

Abstract

The cysteine protease enzyme legumain hydrolyzes peptide bonds with high specificity after asparagine and under more acidic conditions after aspartic acid [Baker E. N.J. Mol. Biol. 1980, 141, 441−484; Baker E. N.; et al. J. Mol. Biol. 1977, 111, 207–210; Drenth J.; et al. Biochemistry 1976, 15, 3731–3738; Menard R.; et al. J. Cell. Biochem. 1994, 137; Polgar L.Eur. J. Biochem. 1978, 88, 513–521; Storer A. C.; et al. Methods Enzymol. 1994, 244, 486–500. Remarkably, legumain additionally exhibits ligase activity that prevails at pH > 5.5. The atomic reaction mechanisms including their pH dependence are only partly understood. Here we present a density functional theory (DFT)-based quantum mechanics/molecular mechanics (QM/MM) study of the detailed reaction mechanism of both activities for human legumain in solution. Contrasting the situation in other papain-like proteases, our calculations reveal that the active site Cys189 must be present in the protonated state for a productive nucleophilic attack and simultaneous rupture of the scissile peptide bond, consistent with the experimental pH profile of legumain-catalyzed cleavages. The resulting thioester intermediate (INT1) is converted by water attack on the thioester into a second intermediate, a diol (INT2), which is released by proton abstraction by Cys189. Surprisingly, we found that ligation is not the exact reverse of the proteolysis but can proceed via two distinct routes. Whereas the transpeptidation route involves aminolysis of the thioester (INT1), at pH 6 a cysteine-independent, histidine-assisted ligation route was found. Given legumain’s important roles in immunity, cancer, and neurodegenerative diseases, our findings open up possibilities for targeted drug design in these fields.
Keywords: legumain, protease, ligase, mechanism, QM/MM, DFT, simulations
Introduction
The most common cysteine proteases are papain, cathepsin, and caspases, which can be found in a series of living organisms1−7 and play significant roles in proteolytic signaling. Therefore, deficiency as well as uncontrolled activity of cysteine proteases may cause many diseases such as cancer,8 muscular dystrophy,9 and Alzheimer’s disease.10 The cysteine protease legumain is overexpressed in several types of cancer and may be displaced from the lysosomes to the cell surface during malignant progression. Because the extracellular microenvironment in many tumors is acidic, it may allow cysteine protease activity also outside of the lysosomes. Legumain11−16 has therefore been utilized for experimental pro-drug activation ensuring tumor-targeted delivery of chemotherapeutic drugs.17
Moreover, legumain has been proposed as a marker for certain cancers and a potential therapeutic agent.18−20 Besides, protease inhibitors could also be employed as therapeutic targets (e.g., MMP inhibitors). Because the most successful inhibitors are usually transition-state-like, it is indispensable to fully understand the catalytic mechanism (intermediate and transition states), protonation states, and electronic properties in detail.
Proteases can also ligate peptide chains, generating cyclic, new, or alternatively spliced peptides. Especially in plants, cyclic peptides (like cyclotides) and protein variants play important roles in biology21 and medicine.22 Moreover, cyclic peptides find broad application in peptide drug engineering.23 However, in vitro cyclization of synthetic peptides is limited by the availability of ligase/transpeptidase enzymes.24−27 Importantly, at more neutral pH, human28 and mouse legumain29 has been shown to exhibit ligase activity.
Particularly for plant legumains, transpeptidation was suggested by Bernath-Levin et al.31 and Harries et al.,25 who performed macrocyclization reaction of SFTI and Kalata B1 in isotope-labeled H2O18 and found that when analyzing the proteolytic or ligated products by mass spectrometry no incorporation of O18 was detectable in the ligation product, which indicates that cyclization was achieved by direct transpeptidation and not through hydrolysis followed by ligation.
However, the exact mechanism of cleavage and ligation is not known. Therefore, within the scope of this project, we intend to investigate the legumain-catalyzed amide cleavage and ligation procedure in atomistic details using high-level (density functional theory (DFT)-based, quantum mechanics/molecular mechanics (QM/MM)) computational methods. Cystein proteases can be divided into two major groups based on their substrate binding. In papain-like enzymes, there is a direct proton transfer between the catalytic Cys and His possible; however, in caspase-like enzymes, the substrate is located between these catalytic residues. In caspase-like cysteine proteases, the Cys-His-Asp catalytic triad in the active site is responsible for the proteolytic activity, whereas the protonation state of these residues is highly debated30,32−36 (neutral or zwitterionic form). According to the commonly accepted mechanism, the catalysis takes place in two steps.30 When the substrate binds, the carbonyl of the scissile peptide bond is buried in the oxy anion hole, which comprises the backbone NH of Cys and Gly. The first step starts with the nucleophilic attack of the deprotonated cysteine residue on the peptide carbonyl carbon and the first tetrahedral intermediate is formed. Subsequently, the acyl enzyme (thioester intermediate) is generated, and, at the same time, a fragment of the substrate is released with an amine terminus and the histidine residue in the protease is restored to its deprotonated form (Figure 1).
Figure 1.

Putative mechanism of thioester formation (first step of the proteolysis) by papain-like cysteine proteases.30
The second step starts with the attack of a nucleophilic water molecule on the carbonyl carbon of the acyl enzyme (Figure 2). At this stage, a second tetrahedral intermediate is generated and a proton from the water molecule is transferred to His. Consequently, the substrate–Cys bond is split and the remaining S– might be neutralized by the positively charged nitrogen of His whereas the free enzyme is regenerated and the reaction can start over again. Dall and Brandstetter37 have successfully determined the crystal structure of prolegumain (PDB code 4FGU) and could identify the catalytic residues (Cys189, His148, and Asn42) in the active site. In addition, Dall and Brandstetter37 reported the crystal structure of the cysteine protease legumain in complex with different substrate analogues (PDB codes: 4AWA, 4AWB, 4AW9) and also describe its substrate recognition. Inhibitors with Asn or Asp residues at the P1 position were identified to be bound covalently to SG(Cys189). Recently, Dall and Brandstetter successfully elucidated the crystal structure of legumain in complex with cystatin E/M, which is the most potent endogenous inhibitor38 of legumain (PDB code: 4N6O(28)). In this structure, the substrate is positioned similarly to a chloromethylketone-based inhibitor (verified by superposition with 4AW9(37)) but is not covalently bound in the active site. Moreover, the substrate binds canonically and has both primed and nonprimed residues; therefore, it serves as an ideal starting point for our computational studies (Figure 3, right).
Figure 2.

Second step of the enzyme-catalyzed proteolytic cleavage (hydrolysis) by papain-like cysteine protease.30
Figure 3.

Active site of papain (1KHP44) (left), caspase-3 (1PAU45) (middle), and human legumain (4AW937) (right). The active site residues are represented as sticks; the enzyme carbons are colored gray. The substrate carbons are colored orange, and the relevant crystal structure waters are depicted as red spheres. Dotted lines denote hydrogen bonds.
Because this was the first time that the structure of legumain has been presented, there is no computational work in the literature on this enzyme. However, a few research groups performed calculations at different levels to elucidate the mechanism of two other members of the cysteine protease family, namely, papain32,33,35,36,39,40 and caspase.41−45
In the active site of papain, the His159–Cys25 distance is around 3.6 Å, and these are ideally positioned for proton transfer between them. However, in the case of legumain, the corresponding His148 is over 6 Å far from Cys189, and therefore, a direct proton transfer is unlikely. In contrast, in legumain (and caspase), the substrate binds between the catalytic cysteine and histidine residues. Therefore, the most relevant theoretical work with respect to our studies are the investigations of Sulpizi et al.,42 who applied DFT-based QM/MM methods to calculate the hydrolysis of the acyl enzyme complex for caspase-3 starting from a covalently bound inhibitor.
Their calculations suggest that the attack of the nucleophilic water molecule (second step of the cleavage) leads to a geminal diol intermediate and shows thereby a remarkable discrepancy between caspases and papain. In addition, Miscione et al.43 performed DFT calculations in the gas phase to study thioester formation for caspase-7 starting from a covalently bound inhibitor complex. Their model was built up from fragments of the active site residues and the Ac-DEVD inhibitor. After removal of the S–C bond, the catalytic cysteine was terminated by a H atom and the substrate by −NH3CH3. The authors propose a multistep proton-hopping mechanism via deprotonation of the neighboring peptide nitrogen and making use of the substrate aspartic acid COO– side chain in order to activate the cysteine. At the same time, they reject a much simpler one-step mechanism due to a higher calculated barrier; however, the surrounding protein environment and the solvent are not considered in their gas-phase simulations.
Moreover, although the active site of caspase-3 shows more similarity with legumain, still there are several differences left. Because legumain cleaves essentially behind asparagine, proton hopping with substrate side chain participation is unfeasible. In caspase-3, the active site water position is rather an analogue of papain; and in legumain, there is an acidic residue (Glu190) close to the catalytic Cys189 that is absolutely missing in both papain and caspase-3 (Figure 3). Besides, legumain is the only protease in which an aspartic acid residue next to the catalytic histidine is ring-closed to a succinimide. Therefore, there remains a number of open questions regarding the detailed reaction mechanism of legumain, particularly the protonation state of the active site residues, the activation of Cys189, and the role of the residues Glu190, His148, and SNN147 and the water molecules.
In the present work, we studied the mechanism of both the protease and the ligase activity of the human legumain in atomistic detail in solution. We employed a comprehensive QM/MM approach at the B3LYP/DFT level of theory using the extensive functionality provided by the recently developed QM/MM46,47 modules in the NWChem48 software package to investigate the attack of the Cys189 on the scissile peptide bond, the possible proton transfer pathways, the water attack, and the product release. Afterward, free energy calculations over the reaction coordinates were employed to determine the rate-limiting step of the proteolysis reaction. In addition, different ligation/transpeptidation mechanisms were studied that were in good agreement with experimental findings. Nevertheless, we would like to emphasize that detailed experimental work is not part of the present paper.
Results and Discussion
The theoretical tools that have been used in these calculations are discussed in the methods section (see the Supporting Information (SI)) and in prior publications.46,49−53 Important to the discussion here is that they are applied to a system containing the protein/substrate complex solvated in aqueous background. To develop a reaction mechanism for the cleavage step, it is necessary to have a reliable initial structure (RS structure) that is based on a good resolution X-ray structure of the legumain/cystatin complex (PDB code: 4N6O(28)) followed by system preparation and optimization, as discussed in the “Computational Details and Methods” section of the SI. Because the protonation state of the Cys189-His148 ion pair has a large influence on the predicted mechanism, it has to be chosen very carefully. Due to the fact that in the cystatin–legumain complex the NE(His148)–SG(Cys189) distance is over 6 Å and in prolegumain these residues are only 4.14 Å from each other, first we supposed that the proton shuttle between them might occur, before the substrate enters the active site, and as soon as the catalytic cycle is completed and the substrate leaves the pocket, the zwitterion regenerates. Hence, first we tried to simulate the acylation pathway starting with a positively charged His148 and a negatively charged Cys189-thiolate using the spring method, as described above. However, all of our attempts to generate this reaction path failed. Relaxation of the system after removal of the constraints resulted in the starting geometry.
Afterward, systematic titration calculations using MOE2016.08 pointed out that at the pH of the protease activity (around pH 5.0) of both Cys189 and His148 is neutral in the reactant state. Therefore, an alternative reaction pathway was necessary, one that initiates the deprotonation of the catalytic cysteine.
