

Abstract
Traumatic brain injury (TBI) results in severe neurological impairments without effective treatments. Inflammation appears to be an important contributor to key pathogenic events such as secondary brain injury following TBI and therefore serves as a promising target for novel therapies. We have recently demonstrated the ability of a molecular construct comprised of the human leukocyte antigen (HLA)-DRαl domain linked covalently to mouse (m)MOG-35-55 peptide (DRα1-MOG-35-55 construct) to reduce CNS inflammation and tissue injury in animal models of multiple sclerosis and ischemic stroke. The aim of the current study was to determine if DRα1-MOG-35-55 treatment of a fluid percussion injury (FPI) mouse model of TBI could reduce the lesion size and improve disease outcome measures. Neurodeficits, lesion size, and immune responses were determined to evaluate the therapeutic potential and mechanisms of neuroprotection induced by DRα1 -MOG-35-55 treatment. The results demonstrated that daily injections of DRα1-MOG-35-55 given after FPI significantly reduced numbers of infiltrating CD74+ and CD86+ macrophages and increased numbers of CD206+ microglia in the brain concomitant with smaller lesion sizes and improvement in neurodeficits. Conversely, DRα1-MOG-35-55 treatment of TBI increased numbers of circulating CD11b+ monocytes and their expression of CD74 but had no detectable effect on cell numbers or marker expression in the spleen. These results demonstrate that DRα1-MOG-35-55 therapy can reduce CNS inflammation and significantly improve histological and clinical outcomes after TBI. Future studies will further examine the potential of DRα1-MOG-35-55 for treatment of TBI.
Keywords: Traumatic brain injury, DRα1-MOG-35-55 therapy, neurological deficits, CD74, infiltrating macrophages/microglia
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity with a lack of effective treatments. TBI can often result in long-term physical and cognitive deficits that involve primary and secondary injuries. The primary injury occurs at the moment of TBI, characterized by the disruption of blood brain barrier and blood vessels that contribute to brain edema (McKee and Lukens 2016; Pop and Badaut 2011). This primary injury precedes downstream events contributing to a secondary injury cascade that includes the activation of brain-resident microglia and astrocytes and recruitment of peripheral immune cells into the brain (Kumar and Loane 2012; McKee and Lukens 2016; Raghupathi 2004; Ramlackhansingh et al. 2011). The inflammatory responses may start minutes after TBI onset and persist for weeks to months (Kumar and Loane 2012; McKee and Lukens 2016; Raghupathi 2004; Ramlackhansingh et al. 2011). As a result, these immune components may promote cell death during the early phase after TBI impact and contribute to subsequent neurological impairments during the later stage (Kumar and Loane 2012; McKee and Lukens 2016; Raghupathi 2004; Ramlackhansingh et al. 2011). Thus, targeting inflammation after TBI may serve as a promising strategy for the development of novel therapy (Corps et al. 2015).
The human leukocyte antigen (HLA)-DRα1 domain linked to mouse (m)MOG-35-55 peptide (DRα1 -MOG-35-55) is a partial major histocompatibility complex (MHC) class II construct that can inhibit neuroantigen-specific T cells and block the binding of the cytokine/chemokine, macrophage migration inhibitory factor (MIF) to its CD74 receptor on monocytes and macrophages (Benedek et al. 2016; Meza-Romero et al. 2014; Wang et al. 2016). Our recent studies have demonstrated the benefit of DRα1-MOG-35-55 in animal models of multiple sclerosis (experimental autoimmune encephalomyeliltis, EAE) and ischemic stroke (middle cerebral artery occlusion, MCAO). The current study aims to evaluate the therapeutic effect of DRα1-MOG-35-55 on functional outcomes, lesion volume and immune changes in the periphery and CNS in a mouse model of TBI.
Materials and Methods
Animals
Male C57BL/6 mice (7–8 weeks old, 20–25g body weight) were housed in animal facilities at Tianjin Neurological Institute (Tianjin, China) under standardized light-dark cycle, and provided with access to food and water. All animal experiments were performed in accordance with the institutional animal care guidelines approved by the Chinese Small Animal Protection Association. All animal studies were approved by the Animal Care and Use Committees of Tianjin Neurological Institute. Animals were randomly assigned to experimental groups. The experimental groups were as follows: FPI+ vehicle and FPI+DRα1-MOG-35-55.
