC2 expression quantified by Western blot across our neuroblastoma cell panel (Figure 3C; Table S1) correlated strongly with GPC2 expression measured by RNA sequencing (Pearson r = 0.57, p < 0.01) (Harenza et al., 2017). We additionally performed immunohistochemistry (IHC) on eight of these neuroblastoma cell lines, showing dense GPC2 staining in a membranous pattern and in exact accordance with our Western blotting data (Figures 3C, 3D, and S3B; Table S1). Next, we performed flow cytometry analysis on two neuroblastoma cell lines, the GPC2 isogenic engineered neuroblastoma cell line Kelly (Kelly GPC2), along with several non-neuroblastoma, low-GPC2 expressing control cell lines, and these data further confirmed neuroblastoma specific cell surface localization of both endogenously and exogenously over-expressed GPC2 (Figure 3E). Additionally, Western blot of membrane extractions from seven neuroblastoma cell lines confirmed that both the core GPC2 (62 kDa) and glycanated GPC2 protein species (80-100 kDa) are associated with the cell membrane (Figure 3F). Next, immunofluorescence studies in neuroblastoma cell lines showed that GPC2 co-localized with the abundantly expressed neuroblastoma cell surface family of cadherin proteins (Figures 3G and S3C). Finally, we performed IHC on 98 neuroblastoma tumors (all stages represented) and 32 PDXs from high-risk human primary tumors in tissue microarrays (Figures 3H and 3I). GPC2 was found to be expressed in 93% (91/98) of neuroblastoma tumors (1/1 recurrent, 4/4 post-treatment, and 86/93 tumors at diagnosis), and 94% (30/32) of PDXs. Further, 73% (95/130) of tumors (human or PDX) had a membrane staining H-score of 100 or greater, confirming high-level GPC2 cell surface expression in a majority of neuroblastoma tissues (Figures 3H and 3I). Taken together with our discovery RNA sequencing neuroblastoma tumor profiling, these data show that a majority of high-risk neuroblastoma tumors have elevated GPC2 mRNA and cell surface protein expression.
Figure 3. GPC2 is expressed in most neuroblastomas and is plasma membrane localized.
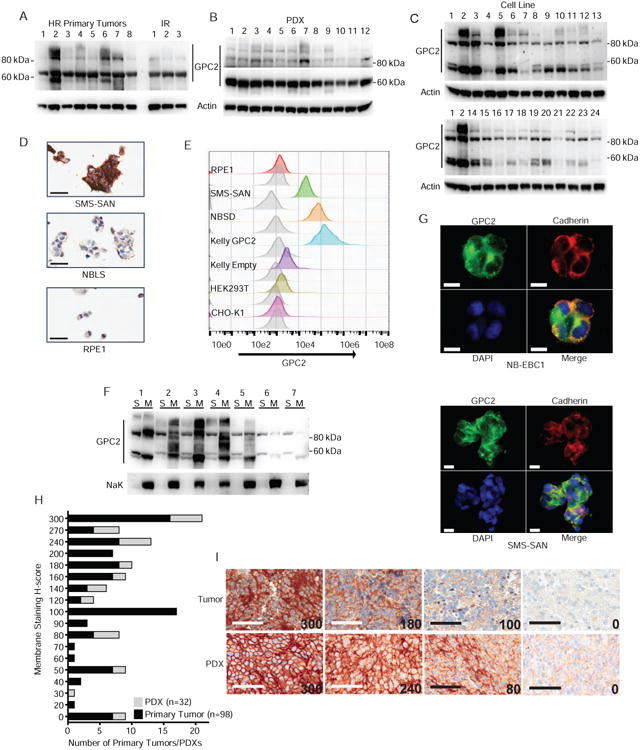
(A-C) Western blots of GPC2 in a panel of neuroblastoma primary tumors (n = 11; A), PDXs (n = 12; B) and cell lines (n = 24; C).
(D) GPC2 IHC staining of neuroblastoma cell lines (high GPC2 expression - SMS-SAN,moderate - NBLS, and very low - RPE1).
(E) GPC2 flow cytometry analysis of a panel of cell lines (high GPC2 expression - SMS-SAN,NBSD, Kelly GPC2, low – Kelly Empty, and very low - RPE1, HEK293T, and CHo-K1). Greyplot represents secondary only staining and colored plots represent staining with D3-GPC2-scFv-FLAG.
(F) Western blot of GPC2 following differential membrane extraction experiments in a panel of neuroblastoma cell lines (n = 7). Western blot of Na, K-ATPase protein also shown as a positive plasma membrane control.
(G) GPC2 immunofluorescence staining in the neuroblastoma cell lines NB-EBC1 and SMS-SAN. Green, GPC2; Red, pan cadherin membrane staining control; Blue, DAPI; Bottom right, merged figure.
(H) Summary of membrane staining H-score of GPC2 IHC of neuroblastoma PDX and primary tumor TMAs.
(I) Representative membrane staining H-score examples (H-score displayed in lower right corner).
Scale bars represent 30 μM (D), 10 μM (G), 60 μM (I)
HR, high-risk; IR, intermediate risk; S, soluble (non-membrane) protein extract; M, membrane protein extract.
See also Figure S3; Table S1.
GPC2 expression is restricted in normal tissues
To be a safe immunotherapeutic target, a cell surface molecule must have limited cell surface expression on normal human tissues. Additionally in pediatric cancers, special attention to normal childhood tissues is critical for these embryonal neoplasms that deregulate developmental pathways. Here we first show that GPC2 mRNA expression is highly restricted in normal human tissues (Figure 1C), including normal neural crest cells (Figure S4A). Next, to confirm this robust differential GPC2 expression at the protein level, we utilized a comprehensive normal pediatric tissue microarray (TMA) that covers all of the major human organs (n = 36 unique pediatric normal tissues), in addition to staining additional neural and neural crest derived tissues considering the embryonal origins of neuroblastoma (Figures 4A and S4; Table S2). By IHC analysis, we confirmed highly restricted GPC2 protein expression in normal tissues (Figures 4A and S4; Table S2). Only 16% (6/36) of the pediatric tissues showed weakly positive and cytoplasmic localized GPC2 staining, and only 2% (1/36; thyroid gland) showed strongly positive cytoplasmic staining (Figure S4; Table S2). Further, only 5% (2/36; skin and esophagus) of pediatric tissues showed possible weak membrane-associated (cytoplasmic/membranous) staining, however with a membrane staining H-score significantly lower than that in neuroblastoma primary tumors or PDXs (Figures 4A and S4; p < 0.01). This limited normal tissue GPC2 protein expression was further confirmed by querying mass spectrometry based Human Proteome Map data (n = 30 normal tissues; http://www.humanproteomemap.org), which showed low level GPC2 expression in the adult testes and the developing fetal brain consistent with our findings here (Kim et al., 2014).
Figure 4. Restricted normal tissue expression of GPC2 and alternative GPC2 transcript expression.
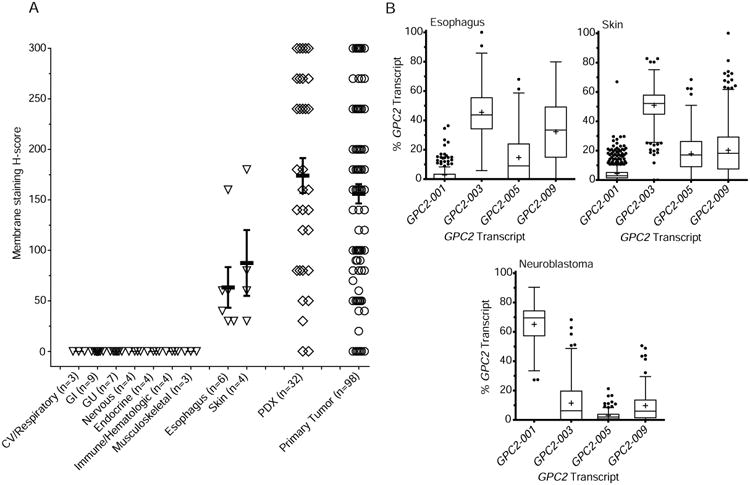
(A) Summary of GPC2 membrane staining H-scores from a pediatric normal tissue array (total n = 36 unique normal tissues, n of each organ system or individual tissue indicated on X-axis). Summary of GPC2 membrane H-scores from neuroblastoma primary tumors/PDXs shown for comparison.
(B) RNA transcript specific analysis of GPC2 expression in primary neuroblastomas and the low-level GPC2 expressing normal tissues skin and esophagus (n = 126 high-risk neuroblastomas, TARGET; n = 201 esophagus samples and 684 skin samples, GTEx).
Data in A represent means ± SEM. Box plots in B extend from the first to the third-quartile, the horizontal line is the median, “+” represents the mean and the error bars represent the 1.5 interquartile range from the first-and third-quartile.
CV, cardiovascular; GI, gastrointestinal; GU, genitourinary.
See also Figure S4; Table S2.
We additionally sought to investigate the possibility of tumor specific GPC2 epitope expression potentially driven by unique tumor derived GPC2 mRNA variants that may further enhance specific GPC2 targeting with immune-based therapies (Casucci et al., 2013). Comparing RNA sequencing data from 126 high-risk neuroblastoma tumors (TARGET) to available GTEx normal tissue RNA sequencing data, we identified a tumor-predominant and specific GPC2 mRNA variant (GPC2-001; ENST00000292377.2) with very restricted normal tissues expression (Figure 4B; Table 1). Alternatively, most normal tissues preferentially express three alternative GPC2 transcripts (GPC2-003 > GPC2-005 ≫ GPC2-009) with very low levels of the neuroblastoma predominant GPC2-001 transcript, including the esophagus and skin (Figure 4B; Table 1). GPC2-001 and GPC2-003 have large open reading frames (ORFs) encoding 62 kDa and 34 kDa GPC2 protein isoforms, respectively, utilizing the same stop codon, however GPC2-003 is predicted to undergo nonsense mediated decay (Table 1). Further, the tumor specific GPC2-001, but not the normal tissue predominant GPC2-003, translates to a protein with an intact secretory signaling peptide essential to GPC2 transport and localization to the cell surface (Table 1). The other predominant normal tissue GPC2 mRNA variants, GPC2-005 and GPC2-009, have much smaller predicted ORFs and are either not predicted to encode a signaling peptide or the C-terminus GPI and heparin sulfate protein domains, or both in the case of GPC2-009 (Figure 4B; Table 1) (Filmus et al., 2008). Taken together, GPC2 is not only a robustly differentially expressed cell surface molecule, but there may also be tumor specific GPC2 epitopes that could be exploited by GPC2-directed immunotherapies.
Table 1.
Identification of tumor specific GPC2 transcripts.
| Name | Ensemble Transcript ID | BPs | Exons | Amino Acids (molecular mass) | Predominant isoform? | PredictedSPa | GPI / HS domains |
|---|---|---|---|---|---|---|---|
| GPC2-001 (GPC2) | ENST00000292377.2 | 2532 | 1-10 | 579 (62 kDa) | Neuroblastomas, medulloblastomas, testes | Yes | Yes |
| GPC2-003 | ENST00000471050.1 | 1652 | 4-10 | 325 (34 kDa) | All normal tissues (non-testes) | No | Yes |
| GPC2-005 | ENST00000480087.1 | 767 | 1-2, 4-6 | 134 (14 kDa) | - | Yes | No |
| GPC2-009 | ENST00000482569.1 | 473 | 1-2 | 98 (10 kDa) | - | No | No |
Signal peptide (SP) predictions done via SignalP 4.0 and PrediSi located at http://www.cbs.dtu.dk/services/SignalP and http://www.predisi.de/, respectively. BPs, base pairs; GPI, glycosylphosphatidylinositol; HS, heparan sulfate.
