

Abstract
B cell activating factor from the TNF family (BAFF) stimulates B-cell proliferation and survival, but excessive BAFF promotes the development of aggressive B cells leading to malignant and autoimmune diseases. Recently, we have reported that rapamycin, a macrocyclic lactone, attenuates human soluble BAFF (hsBAFF)-stimulated B-cell proliferation/survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway. Here, we show that the inhibitory effect of rapamycin on hsBAFF-promoted B cell proliferation/survival is also related to blocking hsBAFF-stimulated phosphorylation of Akt, S6K1 and 4E-BP1, as well as expression of survivin in normal and B-lymphoid (Raji and Daudi) cells. It appeared that both mTORC1 and mTORC2 were involved in the inhibitory activity of rapamycin, as silencing raptor or rictor enhanced rapamycin’s suppression of hsBAFF-induced survivin expression and proliferation/viability in B cells. Also, PP242, an mTORC1/2 kinase inhibitor, repressed survivin expression and cell proliferation/viability more potently than rapamycin (mTORC1 inhibitor) in B cells in response to hsBAFF. Of interest, ectopic expression of constitutively active Akt (myr-Akt) or constitutively active S6K1 (S6K1-ca), or downregulation of 4E-BP1 conferred resistance to rapamycin’s attenuation of hsBAFF-induced survivin expression and B-cell proliferation/viability, whereas overexpression of dominant negative Akt (dn-Akt) or constitutively hypophosphorylated 4E-BP1 (4EBP1-5A), or downregulation of S6K1, or co-treatment with Akt inhibitor potentiated the inhibitory effects of rapamycin. The findings indicate that rapamycin attenuates excessive hsBAFF-induced cell proliferation/survival via blocking mTORC1/2 signaling in normal and neoplastic B-lymphoid cells. Our data underscore that rapamycin may be a potential agent for preventing excessive BAFF-evoked aggressive B-cell malignancies and autoimmune diseases.
Keywords: Rapamycin, BAFF, B cells, Proliferation, mTOR
Introduction
B cell activating factor from the TNF family (BAFF), a type II membrane protein that exists in both membrane-bound and soluble forms, also known as B lymphocyte stimulator (Blys), TNF and apoptosis ligand-related leukocyte-expressed ligand-1 (TALL-1), TNF homologue that activates apoptosis, nuclear factor κB, and c-Jun NH2-terminal kinase (THANK) and zTNF4, is a well-known cytokine that spurs development, maturation and homeostasis of B lymphocytes (Henley et al., 2008; Moore et al., 1999; Mueller et al., 2007; Schneider et al., 1999). It is mainly secreted by monocytes and dendritic cells and acts as a ligand for three TNF-receptor family members: BAFF-R (BR3), B cell maturation antigen (BCMA) and transmembrane activator and cyclophilin ligand interactor (TACI) (Fu et al., 2009; Mackay et al., 2003; Mackay et al., 2007; Schneider and Tschopp, 2003). Multiple reports have shown that abnormal level of BAFF is the culprit of immune-related diseases (Batten et al., 2000; Schweighoffer et al., 2013), and excessive expression of BAFF can cause various autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and SjÖgren’s syndrome (SS) (Bosello et al., 2008; Mackay and Ambrose, 2003; Moisini and Davidson, 2009; Zhang et al., 2001). Patients with these diseases are often detected to have high levels of BAFF in the blood, causing elevated secretion of superfluous autoantibodies (Moisini and Davidson, 2009; Sanz and Lee, 2010). A series of studies have focused on B cells as therapeutic targets in autoimmune diseases (Cornec et al., 2012; Ramanujam and Davidson, 2008; Sasaki et al., 2006). However, how excessive BAFF induces aggressive B-cell proliferation and survival is not well understood.