Proteolysis Pathway
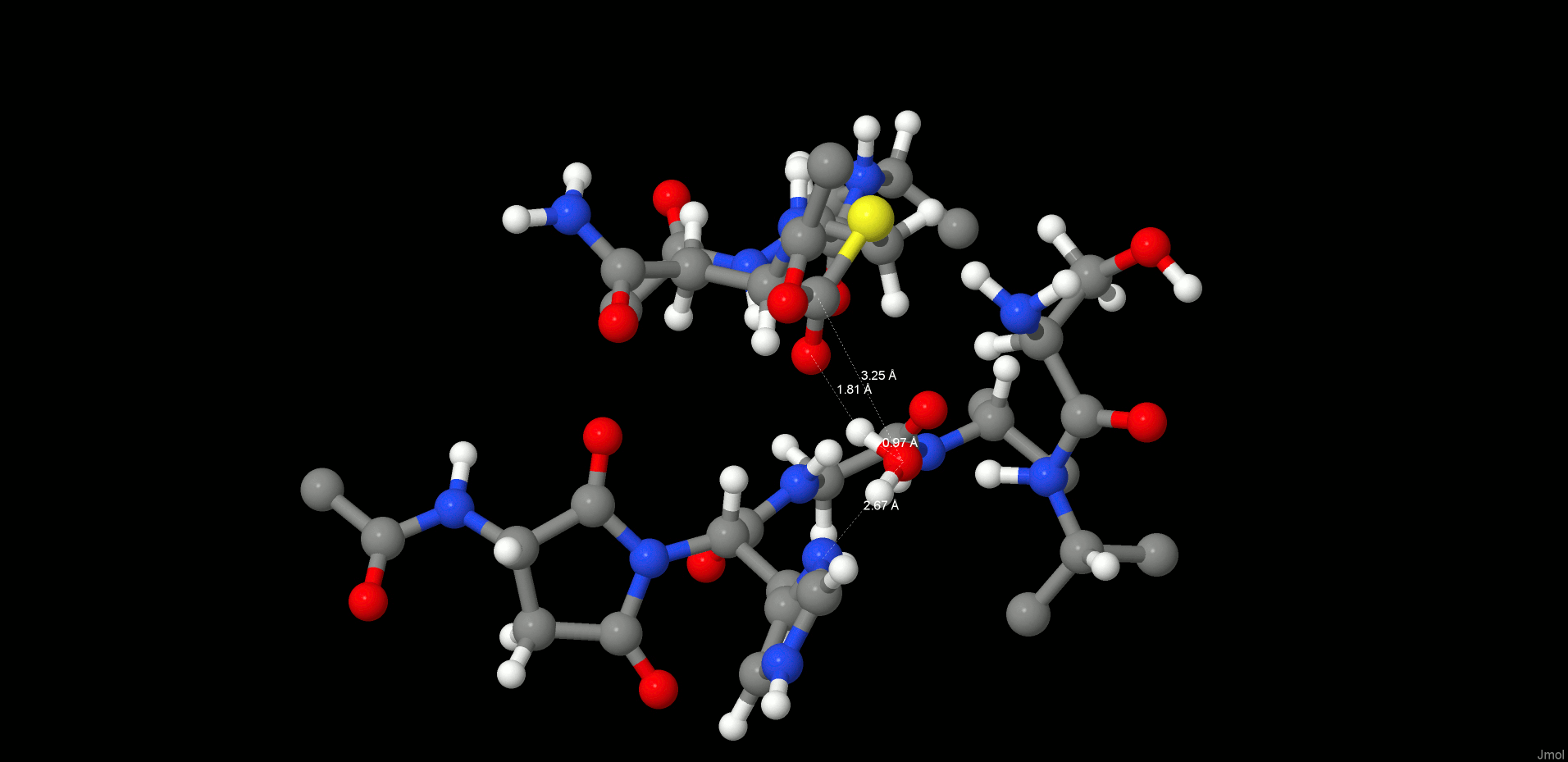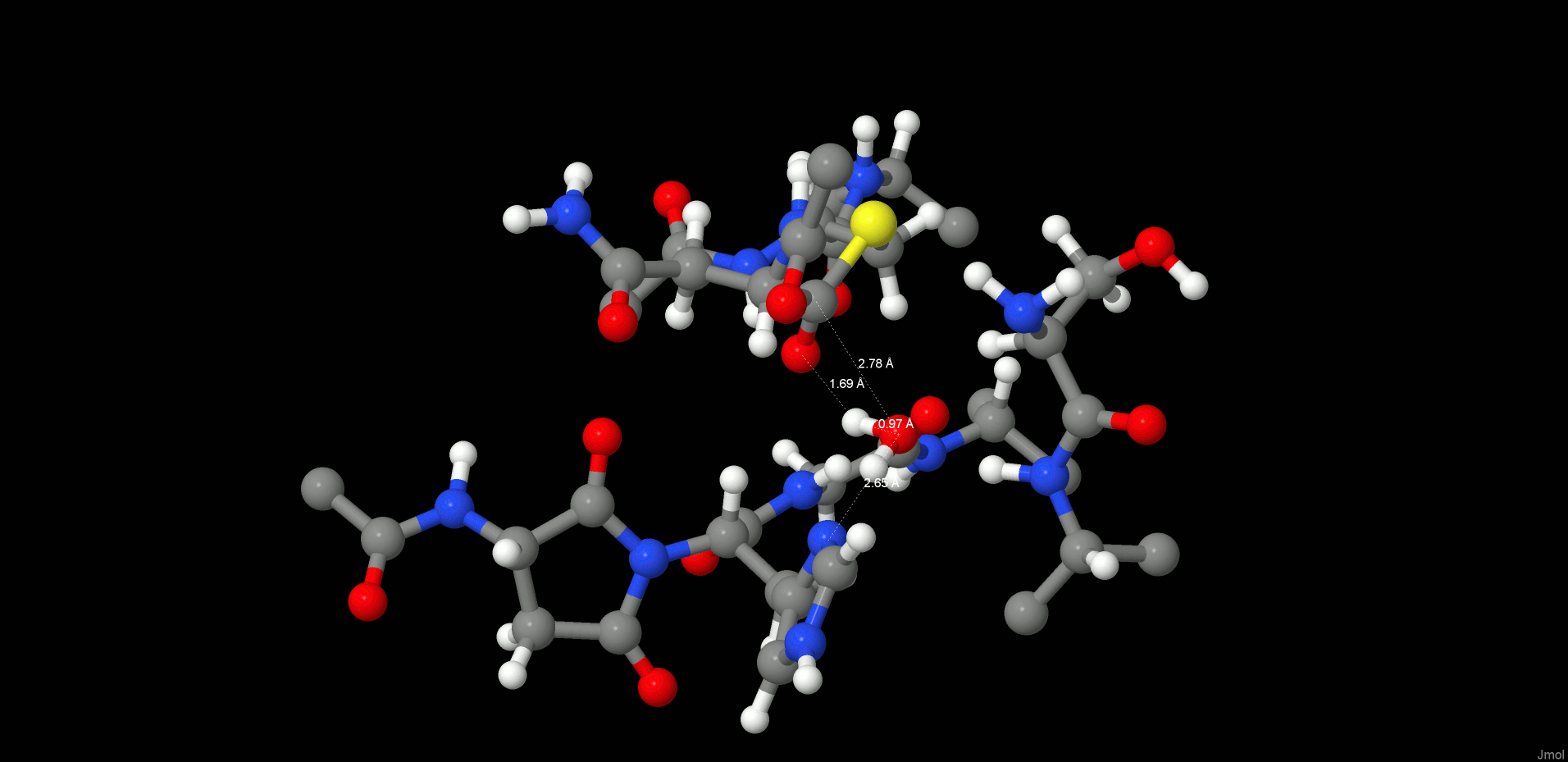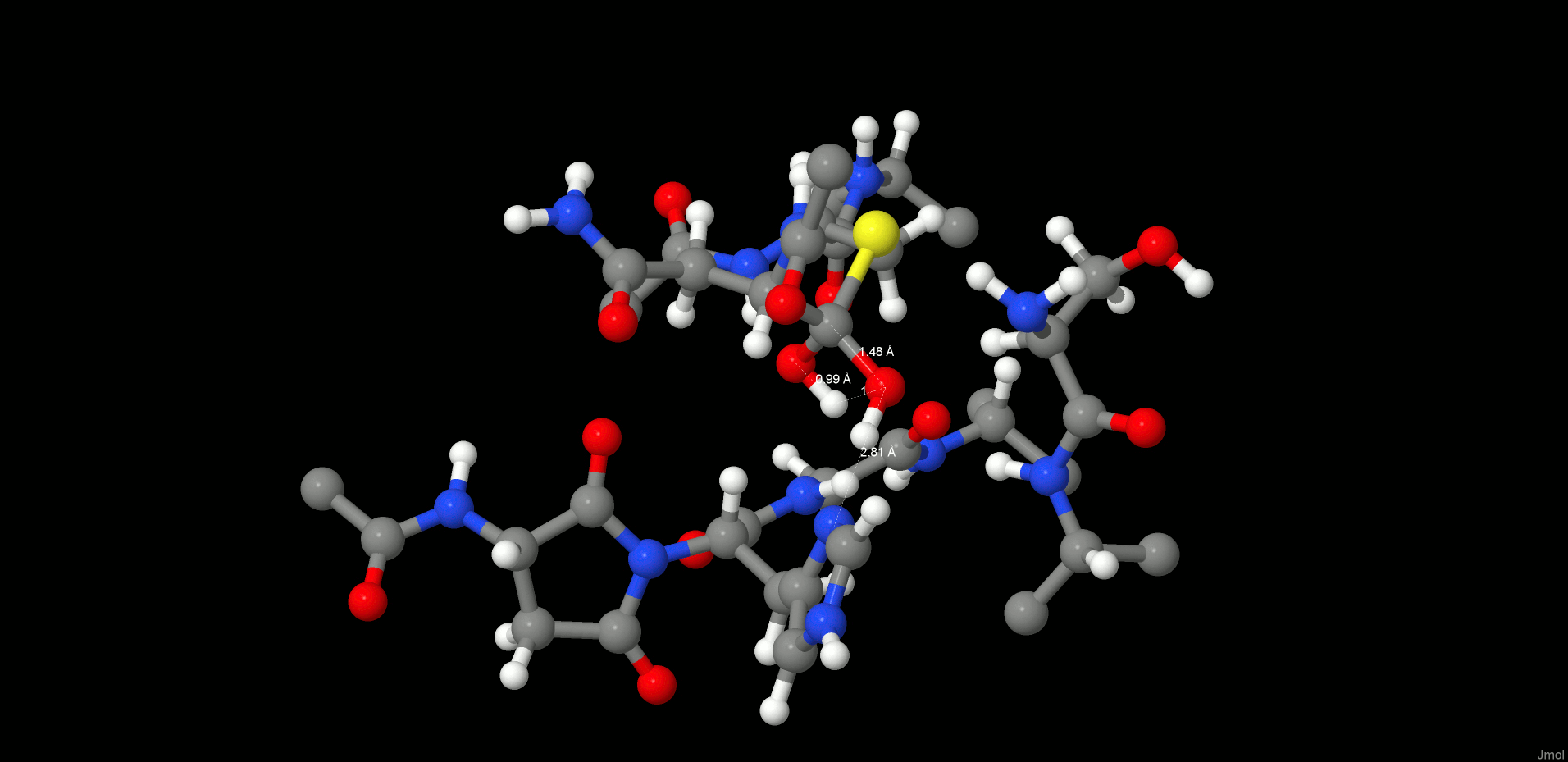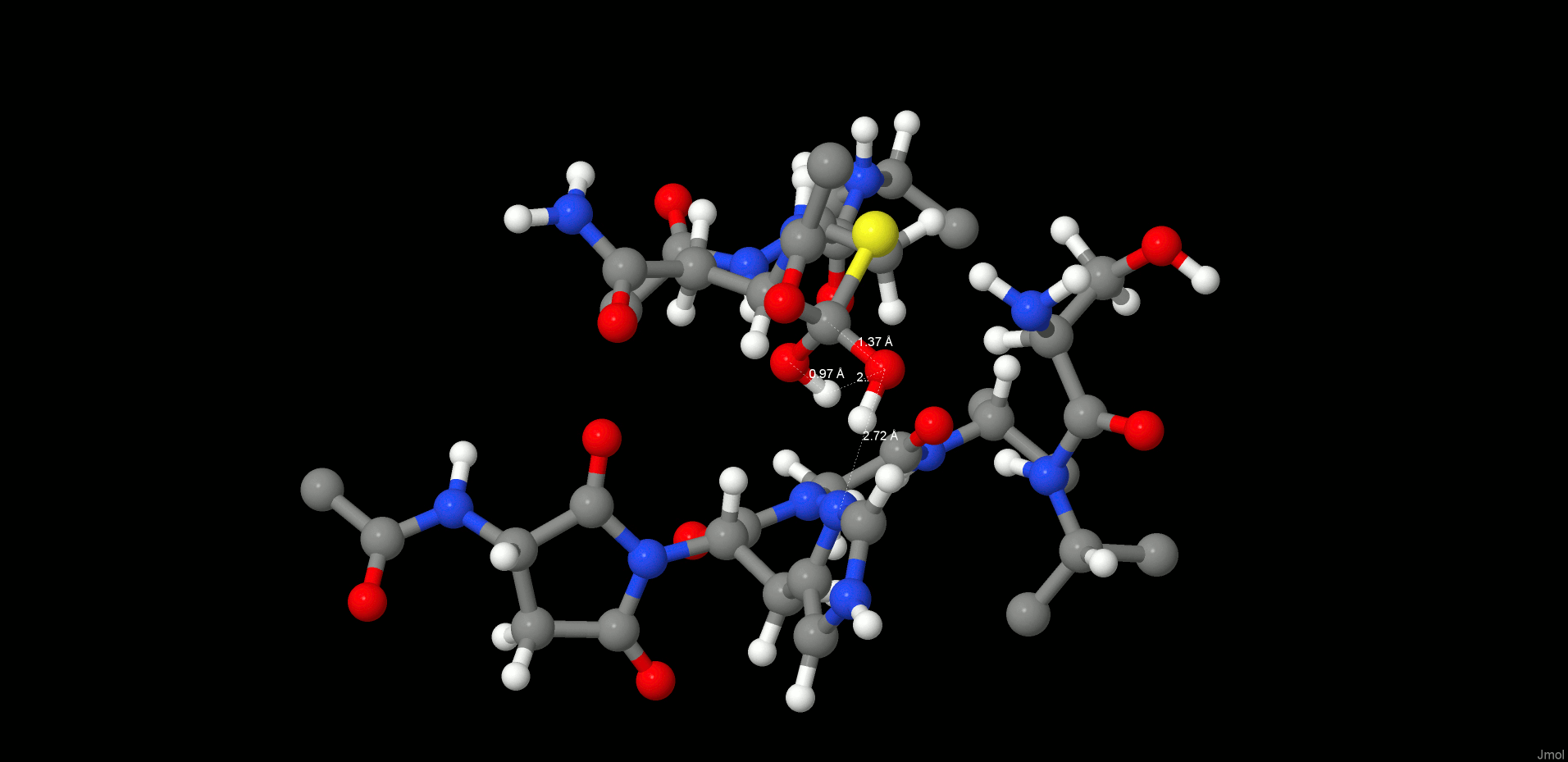
Formation of the First Intermediate (INT1)
In the reactant state (Figure 4, left), the P1 substrate carbonyl is tightly anchored into the oxy anion hole of the active site by strong interactions with the N(Gly149) and N(Cys189) backbone nitrogens. The role of the oxy anion hole is very important. On the one hand, it polarizes the P1 C=O and weakens the C–N peptide bond, and on the other hand, it stabilizes the carbonyl oxygen and O–, which is generated during the reaction. The SG(Cys189) is ideally positioned for a nucleophilic attack, whereas the catalytic water, which participates in the hydrolysis, is located close to the substrate carbonyl and NE(His148) and is also strongly hydrogen bonded between them.