GPC2 is required for neuroblastoma cell proliferation
A tumor's ability to escape from immunotherapy via down-regulation of the cell surface target has arisen as a critical vulnerability of immunotherapeutic treatment approaches (Sotillo et al., 2015). Thus, here we sought to identify cell surface molecules that are not only differentially expressed but also required for tumor sustenance. Accordingly, we next sought to determine if neuroblastoma cells were dependent on GPC2 for cellular growth by using complimentary genetic gain and loss of function studies in neuroblastoma preclinical models across an array of phenotypic assays. We first depleted GPC2 using 2 unique lentiviral shRNA constructs in the neuroblastoma cell line NB-EBC1 and found that GPC2 depletion resulted in programmed cell death accompanied by significant decreases in NB-EBC1 cell growth, both in transient and in longer term growth assays (Figures 5A-F). We next extended these observations to a panel of ten additional neuroblastoma cell lines with a broad range of GPC2 expression. GPC2 depletion resulted in the induction of apoptosis and an associated decrease in cell proliferation in a majority, but not all, of the cell lines we tested both in these transient and more extended growth assays (Figures 5G and S5). To complement these GPC2 loss of function studies, we also stably over-expressed GPC2 in two low GPC2-expressing neuroblastoma cell lines, Kelly and SKNDZ, which resulted in significantly increased cell proliferation (Figures 5B and 5H). The neuroblastoma cell line Kelly has a heterozygous deletion of a large portion of chromosome 7q including the GPC2 locus. Finally, to begin to explore GPC2's role in neuroblastoma tumorigenesis, we performed an unbiased pathway analysis using the MetaCore™ algorithm in two large cohorts of high-risk neuroblastoma RNA sequencing data (TARGET and SEQC), stratified by MYCN amplification status, comparing the GPC2 expression top and bottom 10th percentiles. These analyses showed that, similar to GPC3 in other malignancies (Gao et al., 2015), GPC2 may play an important role in the WNT signaling pathway in neuroblastomas as this was among the top 10 GPC2 associated pathways in all four of these independent analyses (rank of 1st, 3rd, 5th, and 8th, respectively; false discovery rate (FDR) = 2.57 × 10-2 - 1.48 × 10-9; Figure S5K). Collectively, these functional data, taken together with high GPC2 expression being associated with high-risk disease and worse overall survival in patients with neuroblastoma (Figure S2C and S2D), suggest that GPC2 is a neuroblastoma lineage restricted oncoprotein and a critical regulator of neuroblastoma cellular growth. These properties may make GPC2 a cell surface molecule that is less susceptible to immune escape mechanisms.
Figure 5. GPC2 is required for neuroblastoma cell growth.
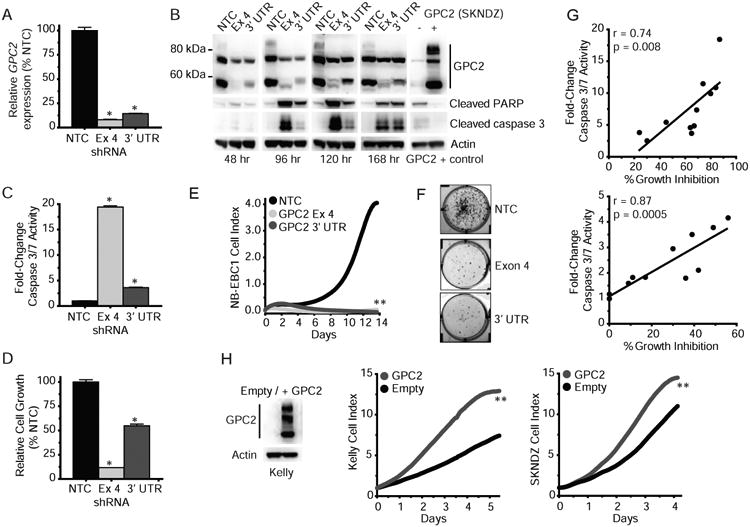
(A) GPC2 quantitative PCR following lentiviral transduction of 2 unique shRNA constructs targeting GPC2 exon 4 and the GPC2 3′ UTR in the neuroblastoma cell line NB-EBC1.
(B) Top, GPC2 Western blot analysis of samples in (A). Bottom, Western blot of cleaved PARP and caspase 3 after GPC2 depletion in NB-EBC1. Positive GPC2 Western blot control (SKNDZ) was run on the same blot as 120/168 hour NB-EBC1 time points.
(C) Caspase 3/7 activity measured after GPC2 depletion in NB-EBC1.
(D-F) NB-EBC1 cell growth following GPC2 depletion shown by CellTiter-Glo® assay (D), RT-CES (E), and colony formation assay (F).
(G) Plot of cell growth measured by CellTiter-Glo® assay and caspase 3/7 activity following GPC2 depletion with 2 unique shRNA constructs targeting GPC2 exon 4 (top) and the GPC2 3′ UTR (bottom) across an extended panel of neuroblastoma cell lines (n = 10 + NB-EBC1). r, Pearson correlation coefficient and p values shown for each GPC2 shRNA.
(H) Neuroblastoma cell growth after forced GPC2 overexpression in Kelly and SKNDZ. Western blot of GPC2 overexpression in Kelly is shown on left and for SKNDZ cells is shown in B. Data in A, C, D are represented as mean ± SEM.
Data in E and H are represented as means of at least a triplicate plating. Data in G are means of 2 separate biological replicates plated in triplicate. Each experiment was done 2-3 independent times with similar results.
*p < 0.0001; **p < 0.001 derived via unpaired t tests.
NTC, non-targeting shRNA control; Ex 4, GPC2 Exon 4 targeting shRNA; 3′ UTR, GPC2 3′ UTR targeting shRNA; Empty, empty pLenti CMV Puro vector; GPC2, GPC2 pLenti CMV Puro vector; hr, hours.
See also Figure S5.
GPC2 is highly expressed in other aggressive pediatric embryonal malignancies
To determine if high GPC2 expression was also present in additional pediatric cancers, we next examined available GPC2 RNA sequencing and gene expression profiling data across an array of pediatric malignancies, including data from the TARGET project, the St. Jude Children's Research Hospital Pediatric Cancer Data Portal (PeCan; https://pecan.stjude.org), and the Genomics Analysis and Visualization Platform (R2; http://r2.amc.nl). Both the embryonal pediatric brain tumor medulloblastoma and the ocular tumor retinoblastoma showed GPC2 expression levels comparable to high-risk neuroblastoma (medulloblastoma total n = 612; retinoblastoma total n = 67 versus neuroblastoma total n = 544; Figures S6A and S6B). Additionally, subsets of other pediatric malignancies (ALL, HGG, and RHB) also express high levels of GPC2 (Figure S6A). We further validated the medulloblastoma observation in an additional set of 97 medulloblastomas subjected to RNA sequencing, which showed significantly higher GPC2 expression in group 4 tumors (Figure 6A, left). Furthermore, similar to our results in neuroblastoma, we also found that GPC2 expression positively correlated with both genomic gain of MYCN or MYC and the GPC2 locus on chromosome 7q in a synergistic manner, however the correlation of MYC(N) gain alone with GPC2 expression in GPC2 diploid tumors is more modest than the correlations we observed in neuroblastoma tumor data sets (Figure 6A, right). Of note, one tumor had a focal GPC2 locus gain and was found to have the highest GPC2 expression in this data set (red circle; Figure 6A). Further, similar to neuroblastoma and distinct from most normal tissues, medulloblastomas primarily express a GPC2 transcript with 10 exons (GPC2-001; Figure 6B; Table 1). Importantly, we next confirmed high-level GPC2 cell surface expression in a majority of diagnostic medulloblastoma tissues via a TMA, showing that 90% (57/63) of tumors stained positive for GPC2 via IHC, with 63% (40/63) of tumors having a membrane staining H-score of 100 or greater (Figures 6C and 6D). Finally, we found that high-level GPC2 expression is maintained in the metastatic medulloblastoma tumor compartment as GPC2 RNA expression is similar in human primary and paired metastatic medulloblastoma samples (p > 0.05; Figure S6C) (Wang et al., 2015), and 4 of 4 distinct human metastatic medulloblastoma xenograft murine models robustly express GPC2 in both CNS, spinal and non-CNS (liver) metastatic lesions (Figure 6E; Table S3). Finally, for retinoblastomas we confirmed dense cell surface GPC2 expression by IHC staining in 3 of 3 primary tumors, but absence of significant GPC2 staining in the accompanying sclera, retina or optic nerve (Figures 6F and S4E; Table S2). We also confirmed retinoblastoma associated nuclear MYCN expression with IHC staining of these 3 specimens as MYCN is also amplified and/or highly expressed in a subset of these tumors (Figure 6F).
Figure 6. GPC2 is expressed in other high-risk neural-derived embryonal cancers.
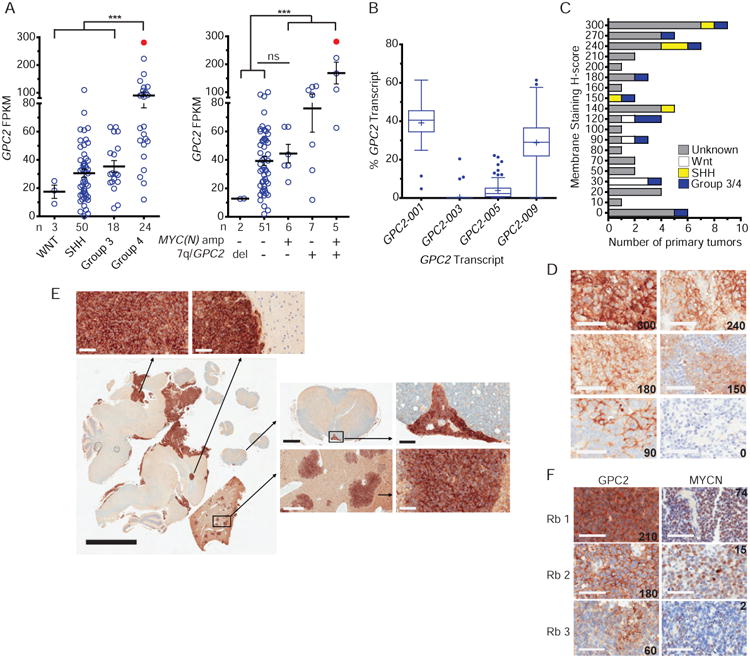
(A) GPC2 RNA sequencing data of medulloblastomas stratified by clinical grouping (n = 95; left) and amplification status at chromosome 7q/GPC2, MYC and MYCN loci (n = 71; right). n indicates number of tumors in each group. Tumor with focal GPC2 locus gain colored in red.
(B) mRNA transcript specific analysis of GPC2 in primary medulloblastomas (n = 96).
(C) Summary of GPC2 membrane staining H-scores of medulloblastoma TMA (n = 63).
(D) Representative membrane staining H-score examples from medulloblastoma TMA (H-score displayed in lower right).
(E) GPC2 IHC staining in a human metastatic medulloblastoma xenograft murine model, withhigher power images of GPC2 staining central nervous system (left), spinal (top right), and livermetastases (bottom right).
(F) GPC2 and MYCN IHC staining of three retinoblastomas. GPC2 membrane staining H-scoreshown in lower right and MYCN copy number shown in upper right corners.
Mean in A indicated by horizontal line ± SEM. Box plots in B extend from the first to the third-quartile, the horizontal line is the median, “+” represents the mean and the error bars represent the 1.5 interquartile range from the first-and third-quartile. Scale bars in D, E (top and right), and F represent 60 μM, E (left) represents 5 μM, E (top middle; spinal cord) represents 500 μM and D (bottom middle; liver) represents 300 μM.
***p < 0.0001 derived via unpaired t test. ns, not significant; Del, deletion; Rb, retinoblastoma.
See also Figure S6; Table S3.