Induction of Fluid percussion injury (FPI) in mice
The mice were anesthetized with 10% chloral hydrate (3 mg/kg, intraperitoneal injection), cleaned and shaved, then placed in a stereotaxic instrument with an attachment for mouse surgery (Stoelting, Inc., IL, USA). A 2mm hole was drilled, with dura intact, in the skull over the left parietal cortex (antero-posterior: +1.5 mm; medio-lateral: −1.2 mm). A 12–16 ms FPI was delivered at a pressure of 1.3–1.5atm. After injury, the scalp incision was closed with interrupted 4-0 silk sutures. After surgery, mice were treated immediately with Vehicle or DRα1-MOG-35-55 by subcutaneous injection and daily thereafter for 9 consecutive days in housing with a 12-h light–dark cycle with food and water.
DRα1-MOG-35-55 cloning, production and purification
Cloning, production, and purification of the DRα1 -mMOG-35-55 construct have been described previously (Meza-Romero et al. 2014). Briefly, DRα1 -mMOG-35-55 was designed as a single gene joining the mouse (m)MOG-35-55-encoding DNA sequence upstream of the HLA-DRα1 domain with a flexible linker (containing a thrombin cleavage site) between both elements. This single exon was cloned between the NcoI and XhoI restriction sites of the pET21d(+) vector, expressed in Escherichia coli, and the protein purified following standard purification techniques including anion exchange and size exclusion chromatography in the presence of 6 M urea. Protein was refolded after extensive dialysis in 20 mM Tris, pH 8.5, then concentrated to 1mg/ml and flash frozen.
DRα1-MOG-35-55 administration
After FPI surgery, mice were randomly divided into two groups, one receiving 0.1 ml (100 μg) DRα1-MOG-35-55, the other receiving 0.1 ml Vehicle (5% dextrose in Tris–HCl, pH 8.5) by subcutaneous injection. DRα1-MOG-35-55 or Vehicle was administrated by s.c. injection immediately after FPI and was given on 9 consecutive days until the experiment ended. Both Vehicle and DRα1-MOG-35-55 treated mice were evaluated for neurological deficits and lesion size using histopathological staining on days 1, 3, 7 and 10. On day 3 after FPI, brain, spleen and blood were harvested for analysis of immune cell counts.
Neurological Assessment
Neurological deficits were assessed using the modified Neurological Severity Score (mNSS) and corner turning tests at day 1, day 3, day 7 and day 10 after FPI by at least two investigators blinded to the treatments of FPI mice in each experiment. The mNSS rates neurological functioning and includes a composite of motor, sensory, reflex and balance tests. The corner turning test was used to assess sensorimotor and postural asymmetries. Each mouse being tested was allowed to enter a corner with an angle of 30 degrees and was required to turn either to the left or the right to exit the corner. This was repeated and recorded 10 times, with at least 5 minutes between trials, and the percentage of left turns out of total turns was calculated.
Lesion size measurement
Lesion volume was measured as previously described (Basrai et al. 2016; Meng et al. 2014; Tobin et al. 2014). At day 1, 3, 7 or 10 after surgery, mice were transcardially perfused with PBS, followed by 4% paraformaldehyde to fix the brain tissue. After embedding with paraffin, serial 6 μm thick sections were cut through the injury site (bregma −1.5mm to 1.50 mm). For each mouse, every 20th brain section (120 μm apart, ~25 sections per brain) were collected for Hematoxylin and Eosin (H&E) staining. To determine lesion area of each slice from H&E, bright field images at 4× magnification were obtained of the lesion site, and two investigators blinded to the experiment calculated the lesion area by tracing damaged or abnormal looking tissue in the ipsilateral cortex using Image J 1.38 (National Institutes of Health, Bethesda, MD, USA). The down boundary of the lesion outline was determined by drawing the area in which the immune cell infiltration was observed, as previously described (Basrai et al. 2016). For upper boundary of the lesion area, we outlined the expanse of the lesion using a mouse atlas (Tobin et al. 2014; Tsukano et al. 2016). In order to confirm superficial lesion measurements using a digital caliper, we assessed the margins of the lesion in the anterior posterior plane. The lesion volumes were computed by integrating the lesion area of each slice measured at each coronal level and the distance between two sections.