The mammalian target of rapamycin (mTOR) plays an important role in regulating nutrient metabolism and promoting the proliferation and survival of lymphocyte cells (Cornu et al., 2013; Laplante and Sabatini, 2012; Zeng and Chi, 2013), and has been extensively evaluated as a therapeutic target in multiple lymphoid malignancies (Yun et al., 2016). As a serine/threonine (Ser/Thr) protein kinase, mTOR functions at least as two complexes, mTORC1 and mTORC2, with distinct substrate specificities (Laplante and Sabatini, 2012). mTORC1 comprises mTOR, raptor (regulatory-associated protein of mTOR), mLST8 (also termed G protein β-subunit-like protein, GβL, a yeast homolog of LST8), PRAS40 (proline-rich Akt substrate 40 kDa) and DEPTOR (DEP-domain-containing mTOR-interacting protein). mTORC2 is composed of mTOR, rictor (rapamycin insensitive companion of mTOR), mLST8, mSin1 (mammalian stress-activated protein kinase-interacting protein 1), protor (protein observed with rictor) and DEPTOR (Cornu et al., 2013; Laplante and Sabatini, 2012; Peterson et al., 2009; Zhou et al., 2010). mTORC1 mediates the phosphorylation of two best-characterized downstream effector molecules, p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E (eIF4E) binding protein 1 (4E-BP1), and controls lipid/protein synthesis, cell growth/proliferation, survival and motility (Cornu et al., 2013; Laplante and Sabatini, 2012). mTORC2 regulates the phosphorylation or activity of protein kinase B (Akt/PKB), serum and glucocorticoid-induced kinase 1 (SGK1), protein kinase C α (PKCα), focal adhesion proteins and small GTPases, and controls cell survival and actin cytoskeleton (Cornu et al., 2013; Laplante and Sabatini, 2012). Mounting evidence has demonstrated that hyperactivation of mTORC1/2 is responsible for the pathogenesis of many cancers (Guertin et al., 2009; Hietakangas and Cohen, 2008; Yang and Klionsky, 2010). As abnormal activity of mTOR has been observed in several autoimmune diseases (Fernandez and Perl, 2010; Laragione and Gulko, 2010; Malemud, 2013), targeting mTOR has become a promising strategy for prevention and treatment of autoimmune diseases (Weichhart and Saemann, 2009). We have recently found that excessive BAFF activates mTOR pathway contributing to proliferation and survival in cultured B lymphocytes (Ke et al., 2013). Therefore, drugs that inhibit this specific pathway may be an effective intervention for BAFF-induced autoimmune disorders.
Rapamycin, a lipophilic macrolide antibiotic, has been used in renal transplantation as an immunosuppressant (Zhou et al., 2010). Traditional view considers rapamycin as a potent and specific inhibitor of mTORC1 (Ballou and Lin, 2008; Zhou et al., 2010). Extensive studies have revealed that rapamycin can also inhibit mTORC2-mediated Akt phosphorylation, depending on cell lines or treatment time/dose (Laplante and Sabatini, 2012; Sarbassov et al., 2006). Recently, rapamycin analogs (termed rapalogs), such as Temsirolimus and Everolimus, have been documented to be effective in treatment of certain autoimmune diseases (Blachly and Baiocchi, 2014; Eyre et al., 2014; Mohindra et al., 2014). However, the underlying mechanism of rapamycin’s action is not fully elucidated. Our recent studies have shown that human soluble BAFF (hsBAFF) stimulates the phosphorylation of S6K1, 4E-BP1 and Akt contributing to B-cell proliferation and survival (Ke et al., 2013; Zeng et al., 2015), which can be blocked by a short time (2 h) pretreatment with rapamycin (Zeng et al., 2015). These findings prompted us to determine whether rapamycin inhibits BAFF-stimulated cell proliferation and survival via targeting mTORC1 and/or mTORC2 signaling in normal and neoplastic B-lymphoid cells.
Here, for the first time, we show that rapamycin attenuates excessive hsBAFF-induced cell proliferation and survival, not only by targeting mTORC1-mediated S6K1/4E-BP1 pathways, but also via targeting mTORC2-mediated Akt pathway in normal and neoplastic B-lymphoid cells. Our findings underscore that rapamycin may be a potential agent for preventing excessive BAFF-evoked aggressive B-cell malignancies and autoimmune diseases.
Materials and Methods
Materials
Anti-CD19 magnetic fluorobeads-B was purchased from One Lambda (Canoga Park, CA, USA). PP242 (mTORC1/2 kinase inhibitor) and Akt inhibitor X were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Rapamycin (mTOR inhibitor) was from ALEXIS (San Diego, CA, USA). Refolded human soluble BAFF (hsBAFF) was a recombinant form of the extracellular domain of the BAFF produced in Escherichia coli from this group (Cao et al., 2005). RPMI 1640 was from Gibco (Rockville, MD, USA). Fetal bovine serum (FBS) was supplied by Hyclone (Logan, UT, USA). CellTiter 96® AQueous One Solution Cell Proliferation Assay kit was from Promega (Madison, WI, USA). Other chemicals were purchased from local commercial sources and were of analytical grade.