A GPC2 targeting antibody-drug conjugate (ADC) is cytotoxic to GPC2 expressing neuroblastoma cells
Other glypican family members have been well credentialed as suitable cell surface targets for antibody-toxin conjugates facilitating impressive rates of antibody internalization allowing efficient delivery of cytotoxic drugs specifically and safely to antigen expressing tumor cells (Gao et al., 2015). Thus, with an aim to develop a GPC2-directed ADC we first identified D3-GPC2-Fab from a human naïve Fab phage display library through panning and screening with recombinant full-length GPC2 protein. D3-GPC2-Fab showed high affinity for GPC2 with a dissociation rate constant (KD) of 9.5 × 10-11 M. We next converted D3-GPC2-Fab to both a D3-GPC2-scFv and a fully human IgG1, D3-GPC2-IgG1, both that also had high specificity for neuroblastoma cell surface GPC2 in flow cytometric analyses (Figures 3E, S7A and S7B). Additionally, D3-GPC2-IgG1 was robustly internalized into the lysosome by binding with extracellular GPC2 on neuroblastoma cells and was found to bind equally to human and mouse GPC2, but not human GPC1 or GPC3 via ELISA (Figures 7A, 7B, S7C and S7D). We next developed a GPC2 directed ADC, D3-GPC2-PBD, by conjugating D3-GPC2-IgG1 with pyrrolobenzodiazepine (PBD) dimers, achieving a drug-antibody ratio (DAR) of 2.6 (Figure 7C). PBD dimers are a potent cytotoxic DNA minor groove interstrand crosslinking agent that are increasingly being used for anticancer therapy (Mantaj et al., 2017; Saunders et al., 2015; Seaman et al., 2017), and was selected here because of the relatively higher potency of DNA damaging agents compared to tubulin binding agents in this disease.
Figure 7. A GPC2 targeting ADC is cytotoxic to GPC2 expressing neuroblastoma cells.
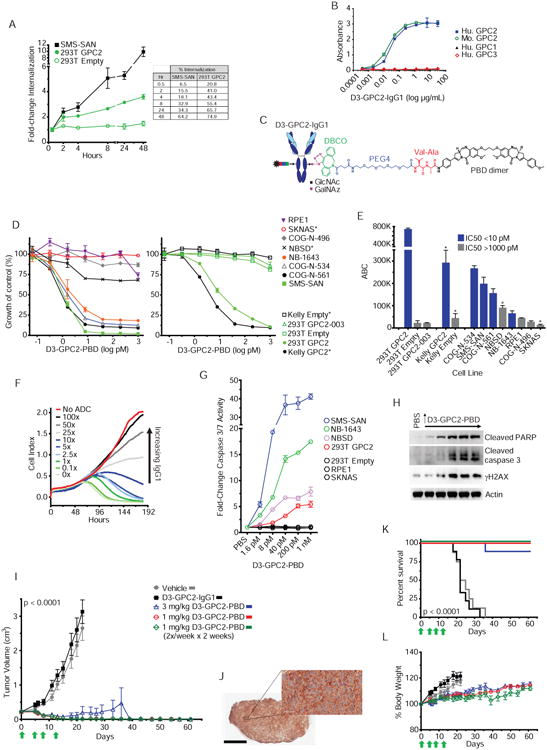
(A) Internalization of D3-GPC2-IgG1 in SMS-SAN and HEK293T GPC2 cells shown as fold-change from baseline (left; 30 minutes) and percent of D3-GPC2-IgG1 internalized (right).
(B) Plot showing D3-GPC2-IgG1 binding to human and mouse GPC2 but not to human GPC1 and GPC3 via ELISA.
(C) Schematic of D3-GPC2-PBD ADC.
(D) Growth plots of endogenous GPC2 expressing neuroblastoma cells (left) and GPC2 isogenic Kelly and HEK293T cells (right) treated with different concentrations of D3-GPC2-PBD.
(E) Summary of IC50 values and semi-quantitative flow cytometry analysis (ABC) with D3-GPC2-IgG1 of the cell lines treated with D3-GPC2-PBD in D.
(F) RT-CES growth plot of GPC2 expressing SMS-SAN cells treated with 200 pM of D3-GPC2-PBD and increasing amounts of D3-GPC2-IgG1 (0× to 100×).
(G) Fold-change caspase 3/7 activity 96 hours after treatment with D3-GPC2-PBD in 7 cell lines with differential GPC2 expression.
(H) Western blot of SMS-SAN cells 72 hours after treatment with D3-GPC2-PBD (1.6, 8, 40, and 200 pm and 1 nm).
(I) NB-1643 patient-derived xenograft (PDX) tumor volumes after treatment with D3-GPC2-IgG1 or D3-GPC2-PBD (n = 8-9 mice per treatment arm).
(J) GPC2 IHC of NB-1643 PDX.
(K) Kaplan-Meier survival analysis of NB-1643 treatment arms in I.
(L) Mean percent body weights from baseline of mice in the NB-1643 PDX treatment arms shown in I.
*, indicates parental cell lines with a TP53 mutation. Scale bars in J represents 2 μM (left) and 60 μM (right). Data in A, B, D, E, and G are represented as mean ± SEM and data in F is represented as a mean. Each experiment was done 2-3 independent times with similar results. Data in I and L represent mean ± SEM for each treatment arm (n = 8-9 mice).
RT-CES, Real-time cell electronic sensing; ABC, Antibody Binding Capacity; GlcNAc, N-Acetylglucosamine; GalNAz, N-azidoacetylgalactosamine tetraacylated; DBCO, dibenzocyclooctyne; PEG4, polyethylene glycol 4; Val-Ala, valine-alanine; PBD, pyrrolobenzodiazepine, Ab, antibody; Hu, human; Mo, mouse; 293T, HEK293T cells.
See also Figures S7, S8; Table S4.
To evaluate for D3-GPC2-PBD induced cytotoxicity, seven human neuroblastoma cell lines, including three recently established (CoG-N-496, CoG-N-534, and CoG-N-561), neural crest control low-GPC2 expressing RPE1 cells, and two sets of paired GPC2 isogenic human cell lines (Kelly and HEK293T GPC2) were treated with ADC concentrations ranging from 0.064 pM to 1 nM (Figures 7D and S8A; Table S4). Comparing these IC50 data to GPC2 cell surface molecule quantitation via antibody binding capacity (ABC) flow cytometric measurements across this same cell line panel showed that D3-GPC2-PBD mediated potent dose dependent cytotoxicity in direct relationship to GPC2 cell surface density (Figure 7E; Table S4). D3-GPC2-PBD responsive cell lines (IC50 ≤10 pM) had a significantly higher GPC2 ABC than less responsive cell lines (IC50 >1 nm; p < 0.05). These estimated ABC values correlated strongly with flow cytometry derived Molecules of Equivalent Soluble Fluorochrome (MESF) values across this cell line panel (Pearson r = 0.99, p < 0.0001; Table S4). To further ensure that neuroblastoma cytotoxicity was a GPC2 and ADC specific finding, we treated SMS-SAN and Kelly GPC2 cells with 200 pM D3-GPC2-PBD in the presence of increasing amounts of D3-GPC2-IgG1 (0× to 100× IgG1), and demonstrated abrogation of D3-GPC2-PBD-induced cytotoxicity in these models (Figures 7F and S8B). Additionally, we also tested a subset of these cell lines with free PBD dimer (0.064 pM to 1 nM) to ensure that this differential cytotoxicity to D3-GPC2-PBD was not due to differential susceptibility to free PBD dimer and we found no difference in free PBD IC50s across ADC susceptibility cohorts (n = 6; p > 0.05); (Figure S8C; Table S4). Considering tumor versus normal tissue differential GPC2 isoform expression (Figures 4 and 6; Table 1), HEK293T cells stably expressing the normal tissue predominate GPC2 isoform, GPC2-003 (ORF within GPC2 exons 5-10), were also engineered, analyzed by flow cytometry and treated with D3-GPC2-PBD in parallel with HEK293T cells overexpressing the neuroblastoma predominate GPC2-001 isoform (GPC2; ORF within GPC2 exons 1-10). These analyses showed that only the neuroblastoma predominant GPC2-001 trafficked to the membrane and was able to mediate D3-GPC2-PBD specific cytotoxicity (Figures 7D, 7E and S7B; Table S4).
Next, to investigate the mechanism of D3-GPC2-PBD induced cytotoxicity, we measured markers of apoptosis and DNA damage after treatment with D3-GPC2-PBD (Figures 7G, 7H and S8D). Treatment with D3-GPC2-PBD induced significant elevation of caspase 3/7 activity across these cellular models in a GPC2-specific fashion (Figures 7G and S8D). Further, elevation of cleaved PARP and cleaved caspase 3, in addition to increased levels of γH2AX, were found in GPC2-expressing SMS-SAN cells by Western blot after D3-GPC2-PBD treatment (Figure 7H). Of note, even at doses up to 1 nm of D3-GPC2-PBD, there was little change in overall GPC2 levels (Figure S8E). Finally, we tested D3-GPC2-PBD in a GPC2-expressing, MYCN amplified, ALK mutant (R1275Q) neuroblastoma NB-1643 PDX murine model, which showed potent in vivo efficacy at even a single 1 mg/kg dose of D3-GPC2-PBD, significantly prolonging survival (p < 0.0001; Figures 7I-L). ADC doses up to 3 mg/kg given once or 1 mg/kg given four times over two weeks were also well tolerated with no discernible toxicities and showed equal efficacy with a total of 26 of 27 mice across these three treatment cohorts with complete and sustained tumor regression over 8 weeks post study enrollment (p < 0.0001; Figures 7I-L). A dose of 7 mg/kg of D3-GPC2-PBD given once was not tolerated in this model. Importantly, the similar mouse and human GPC2 expression profile (Figures 1C and S8F) (Shen et al., 2012), coupled with the equal binding of this ADC to mouse and human GPC2 (Figure 7B), suggests murine models provide a valid assessment of the potential for D3-GPC2-PBD on-target, off-tumor toxicities. Collectively, these results show that D3-GPC2-PBD mediates neuroblastoma cellular cytotoxicity in an antigen and concentration dependent manner in vitro and is similarly efficacious and safe in an in vivo neuroblastoma murine PDX model.
Discussion
The limited landscape of clinically targetable mutated or activated oncogenes in pediatric cancers in general, and neuroblastoma in particular (Pugh et al., 2013), together with the recent success of immune-based therapies (Yu et al., 2010) creates solid rationale for focusing on developing immunotherapeutics for this childhood malignancy. However, a major challenge is identifying cell surface molecules that meet the stringent criteria for optimal immunotherapeutic safety and efficacy. For example, while GD2 has been credentialed as an immunotherapeutic target in neuroblastoma in a recent Phase 3 study of a GD2-directed chimeric antibody (Yu et al., 2010), GD2 expression on nociceptor cells causes significant on-target pain, and while survival is extended, late relapses after or during immunotherapy are frequent. Furthermore, the significant and homogenous GD2 expression in human brain tissues may limit the use of immunotherapeutics, like chimeric antigen receptor (CAR) T cells, that can cross the blood-brain barrier. The robust differential expression of GPC2 coupled with neuroblastoma's dependence on GPC2 may eliminate these immunotherapeutic liabilities. Further defining the GPC2-driven signaling pathways and extracellular binding partners in neuroblastoma, and the effect D3-GPC2-PBD has on these biological processes, will be critical both in understanding the potential of immune escape and also in the possible development of combinatorial therapeutic strategies.
As with any new candidate cell surface molecule for targeted immunotherapies, it is vital to carefully evaluate for the potential for any on-target, off-tumor toxicity. We confirmed very restricted normal tissue GPC2 expression using a multimodal approach, yet clearly close surveillance will still be needed for the limited low-GPC2 expressing normal tissues, in addition to neural-crest derived tissues, as GPC2 directed therapeutics are tested in additional preclinical studies. Recent examples suggest that this limited level of normal tissue cell surface GPC2 expression represents an adequate therapeutic window to allow safe administration of GPC2-directed therapies (Casucci et al., 2013; Kunkele et al., 2017). The two most relevant of these examples are that of CD44v6 and L1CAM, both of which have substantial normal tissue expression comparable to that in the respective tumors being studied, yet each of these cell surface molecules have been safely targeted with potent CAR T cells that have spared antigen expressing normal tissues (Casucci et al., 2013; Kunkele et al., 2017).