Flow cytometry of brain, spleen and blood
At day 3 after FPI, brains and spleens were harvested and homogenized with 40 μm nylon cell strainers (Becton Dickinson, Franklin Lakes, NJ, USA) in PBS. Cell suspensions were cetrifuged at 2,000 rpm for 5 min, and cell pellets were collected. Thereafter, 5 ml of 70% Percoll solution (GE Healthcare Bio Science AB, Uppsala, Sweden) were used to resuspend the brain cell pellet, and a 30% Percoll solution was overlaid. The gradient was centrifuged at 2,000 rpm for 30 min at room temperature. Single cells in the interface between 30% and 70% Percoll were collected for antibody staining. Cells were diluted to 1×106 cells in 100 μl PBS solution with 1% BSA and stained for antibodies and isotype control. For splenocytes, erythrocytes were removed using 1 × RBC lysis buffer (eBioscience, Inc. San Diego, CA USA) as indicated in the instruction manual. Briefly, the cell pellet was resuspended in 5 ml of 1X RBC lysis buffer per spleen and incubated for 5 min at room temperature. Thereafter, 30ml 1x PBS were added to stop the reaction. After centrifugation, the cell pellet was resuspended in 1xPBS solution for cell staining. For blood samples, mononuclear cells were isolated from the whole-blood of angular vein specimens and stained with fluorescent-labeled antibodies. All antibodies were purchased from Biolegend (San Diego, CA, USA) unless otherwise indicated, and the staining protocol folllowed the manual’s instructions. The following antibodies for the flow expriment were used: CD45-Pecy7, CD11b-Percp, CD86-PE, CD206-FITC and CD74-APC. Fluorescence minus one (FMO) controls were stained, respectively. Flow cytometry data were obained on C6 (BD Bioscience, San Jose, CA, USA) and analyzed by Flow Jo version 7.6.1 (Informer Technologies, Walnut Creek, CA,USA).
Statistical analysis
All data are shown as mean ± SEM. Statistical analyses were performed using GraphPad 6.0 software. Two-tailed unpaired Student’s t-test was used to determine significance of differences between two groups. Two-way ANOVA with multiple comparisons followed by the Bonferroni post hoc test were used for comparisons of multi-group data. P values ≤ 0.05 were considered significant.
Results
DRα1-MOG-35-55 attenuates neurodeficits and lesion volume in FPI mice
To assess the therapeutic effects of DRα1 -MOG-35-55 after TBI, we measured neurological impairment scores and lesion volumes in mice subjected to fluid percussion injury (FPI). Mice received nine daily s.c. injections of 100μg DRα1-MOG-35-55 or vehicle starting immediately after FPI (Fig. 1A) and were assessed intermitantly and 24h after after the final treatment (day 10) using the modified Neurological Severity Score (mNSS) and the Corner Turning test. DRα1-MOG-35-55 treatment significantly reduced both neurodeficit measures (Fig. 1B–C) as well as lesion volumes (Fig. 2A–B) at all time points measured after TBI compared to vehicle controls.
Fig. 1. DRα1-MOG-35-55 treatment reduces neurodeficits after TBI.
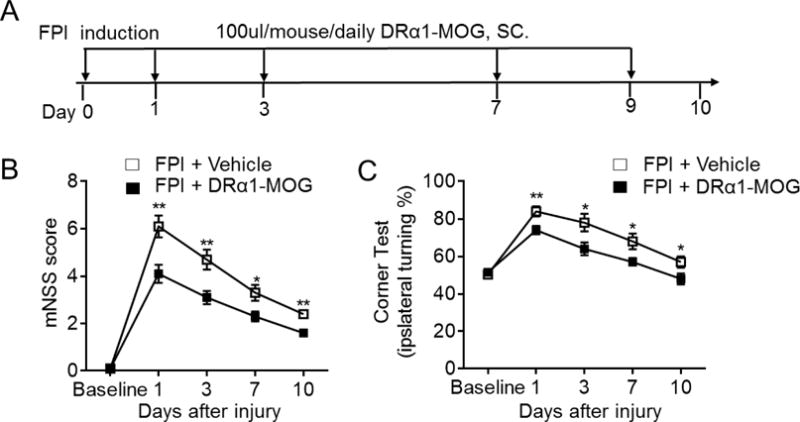
TBI was induced in C57BL/6 mice by FPI. Immediately after FPI, mice received subcutaneous injections of DRα1-MOG-35-55 (100μg in 0.1ml) or Vehicle and treatment was continued daily until the end of experiment (Day 10). A. Flow chart illustrates the regimen of DRα1-MOG-35-55 administration and experimental design. B–C. Neurological assessments were performed to evaluate the motor, sensory and balance functions at indicated time points after TBI in DRα1-MOG-35-55 vs. Vehicle treated mice. n=10 per group. *p <0.05, **p< 0.01. Data are presented as mean ± SEM.
Fig. 2. DRα1-MOG-35-55 treatment reduces lesion size after TBI.
