

Abstract
Aims
Polymorphous low-grade adenocarcinoma (PLGA) is the second most common intra-oral salivary gland malignancy. The vast majority of PLGAs harbor a PRKD1 E710D hotspot somatic mutation or somatic rearrangements of PRKD1, PRKD2 or PRKD3. Given the kinase domain homology among PRKD1, PRKD2 and PRKD3, we sought to define whether PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements would be driven by somatic mutations affecting the kinase domains of PRKD2 or PRKD3.
Methods and results
DNA was extracted from 8 microdissected PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements. Samples were thoroughly centrally reviewed, microdissected and subjected to Sanger sequencing of the kinase domains of PRKD2 and PRKD3 genes. None of the PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements harbored somatic mutations in the kinase domains of PRKD2 or PRKD3 genes.
Conclusion
PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements are unlikely to harbor somatic mutations in the kinase domains of PRKD2 or PRKD3. Further studies are warranted to define the driver genetic events in this subgroup of PLGAs.
Keywords: Sanger sequencing, salivary gland tumors, sequence homology, kinase domain, mutation
INTRODUCTION
Polymorphous low-grade adenocarcinoma (PLGA) represents the second most common salivary gland malignancy of the oral cavity, predominantly affecting the minor salivary glands.1 Despite its characteristic intra-oral presentation, PLGAs can occasionally originate in other anatomical sites, such as the lungs and the breast.2 The age of incidence is usually in the sixth to seventh decades of life; however, PLGAs affecting adolescents and infants have also been reported.3–5 PLGA is a malignant epithelial tumor classically characterized by low-grade cytologic uniformity, architectural diversity (hence the term polymorphous), and infiltrative borders.1 The diverse architectural configurations in which the neoplastic cells can be arranged include tubular, solid, papillary, microcystic, cribriform, fascicular, single file, and strand-like growth patterns.6,7 Despite their locally infiltrative growth and neurotropism, PLGAs have a relatively indolent clinical course.7,8 The wide spectrum of histologic appearances that these tumors display is responsible for major diagnostic challenges in differentiating PLGAs from other more aggressive types of salivary gland neoplasms, such as adenoid cystic carcinoma.9
We have recently identified a recurrent E710D pathognomonic hotspot somatic mutation within the catalytic loop of the kinase domain of PRKD1 in 73% of PLGAs.10 This mutation was found not to be present in other forms of benign or malignant salivary gland tumors10 and in none of the >4000 tumors studied by The Cancer Genome Atlas available on the cBioPortal (www.cbioportal.org).11 PRKD1 encodes the serine/threonine protein kinase D1, which plays a prominent role in many signal transduction pathways related to cell adhesion, cell migration, vesicle transport and cell survival.12 Our previous work demonstrated that the PRKD1 E710D mutation led to significantly increased kinase activity in experimental models, increased cell proliferation indices in vitro and altered glandular architecture in three-dimensional models13 employing non-malignant breast epithelial cells.10 These findings support the contention that the PRKD1 E710D hotspot mutation is not only activating and oncogenic, but also a distinguishing feature of PLGAs.10
In addition to PRKD1, the protein kinase D (PRKD) gene family also includes PRKD2 and PRKD3.14 The genes in this family have similar biological functions (i.e. diacylglycerol and protein-kinase C transduction pathways) and share high levels of sequence identity, in particular in the kinase domain.15 Specifically, the PRKD family members show a highly conserved modular structure consisting of an N-terminal region with regulatory domains and a C-terminal region with a kinase domain, the proteins being maintained in an inactive state through auto-inhibition of the kinase domain by its regulatory domains.12 Deletions or mutations of critical residues within these domains result in constitutive activation of the proteins encoded by PRKD genes.10,14 Rearrangements involving the PRKD gene family have recently been described in cribriform adenocarcinoma of the salivary glands (CASG), a neoplastic entity closely related to PLGA.15 Furthermore, RNA sequencing and/or FISH analysis of CASGs, classic PLGAs, and PLGAs with “mixed/indeterminate” features has revealed the presence of genomic rearrangements involving PRKD1, PRKD2 or PRKD3 in 75% of CASGs, 45% of cases with “mixed/indeterminate” features and in one of 18 classic PLGAs.15
Given the high kinase domain sequence identity among PRKD1, PRKD2 and PRKD3 and that a subset of PLGAs lack PRKD1 hotspot somatic mutations or PRKD gene family rearrangements, we posited that alternative oncogenic mechanisms in the PRKD gene family members, other than the PRKD1 E710D mutation, may be involved in the pathogenesis of PLGA. Here we sought to define whether PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements would be driven by somatic mutations affecting the kinase domains of PRKD2 or PRKD3.