Importantly, our translation of these findings to initial preclinical studies utilizing a GPC2 directed ADC, D3-GPC2-PBD, fully credential GPC2 as a potentially safe immunotherapy targetable cell surface molecule in neuroblastoma and lay the foundation for further evaluation of GPC2-directed immune-based therapies. PBD dimers have several advantages over other ADC payloads, including utilizing a unique mechanism of action resulting in rapidly formed and more persistent PBD-DNA adducts with limited DNA helix disruption thus escaping DNA repair mechanisms (Mantaj et al., 2017). Consequently, it is not unexpected that PBD dimer containing ADCs have activity in multidrug resistant malignancies, a fact that is immediately relevant for children with neuroblastoma (Saunders et al., 2015; Seaman et al., 2017). Not surprising, TP53 mutation status may be important in dictating sensitivity to D3-GPC2-PBD. However, TP53 mutations are very rare in both diagnostic and relapsed neuroblastoma tumors (Bosse and Maris, 2016). Importantly, ADCs with the identical linker have recently been found to be stable in human and monkey serum, but exhibit some instability in mouse serum most likely due to selective cleavage by the carboxlesterase 1C enzyme in certain murine models (Seaman et al., 2017). However, the half-life of PBD dimers in vivo is very short and thus the likelihood of free toxin contributing to the in vivo tumor regression observed here is low (Mantaj et al., 2017; Seaman et al., 2017). Finally, by validating dense cell surface expression of GPC2 in medulloblastomas and retinoblastomas we have additionally nominated these lethal pediatric malignancies to also potentially benefit from GPC2-directed therapies.
Of potential interest, the rare neuroblastomas with low levels of GPC2 expression, invariably show high levels of GPC3 mRNA. Furthermore, although subsets of other pediatric malignancies may also overexpress GPC2, several pediatric neoplasms have consistently low GPC2 expression, but do selectively overexpress other glypicans (i.e. GPC3 in rhabdomysosarcoma, GPC4 in osteosararcoma, and GPC5 in low grade gliomas; TARGET, PeCan), again suggesting mutual exclusivity among glypicans. We suggest that the role of not only GPC2 but also the other glypicans should be further explored in pediatric malignancies in addition to their potential suitability as immunotherapeutic targets. Finally, although we focus here on GPC2, clearly the other 8 highly differentially expressed genes discovered in this analysis, and perhaps additional genes in neuroblastoma that don't meet the criteria of having a mean FPKM of >50 in neuroblastoma tumors, may also warrant consideration as immunotherapeutic targets.
Taken together, these data lay firm groundwork for the development of GPC2 directed immune-based therapies, including not only anti-GPC2 ADCs but also CAR T cells directed towards GPC2. Defining the subsets of other pediatric malignancies beyond these neural derived embryonal malignancies that have GPC2 (or other glypican) overexpression and may be susceptible to anti-glypican immunotherapies will also be pursued. Additional preclinical efficacy and toxicity studies could lead to the introduction of an immunotherapeutic for children with high-risk neuroblastoma and other aggressive pediatric malignancies.
Contact For Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, John Maris (maris@chop.edu).
Experimental Model and Subject Details
Clinical Samples
The neuroblastoma tumor microarray (TMA) was constructed with duplicate punches from formalin-fixed paraffin-embedded neuroblastoma tumors archived at the Children's Hospital of Philadelphia (CHOP) between 1974 and 2004. All tumors were reviewed by a pediatric pathologist (B. Pawel) and 1-4 samples (0.6 mm cores) of representative tissue from each case and normal control tissue were included in the TMA using a manual arrayer (Beecher Instruments, Inc). This study was approved by the CHOP Institutional Review Board (IRB). The pediatric normal tissue array was made from duplicate punches of 42 normal tissue types embedded in paraffin which were collected and de-identified at CHOP from 2005 to 2013 under the IRB exemption (IRB-13-010191). The pediatric medulloblastoma TMA was made from duplicate punches of 63 unique primary tumors which were collected and de-identified at CHOP from 2005-2014 as part of the Children's Brain Tumor Tissue Consortium (CBTTC) under the CHOP IRB protocols 09-007316 and 15-011800. Twenty medulloblastoma tumors also had known subgrouping classification (5 WNT, 5 SHH, 2 G3, 1 G4, and 7 G3/G4). The three unilateral retinoblastoma samples were submitted to the Genetic Diagnostic Laboratory (http://www.med.upenn.edu/genetics/gdl/), Department of Genetics, University of Pennsylvania, for clinical genetic testing of the RB1 gene for presence of coding mutations, methylation status of the RB1 gene promoter region, and determination of MYCN copy number status. Written informed consent for use of the de-identified tissue and data for research was obtained from the parents or legal guardians and this research was approved by the IRB at the University of Pennsylvania (Protocol #706577). Primary neuroblastoma tumors for Western blot were obtained from the Children's oncology Group (COG) neuroblastoma tumor bank.
Animals
Metastatic medulloblastoma models
The metastatic medulloblastoma models (Med-1712FH, Med-211FH, Med-411FH and Med-610FH) were purchased from the Brain Tumor Resource Laboratory (BTRL; http://www.btrl.org) at the Fred Hutchinson Cancer Center. Human tumor cells were prepared as a single cell suspension under standard tissue culture conditions. The cells were then orthotopically injected (100,000 cells in 3 μL) into the cerebellum of 6 to 10-week old NOD SCID gamma (NSG) mice using a stereotaxic frame, Hamilton syringe and pre-determined coordinates. Mice were monitored daily for clinical brain tumor symptoms. Once signs of late-stage brain tumors were observed, mice were euthanized and the brain, spinal cord and liver were harvested. Permission for animal experiments was obtained from the Institutional Animal Care Committee at the Hospital for Sick Children. All animal experiments were performed in accordance with national guidelines and regulations. The brain, liver and spinal cord of sacrificed mice were fixed immediately in 10% formalin prior to paraffin embedding. The extent and location of tumors were initially evaluated by standard hematoxylin and eosin (H&E) staining. The Pathology Research Program (PRP) at Toronto General Hospital (Canada) performed specimen sectioning (at 4 μM) and routine H&E staining.
D3-GPC2-PBD in vivo neuroblastoma PDX models
In vivo murine efficacy studies were designed to assess the activity of D3-GPC2-PBD in the patient-derived xenograft (PDX) neuroblastoma tumor model NB-1643. The NB-1643 PDX tumor model was a generous gift from Dr. Peter Houghton. NB-1643 PDX tumors were implanted into the flanks of 5 to 6-week old female C.B-17 scid mice (C.B-Igh-1b/IcrTac-Prkdcscid; Taconic Biosciences; weight range of 15-20 grams). Animals bearing engrafted tumors between 0.15 cm3 to 0.35 cm3 were then randomly assigned into 6 cohorts of n = 8-9 mice per group (Vehicle (PBS), 7 mg/kg D3-GPC2-IgG1, or 7 mg/kg, 3 mg/kg, 1 mg/kg, or 1 mg/kg × 4 of D3-GPC2-PBD), ensuring each group had a similar mean tumor volume at study enrollment (range mean tumor volume 0.21-0.23 cm3). ADC was dissolved in PBS and equal volumes were given to each mouse across treatment groups. All mice were given a single dose of their respective treatments at enrollment (Day 0) via intraperitoneal (IP) injection. one cohort of mice received 3 subsequent 1 mg/kg IP injections over the following 2 weeks (1 mg/kg × 4 cohort) and the control arm was similarly treated with vehicle (PBS) at these identical time points. Tumor volumes were measured at least twice weekly using calipers and tumor volumes were calculated as volume = ((diameter1/2 + diameter2/2)3*0.5236)/1000. All mice weights were also measured at least twice weekly and mice were monitored daily for signs of clinical toxicity. Mice were maintained in cages of up to 5 mice under barrier conditions and experiments were conducted using protocols and conditions approved by the CHOP Institutional Animal Care and Use Committee (IACUC; Approved IACUC Protocol #643) in a pathogen-free facility fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). Mice were euthanized when tumor volumes reached/exceeded 3 cm3 or an animal displayed signs of clinical toxicity including excessive weight loss. A linear mixed-effects model was used to assess tumor volume over time between ADC treated animals and vehicle controls. Statistical analysis was performed using the nlme package in the R statistical programming language. Significance of survival analysis was computed with a log-rank test.
Cell Lines
Human-derived neuroblastoma cell lines were obtained from the Children's Hospital of Philadelphia (CHOP) cell line bank, the Children's Oncology Group or the American Type Culture Collection (ATCC) and most were cultured in RPMI containing 10% FBS, 2 mM L-Glutamine and 1% streptomycin/penicillin at 37°C under 5% CO2. It is important to note that the SY5Y cell line is a sub-clone of the SKNSH line. SHEP cells were a kind gift from the laboratory of Dr. Michael Hogarty. RPE1 cells are a human retinal pigment epithelium cell line immortalized through the retroviral insertion of human telomerase reverse transcriptase (hTERT), were regularly passaged in the same media and were a kind gift from the laboratory of Dr. Michael Hogarty. Neuroblastoma cell lines COG-N-561, COG-N-534, and COG-N-496 were cultured in IMDM containing 20% FBS, 1% ITS, and 2 mM L-Glutamine. HEK293T cells were obtained from ATCC and grown in DMEM supplemented similarly. CHO-K1 cells were grown in DMEM supplemented similarly. The genomic identity of each line was regularly confirmed using the AmpFLSTR Identifiler kit (Thermo Fischer Scientific), and cell lines were routinely tested to confirm the lack of mycoplasma contamination. The sex of each neuroblastoma cell line is as follows, male sex: CHP134, SKNBE2, SKNBE2C, SKNFI, IMR5, NB-1643, LAN5, NLF, NGP, NB-EBC1, NBLS, COG-N-534, and COG-N-453 and female sex: NB1, SMS-SAN, NB-1691, SY5Y, NB16, NB69, NBSD, Kelly, SKNDZ, SMS-KAN, SKNSH, SKNAS, COG-N-415, COG-N-561, COG-N-496, RPE1, COG-N-440, and COG-N-471.
Method Details
Tumor and normal tissue expression profiling
RNA sequencing data for high-risk neuroblastoma tumors (n = 126), primary tumor-relapsed paired neuroblastoma samples (n = 7 paired samples), and other pediatric tumors (Wilms tumor, n = 132; ALL, n = 194; AML, n = 228; and rhabdoid tumor, n = 5) was generated by the Therapeutically Applicable Research to Generate Effective Treatments project (TARGET data matrix; https://ocg.cancer.gov/programs/target/data-matrix). Informed consent for the Children's oncology Group (COG) Neuroblastoma Biology study was obtained at each CoG institution where respective patients were treated. Tumor samples without identifiers were sent to the Children's Hospital of Philadelphia (CHOP) and thus these studies are exempt from human subject's research guidelines, as confirmed by the CHOP Institutional Review Board. Normal tissue RNA sequencing data was generated by the Genotype-Tissue Expression (GTEx) Project (n = 7,859 samples across 31 unique normal tissues, n = 5 - 1,152 samples per tissue; http://www.gtexportal.org). The GTEx Project was supported by the Common Fund of the office of the Director of the National Institutes of Health. Additional funds were provided by the NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. Donors were enrolled at Biospecimen Source Sites funded by NCI\SAIC-Frederick, Inc. (SAIC-F) subcontracts to the National Disease Research Interchange (10XS170), Roswell Park Cancer Institute (10XS171), and Science Care, Inc. (X10S172). The Laboratory, Data Analysis, and Coordinating Center (LDACC) was funded through a contract (HHSN268201000029C) to The Broad Institute, Inc. Biorepository operations were funded through an SAIC-F subcontract to Van Andel Institute (10ST1035). Additional data repository and project management were provided by SAIC-F (HHSN261200800001E). The Brain Bank was supported by a supplements to University of Miami grants DA006227 & DA033684 and to contract N01MH000028. Statistical Methods development grants were made to the University of Geneva (MH090941 & MH101814), the University of Chicago (MH090951, MH090937, MH101820, MH101825), the University of North Carolina - Chapel Hill (MH090936 & MH101819), Harvard University (MH090948), Stanford University (MH101782), Washington University St Louis (MH101810), and the University of Pennsylvania (MH101822).