Cell culture
Neoplastic B-lymphoid Raji and Daudi cell lines (American Type Culture Collection, Manassas, VA, USA) were maintained in RPMI 1640 medium supplemented with 10% FBS, 100 U/ml penicillin, 100 U/ml streptomycin at 37°C in a humidified incubator containing 5% CO2. Primary B lymphocytes were purified from fresh splenic cells of healthy mice using anti-CD19 magnetic fluorobeads and cultured as described previously (Ke et al., 2013). All procedures used in this study were approved by the Institutional Animal Care and Use Committee, and were in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Recombinant adenoviral constructs and infection of cells
Recombinant adenoviral vectors encoding green fluorescence protein (Ad-GFP), hemagglutinin (HA)-tagged constitutively hypophosphorylated 4E-BP1 (Ad-4EBP1-5A), wild-type S6K1 (Ad-S6K1-wt) and constitutively active S6K1 (Ad-S6K1-ca) were described previously (Liu et al., 2008; Liu et al., 2006; Liu et al., 2010). Recombinant adenoviral vectors encoding HA-tagged dominant negative Akt (Ad-dn-Akt, T308A/S473A) and constitutively active Akt (Ad-myr-Akt) were generously provided by Dr. Kenneth Walsh (Boston University, Boston, MA). For experiments, Raji cells were grown in the growth medium and infected with the individual adenovirus for 24 h at 1 of multiplicity of infection (MOI = 1). Subsequently, cells were used for experiments. Ad-GFP alone served as a control. Expression of HA-tagged 4EBP1-5A, S6K1-wt, S6K1-ca, myr-Akt and dn-Akt was determined by Western blot analysis with antibodies to HA.
Lentiviral shRNA cloning, production and infection of cells
Lentiviral shRNAs to raptor, rictor, S6K1, 4E-BP1 and GFP (for control) were constructed and infected as described previously (Liu et al., 2008; Liu et al., 2006; Liu et al., 2010). For use, Raji cells, when grown to about 70% confluence, were infected with above lentivirus containing medium in the presence of 8 μg/ml polybrene for 12 h twice at an interval of 6 h. Uninfected cells were eliminated by exposure to 2 μg/ml puromycin for 48 h before use. After 5 days of culture, cells were used for experiments.
Assays for cell proliferation, cell viability, and live cell number
Purified mouse B lymphocytes, Raji and/or Daudi cells, or Raji cells infected with Ad-4EBP1-5A, Ad-S6K1-wt, Ad-S6K1-ca, Ad-dn-Akt, Ad-myr-Akt and Ad-GFP, respectively, or Raji cells infected with lentiviral shRNAs to raptor, rictor, raptor/rictor, 4E-BP1, S6K1 and GFP, respectively, were seeded in 24-well plates (3×105 cells/well, for cell proliferation assay and live cell assay) or 96-well plates (3×104 cells/well, for cell viability assay) and cultured for overnight in a humidified incubator of 5 % CO2 at 37 °C. Next day, cells were treated with/without hsBAFF (2.5 μg/ml) for 48 h following pre-incubation with/without rapamycin (100 ng/ml) or PP242 (1 μM) for 2 h, or treated with/without Akt inhibitor X (20 μM) for 1 h and then hsBAFF (2.5 μg/ml) for 48 h following pre-incubation with/without rapamycin (100 ng/ml) for 2 h with 5 replicates of each treatment. Subsequently, the proliferation and the viability of the cells were assessed using a Coulter Counter (Beckman Coulter, Fullerton, CA, USA) and a Synergy™ 2 Multi-function Microplate Reader (Bio-Tek Instruments, Winooski, Vermont, USA), respectively, as described previously (Zeng et al., 2015). Live cells were estimated by counting viable cells using trypan blue exclusion.