MATERIALS AND METHODS
Patients and sample selection
After obtaining Institutional Review Board (IRB) approval from the respective authors’ institutions, representative histologic blocks of PLGAs previously found not to harbor the PRKD1 E710D hotspot mutation10 were selected and retrieved from the pathology departments of the Charles University (Plzen, Czech Republic) and Memorial Sloan Kettering Cancer Center (MSKCC, New York, NY). All cases were reviewed by the contributing pathologists and centrally reviewed by at least two pathologists with an interest and expertise in head and neck cancer pathology (I.W., A.S. and/or N.K.). Only cases classified as classic PLGA according to the World Health Organization criteria1 and lacking the PRKD1 E710D mutation were included in this study.1 Cases that fulfilled the diagnostic criteria of CASG, PLGAs with “mixed/indeterminate” features or adenocarcinoma not otherwise specified were excluded, regardless their mutational status.1
Classic PLGAs lacking the PRKD1 E710D somatic mutation were subjected to fluorescence in situ hybridization (FISH) to rule out rearrangements of PRKD1, PRKD2 and PRKD3, and the results of these analyses were reported elsewhere.15 Only classic PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements were included in this study.15
Patient gender, age at diagnosis and the anatomic site affected by the PLGA were obtained from the medical records from the patients whose tumors were included in this study. After the retrieval of these clinical details, samples were anonymized. Informed consent was obtained when appropriate, following the IRB protocol specifications.
Microdissection and DNA extraction
Representative 8-μm-thick sections of formalin-fixed paraffin-embedded (FFPE) blocks of tumor and adjacent normal tissue were stained with nuclear fast red and microdissected using a sterile needle under a stereomicroscope (Olympus SZ61, Center Valley, PA), to ensure >80% of tumor cell content, as previously described.16 Genomic DNA was extracted from each tumor and matched normal tissue using the DNeasy Blood and Tissue Kit (Qiagen, Valencia, CA), and quantified using the Qubit Fluorometer assay (Invitrogen, Life Technologies, Norwalk, CT), following the manufacturers’ protocols.17,18 All samples had sufficient quantity and quality of DNA for Sanger sequencing analysis.
Amino acid sequence alignment of PRKD1, PRKD2 and PRKD3 kinase domains
The ‘canonical’ amino acid sequences of the kinase domains of PRKD1, PRKD2 and PRKD3 were retrieved from UniProt (http://www.uniprot.org; PRKD1: accession Q15139, positions 583–839; PRKD2: accession Q9BZL6, positions 551–807; PRKD3: accession O94806, positions 576–832). Multiple sequence alignment was performed using the ClustalW2 tool on the EMBL-EBI website (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
PCR amplification and Sanger sequencing
We employed primer sets that amplify the entire kinase domains of PRKD1, PRKD2 and PRKD3. The primers pairs were designed as previously described10 (Supplementary Figure 1 and Supplementary Table 1) and the specificity was also tested using in a previously described in silico method19 (https://genome.ucsc.edu/cgi-bin/hgPcr). PCR amplification of 10ng of genomic DNA was performed using the AmpliTaq 360 Master Mix Kit (Life Technologies, Norwalk, CT) on a Veriti Thermal Cycler (Life Technologies). The thermocycling protocol consisted of an initial incubation step of 95°C for 5 min and then 35 cycles of 95°C for 30 sec, 56°C for 30 sec, 72°C for 30 sec, and one final extension step of 72°C for 10 min. PCR fragments were purified with ExoSAP-IT (Affymetrix, Santa Clara, CA), and the sequencing reactions were performed on an ABI 3730 capillary sequencer using ABI BigDye Terminator chemistry (v3.1, Life Technologies) according to the manufacturer’s instructions. Sequences of the forward and reverse strands were analyzed using MacVector software (MacVector, Inc, Cary, NC).10 All analyses were performed in duplicate.