The TARGET neuroblastoma, other pediatric tumor RNA sequencing data and GTEx RNA sequencing data was downloaded from S3 buckets (Amazon; s3://cgl-rnaseq-recompute-fixed/target/ and s3://cgl-rnaseq-recompute-fixed/gtex/) on 8/5/2016 from prior processed data as described in detail below from the UCSC Computational Genomics Laboratory (Vivian et al., 2016).
RNA sequencing data from primary medulloblastoma tumors (n = 97) can be accessed via the European Genome-phenome Archive (EGA; https://www.ebi.ac.uk/ega/home) accession number EGAD00001001899. Fastq files were processed using the STAR alignment tool (Dobin et al., 2013) and subsequently normalized using the RSEM package (Li and Dewey, 2011) based upon the hg19 reference genome and the GENCODE v19 gene annotation. GPC2 transcript level expression was quantified in units of TPM and GPC2 gene level expression was quantified in units of FPKM.
Additional neuroblastoma tumor RNA sequencing data was profiled via the SEQC project and accessed via the GEO database accession number GSE62564 (Zhang et al., 2015). Additional GPC2 mRNA expression data for neuroblastoma primary tumors (n = 249) profiled with Human Exon arrays (Affymetrix) was also obtained from the TARGET data matrix and processed with Robust Multichip Average (RMA) normalization analysis implemented in the Affymetrix Power Tools (Affymetrix, Inc.). Additional comprehensive normal tissue RNA sequencing data was queried using the Human Protein Atlas portal (n = 32 unique normal tissues; http://www.proteinatlas.org/) (Uhlen et al., 2015). Additional pediatric tumor RNA sequencing data was downloaded on 9/9/2016 from the St. Jude Children's Research Hospital Pediatric Cancer Data Portal (PeCan; https://pecan.stjude.org). GPC2 mRNA expression data from paired primary and metastatic medulloblastomas was downloaded from the GEO database accession number GSE63670 (Wang et al., 2015). RNA sequencing data from normal mouse (C57Bl/6) tissues was accessed via the GEO database accession number GSE29184 (Shen et al., 2012). Finally, additional GPC2 mRNA expression data from neuroblastomas, medulloblastomas, retinoblastomas and neural crest cells was accessed via the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl).
Differential expression analysis
For the neuroblastoma tumor and normal tissue gene level differential expression analysis, we utilized RNA sequencing data from the 126 high-risk neuroblastoma tumors (TARGET) and normal tissues (GTEx) as described above and downloaded gene level data that was previously processed by the UCSC Computational Genomics laboratory using STAR alignment and RSEM normalization using hg38 as the reference genome and GENCODE v23 gene annotation (Vivian et al., 2016). The voom procedure was used to normalize the RSEM generated expected counts followed by differential expression testing using the R package limma to obtain p values and Log-fold changes (LogFCs) (Law et al., 2014; Ritchie et al., 2015). Specifically, a total of 60,498 genes were tested for differential expression between the neuroblastoma tumors and normal tissues using this RNA sequencing data, a total of 1,889,388 (31 normal tissues × 60,498) computations. only genes which were differentially expressed in all 31 normal tissue comparisons were considered for subsequent interrogation. In total, 296 of 60,498 (0.48%) genes were defined as differentially expressed using the stringent cutoff of minimum LogFC > 1 and maximum p < 0.01. These genes were then further analyzed for prediction to be associated or bound with the plasma membrane using the COMPARTMENTS database (http://compartments.jensenlab.org). For plasma membrane designation, we designated only those genes to be potentially associated with the plasma membrane where the max confidence score across all GO categories was associated with Plasma Membrane or Cell Surface and was ≥3. Finally, genes were further filtered by an absolute neuroblastoma tumor RNA expression threshold of a mean FPKM > 50.
overall survival analysis
We performed overall survival analyses utilizing the Kaplan Meier Scanner function with three large neuroblastoma tumor data sets with available survival endpoints via the Genomics Analysis and Visualization Platform (R2; http://r2.amc.nl; Kocak; n = 649, SEQC; n = 498, and Versteeg; n = 88 (Kocak et al., 2013; Valentijn et al., 2012; Zhang et al., 2015)). The Kaplan Scan function segregates a cohort into 2 groups based on gene expression and scanning yields the cutoff where the difference is most statistically significant.
Neuroblastoma cell line profiling
Neuroblastoma cell lines were profiled by RNA and targeted DNA sequencing as previously described (Harenza et al., 2017; Hart et al., 2017) and neuroblastoma cell line GPC2 FPKM, MYCN amplification status, and TP53 and ALK mutation status was queried from these data sets.
Tumor copy number analysis
Neuroblastoma GPC2 copy number analysis from whole genome sequencing (Figure 2A, left) was performed using segmentation data provided by Complete Genomics, Inc. (CGI) for a cohort of the 126 high-risk primary neuroblastoma tumors available through the TARGET data matrix (https://ocg.cancer.gov/programs/target/data-matrix). Gene level copy number for GPC2 was obtained by processing segmentation data with GISTIC2.0. Neuroblastoma GPC2 copy number analysis from SNP arrays (Figure 2A, right) was done utilizing neuroblastoma tumor SNP genotyping data from 266 primary tumor samples previously deposited in the TARGET data matrix. Gene level copy number for GPC2 was also calculated with GISTIC2.0 as above. SNP genotyping of primary medulloblastoma tumors and genomic copy number processing and analysis was queried from data previously described (Northcott et al., 2012). The thresholds for genomic gains and losses and broad versus focal events were as previously described (Northcott et al., 2012).
GPC2 transcript level expression analyses
For normal tissue GPC2 transcript level expression analysis, data was downloaded directly from the GTEx website (n = 8,555 tissue samples, 30 unique normal tissues), that had been processed using Tophat alignment tool and quantified as RPKM using the Flux Capacitor tool on the hg19 reference genome (Griebel et al., 2012). For neuroblastoma tumor GPC2 transcript level expression, RNA sequencing data from 126 high-risk neuroblastomas tumors (TARGET) detailed above was processed using the STAR alignment tool (Dobin et al., 2013) and subsequently normalized using the RSEM package (Li and Dewey, 2011) based on the hg19 reference genome and GENECODE v19 gene annotation to get FPKM quantification for the 7 predicted GPC2 transcripts (Ensembl GRCh37). A percent (%) transcript for each of the 7 GPC2 transcripts was calculated in tumors and normal tissues by calculating FPKM transcript/total FPKM for each of the 7 predicted GPC2 transcripts.
MetaCore™ pathway analysis
To identify pathways significantly associated with high GPC2 expression in high-risk neuroblastoma, we performed a pathway enrichment analysis on MYCN amplified and non-amplified tumor cohorts from 2 unique neuroblastoma tumor data sets (TARGET, n = 126; 33 MYCN amplified, 92 MYCN non-amplified, 1 unknown and SEQC, n = 176; 92 MYCN amplified, 83 MYCN non-amplified, 1 unknown; GEO accession number GSE62564). For each of the four sample sets, GPC2 low and high expression samples were identified using the 10th and 90th percentile cutoff distribution of GPC2 FPKM. The voom procedure was used to normalize the RSEM generated expected counts followed by differential expression testing using the R package limma to obtain p values (Law et al., 2014; Ritchie et al., 2015). An FDR cutoff of 0.05 was used to identify significantly differentially expressed genes between high and low GPC2 expressing tumors in each dataset. These gene sets were used as input for MetaCore™ Functional Enrichment by Ontology using pathway maps in order to identify biological pathways that are associated with high GPC2 expression in high-risk neuroblastoma tumors (https://portal.genego.com/cgi/data_manager.cgi). An FDR cutoff of 0.05 was used to identify pathway maps significantly associated with GPC2 expression which was further limited to the top 50 pathway maps per independent dataset.
MYCN chromatin immunoprecipitation sequencing
Chromatin immunoprecipitation was performed on adherent cells using an N-Myc Antibody (sc-53993, Santa Cruz Biotechnology, B8.4.B). MYCN amplified neuroblastoma cell lines (Kelly, NGP, and NB-1643) were grown to 80% confluence in 15 cm tissue culture plates in 20 mL of media. 415 μL of 37% formaldehyde (final concentration 0.75%) was added to the media and the plate was rocked for 10 minutes to fix cells. 1.5 mL of 2.5 M glycine (final concentration 0.18 M) was then added to quench the formaldehyde and the plate was rocked for an additional 5 minutes. Cells were lysed in 5 pellet volumes of FA lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1 mM EDTA pH 8.0, 1.0 % Triton-X-100, 0.1 % SDS, 0.1 % Deoxycholate), supplemented with fresh protease inhibitors and DTT. Beads were washed three times in ChIP Wash Buffer (0.1 % SDS, 1.0 % Triton-X-100, 2 mM EDTA pH 8.0, 150 mM NaCl, 20 mM Tris-HCL pH 8.0) and once with Final Wash Buffer (0.1 % SDS, 1.0 % Triton-X-100, 2 mM EDTA pH 8.0, 500 mM NaCl, 20 mM Tris-HCL pH 8.0). Libraries were constructed using the NEB ultra-kit according to the manufacturer's instructions.
We calculated phred quality scores using the perl script (phredDetector.pl;https://raw.githubusercontent.com/douglasgscofield/bioinfo/master/scripts/phredDetector.pl) for each sample to describe the confidence of each base call in each sequence tag to filter low-quality reads. We used the phred scores in the wrapper tool Trim Galore (http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) which uses the Cutadapt tool for quality trimming and the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) to provide quality control metrics on the trimmed sequences. Following trimming and quality control, we next used Bowtie (Langmead et al., 2009) to align the trimmed reads to the hg19 reference genome and the Samtools rmdup tool (Li et al., 2009) to remove duplicates from the aligned reads. After removing the duplicates, we used the MACS2 callpeak tool (Zhang et al., 2008) to call significant peaks using a q value (minimum False Discovery Rate) cutoff of 0.05. Results were returned in units of signal per million reads (using the --SPMR option) to get normalized peak values. We then filtered the called peaks to remove artifacts that tend to show high signal such as centromeric, telomeric and satellite repeats using the previously defined genomic blacklisted regions (https://sites.google.com/site/anshulkundaje/projects/blacklists). The output files were converted to bigWig format for visualization in the Integrative Genomics Viewer (IGV).
Real-Time PCR analysis
Total RNA was isolated from neuroblastoma cells utilizing Qiashredder and RNeasy mini kits (Qiagen) and mRNAs were converted to cDNA using SuperScript™ First-Strand Synthesis System for RT-PCR (Thermo Fischer Scientific). Taqman® gene expression assays (Thermo Fischer Scientific) were used to quantitate MYCN (Hs00232074_m1), GPC2 (Hs00415099_m1), UBC (Hs00824723_m1), and HPRT1 (Hs02800695_m1) mRNA levels. RT-PCR analysis was performed on an Applied Biosystems 7900HT Sequence Detection System using standard cycling conditions and mRNA expression values were quantified with corresponding standard curves. Ratios of mRNA quantities were normalized by comparing mRNA expression of interest to the geomean of mean UBC and HPRT1 mRNA expression values.