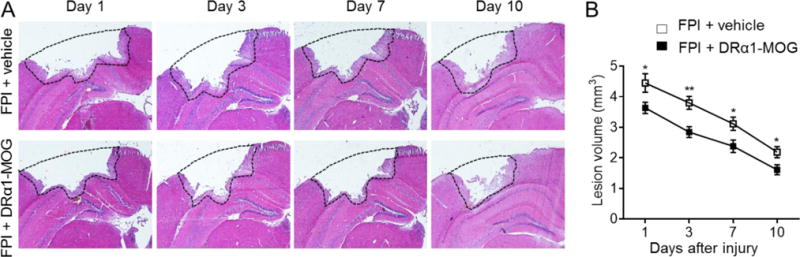
TBI was induced in C57BL/6 mice by FPI. Immediately after FPI, mice received subcutaneous injections of DRα1-MOG-35-55 (100μg in 0.1ml) or Vehicle. Treatment was continued daily until the end of experiment. A. H&E images show lesion area in coronal brain tissue sections of mice receiving Vehicle or DRα1-MOG-35-55 after TBI. B. Quantification of lesion size at indicated time points after TBI in mice receiving Vehicle or DRα1-MOG-35-55 treatment. *P<0.05, **P<0.01. n=8/group. Data are presented as mean ± SEM.
DRα1-MOG-35-55 reduces brain infiltration of CD11b+ cells and their expression of CD74
To determine whether DRα1 -MOG-35-55 treatment of brain injury could affect the cell numbers and CD74 expression of CD11b+ cells in the brain, we assessed numbers of non-activated brain-intrinsic microglia (CD11b+CD45int) and brain-infiltrating macrophages/activated microglia (CD11b+CD45hi) after TBI. We found that DRα1-MOG-35-55 treatment significantly reduced the total number of brain-infiltrating cells [vehicle: 6.83 ± 0.64 versus DRα1-MOG-35-55: 3.95 ± 0.38, (×106/per brain), p=0.01] (Fig. 3A–B), with predominant effects on CD11b+CD45hi macrophages/activated microglia (Fig. 3C) that expressed CD74 [vehicle: 64.93 ± 11.62 versus DRα1-MOG-35-55: 33.28 ± 6.64, (×103/per brain), p=0.03] (Fig. 3D). In contrast, no changes were observed in total numbers or CD74 expression of non-activated CD11b+CD45int microglia after treatment with DRα1-MOG-35-55 [vehicle: 17.57 ± 2.81 versus DRα1-MOG-35-55: 11.71±2.31, (×103/per brain), p=0.12]. These results thus demonstrate that treatment with the DRα1-MOG-35-55 construct can reduce the infiltration and local activation of CD11b+CD45hi cells in the injured brain and their expression of CD74 after TBI.
Fig. 3. DRα1-MOG-35-55 treatment reduces activation of CD11b+CD45hi cells in brain after TBI.
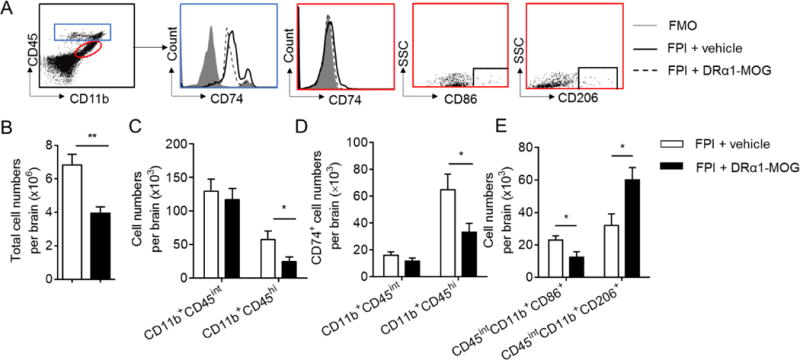
TBI was induced in C57BL/6 mice by FPI. Immediately after FPI, mice received a subcutaneous injection of DRα1-MOG-35-55 (100μg in 0.1ml) or Vehicle. Treatment was continued daily until day 3 after TBI. A. The flow gating strategy for microglia (CD11b+CD45int), brain-infiltrating macrophages/activated microglia (CD11b+CD45hi), and their surface expression of CD74, CD86 and CD206 at day 3 after TBI. B. Bar graph shows total numbers of microglia and brain-infiltrating macrophages per brain at day 3 after TBI in mice receiving DRα1 -MOG-35-55 or Vehicle. C. Bar graph shows total numbers of microglia or brain-infiltrating macrophages per brain at day 3 after TBI in mice receiving DRα1 -MOG-35-55 or Vehicle. D. Bar graph shows the expression of CD74 on microglia or brain-infiltrating macrophages at day 3 after TBI in mice receiving indicated treatments. In B–D, n =10 for Vehicle group; n =11 for DRα1-MOG-35-55 group. E. Bar graph shows the expression of CD86 or CD206 in microglia at day 3 after TBI in mice receiving indicated treatments. n = 7 mice per group. *p <0.05, **p< 0.01. Data are presented as mean ± SEM
Polarization of CD11b+CD45int cells toward a pro-inflammatory phenotype may promote brain injury after TBI. CD86 and CD206 are two markers routinely used to respectively characterize the pro-inflammatory versus anti-inflammatory phenotypes of CD11b+CD45int cells. We therefore determined the expression of CD86 and CD206 by brain-infiltrating CD11b+CD45int cells. The results clearly demonstrated a reduction of CD86-expressing inflammatory CD11b+CD45int cells [vehicle: 22.96 ± 2.62 versus DRα1-MOG-35-55: 12.44 ± 3.36, ×103/per brain), p=0.031] and an increase of anti-inflammatory CD206-expressing CD11b+CD45int cells after treatment with DRα1-MOG-35-55 [vehicle: 32.03 ± 7.08 versus DRα1-MOG-35-55: 60.09 ± 7.59, ×103/per brain), p=0.02] (Fig. 3E).