RESULTS
Histopathologic review
Eight classic PLGAs affecting 1 male and 7 female patients, with a median age at the diagnosis of 66 years (range 32 – 77 years, Table 1), were confirmed to lack the PRKD1 E710D hotspot mutation (Table 1). All cases displayed the bona fide histologic features of classic PLGA.20 The neoplastic cells showed low-grade features with the typical cytologic uniformity described in this specific histologic type of salivary gland malignancy (Figure 1). Neoplastic cells were arranged in small nests with fascicular and targetoid appearance, cords and tubules/duct-like structures with infiltrating borders. At higher magnification, these structures were composed of cuboidal, columnar, and/or spindled cells with small bland and uniform ground-glass nuclei with indistinct nucleoli and eosinophilic cytoplasm. The short fascicles of tumor cells occasionally displayed a targetoid arrangement around vessels and/or nerves. Mitotic figures were vanishingly rare, with an average mitotic count of <1/10 high-power fields. The tumor stroma was composed of fibrous tissue with varying degrees of hyalinization and myxoid change. No chondroid differentiation or necrosis was observed. Stromal mucinosis and elastosis could be rarely observed along with areas of intra-lesional hemorrhage. Interestingly, focal apocrine differentiation was observed in case PLGA_04. Taken together, the eight PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements displayed histologic features similar to those documented in PLGAs harboring PRKD1 E710D pathognomonic somatic mutations10 or PRKD gene family rearrangements.15
Table 1.
Clinicopathologic and molecular features of the PLGAs included in this study.
| Sample ID | Gender | Age (years) | Location | Histologic grade | PRKD family genes status | |
|---|---|---|---|---|---|---|
| Rearrangements (FISH) | PRKD1, PRKD2 and PRKD3 Kinase domains (Sanger sequencing) | |||||
| PLGA_01 | Female | 32 | Maxillary sinus | 1 | Not present | Wild-type |
| PLGA_02 | Male | 73 | Palate | 1 | Not present | Wild-type |
| PLGA_03 | Female | 77 | Buccal mucosa | 1 | Not present | Wild-type |
| PLGA_04 | Female | 77 | Buccal mucosa | 1 | Not present | Wild-type |
| PLGA_05 | Female | 70 | Upper lip | 1 | Not present | Wild-type |
| PLGA_06 | Female | 63 | Lip | 1 | Not present | Wild-type |
| PLGA_07 | Female | 67 | Soft palate | 1 | Not present | Wild-type |
| PLGA_12 | Female | 50 | Maxilla | 1 | Not present | Wild-type |
FISH, fluorescence in situ hybridization; PLGA, polymorphous low-grade adenocarcinoma.
Figure 1. Representative micrographs of polymorphous low-grade adenocarcinoma of the salivary glands included in this study.
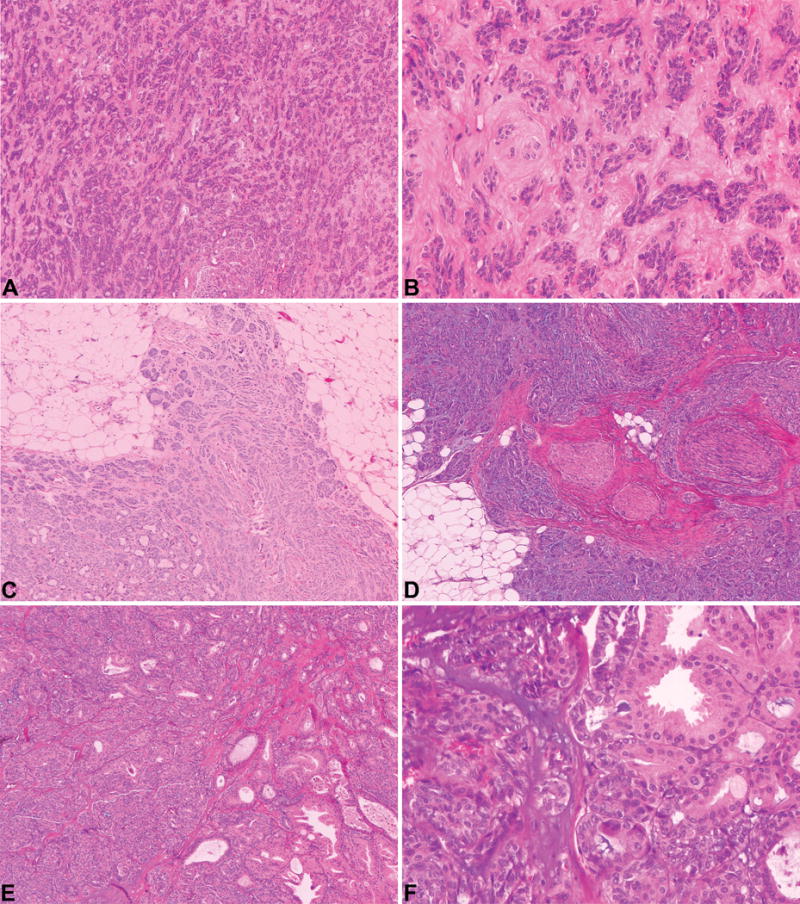
Classic polymorphous low-grade adenocarcinomas with neoplastic cells arranged in nests, cords and duct-like structures (A; PLGA_05) composed of neoplastic cells with small, bland and uniform ground-glass nuclei with indistinct nucleoli and eosinophilic cytoplasm (B; PLGA_05). Tumors displayed infiltrating borders (C; PLGA_06) and foci of perineural invasion were observed (D; PLGA_03). PLGA_04 displayed the architectural diversity characteristic of polymorphous low-grade adenocarcinoma (E). In this case, a focus of apocrine differentiation was observed (E and F). Original magnification ×50 - A, C, D and E; x200 – B and F.