Neuroblastoma PDX TMA and cell line IHC
The human neuroblastoma patient-derived xenograft (PDX) array was constructed from duplicate punches from murine PDXs removed surgically at 3 cm3 size and subsequently fixed with 10% buffered formalin and embedded in paraffin. Samples for neuroblastoma cell line immunohistochemistry were derived from suspending 10-20 million live neuroblastoma cells in 50 μL of RPMI media and mixing with 10× the amount of 1% agarose in PBS after boiling and subsequently cooling and fixing in 10% buffered formalin.
Immunohistochemistry (IHC)
Glypican-2 antibody (1:500; F-5, sc-393824, Santa Cruz Biotechnology) was used to stain formalin fixed, paraffin embedded TMA slides and slides from metastatic medulloblastoma xenograft models. Staining was performed on a Bond Max automated staining system (Leica Biosystems) using the Bond Refine polymer staining kit (Leica Biosystems) according to the standard protocol with the exception that the primary antibody incubation was extended to 1 hour at room temperature. Antigen retrieval was performed with E2 retrieval solution (Leica Biosystems) for 20 minutes. Slides were rinsed, dehydrated through a series of ascending concentrations of ethanol and xylene, and then cover slipped. Stained slides were then digitally scanned at 20× magnification on an Aperio CS-O slide scanner (Leica Biosystems). Each tumor staining was graded by an experienced pathologist with expertise in the tumor/tissue being stained and evaluated for staining percentage and intensity (0/none to 3/intense) of staining. A membrane staining H-score was calculated (intensity × % cells; 0 to 300) for each tumor or normal tissue. Representative pictures were taken on scanned IHC slides with Aperio ImageScope program v12.2.2.5015.
Flow cytometry
We performed initial flow cytometry analysis (Figure 3E) to confirm GPC2 expression on neuroblastoma cells with D3-GPC2-scFv-FLAG (10 μg/ml). Neuroblastoma cell lines with differential GPC2 expression, Kelly GPC2 isogenic cells, and RPE1, HEK293T, and CHO-K1 cells were analyzed in duplicate. Cells were washed twice with PBS and reconstituted in PBS at a concentration of 1 million cells/mL after which 1 μL of fixable viability dye was added (1:1,000). Cells were then incubated for 30 minutes on ice in the dark, washed twice with FACS buffer (500 mL PBS, 10 mL FBS, 2 mL 0.5 M EDTA), and then resuspended in D3-GPC2-scFv-FLAG (10 μg/ml) in FACS buffer. After 30 minutes on ice in the dark, cells were washed twice with FACS buffer and then resuspended in secondary antibody (4 μg/mL APC anti-DYKDDDDK Tag Antibody, BioLegend #637308) in FACS buffer. After 20 minutes of incubating in the dark, cells were washed twice with FACS buffer and analyzed on a CytoFLEX Flow Cytometer (Beckman Coulter). Data was analyzed using FlowJo software.
To further semi-quantitate GPC2 cell surface expression on a panel of neuroblastoma cell lines we utilized in D3-GPC2-PBD cytotoxicity studies, we analyzed each cell line by flow cytometry staining with D3-GPC2-IgG1 in duplicate. Neuroblastoma cells with differential GPC2 expression, Kelly/HEK293T GPC2 isogenic cells, and RPE1 cells were plated and grown in cell culture for 48 hours, washed twice with cold PBS, and reconstituted and aliquoted in FACS buffer (500 mL PBS, 10 mL FBS, 2 mL 0.5 M EDTA). D3-GPC2-IgG1 was added at a final concentration of 10 μg/ml in FACS buffer for 30 minutes on ice in the dark. Cells were then washed twice with FACS buffer and resuspended in secondary antibody (10 μg/mL, Mouse anti-Human IgG1 Fc Secondary Antibody, Alexa Fluor 488) in FACS buffer for 20 minutes on ice in the dark. Following incubation, cells were washed twice with FACS buffer and analyzed on an Attune Acoustic Focusing Flow Cytometer (Applied Biosystems). The mean fluorescence intensity was converted into MESF using Quantum™ Alexa Fluor 488 MESF beads (Bangs laboratories, 488) and ABC values using Quantum™ Simply Cellular anti-Human IgG (Bangs Laboratories, 816) following the manufacturer's protocols. Data was analyzed using FlowJo software.
Immunofluorescence
NB-EBC1 and SMS-SAN neuroblastoma cells were plated on glass coverslips in 48-well tissue culture plates or in chamber slides (Lab-Tek II CC chamber slides; VWR). After 48-72 hours, cells were washed with cold PBS × 2, fixed with 4% formaldehyde for 15 minutes at room temperature, washed with PBS × 3, blocked with 5% normal goat serum in PBS for 1 hour and then incubated at 4 °C overnight in primary antibody. Primary antibodies included Glypican-2 (1:5,000; F-5, sc-393824, Santa Cruz Biotechnology) and Anti-pan Cadherin (1:500; Abcam ab6529) and were diluted in 1% BSA in PBS. After overnight incubation at 4°C, cells were washed with PBS × 3 and incubated with secondary antibody (1:1,000 of Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 or Goat anti-Rabbit IgG (H+L) cross-Adsorbed secondary Antibody, Alexa Fluor 594) for 1 hour at room temperature and then washed with PBS × 3. Cells were mounted with ProLong gold with DAPI (Thermo Fisher Scientific, #P36931) and visualized with a Leica DM5000B microscope and photographed with a Leica DFC365 FX camera at 100×. Secondary antibody only incubations and low GPC2 expressing cells (RPE1 cells) were done in parallel to ensure antibody specificity.
Immunofluorescence internalization assays
SMS-SAN neuroblastoma cells were plated (50,000 cells/well) on glass coverslips in a 24-well tissue culture plate and grown as above. After 48 hours, cells were treated with 10 μg/mL of D3-GPC2-IgG1 in FACS buffer for 30 minutes on ice. Cells were then washed with cold PBS × 2 and incubated in RPMI growth media for either 30 minutes or 24 hours. After the indicated incubation period, cells were fixed with 4% formaldehyde for 15 minutes at room temperature and washed with PBS × 3. Cells incubated with D3-GPC2-IgG1 for 24 hours were also permeabilized with cold 100% methanol for 10 minutes at -20°C and washed with PBS × 3. All specimens were then blocked with 5% normal goat serum in PBS for 1 hour and incubated at 4 °C overnight in primary antibody. Primary antibodi es included Glypican-2 (1:5,000; F-5, sc-393824, Santa Cruz Biotechnology), Anti-pan Cadherin (1:500; Abcam ab6529) and LAMP1 (D2D11) XP® (1:1,000; #9091S, Cell Signaling) diluted in 1% BSA 0.3% Triton™ X-100 in PBS. After overnight incubation at 4°C, cells were washe d with PBS × 3 and incubated with secondary antibody (1:1,000 of Goat anti-Mouse IgG (H+L) cross-Adsorbed Secondary Antibody, Alexa Fluor 488, Goat anti-Human IgG (H+L) cross-Adsorbed secondary, Alexa Fluor 488, or Goat anti-Rabbit IgG (H+L) cross-Adsorbed secondary Antibody, Alexa Fluor 594) for 1 hour at room temperature and then washed with PBS × 3. Specimens were mounted and imaged as described above.
Western blotting
Whole-cell lysates were prepared with cell lysis buffer (#9803, Cell Signaling Technology) and phenylmethyl sulfonyl fluoride (Sigma-Aldrich), briefly sonicated, rotated for 15 minutes at 4°C, and then lysates were centrifuged for 10 minutes, supernatant removed and protein concentration quantified via Bradford assays. Lysates (10-50 μg) were separated on 10% Bis-Tris gels (Life Technologies), transferred to a PVDF membrane (Bio-Rad), blocked in 5% nonfat milk in Tris-buffered saline and Tween-20 (TBS-T), and blotted using standard protocols. Membranes were incubated at 4 °C overnight in primar y antibody, washed × 3 in TBS-T, then incubated in 1:2,000 diluted HRP-labeled secondary antibody (Santa Cruz Biotechnology) at room temperature for 1 hour, washed an additional × 3 with TBS-T, and then developed with chemiluminescent reagents (Thermo Fischer Scientific, SuperSignal West Femto). The following primary antibodies were used: β-Actin (1:5,000; #4967S, Cell Signaling Technology), N-MYC (1:1,000; #9405S, Cell Signaling Technology), Glypican-2 (1:500; F-5, sc-393824, Santa Cruz Biotechnology), Cleaved Caspase-3 (1:1,000; #9664S, Cell Signaling Technology), Cleaved PARP1 (Asp214) (1:1,000; #9541S, Cell Signaling Technology), and Na, K-ATPase (1:1,000; #3010S, Cell Signaling Technology).
Membrane protein fractionation
Membrane protein fractionation was done using a ProteoExtract® Native Membrane Protein Extraction kit (Millipore Sigma) according to the manufacturer's instructions.
Lentiviral preparation and transduction
pLKo.1 lentiviral shRNA plasmids targeting GPC2 and MYCN or a non-targeting control (NTC) were obtained from Sigma MISSIoN shRNA consortium: shGPC2 Exon 4, TRCN0000220037; shGPC2 3′UTR, TRCN0000147974; shMYCN-1, TRCN00000358381; shMYCN-5, TRCN0000020695; shMYCN-6, TRCN0000020696; shMYCN-7, TRCN0000020697; and shNTC (SHC002, MISSIoN® pLKO.1-puro non-mammalian shRNA control plasmid) were transfected along with pMD2.G (encoding envelope plasmid VSV-G) and psPAX2 (packaging plasmid) into HEK293T cells utilizing FuGENE® 6. The pMD2.G and psPAX2 plasmids were a kind gift from the laboratory of Dr. Robert Schnepp. The virus-containing supernatant was collected 48 and 72 hours later and filtered with 0.45 μM nitrocellulose membranes. To increase transduction efficiency, virus was added to cells in the presence of 8 μg/ml polybrene (Sigma). Media was changed the next day and puromycin (Sigma) was added for selection for at least 48 hours prior to plating for cell growth assays as below and/or cells were pelleted for Western blot or RNA extraction for GPC2 expression analysis. For GPC2 isogenic cell line engineering, lentiviruses were prepared and utilized as above. GPC2 transduced cells (Kelly and HEK293T GPC2 cells) were also group selected in puromycin prior to using.
Cell proliferation and apoptosis assays
Neuroblastoma cells were transduced with GPC2 targeting shRNA as above and after 48-72 hours of selection in puromycin, GPC2 depleted or control shNTC transduced cells were plated in at least triplicate in a Real-time Excelligence system (RT-CES; F Hoffman La-Roche, Basal, Switzerland) and growth was monitored every 30 minutes. For CellTiter-Glo® Luminescent Cell Viability Assays (Promega) or Caspase Glo® 3/7 Assays (Promega), shRNA transduced neuroblastoma cells were seeded in 96-well plates in parallel to the above after 48-72 hours of selection in puromycin and assayed after 72 hours of additional growth. Luminescence was measured with the respective luminescence assay according to manufacturer's instructions (Promega) and quantified relative to shNTC transduced cells. For the Caspase Glo® 3/7 Assays, luminescence values were quantified relative to shNTC transduced cells and further normalized to the cell number using the CellTiter-Glo® Assay luminescence values performed in parallel. Data presented are representative of at least two independent experiments.
Foci formation assay
Neuroblastoma cells were transduced as above with either shGPC2 exon 4, shGPC2 3′ UTR or shNTC containing lentivirus and after 48-72 hours of puromycin selection were plated in 6 well dishes, grown for 2-5 weeks, fixed, stained with 0.4% crystal violet and photographed. Data presented are representative of at least two independent experiments.