Effects of DRα1-MOG-35-55 treatment on peripheral CD11b+ cells
To evaluate the effects of DRα1-MOG-35-55 treatment on peripheral CD11b+ cells, we measured the number of CD11b+ cells and their expression of CD74 in the spleen or peripheral blood on day 3 after TBI induction. As shown in Fig. 4, no changes were observed in total [vehicle: 4.11±0.55 and DRα1-MOG-35-55: 5.35±0.43, (× 107/per spleen), p=0.11] or CD11b+ cell numbers [vehicle: 4.925±1.16 and DRα1-MOG-35-55: 7.06±1.66, (× 106/per spleen), p=0.32], or CD74+, CD86+ or CD206+ cells in the spleen after DRα1-MOG-35-55 treatment [CD74, vehicle: 5.21 ± 1.32 versus DRα1-MOG-35-55: 4.92 ± 1.38, (× 106/per spleen), p=0.88 ; CD86, vehicle: 5.84 ± 1.81 versus DRα1-MOG-35-55: 6.98±1.72, (× 106/per spleen), p=0.66; CD206, vehicle: 2.36 ± 0.44 versus DRα1-MOG-35-55: 2.56 ± 0.72, (× 106/per spleen), p=0.82] (Fig. 4A–D). In contrast, treatment with DRα1-MOG-35-55 induced significant increases in total circulating cell numbers as well as in CD11b+ [vehicle: 58.33 ± 10.47 versus DRα1-MOG-35-55: 122.60 ± 20.05, (× 106/ml), p=0.01] and CD74+ cells [vehicle: 47.53 ± 9.39 versus DRα1-MOG-35-55: 97.32 ± 14.11, (× 103/ml), p=0.01] in the peripheral blood (Fig. 5A–D).
Fig. 4. Immune cell subsets in spleens of mice receiving DRα1-MOG-35-55 after TBI.
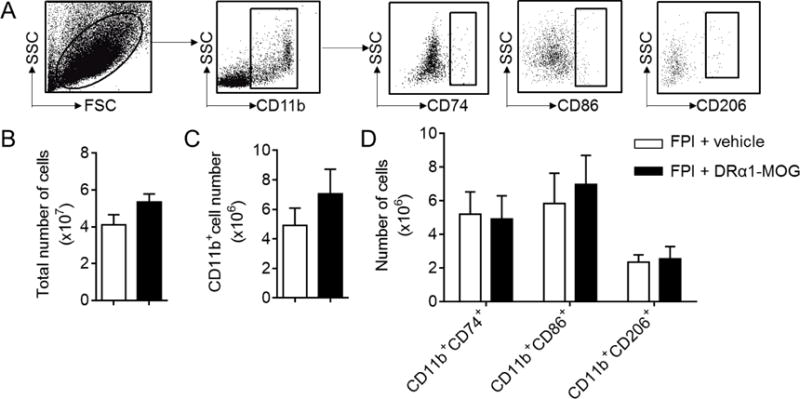
TBI was induced in C57BL/6 mice by FPI. Immediately after FPI, mice received a subcutaneous injection of DRα1-MOG-35-55 (100μg in 0.1ml) or Vehicle. Treatment was continued daily until day 3 after TBI. A. Gating strategy shows the expression of CD74, CD86 and CD206 in CD11b+ cells obtained from spleen. B. Bar graph shows the total numbers of splenocytes at day 3 after TBI in mice receiving DRα1 -MOG-35-55 or Vehicle. C. Bar graph shows the numbers of CD11b+ cells obtained from spleen at day 3 after TBI in mice receiving DRα1-MOG-35-55 or Vehicle. D. Bar graph shows the numbers of CD11b+ cells expressing CD74, CD86 or CD206 obtained from spleen at day 3 after TBI in mice receiving DRα1 -MOG-35-55 or Vehicle. n = 4-5 mice per group. Data are presented as mean ± SEM
Fig. 5. Assessment of immune cell subsets in blood of mice receiving DRα1-MOG-35-55 after TBI.