Sequencing analysis
To define the sequence similarity of the kinase domains of the PRKD gene family members, we performed a multiple sequence alignment of the 257 amino acid residues that constitute the kinase domains of PRKD1, PRDK2 and PRKD3. This analysis revealed that their median sequence identity was 92.2% (range 91.1%–94.2%, Figure 2). Given the high degree of sequence identity among the kinase domains of PRKD1, PRKD2 and PRKD3 genes, we sequenced the kinase domains of PRKD2 and PRKD3. Sanger sequencing analysis of the entire kinase domains of PRKD2 and PRKD3 revealed no somatic mutations (Table 1 and Figure 3) in any of the PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements analyzed in this study (Figure 3).
Figure 2. Multiple-sequence alignment (ClustalW) of PRKD1, PRKD2 and PRKD3 kinase domains.
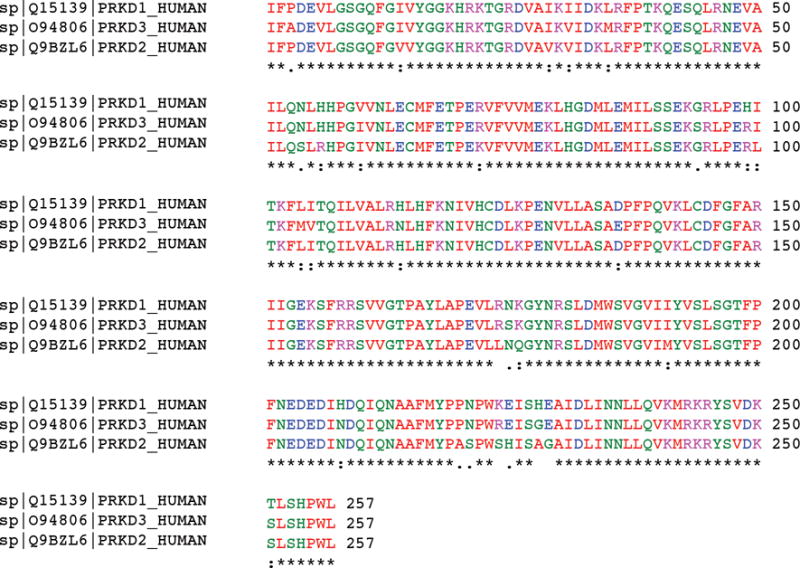
Conserved amino acids are highlighted in similar colors.
Figure 3. Polymorphous low-grade adenocarcinomas (PLGAs) lacking PRKD1 somatic mutations or PRKD gene family rearrangements lack PRKD2 and PRKD3 kinase domain mutations.