Figure 4.

Reactant (left) and INT1 (right) structure along the reaction path of the substrate cleavage showing a zoom of the active site of the legumain–pentapeptide substrate complex. Enzyme residues are represented by gray carbons, and the substrate is represented by orange ones. Dotted lines denote H-bonds.
Note that there is no catalytic water present in the active site of the legumain–cystatin complex (PDB code 4N6O(28)) because the position of the water is occupied by C=O of the P2′ amino acid. However, after shortening the cystatin to a pentamer (see SI) and performing molecular dynamics simulations, the catalytic water (Wat305) could enter the pocket and take the position where it is excellently oriented for the reaction. In order to initiate the cleavage process, a harmonic restraint of 1.8 Å was imposed on the SG(Cys189)–C(Asn302) distance to simulate the attack of the sulfur. Upon optimization, we could observe a coordinated attack of SG(Cys189) on the P1 carbonyl carbon and a proton transfer of HG(Cys189) to the P1′ scissile peptide nitrogen, leading to an intermediate disruption of the peptide bond and generation of the acyl enzyme (INT1, Figure 4, right). Unexpectedly, no tetrahedral intermediate (Figure 1) was generated but a thioester, which would rather correspond to the second intermediate. This remained stable even after removal of the constraint and subsequent relaxation. In this first intermediate state (INT1), the position of the active site water, histidine, succinimide, and serine residues barely changes and also the participating carbonyl remains in the oxy anion hole. The largest movement can be associated with the substrate and the catalytic cysteine (Cys189).
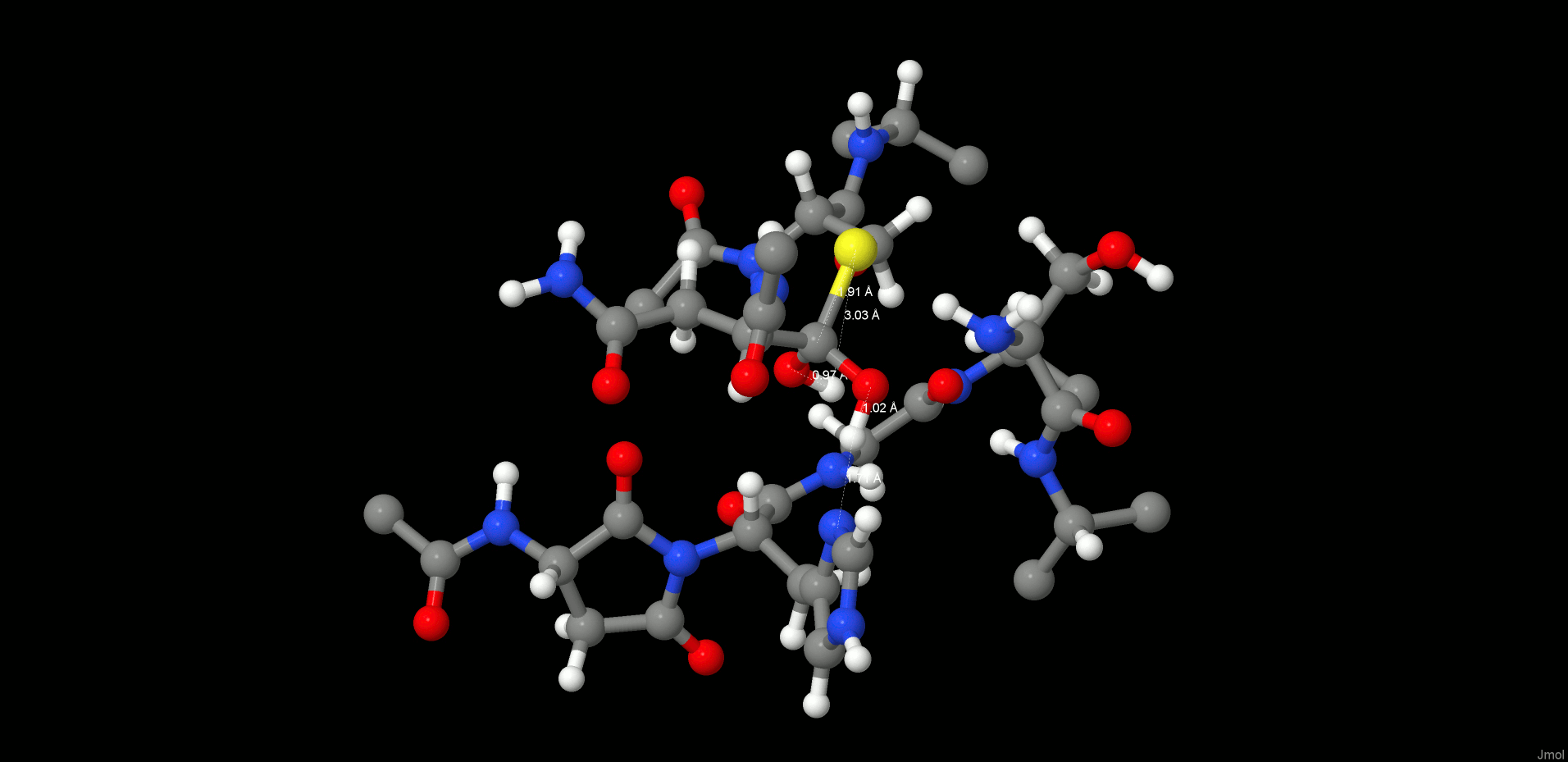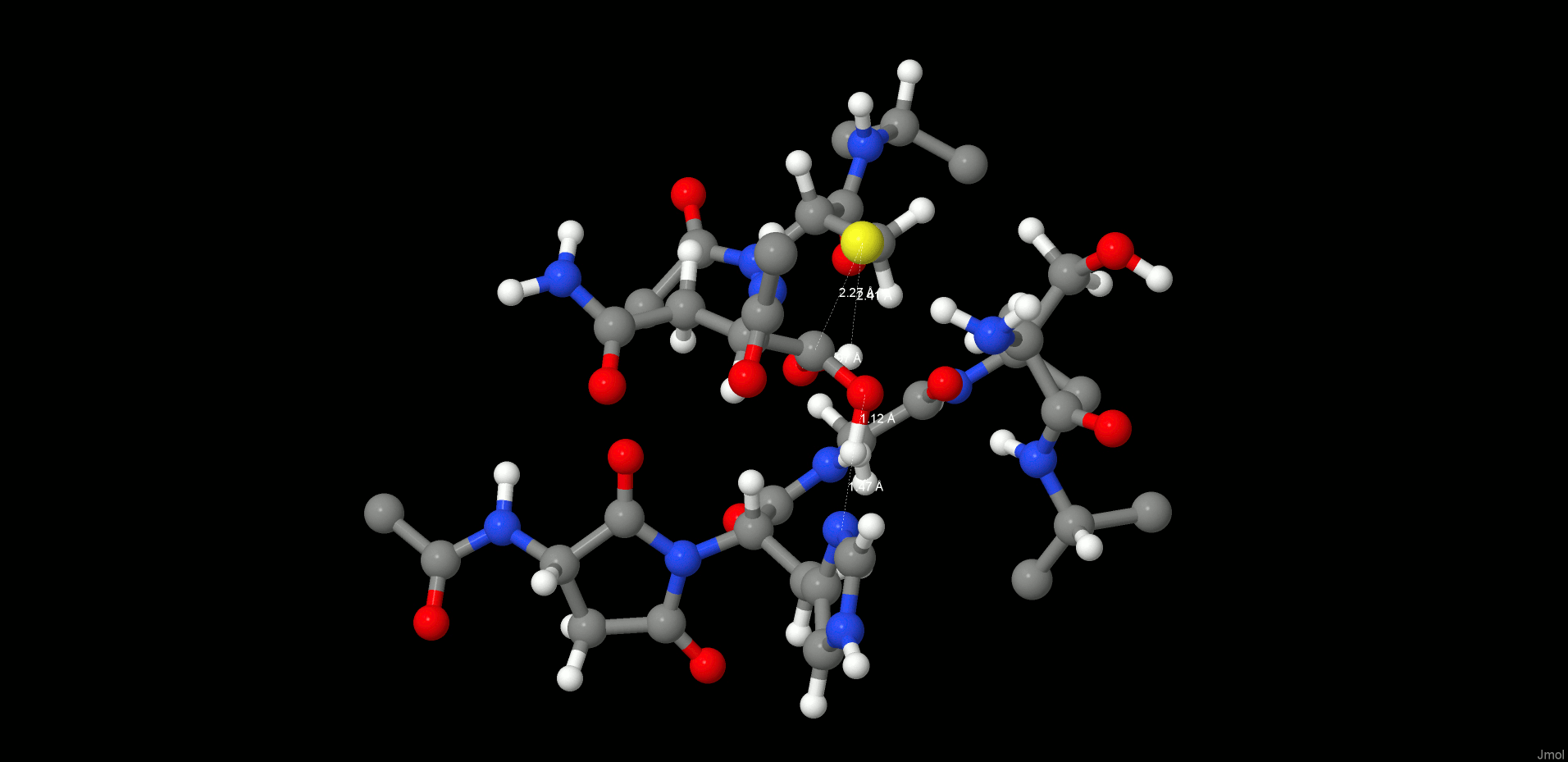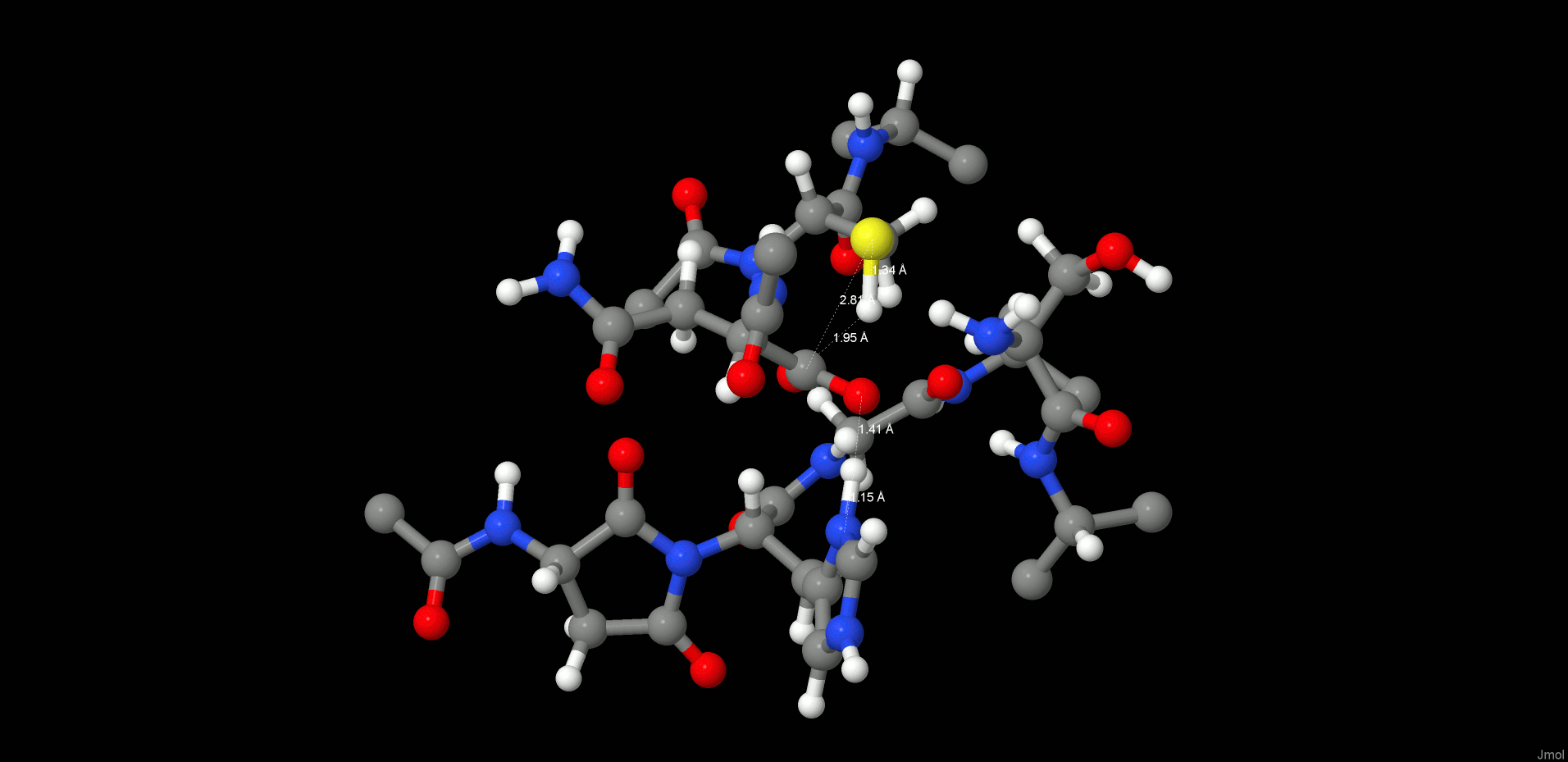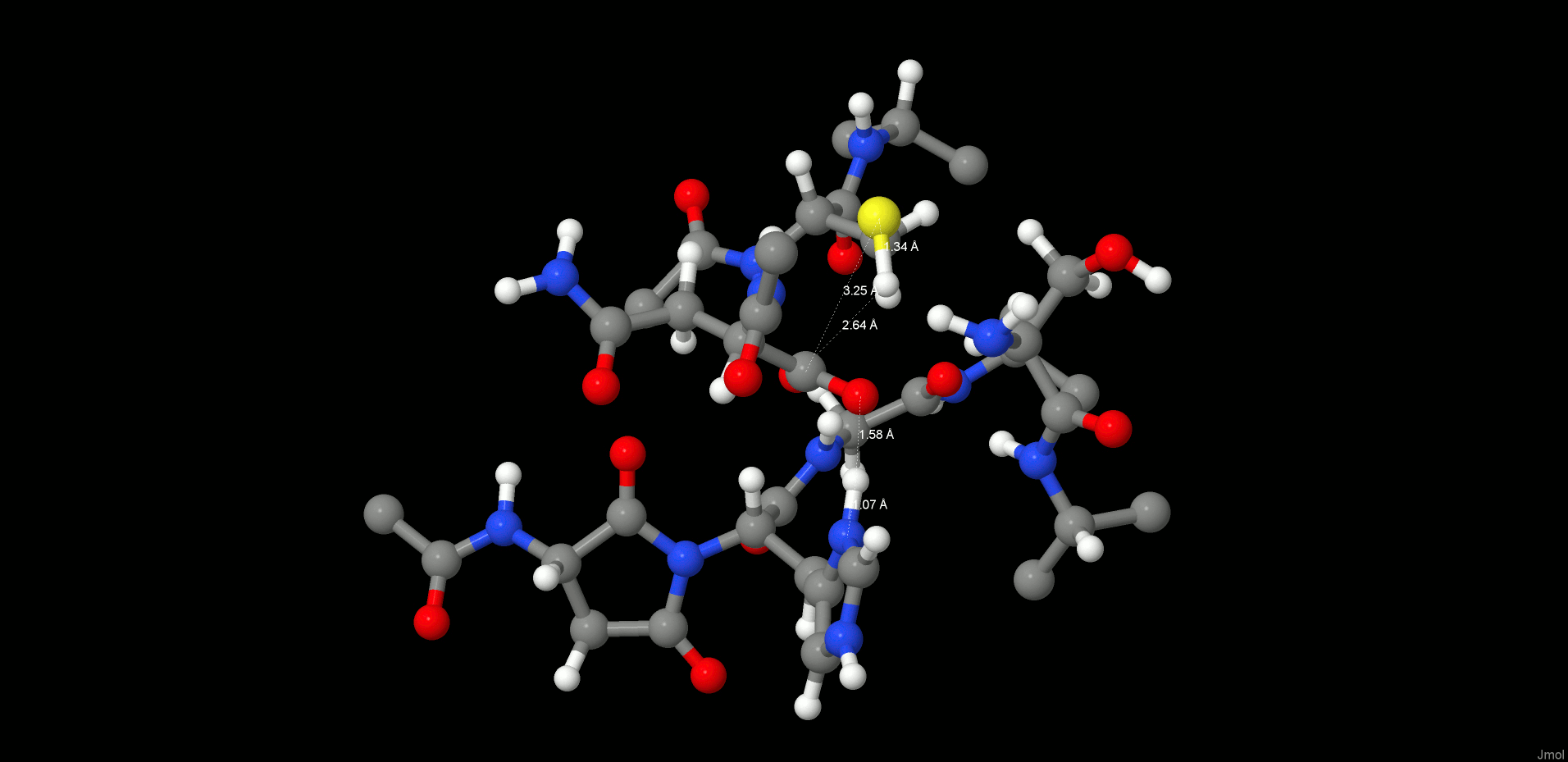
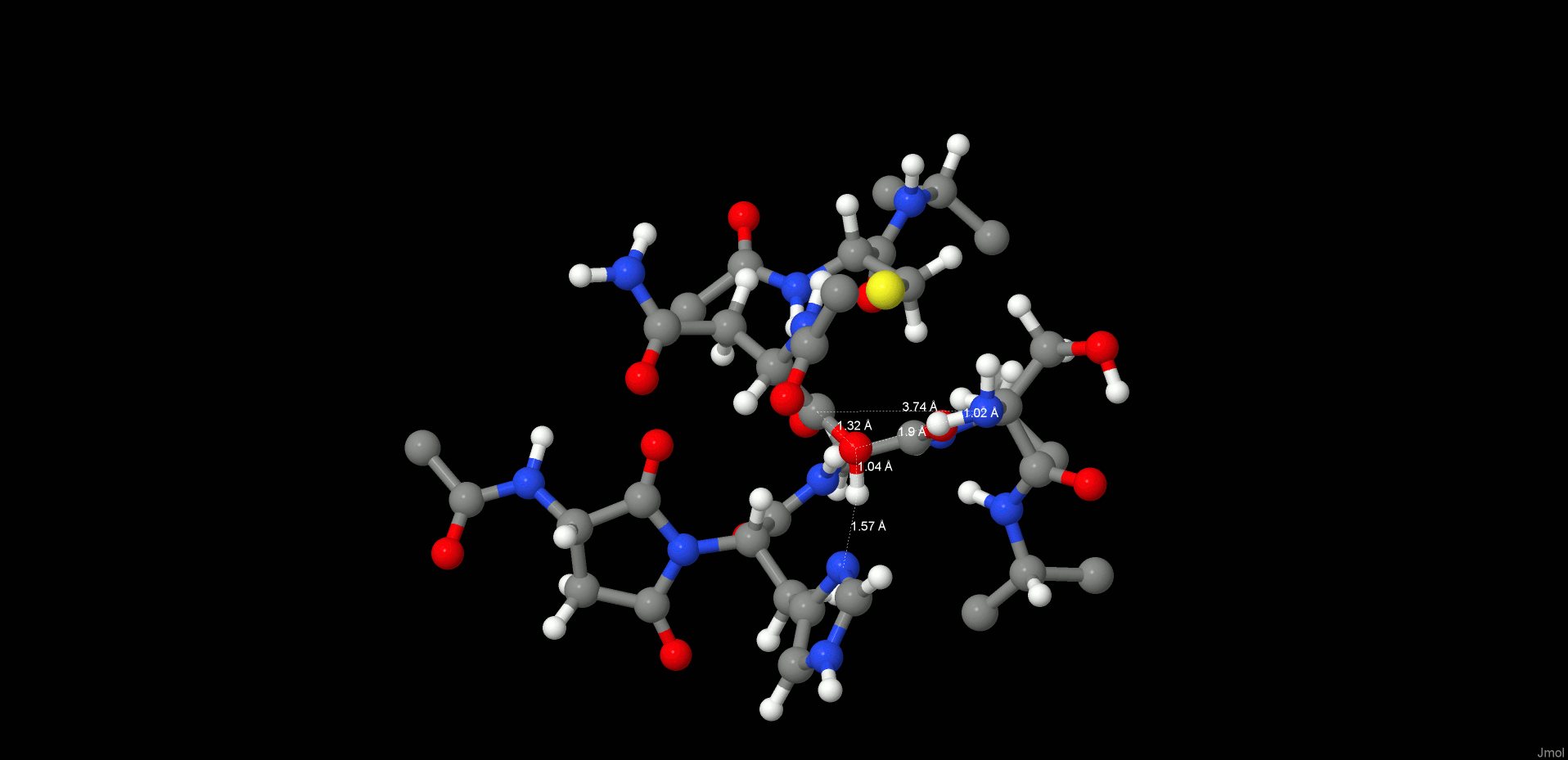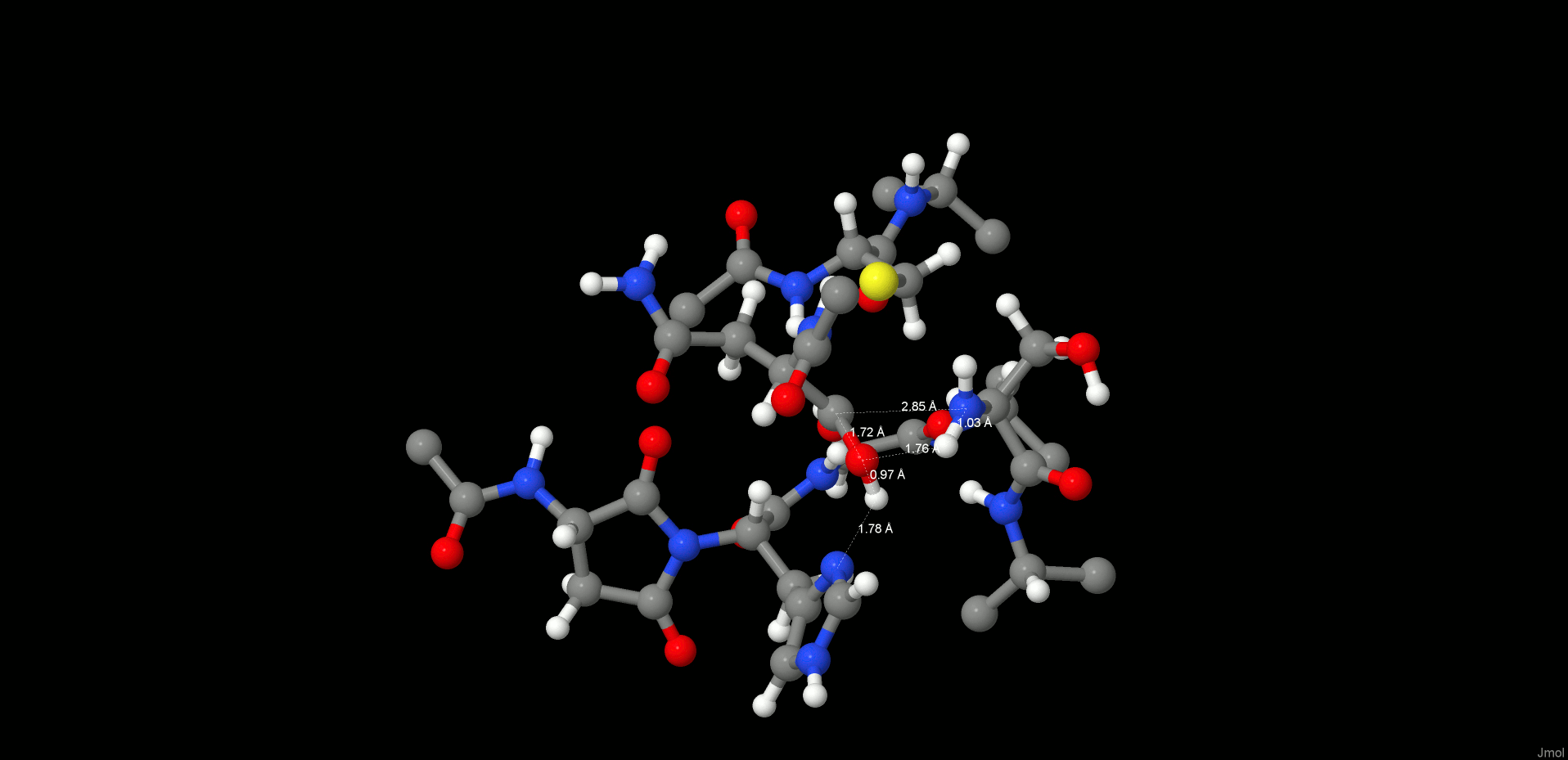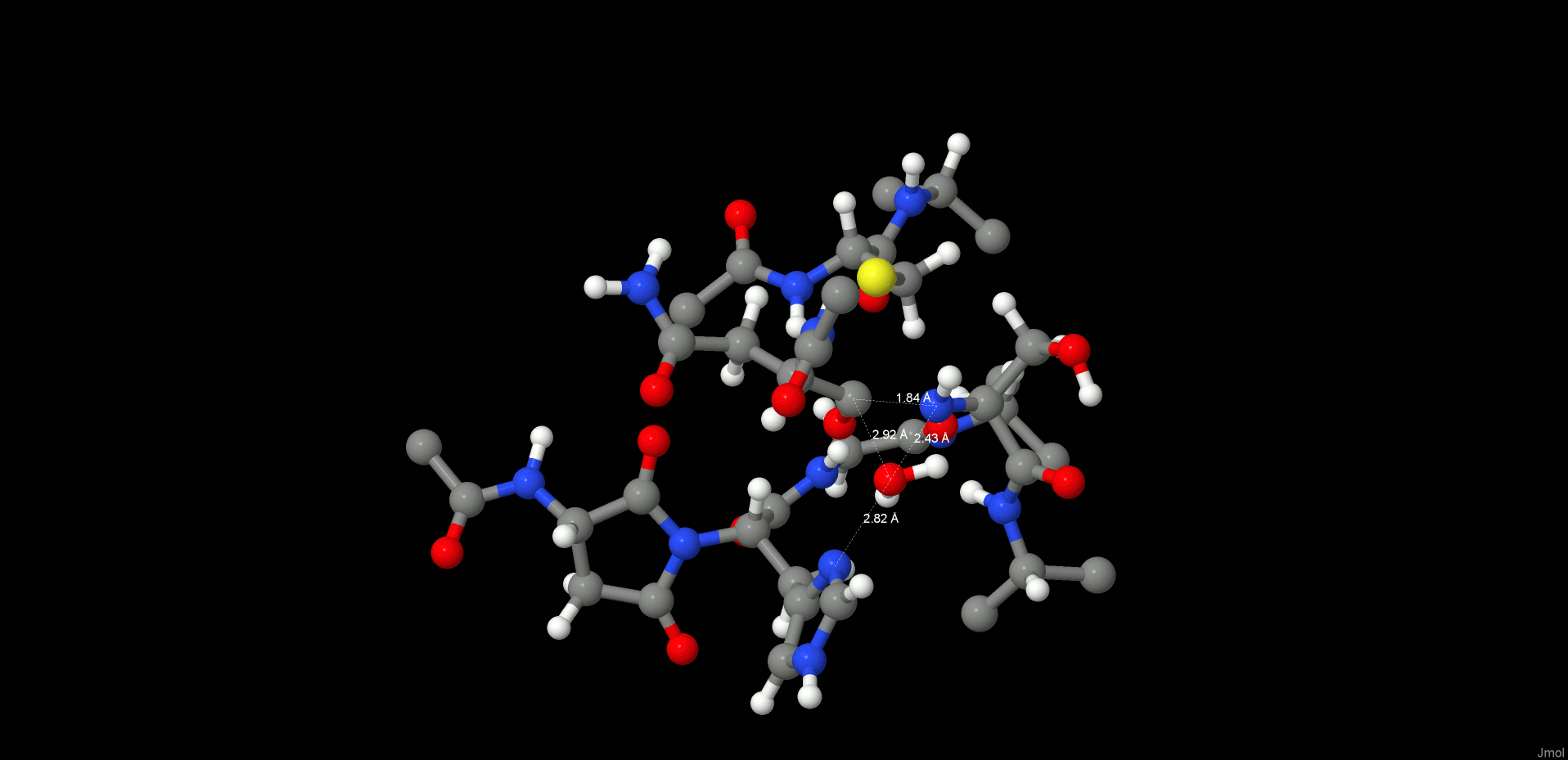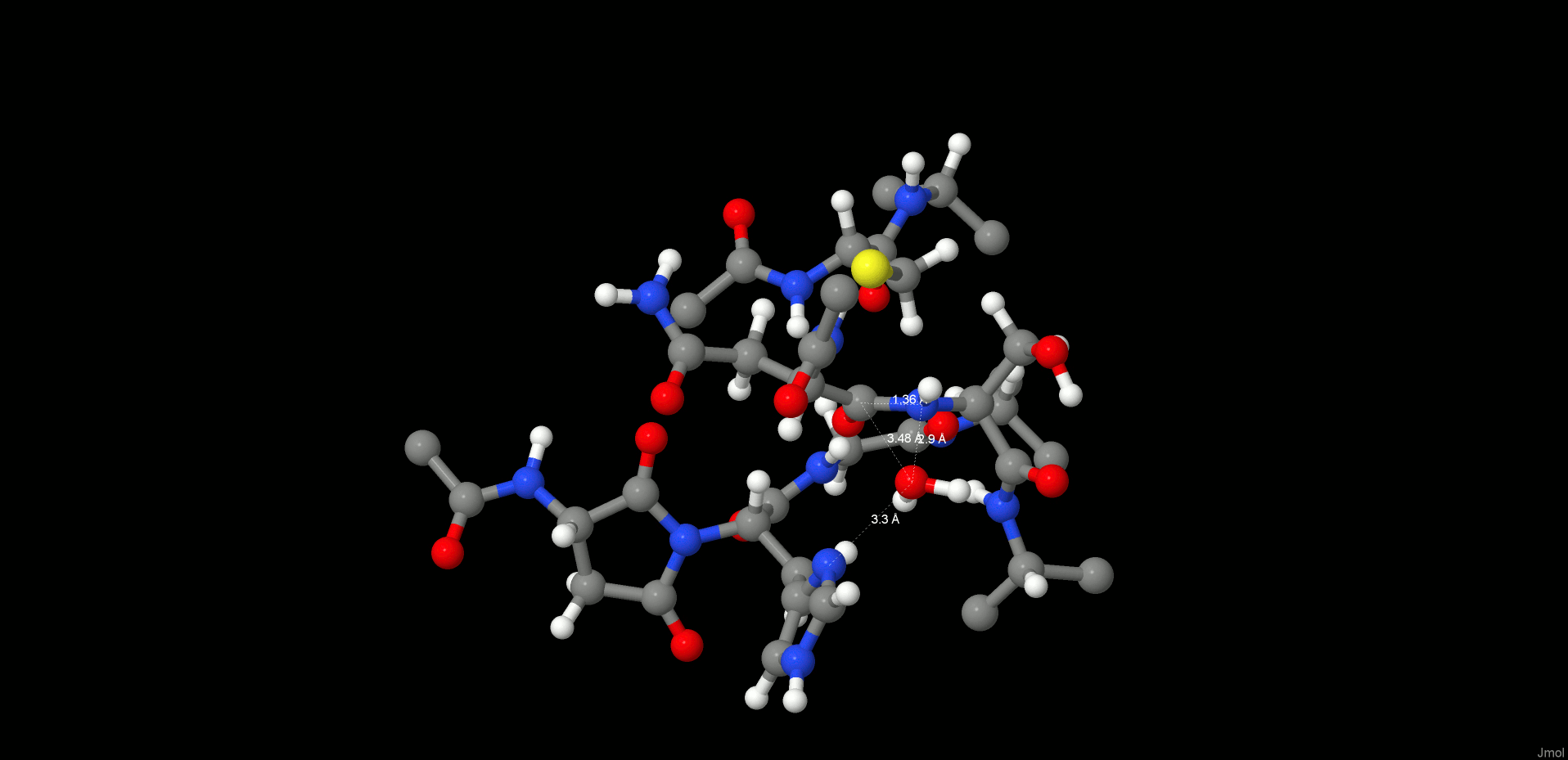
Formation of the Second Intermediate (INT2)
The cleavage reaction proceeds with hydrolysis of the thioester. The position and orientation of the catalytic water are optimal for nucleophilic attack.