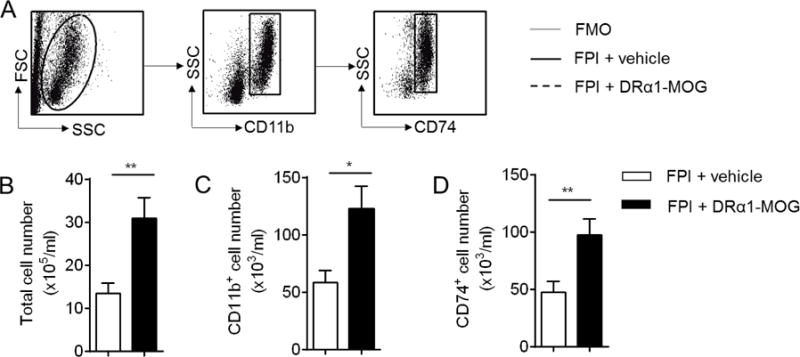
TBI was induced in C57BL/6 mice by fluid percussion injury (FPI). Immediately after FPI, mice received a subcutaneous injection of DRα1-MOG-35-55 (100μg in 0.1ml) or Vehicle. Treatment was continued daily until day 3 after FPI. A. Gating strategy shows the expression of CD74 on CD11b+ cells in peripheral blood. B. Bar graph shows the total number of blood cells at day 3 after FPI in mice receiving RTL DRα1 -MOG or Vehicle. C. Bar graph shows the numbers of circulating CD11b+ cells at day 3 after FPI in mice receiving DRα1-MOG-35-55 or Vehicle. D. Bar graph shows the number of circulating CD11b+ cells expressing CD74 at day 3 after FPI in mice receiving DRα1-MOG-35-55 or Vehicle. *p <0.05, **p< 0.01. n=9 per group. Data are presented as mean ± SEM
Discussion
This study provides the first evidence that DRα1 -MOG-35-55 treatment improves the outcome of TBI. Our results demonstrate that daily s.c. administration of DRα1-MOG-35-55 can significantly enhance functional outcomes, reduce brain lesion size, block infiltration of CD11b+ cells into the injured brain, and promote an anti-inflammatory phenotype in activated CD11b+CD45hi cells that infiltrate from the periphery or that result from local activation of resident CD11b+CD45int microglia after TBI. These changes observed in the injured brain were in stark contrast with increased numbers of activated monocytes in the blood and the lack of any demonstrable changes in the spleen.
Recombinant T-cell receptor (TCR) ligands (RTL) are molecular constructs comprised of covalently linked α1 and β1 domains of MHC II molecules with an attached antigenic peptide (Pan et al. 2014; Zhu et al. 2015; Zhu et al. 2014). Unlike four-domain MHC II molecules that induce T cell activation, RTLs are partial agonists of TCR and can deviate autoreactive T cell responses towards an anti-inflammatory phenotype (Burrows et al. 2001; Wang et al. 2003). Moreover, RTL constructs can block binding and downstream signaling of the cytokine/chemokine, macrophage migration inhibitory factor (MIF) through its CD74 receptor on monocytes and macrophages (Vandenbark et al. 2013) (featured on MDLinx.com;). It was previously demonstrated that neurodegeneration after TBI is dependent upon CD74 expression on antigen presenting cells (Tobin et al. 2014). RTL treatment of stroke has been shown to inhibit brain-reactive T cells without inducing general immunosuppression (Dziennis et al. 2011). Previous studies demonstrated that RTL treatment can protect against EAE and ischemic brain injury in mice (Dziennis et al. 2011; Pan et al. 2014; Subramanian et al. 2009; Zhu et al. 2015; Zhu et al. 2014) However, the clinical translation of RTL for treatment of acute thrombolytic or TBI events is restricted by the requirement for rapid matching of recipient MHC II with the β1 domains of the RTL construct. Thus, we developed a second generation partial MHC construct, DRα1-MOG-35-55 that was shown to reverse EAE (Meza-Romero et al. 2014) as well as treat ischemic brain injury after transient MCAO (Benedek et al. 2014). Like, RTLs, DRα1-MOG-35-55 can also bind with high affinity to CD74 that we show here to be increased in the CNS of mice after TBI. We further showed that DRα1-MOG-35-55 treatment not only reduced the influx of T cells into the ischemic brain, but also shifted microglia/macrophage polarization toward a beneficial M2-like phenotype (increased expression of CD206). These findings suggest that DRα1-MOG-35-55 may thus act as a competitive inhibitor of MIF binding to CD74 and blockade of its downstream chemotactic and inflammatory activities. (Meza-Romero et al. 2016) Such mechanisms could account for the clinical and histological improvement after treatment with DRα1-MOG-35-55 of mice after TBI. Because the DRα1 domain is expressed in all humans, DRα1-MOG-35-55 treatment does not require HLA screening of potential recipients and could be administered immediately after TBI in human subjects.