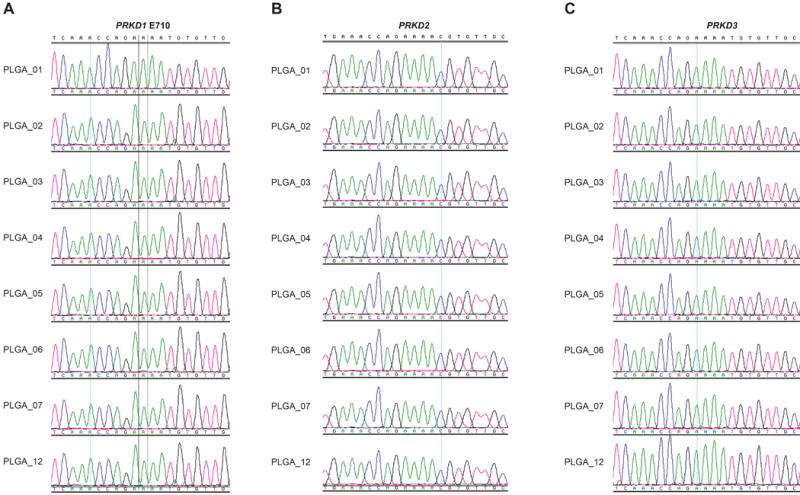
(A) Representative sequence electropherograms of the PRKD1 E710 residue (catalytic loop) from the 8 PLGAs included in our study. (B) Representative sequence electropherograms of PRKD2 catalytic loop from the 8 PLGAs included in our study. (C) Representative sequence electropherograms of the PRKD3 catalytic loop from the 8 PLGAs included in our study.
DISCUSSION
Salivary gland tumors constitute a relatively rare and heterogeneous group of lesions that encompasses entities with different morphologies, outcomes, and biological characteristics.21 In recent decades, a remarkable number of pathognomonic somatic genetic alterations underpinning specific types of salivary gland tumors have been identified. Recently, our group demonstrated that 73% of PLGAs harbor a novel E710D PRKD1 activating hotspot somatic mutation, which likely constitutes a major driver of this disease.10 Importantly, this mutation was not found in 312 non-PLGA salivary gland tumors. This hotspot mutation is not only of biological interest, but also constitutes an ancillary molecular marker to differentiate PLGAs from their more aggressive mimics, including adenoid cystic carcinomas.10 Interestingly, genomic rearrangements involving PRKD gene family members, namely PRKD1, PRKD2 and PRKD3, have been documented in 75% of CASG, a salivary gland tumor closely related to classic PLGAs.15 Importantly, Weinreb et al.15 reported on the presence of a PRKD2 somatic rearrangement in one of 18 classic PLGAs,15 and a PRKD1 wild-type PLGA reported by our group expressed an ACTN4-PRKD2 somatic rearrangement.10 Taken together, these observations are consistent with the notion that PLGAs may constitute a convergent phenotype22,23 and be driven by activating somatic genetic alterations affecting PRKD1, PRKD2 and PRKD3.
Sequence alignment of the kinase domains of the PRKD gene family members confirmed that the domains are composed of highly conserved sequences, with sequence identity >90% (Figure 2). The three members of the PRKD family are structurally similar, supporting the notion that they likely harbor homologous functions in the diacylglycerol and protein kinase C signal transduction pathway.12,24 To determine whether the minority of PLGAs that neither harbor the PRKD1 E710D activating mutation nor somatic rearrangements of PRKD1, PRKD2 or PRKD3 would be driven by somatic mutations affecting the kinase domains of PRKD2 or PRKD3, we sequenced the kinase domains of these genes in eight PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements. None of the cases displayed somatic mutations in PRKD2 or PRKD3. These PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements had morphological features entirely consistent with those of classic PLGAs driven by the PRKD1 E710D somatic mutation. Taken together, our findings demonstrate that a subset of PLGAs may be driven by genetic or epigenetic alterations affecting genes other than the three members of the PRKD gene family.
Apocrine differentiation, which is not uncommonly found in other types of malignant salivary gland tumors (e.g. salivary duct carcinoma),25,26 has rarely been documented in PLGAs. One of the cases included in this study displayed focal apocrine differentiation (Figures 1E, F). This prompted us to perform a review of the cases included in Weinreb et al.10 for the presence of apocrine differentiation in the 43 PLGAs harboring the E710D PRKD1 hotspot mutation. Of these cases, only one (2%) displayed areas of apocrine differentiation. Further studies are warranted to define the frequency of apocrine differentiation in PLGAs and other histologic features that may distinguish PLGAs harboring somatic genetic alterations affecting PRKD1, PRKD2 or PRKD3 from those displaying wild-type PRKD1, PRKD2 or PRKD3.