Western blot analysis
Purified mouse B lymphocytes, Raji and/or Daudi cells, or Raji cells infected with Ad-4EBP1-5A, Ad-S6K1-wt,Ad-S6K1-ca, Ad-dn-Akt, Ad-myr-Akt and Ad-GFP, respectively, or Raji cells infected with lentiviral shRNAs to raptor, rictor, raptor/rictor, 4E-BP1, S6K1 and GFP, respectively, after treatments, were subjected to Western blotting as described (Chen et al., 2010). In brief, lysates containing equivalent amounts of protein were separated on 7–12% SDS–polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were incubated with PBS containing 0.05% Tween 20 and 5% nonfat dry milk to block nonspecific binding, and then with primary antibodies against phosphorylated Akt (p-Akt) (Ser473), p-S6K1 (Thr389), p-S6 (Ser235/236), p-4E-BP1 (Thr70), 4E-BP1 (Cell Signaling Technology, Danvers, MA, USA), β-actin, S6K1, Akt, S6, survivin, GSK3β (Santa Cruz Biotechnology, Santa Cruz, CA, USA), raptor, rictor (Bethyl Laboratories, Montgomery, TX, USA), phospho-GSK3β (Ser9) (Epitomics, Burlingame, CA, USA), phospho-Akt (Thr308), HA (Sigma, St. Louis, MO, USA) overnight at 4°C, respectively, followed by incubating with appropriate secondary antibodies including horseradish peroxidase-coupled goat anti-rabbit IgG, goat anti-mouse IgG, or rabbit anti-goat IgG (Pierce, Rockford, IL, USA) overnight at 4°C. Immunoreactive bands were visualized by using enhanced chemiluminescence solution (Millipore, Billerica, MA, USA). The blots for detected proteins were semi-quantified using NIH Image J software (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Results were expressed as mean ± standard error of the mean (SEM). Analysis of statistical significance was performed using Student’s t-test for non-paired replicates. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-test to compare replicate means. P-Value of < 0.05 was considered significant.
Results
Rapamycin inhibition of cell proliferation and survival involves blocking hsBAFF-stimulated Akt and S6K1/4E-BP1 pathways in B cells
We have recently shown that excessive hsBAFF induces B-cell proliferation and survival in part through activation of mTOR signaling pathway (Ke et al., 2013; Zeng et al., 2015). Rapamycin inhibits hsBAFF-induced B-cell proliferation/viability in an mTOR kinase activity-dependent manner (Zeng et al., 2015). In agreement with the above findings, here we also observed that pretreatment of B cells (Raji cells, Daudi cells and normal primary mouse B lymphocytes) with rapamycin (100 ng/ml) for 2 h inhibited cell proliferation and viability stimulated by 48-h exposure to hsBAFF (2.5 μg/ml), as determined by cell counting (Fig. 1A), the MTS assay (Fig. 1B) and trypan blue exclusion assay (Fig. 1C), respectively. Furthermore, our Western blot analysis also showed that hsBAFF-induced robust phospho-Akt (Ser473 and Thr308), phospho-S6K1 (Thr389), phospho-S6 (Ser235/236, a substrate of S6K1), phospho-4E-BP1 (Thr70) and survivin were greatly attenuated by rapamycin (Fig. 1D and E). These results suggest that rapamycin inhibition of cell proliferation and survival involves blocking activation of Akt and S6K1/4E-BP1 pathways in hsBAFF-stimulated B cells.
Fig. 1.
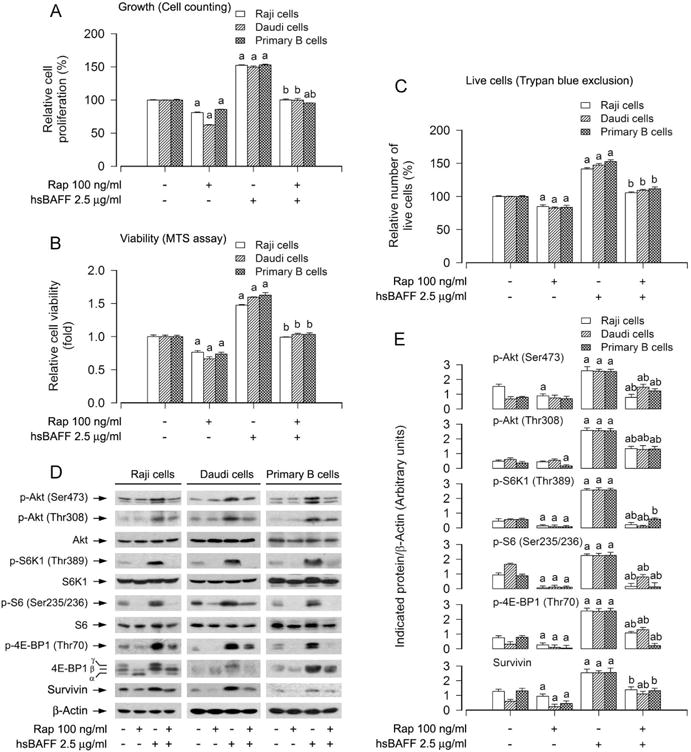
Inhibitory effect of administered rapamycin in Raji cells, Daudi cells and primary B lymphocytes on hsBAFF-stimulated phosphorylation of S6K1/4E-BP1 and Akt as well as cell proliferation and survival. Raji cells, Daudi cells and purified mouse splenic B lymphocytes were pretreated with/without rapamycin (Rap, 100 ng/ml) for 2 h, and then stimulated with/without 2.5 μg/ml hsBAFF for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A) The cell proliferation was estimated by cell counting. (B) The cell viability was determined by the MTS assay. (C) The relative number of live cells was estimated by trypan blue exclusion assay. (D) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in three independent experiments. (E) The blots for p-Akt (Ser473), p-Akt (Thr308), p-S6K1 (Thr389), p-S6 (Ser235/236), p-4E-BP1 (Thr70), and survivin were semi-quantified. Results are presented as mean ± SEM (n = 3–5). aP<0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group.