The H-bond distances between OW(WTR305)–O(Asn302) and OW(WTR305)–ND1(His148) are as short as 2.74 and 2.67 Å, respectively, which facilitates the polarization and thereby activation of the water molecule. To model this reaction step, a spring of 1.35 Å was applied to the OW(WTR305)–C(Asn302) distance, which yielded the second stable intermediate state (INT2, Figure 5, left) along the reaction coordinates of the cleavage procedure. However, the 2HW(WTR305) was not transferred to the ND1(His148) as expected; indeed, a diol was generated, as found also by Sulpizi et al.42 for caspase-3. Importantly, His148 does not serve as a general base during water activation and attack. Transition state search calculations (see the SI and the Reaction Energetics and Transition State Search section) show that the 3HW(WTR305) proton is pulled off by the carbonyl oxygen O(Asn302) and the remaining OH– attacks the carbon to produce a tetrahedral intermediate diol, which is still covalently bound to Cys189.
Figure 5.

INT2 (left) and product state (PS) (right) structures along the reaction path of the substrate cleavage showing a zoom of the active site of the legumain–pentapeptide complex. Enzyme residues are represented by gray carbons, and the substrate is represented by orange ones. Dotted lines denote H-bonds.
The second intermediate state (INT2) shows a tetrahedral structure, where the former water oxygen is coordinated with NE2(His148) and the SG(Cys189) is H-bonded to OG(Ser215), N(Ser216), and the free N-termini of Ser303. In addition, the participating carbonyl, which now forms a diol, remains in the oxy anion hole.
Generation of the Product State (PS)
To complete the proteolysis, the C(Asn302)–SG(Cys189) bond must break to regenerate the enzyme and to release the cleavage products. To achieve bond breaking, the proton (2HW(WTR305)) from the former carbonyl (O(Asn302)) was transferred to SG(Cys189) by using a constraint of 1 Å for the given distance.
Consequently, the thioester was cleaved and the proton (3HW(WTR305)) from the other oxygen (OW(WTR305)) of the prior diol was shifted to NE2(His148). In order to clarify the correct protonation state of the product step, further attempts were carried out to transfer the 3HW(WTR305) proton either to N(Ser303) to generate a zwitterion between the cleaved C- and N-termini or to the carboxyl end of the C-terminus to preserve neutrality at the cleavage site. However, after release of the constraint, the proton of interest always shifted back to NE2(His148) (Figure 5, right). Therefore, in the most stable state, the carboxylate C-terminus is deprotonated and is strongly H-bonded to SG(Cys189) as well as to the positively charged NE2(His148), and the N-terminus is neutral. We suppose that when the N-terminus leaves the pocket, it removes the proton from NE2(His148) probably via a water molecule and thereby regenerates the initial protonation state.
In addition, another alternative pathway has also been considered in which the P1 asparagine first forms a succinimide to enhance the reactivity and upon nucleophilic attack of the SG(Cys189) a tetrahedral intermediate should form. However, this attempt has failed because during relaxation the system always fell back to the reactant state.
Ligation Pathway
As described in the Introduction, legumain exhibits unique pH-dependent dual protease–ligase activity, whereas legumain is a protease at acidic conditions and ligation takes place at more neutral pH.
In contrast to other cysteine proteases like caspase or papain, in the case of legumain, Dall et al. found the peptide ligase activity at pH 6.0 in human AEP when they studied the mechanistic aspects of AEP inhibition by cystatin E/M.28 Due to the higher crystallization pH, the authors suggest that the legumain–hCE complex structure rather corresponds to the ligase state. Moreover, they point out that active site SG(Cys189) was rotated away from the scissile peptide bond, suggesting that it is not directly involved in the ligation reaction. Further experiments modifying the catalytic Cys189 also support this theory. On the one hand, Dall et al. oxidized the Cys189 by adding S-methylmethanethiosulfonate (MMTS) to generate a mixed disulfide Cys189–S–CH3,28 and on the other hand, Cys189 was mutated to Met189 (data not shown, unpublished results).
In both cases, the protease activity was suppressed, as expected; however, the ligase activity was preserved. Their findings put forward that there must be a mechanism without Cys189 participation, which is not the exact reversal of proteolysis via a stable thioester.
Our in silico simulations gave further support to a possible Cys189 independent mechanism because, by using the spring method as described in the SI, we could not generate the exact backward pathway via Cys189–thioester.
Therefore, we turned our attention to an alternative pathway, which comprises the direct attack of the N-terminus on the carbonyl of the C-terminus (Figure 6). Hence, first we started with the product state of the cleavage reaction (protonated Cys189) and applied a restraint of 1.35 Å between C(Asn302) and N(Ser303) to generate the peptide bond. However, surprisingly, the desired reaction path could not be produced. In the case of the cleavage mechanism, we have already seen that the protonation state of the active site residues is very crucial for the reaction mechanism to proceed, and we need to take into consideration that ligation takes place at higher pH than the cleavage reaction. Titration and pKa calculations (the pKa of Cys189 in the enzyme environment was 5.6) therefore suggested that at pH 6.0 the catalytic cysteine, Cys189, is present as a thiolate. Reoptimization of the system with deprotonated Cys189 resulted in a proton shift between NE2(His148) and the carboxylate of the C-terminus, which is comprehensible due to the subsequent charge equalization. The newly generated reactant state (RS, Figure 7 left) was the starting point for the ligation simulations.
Figure 6.

Proposed ligation pathway without Cys189 participation (left) and the transpeptidation mechanism (right).
Figure 7.
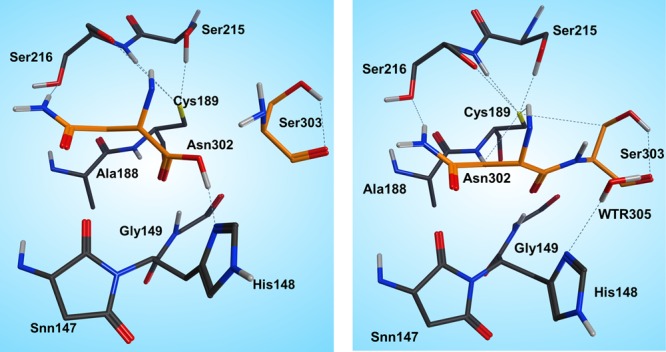
Reactant (left) and product (right) states of the ligation step.
In the reactant state, all participating residues (Asn302, Ser303, His148) are neutral, and Cys189 is deprotonated. The carboxylate proton of the C-terminus is H-bonded to NE2(His148), and the oxygen is coordinated by Ser303, whereas the proton migrates directly toward the OD2(Asn302) and thereby is ideally positioned for transfer and consecutive water formation. Thus, to model this reaction step, a constraint was applied to transfer a proton from the P1′ N(Ser303) to P1 residue OD2(Asn302) to activate both the C- and N-termini. After water formation and release, as expected, the N-terminus attacks the carbonyl and the peptide bond is formed (Figures 6 and 7 right).
Generation of the Product Step Using S–(Cys189)
The calculated product state of the ligase reaction fits very well with the reactant state of the protease action (Figure S1). That is, either the substrate can leave the active site or, upon reduction of the pH, the cleavage procedure can start over again. The carbonyl of the scissile peptide bond points toward the oxy anion hole (N(Gly149) and N(Cys189)), and the catalytic water is again positioned by NE2(His148) and N(Ser303). Moreover, the negatively charged cysteine thiolate is stabilized by the surrounding serine residues (Ser215, Ser216) during the entire reaction pathway.
Further support and confirmation of the above calculated mechanism arises from calculations with MMTS-blocked Cys189 (Cys189–S–CH3) and C189M mutant enzyme (see the SI).
Transpeptidation
The simulations have also shown that, although we cannot generate the exact reverse pathway of the proteolysis, the formation of a thioester and thereby transpeptidation are possible (INT1, Figure 4 right). We suppose that at this stage there is competition between the catalytic water and the N-terminus. The sulfur of the thioester is not strong enough as a base to remove the proton from the N-terminus due to the fact that the water molecule is the stronger nucleophile (Figure 6 right).