Our study has several limitations. First, the size of the cohort was small owing to the rarity of PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements. Therefore, this study should be considered exploratory and hypothesis-generating. Second, we have focused only on the kinase domains of PRKD2 and PRKD3, and somatic genetic alterations affecting other regions of these genes could theoretically result in their activation. A review of the results from The Cancer Genome Atlas of the mutations affecting regions other than the kinase domains of PRKD2 and PRKD3 revealed that mutations affecting these regions are almost exclusively non-pathogenic/passengers (only two out of 208 missense mutations considered as potentially pathogenic based on the results from cBioPortal (www.cbioportal.org)).11 Third, the PRKD family belongs to the larger CAMK group of kinases and the kinase domains of the PRKD genes share high sequence similarity to those of other CAMK members.27 It is thus plausible that PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements may harbor mutations in other genes in the CAMK family. We are currently in the process of acquiring samples of PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements with sufficient germline and tumor DNA of optimal quality for massively parallel sequencing analysis of these genes. Fourth, alternative drivers of PLGAs lacking PRKD1 somatic mutations or PRKD gene family rearrangements may be epigenetic, and if so, these would not be detected through conventional massively parallel sequencing analysis of these tumors. Despite these limitations, our study provides direct evidence that albeit being a convergent phenotype, PLGAs constitute a heterogeneous group of tumors, some of which are not driven by somatic mutations affecting the kinase domains of PRKD1, PRKD2 and PRKD3 or somatic rearrangements affecting PRKD family genes. Further studies to define the drivers of these tumors are warranted.
Supplementary Material
Supplementary Figure 1: Schematic representation of intron/exon primer pairs used to amplify the hotspot region in the catalytic loop of the PRKD1 gene and the kinase domains of PRKD2 and PRKD3.
Supplementary Table 1: List of primers used to amplify the hotspot region in the catalytic loop of the PRKD1 gene and the kinase domains of PRKD2 and PRKD3.
Acknowledgments
SP is funded by a Susan G Komen Postdoctoral Fellowship Grant (PDF14298348). Research reported in this publication was supported in part by a Cancer Center Support Grant of the National Institutes of Health/National Cancer Institute (Grant No. P30CA008748). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of interest: The authors have no conflicts of interest to declare
References
- 1.Barnes L, Eveson JW, Reichart P, Sidransky D, editors. World Health Organization classification of tumours. Pathology and genetics of head and neck tumours. Lyon: IARC Press; 2005. [Google Scholar]
- 2.Foschini MP, Eusebi V. Rare (new) entities of the breast and medullary carcinoma. Pathology. 2009;41:48–56. doi: 10.1080/00313020802563528. [DOI] [PubMed] [Google Scholar]
- 3.Olusanya AA, Akadiri OA, Akinmoladun VI, Adeyemi BF. Polymorphous low grade adenocarcinoma: Literature review and report of lower lip lesion with suspected lung metastasis. J Maxillofac Oral Surg. 2011;10:60–63. doi: 10.1007/s12663-011-0185-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee VK, McCaughan BC, Scolyer RA. Polymorphous low-grade adenocarcinoma in the lung: A case report. Int J Surg Pathol. 2004;12:287–292. doi: 10.1177/106689690401200313. [DOI] [PubMed] [Google Scholar]
- 5.Tanaka F, Wada H, Inui K, et al. Pulmonary metastasis of polymorphous low-grade adenocarcinoma of the minor salivary gland. Thorac Cardiovasc Surg. 1995;43:178–180. doi: 10.1055/s-2007-1013795. [DOI] [PubMed] [Google Scholar]
- 6.Simpson RH, Pereira EM, Ribeiro AC, Abdulkadir A, Reis-Filho JS. Polymorphous low-grade adenocarcinoma of the salivary glands with transformation to high-grade carcinoma. Histopathology. 2002;41:250–259. doi: 10.1046/j.1365-2559.2002.01439.x. [DOI] [PubMed] [Google Scholar]