Rapamycin inhibits hsBAFF-induced B-cell proliferation and viability by targeting both mTORC1 and mTORC2
mTORC1 and mTORC2 are two well-known distinct mTOR complexes: the former regulates the phosphorylation of S6K/4E-BP1 and the latter mediates the phosphorylation of Akt at Ser473 (Cornu et al., 2013; Laplante and Sabatini, 2012). Our current study has found that rapamycin not only profoundly inhibited hsBAFF-induced phosphorylation of S6K1 and 4E-BP1, but also remarkably suppressed hsBAFF-induced phosphorylation of Akt in B cells (Fig. 1C and D), suggesting that rapamycin may prevent B cells from hsBAFF-induced cell proliferation/viability by targeting both mTORC1 and mTORC2. To determine whether this is true, mTORC1 and mTORC2 were disrupted by silencing raptor and rictor, respectively. For this, Raji cells, infected with lentiviral shRNAs to raptor, rictor or GFP (as control), respectively, were pretreated with/without rapamycin (100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h or 48 h. Western blot analysis revealed that lentiviral shRNA to raptor or rictor, but not GFP, downregulated raptor or rictor protein expression by ~90% in the cells (Fig. 2A). Downregulation of raptor substantially inhibited the basal and hsBAFF-induced phosphorylation of S6K1/4E-BP1, whereas downregulation of rictor obviously inhibited the basal and hsBAFF-induced phosphorylation of Akt (Ser 473) (Fig. 2A and B). Importantly, pretreatment with rapamycin for 2 h not only potently inhibited hsBAFF-induced phosphorylation of S6K1/4E-BP1, but also obviously inhibited hsBAFF-induced phosphorylation of Akt in Raji cells infected with lentiviral shRNA to raptor, rictor or GFP (Fig. 2A and B). Of interest, silencing raptor or rictor reinforced the inhibitory effect of rapamycin on the basal and hsBAFF-induced survivin compared to the control (GFP shRNA) (Fig. 2A and B). Consistently, silencing raptor or rictor alone markedly repressed the basal and hsBAFF-induced cell proliferation/viability in Raji cells (Fig. 2C and D). Furthermore, rapamycin was able to potentiate the inhibitory effect of raptor shRNA or rictor shRNA on hsBAFF-induced cell proliferation/viability (Fig. 2C and D).
Fig. 2.