The DRα1 -MOG-35-55 construct was initially developed as a therapy for the MOG-35-55 peptide-induced murine model of multiple sclerosis, but it was also found to be highly effective for treatment of ischemic stroke and methamphetamine addiction in which T cell involvement is less clear. However, these inflammatory CNS conditions have in common increases in brain infiltrating CD11b+CD45high macrophages (Benedek et al. 2014; Meza-Romero et al. 2014; Wang et al. 2016). DRα1-MOG-35-55 treatment has been shown to inhibit the infiltration of both T cells and CD11b+CD45high macrophages in EAE and MCAO (Benedek et al. 2014; Meza-Romero et al. 2014; Wang et al. 2016), and we observed the same phenomenon for macrophage migration inhibition in the current study using a mouse TBI model. However, while the infiltration of CD11b+CD45high macrophages was reduced after DRα1-MOG-35-55 treatment, general T cell responses were largely unaltered (data not shown). This result was not surprising, since involvement of brain specific T cells has not yet been established in TBI.
It is also noteworthy that DRα1 -MOG-35-55 treatment increased the total immune cell numbers in blood similar to what was reported earlier in the spleens of mice with EAE and MCAO. In EAE, spleen cell numbers increased after immunization to induce EAE and increased further with treatment. (Meza-Romero et al. 2014) In MCAO, vessel occlusion reduced spleen cell numbers by ~90%, but again, treatment partially restored spleen cell numbers. (Benedek et al. 2014) Our current data in TBI indicate minimal impact of DRα1-MOG-35-55 on CD11b+ cells in spleen, but a near doubling of total cells and CD74-expressing CD11b+ cells in blood. In contrast, DRα1-MOG-35-55 treatment of TBI strongly inhibits the activation and recruitment of brain-infiltrating CD11b+CD45hi cells and their expression of CD74, as well as reduced expression of the co-stimulatory CD86 molecule but enhanced expression of the anti-inflammatory M2 marker, CD206, on CD11b+CD45int microglial cells within the CNS. These findings suggest that DRα1-MOG-35-55 binding to CD74 resulting in blockade of MIF activity that prevents activation and recruitment of circulating CD11b+ cells into the injured brain after TBI and enhancement of M2-like microglial activity within the CNS.
In summary, this study shows the therapeutic efficacy of DRα1-MOG-35-55 in a mouse model of TBI. Considering the potency of DRα1 -MOG-35-55 to reduce brain injury and neurodeficits after FPI impact, as well as its ability to provide benefit across MHC barriers, it is anticipated that there may be long-term benefit for TBI of continued intermittent treatment with DRα1-MOG-35-55.
Acknowledgments
This study was supported in part by the National Science Foundation of China grant 81471535 (to ML); US National Institutes of Health grants R01NS075887 and R01NS76013 (to HO) and AI 122574 (to AAV); Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Funding: This study was supported in part by the National Science Foundation of China grant 81471535 (to ML); US National Institutes of Health grants R01NS092713 (to HO) and AI 122574 (to AAV); Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
Author Contributions
H.O. and M.L. formulated the study concept and designed the studies; Y.L., Z.L., H.R., L.Z., S.G. and M.L. performed the studies and interpreted the results; Y.L., Z.L., L.R., Z.C., M.L. and H.O. wrote and edited the manuscript; R.M-R, G.B and AAV produced the DRa1-mMOG-35-55 construct and reviewed the manuscript.
Conflict of Interest: Drs. Vandenbark, Offner, Benedek, Meza-Romero and OHSU have a significant financial interest in Artielle ImmunoTherapeutics, Inc., a company that may have a commercial interest in the results of this research and technology. This potential conflict of interest has been reviewed and managed by the OHSU and VA Portland Health Care System Conflict of Interest in Research Committees. The other authors (Liu Yang, Zhijia Liu, Honglei Ren, Lei Zhang, Siman Gao, Li Ren, Zhi Chai, and Minshu Li) declare no conflicts of interest.
Statement on the Welfare of Animals: All applicable international, national and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution or practice at which the studies were conducted. This article does not contain any studies with human participants performed by any of the authors.