- 7.Evans HL, Luna MA. Polymorphous low-grade adenocarcinoma: A study of 40 cases with long-term follow up and an evaluation of the importance of papillary areas. Am J Surg Pathol. 2000;24:1319–1328. doi: 10.1097/00000478-200010000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Perez-Ordonez B, Linkov I, Huvos AG. Polymorphous low-grade adenocarcinoma of minor salivary glands: A study of 17 cases with emphasis on cell differentiation. Histopathology. 1998;32:521–529. doi: 10.1046/j.1365-2559.1998.t01-2-00410.x. [DOI] [PubMed] [Google Scholar]
- 9.Marchio C, Weigelt B, Reis-Filho JS. Adenoid cystic carcinomas of the breast and salivary glands (or ‘the strange case of dr jekyll and mr hyde’ of exocrine gland carcinomas) J Clin Pathol. 2010;63:220–228. doi: 10.1136/jcp.2009.073908. [DOI] [PubMed] [Google Scholar]
- 10.Weinreb I, Piscuoglio S, Martelotto LG, et al. Hotspot activating prkd1 somatic mutations in polymorphous low-grade adenocarcinomas of the salivary glands. Nat Genet. 2014;46:1166–1169. doi: 10.1038/ng.3096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rozengurt E, Rey O, Waldron RT. Protein kinase d signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 13.Weigelt B, Ghajar CM, Bissell MJ. The need for complex 3d culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev. 2014;69–70:42–51. doi: 10.1016/j.addr.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rykx A, De Kimpe L, Mikhalap S, et al. Protein kinase d: A family affair. FEBS Lett. 2003;546:81–86. doi: 10.1016/s0014-5793(03)00487-3. [DOI] [PubMed] [Google Scholar]
- 15.Weinreb I, Zhang L, Tirunagari LM, et al. Novel prkd gene rearrangements and variant fusions in cribriform adenocarcinoma of salivary gland origin. Genes Chromosomes Cancer. 2014;53:845–856. doi: 10.1002/gcc.22195. [DOI] [PubMed] [Google Scholar]
- 16.Marchio C, Iravani M, Natrajan R, et al. Genomic and immunophenotypical characterization of pure micropapillary carcinomas of the breast. J Pathol. 2008;215:398–410. doi: 10.1002/path.2368. [DOI] [PubMed] [Google Scholar]
- 17.Guerini-Rocco E, Hodi Z, Piscuoglio S, et al. The repertoire of somatic genetic alterations of acinic cell carcinomas of the breast: An exploratory, hypothesis-generating study. J Pathol. 2015;237:166–178. doi: 10.1002/path.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Natrajan R, Wilkerson PM, Marchio C, et al. Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol. 2014;232:553–565. doi: 10.1002/path.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng CK, Martelotto LG, Gauthier A, et al. Intra-tumor genetic heterogeneity and alternative driver genetic alterations in breast cancers with heterogeneous HER2 gene amplification. Genome Biol. 2015;16:107. doi: 10.1186/s13059-015-0657-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barnes L. Surgical pathology of the head and neck. third. Taylor & Francis; 2008. [Google Scholar]
- 21.Sreeja C, Shahela T, Aesha S, Satish MK. Taxonomy of salivary gland neoplasm. J Clin Diagn Res. 2014;8:291–293. doi: 10.7860/JCDR/2014/7345.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ashworth A, Lord CJ, Reis-Filho JS. Genetic interactions in cancer progression and treatment. Cell. 2011;145:30–38. doi: 10.1016/j.cell.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 23.Weigelt B, Reis-Filho JS. Epistatic interactions and drug response. J Pathol. 2014;232:255–263. doi: 10.1002/path.4265. [DOI] [PubMed] [Google Scholar]
- 24.Sundram V, Chauhan SC, Jaggi M. Emerging roles of protein kinase d1 in cancer. Mol Cancer Res. 2011;9:985–996. doi: 10.1158/1541-7786.MCR-10-0365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McHugh JB, Visscher DW, Barnes EL. Update on selected salivary gland neoplasms. Arch Pathol Lab Med. 2009;133:1763–1774. doi: 10.5858/133.11.1763. [DOI] [PubMed] [Google Scholar]
- 26.Simpson RH, Desai S, Di Palma S. Salivary duct carcinoma in situ of the parotid gland. Histopathology. 2008;53:416–425. doi: 10.1111/j.1365-2559.2008.03135.x. [DOI] [PubMed] [Google Scholar]
- 27.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase d: A target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1: Schematic representation of intron/exon primer pairs used to amplify the hotspot region in the catalytic loop of the PRKD1 gene and the kinase domains of PRKD2 and PRKD3.
Supplementary Table 1: List of primers used to amplify the hotspot region in the catalytic loop of the PRKD1 gene and the kinase domains of PRKD2 and PRKD3.