GPC2 and MYCN lentiviral constructs
GPC2 cDNA in the gateway donor vector pDoNR221 was purchased from Harvard PlasmID Repository (plasmid ID: HsCD00045342) and cloned into pLenti CMV Puro DEST (w118-1), which was a gift from Eric Campeau (Campeau et al., 2009) and was purchased via Addgene (plasmid #17452), with Gateway® LR clonase enzyme (Invitrogen) via the manufacturer's protocol to make the GPC2 pLenti CMV Puro vector (GPC2=GPC2-001). The GPC2-003 pLenti CMV Puro vector was constructed similarly after PCR amplification of the GPC2-003 coding sequence contained within GPC2 exons 5 to 10 using the following primers (forward: CCAGGCTCTGATGCGTCT and reverse: GGTCCTATCGAGGTCCAAG) from the GPC2 pLenti CMV Puro vector followed by TA cloning into a gateway donor vector using a pCR™8/GW/ToPo™ TA cloning kit according to the manufacturer's protocol. The GPC2-003 cDNA was then cloned into the pLenti CMV Puro DEST vector as above using the Gateway® LR clonase enzyme. MYCN cDNA in a pCMV6-XL4 vector was purchased from origene (SC116780) and PCR amplified to attach attB sites using Phusion High Fidelity Polymerase (New England BioLabs, M0530), 5× GC Buffer, and 3% DMSo, and MYCN-attB1: GGGGACAAGTTTGTACAAAAAAGCAGGCTCCACCATGCCGAGCTGCTCCACG and MYCN-attB2: GGGGACCACTTTGTACAAGAAAGCTGGGTTCTAGCAAGTCCGAGCGTGTTCAAT primers. The DNA was gel purified (Zymo Research, #D4001), recombined with pDONOR221 using Gateway® BP Clonase® II enzyme according to the manufacturer's protocol, and subsequently cloned into a pLenti CMV Puro DEST vector as above to make the MYCN pLenti CMV Puro vector. Vector sequences were confirmed via restriction digest and Sanger sequencing.
GPC2 promoter luciferase reporter assays
GPC2 promoter assays were carried out with the Lightswitch Luciferase Assay System (Switchgear Genomics). On day 0, 5-10,000 SHEP or HEK293T cells were seeded in triplicate in 96-well plates in RPM1 or DMEM, respectively. On day 1, cells were transfected with the GPC2 promoter reporter (Switchgear product ID: S719588), which contains the MYCN binding Ebox motif upstream of the GPC2 promoter, or the empty promoter vector control (Switchgear product ID: S790005), and a MYCN pLenti CMV Puro vector or the pLenti CMV Puro empty vector. For transfection in SHEP cells, the TransFast™ Transfection Reagent (Promega product ID: E2431) was used with a 1:1 TransFast:DNA ratio according to the manufacturer's protocol. For transfection in HEK293T cells, FuGENE® 6 Transfection Reagent was used with a 3:1 FuGENE:DNA ratio according to the manufacturer's protocol. Transfected cells were incubated for 48-72 hours and then harvested for luciferase assays per the manufacturer's instructions using the LightSwitch assay reagent (Switchgear product ID: LS010). Normalization for differences in cell number was achieved by doing a parallel CellTiter-Glo® luminescence assay (Promega) and normalizing GPC2 reporter luciferase values to CellTiter-Glo® luminescence assay values for parallel transfected cells.
Isolation and preparation of D3-GPC2-IgG1
A naive human Fab phage display library constructed from peripheral blood B cells of 50 healthy donors was used for selection of Fabs against purified recombinant GPC2 ectodomain (#2304, R&D Systems, Inc.) as previously described (Zhu et al., 2006). Briefly, the isolated Fabs were expressed, purified and tested for binding to the GPC2 ectodomain through ELISA and the best binder, designated as D3-GPC2-Fab, was converted to a single chain variable fragment (scFv) and a full-length human IgG1. The D3-GPC2-scFv was expressed in HB2151 cells and purified as previously described (Zhu et al., 2006). Briefly, plasmids was transformed into HB2151 cells. A single colony was picked from the plate containing freshly transformed cells, inoculated into 200 mL 2YT medium broth containing 100 μg/ml ampicillin and 0.2% glucose, and incubated at 37°C with shaking at 250 rpm. When the culture OD at 600 nm reached 0.90, isopropyl-β-d-thiogalactopyranoside at a 0.5 mM final concentration was added, and the culture was further incubated overnight at 30°C. The bacterial pellet was collected after centrifugation at 8,000 × g for 20 minutes and resuspended in PBS buffer containing 0.5 mil polymixin B (#P4119, Sigma-Aldrich). After 30 minutes incubation with rotation at 50 rpm at room temperature, the mixture was centrifuged at 25,000 × g for 25 minutes at 4°C, and the supernatant was used for 6× His-tagged scFv purification using Nickel charged resin (Ni-NTA Agarose, #30210, QIAGEN). The full length lgG1 DNA construct was transiently transfected into Freestyle™ 293-F cells (#R79007, Thermo Fisher Scientific) for antibody production and the D3-GPC2-lgG1 was purified on a protein A column.
ELISA binding assay
Recombinant human and mouse GPC2 (R&D systems, Inc.) were coated on Corning EIA/RIA high-binding 96-well plates (Corning, Inc.) at 50 ng per well overnight at 4°C and blocked with 3% nonfat milk in PBS. Five-fold serially diluted D3-GPC2-lgG1 was added and incubated at room temperature for 2 hours. The plates were washed with PBS containing 0.05% Tween-20. Bound D3-GPC2-lgG1 was detected by an Anti-Human IgG (Fc specific)-Peroxidase antibody produced in goat (#A0170, Sigma-Aldrich). The assay was developed at room temperature with ABTS™ substrate (Roche) and the absorbance was quantified at 405 nm.
D3-GPC2-Fab surface plasmon resonance
Binding affinity of D3-GPC2-Fab to recombinant GPC2 was measured using a BIAcore X100 instrument (GE Healthcare). Purified human recombinant GPC2 (R&D Systems) was diluted in 10 mM sodium acetate buffer (pH 5.5) and immobilized on a CM5 biosensor chip using an amine coupling kit. The running buffer was HBS-EP (10 mM HEPES, pH 7.4, 150 mM NaCI, 3 mM EDTA, 0.05% surfactant P20). The D3-GPC2-Fab was diluted in running buffer to desired concentrations. D3-GPC2-Fab was allowed to flow over the chip cells for 3 min after which the chip cells were flown with antibody-free solution for 10 min to allow for Fab dissociation. After Fab dissociation the chip was regenerated using 10 mM acetate and 0.5 M NaCI (pH 4.0). The absorption and desorption isotherms were fitted using a 1:1 binding model and a dissociation rate constant (KD) was estimated with BIAevaluation software.
D3-GPC2-IgGl internalization using pHAb dye
D3-GPC2-lgG1 was conjugated to pHAb amine reactive dye (Promega) to obtain D3-GPC2-lgG1-pHAb dye according to the manufacturer's protocol. Briefly, 1.2 mL of stock amine reactive pHAb dye was added to 100 μg of antibody in PBS and the conjugation reaction was kept at room temperature for 1 hour and excess dye was subsequently removed by washing with PBS (10 × 0.5 mL centrifugal filter units (Millipore)). Kelly GPC2, SMS-SAN, or RPE1 cells were seeded in a 6-well plate for 24 hours and D3-GPC2-lgG1-pHAb dye was then added to a final concentration of 20 nM (3 μg/mL) and cells were incubated at 37 or 4°C (arrested endocytosis) for 6 hours. After incubation, cells incubated with antibody and control cells incubated in just media were detached from the plates, washed and kept at 4°C until FACS measurement. Fluorescence from the pH sensitive dye was measured using a BD FACSCalibur.
D3-GPC2-IgG1 flow cytometry internalization
Briefly, SMS-SAN (750,000 cells/plate), HEK293T (250,000 cells/plate), or HEK293T GPC2 cells (250,000 cells/plate) were plated in 6 × 6 cm tissue culture plates and grown as above. After 48 hours, D3-GPC2-IgG1 (100-200 μg) was conjugated to Alexa Fluor 488 fluorophore with a Dylight 488 Fast Conjugation Kit (Abcam ab201799) and cells were treated with 10 μg/mL 488 fluorophore-conjugated D3-GPC2-IgG1 (D3-GPC2-IgG1-AF488) in RPMI growth media for 30 minutes on ice. Cells were then washed three times with cold RPMI to remove excess D3-GPC2-IgG1-AF488 and incubated at 37°C for the indic ated time periods (0, 2, 4, 8, 24, and 48 hours). At the indicated time period, a 6 cm culture plate was chilled on ice to arrest endocytosis, washed with cold PBS, and cells were reconstituted in FACS buffer, centrifuged at 300 × g for 5 minutes at 4°C, resuspended in FACS buffer, and split into two 1.5 mL Eppendorf tubes. Cells in one Eppendorf tube were treated with 50 μg/mL Alexa Fluor 488 Polyclonal Antibody on ice in the dark for 30 minutes to quench fluorescence on the cell surface (quenched sample), while the other cell sample was left untreated (unquenched sample). Cells in both tubes were then spun at 300 × g for 5 minutes at 4°C, resuspended in FACS buffer, and analyzed on an Attune Acoustic Focusing Flow Cytometer (Applied Biosystems) to obtain the MFI for quenched and unquenched cell samples. Percent internalization was calculated with the equation (MFIquenched / MFIunquenched) × 100% for each time point. The fold-change in internalization was calculated by dividing the percent internalization at each time point by the percent internalization at the 30 minute time point for each cell line.
Preparation of D3-GPC2-PBD
The purified D3-GPC2-IgG1 was directly used for glycan-based site-specific modification and conjugation as previously described (Zhu et al., 2014) except that C2-Azide-Galacotose was used as substrate for the Fc-glycan modification. Briefly, DBCO-PEG4-PBD was used as the payload for the conjugation following the click chemistry based approach. To maintain solubility of the DBCO-linker-PBD, the azide-attached antibody itself was diluted with propylene glycol to a final concentration of 33%. The solution of DBCO-linker-PBD in propylene glycol was added to the antibody solution with a drug-to-antibody mole ratio of approximately 4:1. The final concentration of propylene glycol in the conjugation reaction was 50%. The reaction was allowed to proceed for at least 4 hours at room temperature and purified by size exclusion chromatography and concentrated in PBS.
D3-GPC2-PBD cytotoxicity/apoptosis assays
The cytotoxicity of D3-GPC2-PBD and free PBD dimer was tested in a panel of cell lines including native neuroblastoma cell lines (SMS-SAN, COG-N-561, COG-N-534, NBSD, COG-N-496, NB-1643 and SKNAS), isogenic GPC2 cell lines (Kelly and HEK293T GPC2/Empty vector) and RPE1 cells as a control. 1,000-3,000 cells were plated on Day 1 in a 96-well plate. on Day 2, serial dilutions of D3-GPC2-PBD or free PBD dimer (Levena Biopharma US) (1 nM - 0.064 pm) were added to duplicate or triplicate wells. After 96 hours, cell viability was determined using a CellTiter-Glo® Assay (Promega) in a GloMax (Promega) plate reader according to the manufacturer's instructions. Luminescence values were normalized to vehicle treated wells and data were analyzed and graphed in GraphPad Prism software Version 5.01 and a log (inhibitor) vs. response nonlinear regression model was used to calculate IC50s. 10,000 SMS-SAN and Kelly GPC2 cells were also plated in a 96-well plate on the RT-CES system and after 24 hours they were treated with the same serial dilutions of D3-GPC2-PBD (1 nM - 0.064 pm) and growth was monitored every 60 minutes. For D3-GPC2-IgG1 competition studies, 10,000 Kelly GPC2 and SMS-SAN cells were plated in a 96-well plate on the RT-CES system. After 24 hours, wells were diluted to 200 pM of D3-GPC2-PBD except for the no ADC control, and serial excess D3-GPC2-IgG1 was also added to triplicate wells (0×, 0.1×, 1×, 2.5×, 5×, 10×, 25×, 50×, and 100× excess D3-GPC2-IgG1). Growth was monitored every 60 minutes as above. Finally for caspase 3/7 activity levels, cells were treated as above with D3-GPC2-PBD and Caspase Glo® 3/7 Assays (Promega) were utilized according to the manufacturer's instructions at the indicated time points. Each ADC dose was quantified relative to vehicle treated cells and further normalized to the cell number using CellTiter-Glo® assay luminescence values performed in parallel as above. Each experiment was plated in at least a technical duplicate and data presented are representative of at least two independent experiments.