Therefore, as long as water is present in the active site, hydrolysis is favored over ligation. To prove this theory, we removed the catalytic water from the pocket, optimized the structure, and applied a constraint of 1.35 Å between C(Asn302) and N(Ser303) to generate the peptide bond. At the same time, the proton from N(Ser303) was automatically transferred to SG(Cys189), and the reaction was complete. After removal of the restraint, the generated transpeptidation product remained stable.
Reaction Energetics and Transition State Search
In order to achieve a complete overview of the catalytic pathway and to designate the stationary points and free energy profile of the reaction coordinate, we performed nudged elastic band (NEB) calculations46 between the reactant, intermediate, and product states. The initial pathway for NEB calculations was calculated in three steps for the cleavage reaction by linear interpolation between the reactant, INT1, INT2, and product states of the entire solute–solvent system with 15 or 20 beads/replicas for each segment. In the case of ligation, the reaction energetics were calculated in one step, from reactant to product, because no intermediate was found along the reaction coordinates.
Proteolysis
In the reactant to the intermediate step, there are two important events along the reaction coordinates (Figure 8). The first one is the transfer of the HG(Cys189) proton to the scissile peptide bond nitrogen N(Ser303), and the second one is the attack of the thiolate nucleophile on the carbonyl and thereby the formation of the thioester structure. According to our calculations, the energy of this first step determines the energy barrier and the overall reaction rate. This point occurs at the maximum energy along the reaction path at 19.3 kcal (Table 1, Figure S6), which fits well with the findings of Ma et al.54 for cathepsin K and Wei et al.36 for papain. There are three important structural changes that occur simultaneously as the reaction proceeds. One is elongation of the C(Asn302)–N(Ser303) peptide bond, the second one is transfer of the HG(Cys189) proton to the N-terminus, and the third is nucleophilic attack of the deprotonated Cys189 on the carbonyl to generate the thioester. The transition state (TS1) is rather dissociated because at this point the scissile bonds are already broken; however, neither the SG(Cys189)–C(Asn302) nor the HG(Cys189)–N(Ser303) bond has been generated yet (Figure S6). Next, the thioester is produced and the proton transfer occurs stepwise until the first intermediate state is reached (INT1, Figure 4 right).
Figure 8.

Calculated free energy profile for the ligation.
Table 1. Energetics (kcal/mol) of the Cleavage Procedurea.
| EQM | EQM/MM | |
|---|---|---|
| TS1 | 23.7 | 19.3 |
| INT1 | 2.5 | –1.9 |
| TS2 | 9.8 | 7.0 |
| INT2 | 1.8 | –1.2 |
| TS3 | 9.5 | 7.1 |
| PS | –1.9 | –3.4 |
All energies are referenced to the energy of the reactant complex.
The second step of the reaction shows a rather late transition state (Figure S7, left), with a much lower barrier of 7.0 kcal/mol. First, the catalytic water approaches the carbonyl, accompanied by movement and strong coordination of His148, where the NE2(His148)–OW(Wat305) distance remains between 2.65 and 2.81 Å during the whole diol formation. The transition state search calculations also show that first the 2HW(Wat305) is pulled off from the water oxygen and transferred to carbonyl oxygen and then the remaining OH– attacks the carbonyl to build a diol. This fact also clarifies the nature of the nucleophile. At the transition state (TS2, Figure S7 left), the 2HW(Wat305) is exactly shared between OW(Wat305) and O(Asn302) with a bond distance of 1.21 Å each. Afterward, the OW(Wat305)–C(Asn302) bond and thereby the diol (INT2, Figure 5 left) are generated rapidly, and the system relaxes until a favorable conformation is reached.
To finish the cleavage procedure, the SG(Cys189)–C(Asn302) bond breaks readily with a low barrier of 7.1 kcal/mol. In addition, there are two proton transfers along these coordinates. At the transition state (TS3, Figure S7 right), the SG(Cys189)–C(Asn302) bond is already broken and the 3HW(Wat305) proton is shared between His148 and the substrate carboxylate with a bond length of 1.27 Å, separately. Subsequently, the 2HW(Wat305) proton shuttles to SG(Cys189) to complete the proteolysis, which results in the expected product state (Figure 7 right), where the substrate can either leave the pocket or religate.
Ligation
As described in the Ligation pathway section, ligation can proceed in one step without Cys189 participation, which has also been confirmed both theoretically and experimentally by applying a Met189 mutant and/or disulfide Cys189–S–CH3 of the wild-type enzyme (see also the SI). Figure 8 shows the QM/MM energy profile of the ligation.
The shapes of the free energy curves (Figure 8 for the wild-type enzyme and Figure S3 for the C189M mutant) are quite similar; however, the Met189 variant shows a slightly lower barrier of 12.6 kcal/mol in comparison with the native enzyme (16.4 kcal/mol). However, it was not possible to compare the reaction rates experimentally because at some extent a reverse reaction might always take place for the wild-type enzyme. The small difference in energies might be due to the fact that in the case of the methionine mutant there is no charge on residue 189 and, hence, the overall charge of the active site is 0; in contrast, in the native enzyme, Cys189 bears a negative charge. In addition, ligation with the native enzyme starts with a neutral C- and N-termini and neutral His148; although the mechanism with Cys189–S–CH3 and/or C189M mutant begins with a neutral N-terminus, the deprotonated C-terminus and protonated His148 provide a better charge distribution. Both reactions (native and modified enzyme) are initiated by a shortening of the C–N distance and proton transfer from N(Ser303) to the carboxylate of the C-terminus.
However, for Cys189–S–CH3 and Met189 mutant, an additional proton transfer from His148 to carboxylate is necessary to liberate the water molecule.
Transpeptidation
Transition state search and NEB simulations have shown that the transpeptidation is a one-step procedure, where proton transfer from the N-terminus to SG(Cys189) and attack of the N(Ser303) on the carbonyl and thereby the formation of the peptide bond are concerted. Calculations of the energy along the NEB coordinates give further support that hydrolysis is favored over ligation as long as water is present in the active site. Although the reaction barrier of the transpeptidation is 23.3 kcal/mol (Figures 9, S9) and thus comparable with thioester formation (19.3 kcal/mol), it is much higher than the barrier of hydrolysis of the thioester (7.1 kcal/mol). Consequently, transpeptidation is possible only if the catalytic water either leaves the pocket or has no space to enter the active site (e.g., legumain/cystatin complex).
Figure 9.

Calculated free energy profile for the transpeptidation.
Conclusions
In this publication, we present mechanistic details of the enzymatic protease and ligase activity of human legumain using hybrid QM/MM methods at the DFT/b3lyp level of theory. Our calculations were based on the crystal structure of human legumain in complex with human cystatin E (PDB code 4N6O(28)) and on the experimental findings of Dall et al.28,37,55
In addition, we could clarify the pH dependence as a switch between the unique dual asparaginyl–endopeptidase and ligase activity of legumain. Thus, the protonation state of the catalytic cysteine (Cys189) and histidine (His148) is crucial for the reaction mechanism to proceed toward either cleavage or ligation. Because at lower pH the cysteine is protonated, the calculated proteolysis mechanism starts with protonated Cys189, which transfers its proton to the scissile nitrogen to generate a thiolate, which can then attack the peptide carbonyl. At the same time, His148 is neutral and positions the catalytic water for the hydrolysis step. Importantly, no papain-like classical intermediate was found because the first intermediate is already the acyl enzyme. The second step of the reaction is hydrolysis of the thioester and starts with attack of the water on the carbonyl and results in a diol, similarly to caspases,42 which is the second intermediate along the proteolysis pathway. Finally, a water proton from the diol is translated to Cys189 to complete the reaction, where the role of the catalytic His148 is to serves as a base to the diol during product formation. The calculated reaction energetics have shown that the rate-limiting step of the cleavage step is the formation of the thioester with a barrier of 19.3 kcal/mol, which is a range similar to that of other representatives of the cysteine protease family (papain36 and cathepsin56).
Further support was given by calculations (and experimental results) from point mutations. Replacement of the neighboring residue Glu190 by Lys190 lowers the reaction barrier (see the SI) and speeds up the process because the positively charged Lys190 residue favors deprotonation of Cys189. Moreover, Lys190 reduces the local pKa of Cys189 and shifts the pH activity range for the proteolysis to lower pH.
The ligation step can proceed as a one-step procedure without cysteine participation. This surprising fact has also been proven experimentally both by blocking Cys189 using MMTS and by applying the point mutation C189M. While in both cases no cleavage was possible, the ligation activity remained active. Our simulations have also revealed that at elevated pH the catalytic cysteine becomes deprotonated and the ligation is assisted by the catalytic histidine His148 only, through proton transfer to the C-terminus carboxylate and thereby to the catalytic water. Moreover, we could not generate a reaction path with protonated Cys189; therefore, only legumain with deprotonated Cys189 can act as a ligase, which is consistent with the experimental pH profile of the reaction. The exact reverse reaction of the proteolysis is not possible because in the presence of the catalytic water the sulfur of the thioester is not strong enough as a base to remove the proton from the N-terminus. However, transpeptidation via thioester is possible if there is no catalytic water available at the active site. In addition, both for ligation and transpeptidation, the pH plays a crucial role also for the substrate because the incoming peptide needs to be neutral.
We have demonstrated that experimental findings can be explained and supported by computational studies and could elucidate the complete reaction mechanism of legumain for both the protease and ligase activity in atomistic detail while considering the whole protein and solvent surrounding. While calculations of other groups on further cysteine protease enzymes (papain, caspase, and cathepsin) were based on a covalently bound inhibitor–enzyme complex, which was modified to generate a starting structure, we started our simulations with a perfect reactant state mimic complex of cystatin–legumain.
Moreover, this is first time that the complete reaction pathways of both enzymatic activities are presented and is in excellent agreement with experimental data.
Acknowledgments
This work was supported by the Austrian Science Funds (FWF) under Contract Number M-1901 and W_01213. The QM/MM calculations were carried out using the scientific computing facility “MACH” at the Johannes Kepler University Linz (JKU). The authors are very thankful to Martina Wiesbauer for the preparation of the C189M mutant. Moreover, we also appreciate the assistance of Stuart Bogatko using NWChem and the fruitful discussions with Peter Goettig.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.7b01505.