References
- Basrai HS, Christie KJ, Turbic A, Bye N, Turnley AM. Suppressor of Cytokine Signaling-2 (SOCS2) Regulates the Microglial Response and Improves Functional Outcome after Traumatic Brain Injury in Mice. PloS one. 2016;11:e0153418. doi: 10.1371/journal.pone.0153418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedek G, Vandenbark AA, Alkayed NJ, Offner H. Partial MHC class II constructs as novel immunomodulatory therapy for stroke. Neurochemistry international. 2016 doi: 10.1016/j.neuint.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedek G, et al. A novel HLA-DRalpha1-MOG-35-55 construct treats experimental stroke. Metab Brain Dis. 2014;29:37–45. doi: 10.1007/s11011-013-9440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrows GG, et al. Rudimentary TCR signaling triggers default IL-10 secretion by human Th1 cells. Journal of immunology. 2001;167:4386–4395. doi: 10.4049/jimmunol.167.8.4386. [DOI] [PubMed] [Google Scholar]
- Corps KN, Roth TL, McGavern DB. Inflammation and neuroprotection in traumatic brain injury. JAMA Neurol. 2015;72:355–362. doi: 10.1001/jamaneurol.2014.3558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziennis S, et al. Therapy with recombinant T-cell receptor ligand reduces infarct size and infiltrating inflammatory cells in brain after middle cerebral artery occlusion in mice. Metab Brain Dis. 2011;26:123–133. doi: 10.1007/s11011-011-9241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Loane DJ. Neuroinflammation after traumatic brain injury: opportunities for therapeutic intervention. Brain, behavior, and immunity. 2012;26:1191–1201. doi: 10.1016/j.bbi.2012.06.008. [DOI] [PubMed] [Google Scholar]
- McKee CA, Lukens JR. Emerging Roles for the Immune System in Traumatic Brain Injury. Front Immunol. 2016;7:556. doi: 10.3389/fimmu.2016.00556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Chopp M, Zhang Y, Liu Z, An A, Mahmood A, Xiong Y. ubacute intranasal administration of tissue plasminogen activator promotes neuroplasticity and improves functional recovery following traumatic brain injury in rats. PloS one. 2014;9:e106238. doi: 10.1371/journal.pone.0106238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza-Romero R, Benedek G, Leng L, Bucala R, Vandenbark AA. Predicted structure of MIF/CD74 and RTL1000/CD74 complexes. Metab Brain Dis. 2016;31:249–255. doi: 10.1007/s11011-016-9798-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza-Romero R, et al. HLA-DRalpha1 constructs block CD74 expression and MIF effects in experimental autoimmune encephalomyelitis. Journal of immunology. 2014;192:4164–4173. doi: 10.4049/jimmunol.1303118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J, et al. Novel Humanized Recombinant T Cell Receptor Ligands Protect the Female Brain After Experimental Stroke Transl. Stroke Res. 2014 doi: 10.1007/s12975-014-0345-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop V, Badaut J. A neurovascular perspective for long-term changes after brain trauma Transl. Stroke Res. 2011;2:533–545. doi: 10.1007/s12975-011-0126-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain pathology. 2004;14:215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Subramanian S, et al. Recombinant T cell receptor ligand treats experimental stroke. Stroke; a journal of cerebral circulation. 2009;40:2539–2545. doi: 10.1161/STROKEAHA.108.543991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobin RP, et al. Traumatic brain injury causes selective, CD74-dependent peripheral lymphocyte activation that exacerbates neurodegeneration. Acta Neuropathol Commun. 2014;2:143. doi: 10.1186/s40478-014-0143-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukano H, Horie M, Hishida R, Takahashi K, Takebayashi H, Shibuki K. Quantitative map of multiple auditory cortical regions with a stereotaxic fine-scale atlas of the mouse brain. Scientific reports. 2016;6:22315. doi: 10.1038/srep22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbark AA, et al. A novel regulatory pathway for autoimmune disease: binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance. J Autoimmun. 2013;40:96–110. doi: 10.1016/j.jaut.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, et al. Recombinant TCR ligand induces early TCR signaling and a unique pattern of downstream activation. Journal of immunology. 2003;171:1934–1940. doi: 10.4049/jimmunol.171.4.1934. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. DRalpha1-MOG-35-55 Reduces Permanent Ischemic Brain Injury. Transl Stroke Res. 2016 doi: 10.1007/s12975-016-0514-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Casper A, Libal NL, Murphy SJ, Bodhankar S, Offner H, Alkayed NJ. Preclinical Evaluation of Recombinant T Cell Receptor Ligand RTL1000 as a Therapeutic Agent in Ischemic Stroke. Translational stroke research. 2015;6:60–68. doi: 10.1007/s12975-014-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Libal NL, Casper A, Bodhankar S, Offner H, Alkayed NJ. Recombinant T Cell Receptor Ligand Treatment Improves Neurological Outcome in the Presence of Tissue Plasminogen Activator in Experimental Ischemic Stroke. Transl Stroke Res. 2014 doi: 10.1007/s12975-014-0348-8. [DOI] [PMC free article] [PubMed] [Google Scholar]