Quantification and Statistical Analysis
Differences between groups were presented as the mean ± SEM as noted in the figure legends. Experimental sample numbers (n) are indicated in the figures, figure legends, and results section. All tests were two sided and p values < 0.05 were considered statistically significant, except in Figure 1A where p < 0.01 was used as a cutoff for differentially expressed genes and were corrected for multiple comparisons. All statistical analysis was done with GraphPad Prism Version 5.01 and the R programming language. ImageJ software was utilized for quantitation of select immunoblots.
Data and Software Availability
The MYCN chromatin immunoprecipitation sequencing data have been deposited in the GEO database under the accession number GSE94782.
Supplementary Material
Significance.
Children with high-risk neuroblastoma continue to have poor outcomes despite marked intensification of therapy. With the persistence of lineage specific cell surface molecules on embryonal malignancies, there is clear rationale for developing targeted immunotherapies specific for childhood cancers. Here, we identify and validate GPC2 to be a robustly differentially expressed cell surface oncoprotein that is transcriptionally activated by MYCN and may be less susceptible to immune escape mechanisms given its importance in tumorigenesis. We next develop an antibody-drug conjugate targeting GPC2 that is efficacious in a neuroblastoma patient-derived xenograft model. This work establishes a strong foundation for the development of GPC2-directed immunotherapeutics and for the further exploration of the role of glypicans in neuroblastoma and other cancers.
Highlights.
Neuroblastomas differentially express high-levels of GPC2 versus normal tissues
MYCN and gain of chromosome 7q mediate neuroblastoma GPC2 differentialexpression
GPC2 is integral in neuroblastoma cell proliferation
A GPC2 directed antibody-drug conjugate is cytotoxic to neuroblastoma cells
Acknowledgments
Kristopher R. Bosse is a Damon Runyon Physician-Scientist supported (in part) by the Damon Runyon Cancer Research Foundation (PST-07-16). This research was also supported in part by a Stand Up To Cancer St. Baldrick¹s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113) (J.M.M., C.M., M.T., and D.S.D). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research. This work was also supported by grants T32 CA9615-25 (K.B.) and NIH/NIGMS T32 GM008638 (K.B.), the Alex's Lemonade Stand Foundation (K.B., J.M.M., R.T.S, and M.R.), and the Giulio D'Angio Endowed Chair (J.M.M.). This study was also supported by the intramural research program of the NCI, National Institutes of Health, and Department of Health and Human Services (DHHS). The content of this publication does not necessarily reflect the views or policies of the DHHS nor does mention of trade names, commercial products, or organizations imply endorsement by the US government. This study was also made possible in part due to The Children's Brain Tumor Tissue Consortium (CBTTC). We thank Dr. Bradley St. Croix for help with making the D3-GPC2-PBD figure.
Footnotes
Author Contributions: Conceptualization, K.R.B., P.R. and J.M.M.; Methodology, K.R.B., P.R., Z.Z., D.S.D., and J.M.M; Software, P.R., K.S.R., G.L.G., and J.L.H; Formal Analysis, K.R.B, P.R., Z.Z., S.H., K.S.R., R.T.S., G.L.G., J.L.H., B.P., T.B., M.S.; Investigation, K.R.B., P.R., Z.Z., M.L., D.M., S.H., K.S.R., M.R, L.D., S.M., R.T.S., D.V.Z., Y.F., Y.W., J.H., G.L.G., N.M.K., J.L.H.; Resources, L.D, S.M., J.W., A.G., J.K., M.A.M., M.D.T.; Funding Acquisition, K.R.B, J.M.M, C.M., D.S.D, J.K. and M.D.T.; Writing – Original and Revised Draft, K.R.B. and J.M.M.; Supervision, K.R.B., M.D.T., D.S.D., C.M., and J.M.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bosse KR, Maris JM. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer. 2016;122:20–33. doi: 10.1002/cncr.29706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO, Campisi J, Yaswen P, Cooper PK, Kaufman PD. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS one. 2009;4:e6529. doi: 10.1371/journal.pone.0006529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter EL, Haglund EA, Mace EM, Deng D, Martinez D, Wood AC, Chow AK, Weiser DA, Belcastro LT, Winter C, et al. Antibody targeting of anaplastic lymphoma kinase induces cytotoxicity of human neuroblastoma. Oncogene. 2012;31:4859–4867. doi: 10.1038/onc.2011.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casucci M, Nicolis di Robilant B, Falcone L, Camisa B, Norelli M, Genovese P, Gentner B, Gullotta F, Ponzoni M, Bernardi M, et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood. 2013;122:3461–3472. doi: 10.1182/blood-2013-04-493361. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filmus J, Capurro M, Rast J. Glypicans. Genome Biol. 2008;9:224. doi: 10.1186/gb-2008-9-5-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Tang Z, Zhang YF, Feng M, Qian M, Dimitrov DS, Ho M. Immunotoxin targeting glypican-3 regresses liver cancer via dual inhibition of Wnt signalling and protein synthesis. Nat Commun. 2015;6:6536. doi: 10.1038/ncomms7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griebel T, Zacher B, Ribeca P, Raineri E, Lacroix V, Guigo R, Sammeth M. Modelling and simulating generic RNA-Seq experiments with the flux simulator. Nucleic Acids Res. 2012;40:10073–10083. doi: 10.1093/nar/gks666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harenza JL, Diamond MA, Adams RN, Song MM, Davidson HL, Hart LS, Dent MH, Fortina P, Reynolds CP, Maris JM. Transcriptomic profiling of 39 commonly-used neuroblastoma cell lines. Sci Data. 2017;4:170033. doi: 10.1038/sdata.2017.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart LS, Rader J, Raman P, Batra V, Russell MR, Tsang M, Gagliardi M, Chen L, Martinez D, Li Y, et al. Preclinical Therapeutic Synergy of MEK1/2 and CDK4/6 Inhibition in Neuroblastoma. Clin Cancer Res. 2017;23:1785–1796. doi: 10.1158/1078-0432.CCR-16-1131. [DOI] [PubMed] [Google Scholar]
- Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, et al. A draft map of the human proteome. Nature. 2014;509:575–581. doi: 10.1038/nature13302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocak H, Ackermann S, Hero B, Kahlert Y, oberthuer A, Juraeva D, Roels F, Theissen J, Westermann F, Deubzer H, et al. Hox-C9 activates the intrinsic pathway of apoptosis and is associated with spontaneous regression in neuroblastoma. Cell Death Dis. 2013;4:e586. doi: 10.1038/cddis.2013.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkele A, Taraseviciute A, Finn LS, Johnson AJ, Berger C, Finney O, Chang CA, Rolczynski LS, Brown C, Mgebroff S, et al. Preclinical Assessment of CD171-Directed CAR T-cell Adoptive Therapy for Childhood Neuroblastoma: CE7 Epitope Target Safety and Product Manufacturing Feasibility. Clin Cancer Res. 2017;23:466–477. doi: 10.1158/1078-0432.CCR-16-0354. [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantaj J, Jackson PJ, Rahman KM, Thurston DE. From Anthramycin to Pyrrolobenzodiazepine (PBD)-Containing Antibody-Drug Conjugates (ADCs) Angew Chem Int Ed Engl. 2017;56:462–488. doi: 10.1002/anie.201510610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matas-Rico E, van Veen M, Leyton-Puig D, van den Berg J, Koster J, Kedziora KM, Molenaar B, Weerts MJ, de Rink I, Medema RH, et al. Glycerophosphodiesterase GDE2 Promotes Neuroblastoma Differentiation through Glypican Release and Is a Marker of Clinical outcome. Cancer Cell. 2016;30:986. doi: 10.1016/j.ccell.2016.11.008. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Shih DJ, Peacock J, Garzia L, Morrissy AS, Zichner T, Stutz AM, Korshunov A, Reimand J, Schumacher SE, et al. Subgroup-specific structural variation across 1,000 medulloblastoma genomes. Nature. 2012;488:49–56. doi: 10.1038/nature11327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orentas RJ, Yang JJ, Wen X, Wei JS, Mackall CL, Khan J. Identification of cell surface proteins as potential immunotherapy targets in 12 pediatric cancers. Front oncol. 2012;2:194. doi: 10.3389/fonc.2012.00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders LR, Bankovich AJ, Anderson WC, Aujay MA, Bheddah S, Black K, Desai R, Escarpe PA, Hampl J, Laysang A, et al. A DLL3-targeted antibody-drug conjugate eradicates high-grade pulmonary neuroendocrine tumor-initiating cells in vivo. Sci Transl Med. 2015;7:302ra136. doi: 10.1126/scitranslmed.aac9459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman S, Zhu Z, Saha S, Zhang XM, Yang MY, Hilton MB, Morris K, Szot C, Morris H, Swing DA, et al. Eradication of Tumors through Simultaneous Ablation of CD276/B7-H3-Positive Tumor Cells and Tumor Vasculature. Cancer Cell. 2017;31:501–515 e508. doi: 10.1016/j.ccell.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, Ren B. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings RL, Howard J, Dunlop A, Mullarkey M, McDermott M, Breatnach F, O'Meara A. Are gains of chromosomal regions 7q and 11p important abnormalities in neuroblastoma? Cancer Genet Cytogenet. 2003;140:133–137. doi: 10.1016/s0165-4608(02)00681-7. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Cheung NK. Disialoganglioside GD2 as a therapeutic target for human diseases. Expert opin Ther Targets. 2015;19:349–362. doi: 10.1517/14728222.2014.986459. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Valentijn LJ, Koppen A, van Asperen R, Root HA, Haneveld F, Versteeg R. Inhibition of a new differentiation pathway in neuroblastoma by copy number defects of N-myc, Cdc42, and nm23 genes. Cancer Res. 2005;65:3136–3145. doi: 10.1158/0008-5472.CAN-04-2469. [DOI] [PubMed] [Google Scholar]
- Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans ME, Molenaar JJ, van Nes J, Versteeg R. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc Natl Acad Sci U S A. 2012;109:19190–19195. doi: 10.1073/pnas.1208215109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivian J, Rao A, Nothaft FA, Ketchum C, Armstrong J, Novak A, Pfeil J, Narkizian J, Deran AD, Musselman-Brown A, et al. Rapid and efficient analysis of 20,000 RNA-seq samples with Toil. bioRxiv. 2016:062497. [Google Scholar]
- Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DM, Remke M, Wu X, Garzia L, Luu B, Cavalli F, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta Neuropathol. 2015;129:449–457. doi: 10.1007/s00401-015-1389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu AL, Gilman AL, Ozkaynak MF, London WB, Kreissman SG, Chen HX, Smith M, Anderson B, Villablanca JG, Matthay KK, et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yu Y, Hertwig F, Thierry-Mieg J, Thierry-Mieg D, Wang J, Furlanello C, Devanarayan V, Cheng J, Deng Y, et al. Comparison of RNA-seq and microarray-based models for clinical endpoint prediction. Genome Biol. 2015;16:133. doi: 10.1186/s13059-015-0694-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Dimitrov AS, Bossart KN, Crameri G, Bishop KA, Choudhry V, Mungall BA, Feng YR, Choudhary A, Zhang MY, et al. Potent neutralization of Hendra and Nipah viruses by human monoclonal antibodies. J Virol. 2006;80:891–899. doi: 10.1128/JVI.80.2.891-899.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Ramakrishnan B, Li J, Wang Y, Feng Y, Prabakaran P, Colantonio S, Dyba MA, Qasba PK, Dimitrov DS. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. MAbs. 2014;6:1190–1200. doi: 10.4161/mabs.29889. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.