PDB file of the reactant state (PDB)
PDB file of the INT1 intermediate state (PDB)
PDB file of the INT2 intermediate state (PDB)
PDB file of the product state (PDB)
Computational methods, description of the calculations on the mutants, and transition state structures (PDF)
Movies of the NEB XYZ coordinates for INT1–INT2 (GIF)
Movies of the NEB XYZ coordinates for INT2–PS (GIF)
Movies of the NEB XYZ coordinates for RS–INT1 (GIF)
Movies of the NEB XYZ coordinates for ligation without Cys (GIF)
The authors declare no competing financial interest.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- Baker E. N. J. Mol. Biol. 1980, 141 (4), 441–484. 10.1016/0022-2836(80)90255-7. [DOI] [PubMed] [Google Scholar]
- Baker E. N.; Rumball S. V. J. Mol. Biol. 1977, 111 (2), 207–210. 10.1016/S0022-2836(77)80124-1. [DOI] [PubMed] [Google Scholar]
- Drenth J.; Kalk K. H.; Swen H. M. Biochemistry 1976, 15 (17), 3731–3738. 10.1021/bi00662a014. [DOI] [PubMed] [Google Scholar]
- Menard R.; Plouffe C.; Carmona E.; Storer A. C.; Krantz A.; Smith R. A. J. Cell. Biochem. 1994, 137–137. [Google Scholar]
- Polgar L.; Halasz P. Eur. J. Biochem. 1978, 88 (2), 513–521. 10.1111/j.1432-1033.1978.tb12477.x. [DOI] [PubMed] [Google Scholar]
- Storer A. C.; Menard R. Methods Enzymol. 1994, 244, 486–500. 10.1016/0076-6879(94)44035-2. [DOI] [PubMed] [Google Scholar]
- Polgar L.; Halasz P. Biochem. J. 1982, 207 (1), 1–10. 10.1042/bj2070001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lah T.; Clifford J.; Helmer K.; Day N.; Moin K.; Honn K.; Crissman J.; Sloane B. Biochim. Biophys. Acta, Gen. Subj. 1989, 993 (1), 63–73. 10.1016/0304-4165(89)90144-X. [DOI] [PubMed] [Google Scholar]
- Gopalan P.; Dufresne M. J.; Warner A. H. Biochem. Cell Biol. 1986, 64 (10), 1010–1019. 10.1139/o86-134. [DOI] [PubMed] [Google Scholar]
- Lecaille F.; Kaleta J.; Brömme D. Chem. Rev. 2002, 102 (12), 4459–4488. 10.1021/cr0101656. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Dando P. M.; Rawlings N. D.; Brown M. A.; Young N. E.; Stevens R. A.; Hewitt E.; Watts C.; Barrett A. J. J. Biol. Chem. 1997, 272 (12), 8090–8098. 10.1074/jbc.272.12.8090. [DOI] [PubMed] [Google Scholar]
- Chen J. M.; Dando P. M.; Stevens R. A. E.; Fortunato M.; Barrett A. J. Biochem. J. 1998, 335, 111–117. 10.1042/bj3350111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. M.; Fortunato M.; Barrett A. J. Biochem. J. 2000, 352, 327–334. 10.1042/bj3520327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dando P. M.; Fortunato M.; Smith L.; Knight C. G.; McKendrick J. E.; Barrett A. J. Biochem. J. 1999, 339, 743–749. 10.1042/bj3390743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dando P. M.; Fortunato M.; Strand G. B.; Smith T. S.; Barrett A. J. Protein Expression Purif. 2003, 28 (1), 111–119. 10.1016/S1046-5928(02)00632-0. [DOI] [PubMed] [Google Scholar]
- Rotari V. I.; Dando P. M.; Barrett A. J. Biol. Chem. 2001, 382 (6), 953–959. 10.1515/BC.2001.119. [DOI] [PubMed] [Google Scholar]
- The cystein protease legumain – function and importance in metastases. http://www.ous-research.no/home/malandsmo/Project%20catalogue/13608 (2017).
- Edgington L. E.; Verdoes M.; Ortega A.; Withana N. P.; Lee J.; Syed S.; Bachmann M. H.; Blum G.; Bogyo M. J. Am. Chem. Soc. 2013, 135 (1), 174–182. 10.1021/ja307083b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haugen M. H.; Johansen H. T.; Pettersen S. J.; Solberg R.; Brix K.; Flatmark K.; Maelandsmo G. M. PLoS One 2013, 8, e52980. 10.1371/journal.pone.0052980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R.; Johansen H. T.; Nilsen H.; Haugen M. H.; Pettersen S. J.; Maelandsmo G. M.; Abrahamson M.; Solberg R. Biochimie 2012, 94 (12), 2590–2599. 10.1016/j.biochi.2012.07.026. [DOI] [PubMed] [Google Scholar]
- Craik D. J. Toxins 2012, 4 (2), 139–156. 10.3390/toxins4020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould A.; Ji Y. B.; Aboye T. L.; Camarero J. A. Curr. Pharm. Des. 2011, 17 (38), 4294–4307. 10.2174/138161211798999438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesner A.; Legowska A.; Wysocka M.; Rolka K. Curr. Pharm. Des. 2011, 17 (38), 4308–4317. 10.2174/138161211798999393. [DOI] [PubMed] [Google Scholar]
- Clancy K. W.; Melvin J. A.; McCafferty D. G. Biopolymers 2010, 94 (4), 385–396. 10.1002/bip.21472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K. S.; Durek T.; Kaas Q.; Poth A. G.; Gilding E. K.; Conlan B. F.; Saska I.; Daly N. L.; van der Weerden N. L.; Craik D. J.; Anderson M. A. Nat. Commun. 2015, 6, 10199. 10.1038/ncomms10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehnke J.; Bent A.; Houssen W. E.; Zollman D.; Morawitz F.; Shirran S.; Vendome J.; Nneoyiegbe A. F.; Trembleau L.; Botting C. H.; Smith M. C. M.; Jaspars M.; Naismith J. H. Nat. Struct. Mol. Biol. 2012, 19 (8), 767–772. 10.1038/nsmb.2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen G. K. T.; Wang S. J.; Qiu Y. B.; Hemu X.; Lian Y. L.; Tam J. P. Nat. Chem. Biol. 2014, 10 (9), 732–738. 10.1038/nchembio.1586. [DOI] [PubMed] [Google Scholar]
- Dall E.; Fegg J. C.; Briza P.; Brandstetter H. Angew. Chem., Int. Ed. 2015, 54 (10), 2917–2921. 10.1002/anie.201409135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L. X.; Hua T.; Crowley C.; Ru H.; Ni X. M.; Shaw N.; Jiao L. Y.; Ding W.; Qu L.; Hung L. W.; Huang W.; Liu L.; Ye K. Q.; Ouyang S. Y.; Cheng G. H.; Liu Z. J. Cell Res. 2014, 24 (3), 344–358. 10.1038/cr.2014.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentes-Prior P.; Salvesen G. S. Biochem. J. 2004, 384, 201–232. 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernath-Levin K.; Nelson C.; Elliott A. G.; Jayasena A. S.; Millar A. H.; Craik D. J.; Mylne J. S. Chem. Biol. 2015, 22 (5), 571–582. 10.1016/j.chembiol.2015.04.010. [DOI] [PubMed] [Google Scholar]
- Harrison M. J.; Burton N. A.; Hillier I. H. J. Am. Chem. Soc. 1997, 119 (50), 12285–12291. 10.1021/ja9711472. [DOI] [Google Scholar]
- Harrison M. J.; Burton N. A.; Hillier I. H.; Gould I. R. Chem. Commun. 1996, 24, 2769–2770. 10.1039/cc9960002769. [DOI] [Google Scholar]
- Shokhen M.; Khazanov N.; Albeck A. Proteins: Struct., Funct., Genet. 2009, 77 (4), 916–926. 10.1002/prot.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokhen M.; Khazanov N.; Albeck A. Proteins: Struct., Funct., Genet. 2011, 79 (3), 975–985. 10.1002/prot.22939. [DOI] [PubMed] [Google Scholar]
- Wei D. H.; Huang X. Q.; Liu J. J.; Tang M. S.; Zhan C. G. Biochemistry 2013, 52 (30), 5145–5154. 10.1021/bi400629r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E.; Brandstetter H. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (27), 10940–10945. 10.1073/pnas.1300686110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Fernandez M.; Barrett A. J.; Gerhartz B.; Dando P. M.; Ni J. A.; Abrahamson M. J. Biol. Chem. 1999, 274 (27), 19195–19203. 10.1074/jbc.274.27.19195. [DOI] [PubMed] [Google Scholar]
- Arad D.; Langridge R.; Kollman P. A. J. Am. Chem. Soc. 1990, 112 (2), 491–502. 10.1021/ja00158a004. [DOI] [Google Scholar]
- Welsh W. J.; Lin Y. J. Mol. Struct.: THEOCHEM 1997, 401 (3), 315–326. 10.1016/S0166-1280(97)00025-0. [DOI] [Google Scholar]
- Brady K. D.; Giegel D. A.; Grinnell C.; Lunney E.; Talanian R. V.; Wong W.; Walker N. Bioorg. Med. Chem. 1999, 7 (4), 621–631. 10.1016/S0968-0896(99)00009-7. [DOI] [PubMed] [Google Scholar]
- Sulpizi M.; Laio A.; VandeVondele J.; Cattaneo A.; Rothlisberger U.; Carloni P. Proteins: Struct., Funct., Genet. 2003, 52 (2), 212–224. 10.1002/prot.10275. [DOI] [PubMed] [Google Scholar]
- Miscione G. P.; Calvaresi M.; Bottoni A. J. Phys. Chem. B 2010, 114 (13), 4637–4645. 10.1021/jp908991z. [DOI] [PubMed] [Google Scholar]
- Janowski R.; Kozak M.; Jankowska E.; Grzonka Z.; Jaskolski M. J. Pept. Res. 2004, 64 (4), 141–150. 10.1111/j.1399-3011.2004.00181.x. [DOI] [PubMed] [Google Scholar]
- Rotonda J.; Nicholson D. W.; Fazil K. M.; Gallant M.; Gareau Y.; Labelle M.; Peterson E. P.; Rasper D. M.; Ruel R.; Vaillancourt J. P.; Thornberry N. A.; Becker J. W. Nat. Struct. Biol. 1996, 3 (7), 619–625. 10.1038/nsb0796-619. [DOI] [PubMed] [Google Scholar]
- Valiev M.; Garrett B. C.; Tsai M. K.; Kowalski K.; Kathmann S. M.; Schenter G. K.; Dupuis M. J. Chem. Phys. 2007, 127 (5), 051102. 10.1063/1.2768343. [DOI] [PubMed] [Google Scholar]
- Valiev M.; Kawai R.; Adams J. A.; Weare J. H. J. Am. Chem. Soc. 2003, 125 (33), 9926–9927. 10.1021/ja029618u. [DOI] [PubMed] [Google Scholar]
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma T. P.; Van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; de Jong W. A. Comput. Phys. Commun. 2010, 181 (9), 1477–1489. 10.1016/j.cpc.2010.04.018. [DOI] [Google Scholar]
- Elsaesser B.; Fels G. J. Mol. Model. 2011, 17 (8), 1953–1962. 10.1007/s00894-010-0900-8. [DOI] [PubMed] [Google Scholar]
- Henkelman G.; Jonsson H. J. Chem. Phys. 2000, 113 (22), 9978–9985. 10.1063/1.1323224. [DOI] [Google Scholar]
- Valiev M.; Bylaska E.; Tsemekman K.; Bogatko S.; Weare J. Geochim. Cosmochim. Acta 2005, 69 (10), A511–a511. [Google Scholar]
- Zhang Y. K.; Lee T. S.; Yang W. T. J. Chem. Phys. 1999, 110 (1), 46–54. 10.1063/1.478083. [DOI] [Google Scholar]
- Zhang Y. K.; Liu H. Y.; Yang W. T. J. Chem. Phys. 2000, 112 (8), 3483–3492. 10.1063/1.480503. [DOI] [Google Scholar]
- Ma S.; Devi-Kesavan L. S.; Gao J. J. Am. Chem. Soc. 2007, 129 (44), 13633–13645. 10.1021/ja074222+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dall E.; Brandstetter H. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2012, 68, 24–31. 10.1107/S1744309111048020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresso A. W.; Wu R. Y.; Kern J. W.; Zhang R. G.; Janik D.; Missiakas D. M.; Duban M. E.; Joachimiak A.; Schneewind O. J. Biol. Chem. 2007, 282 (32), 23129–23139. 10.1074/jbc.M701857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.