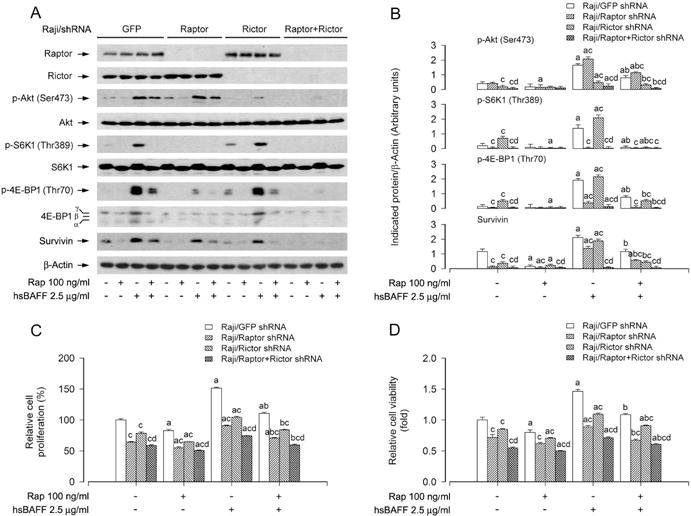
Effects of down-regulation of raptor, rictor, raptor/rictor in Raji cells on rapamycin’s prevention from hsBAFF-induced activation of mTORC1 and mTORC2 pathways as well as cell proliferation/viability. Raji cells, infected with lentiviral shRNAs to raptor, rictor, raptor/rictor or GFP (as control), were pretreated with/without rapamycin (Rap, 100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-S6K1 (Thr389), p-4E-BP1 (Thr70), and survivin were semi-quantified. (C) The cell proliferation was evaluated by cell counting. (D) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP<0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group; cP<0.05, Raptor shRNA group, Rictor shRNA group or Raptor/Rictor shRNA group vs GFP shRNA group; dP< 0.05, Raptor/Rictor shRNA group vs RaptorshRNA group or Rictor shRNA group.
To substantiate the role of mTORC1 and mTORC2 in hsBAFF-induced cell proliferation/viability, next, we performed double knockdown of raptor and rictor. Western blot results showed that co-infection with lentiviral shRNAs to raptor and rictor silenced raptor/rictor protein expression by ~90% in Raji cells (Fig. 2A). Of interest, the double knockdown of raptor/rictor displayed a more potent inhibitory effect on hsBAFF-induced phosphorylation of Akt, S6K1 and 4E-BP1, expression of survivin, as well as increase of cell proliferation/viability than the single knockdown of raptor or rictor (Fig. 2A–D). Furthermore, the double knockdown of raptor/rictor also more effectively strengthened the inhibitory effect of rapamycin on hsBAFF-induced expression of survivin and increase of cell proliferation/viability, compared to the single knockdown of raptor or rictor (Fig. 2A–D).
To further corroborate rapamycin’s inhibitory effect on hsBAFF-induced B-cell proliferation/viability is indeed by suppressing both mTORC1 and mTORC2, PP242, an mTORC1/2 inhibitor, was used. As predicted, PP242 inhibited both mTORC1-mediated phosphorylation of S6K1/4E-BP1 and mTORC2-mediated phosphorylation of Akt in Raji cells and primary B lymphocytes (Fig. 3A and B). Interestingly, PP242 (inhibition of mTORC1/2) suppressed the basal or hsBAFF-stimulated survivin expression and cell proliferation/viability in the cells more potently than rapamycin (inhibition of mTORC1) (Fig. 3A–D). Taken together, the above results demonstrate that both mTORC1 and mTORC2 are involved in rapamycin’s inhibition of hsBAFF-induced proliferation/viability in B cells.
Fig. 3.
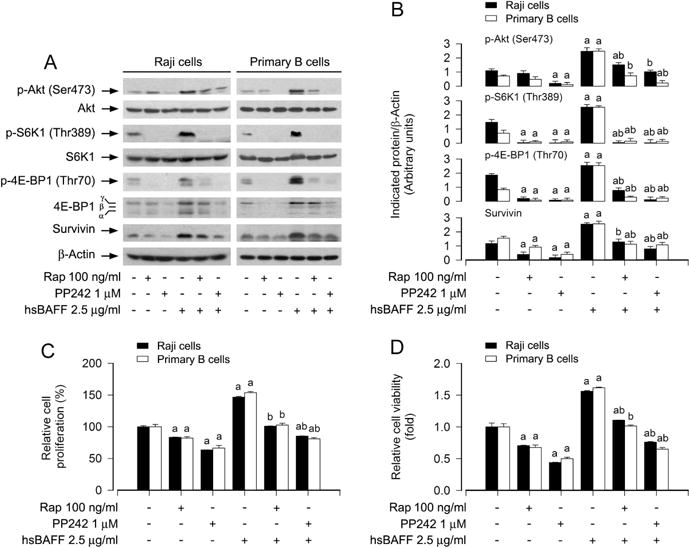
Block effect of inhibition of mTORC1/2 by PP242 in Raji cells and primary B lymphocytes on hsBAFF-stimulated cell proliferation/viability. Raji cells and purified mouse splenic B lymphocytes were pretreated with/without rapamycin (Rap, 100 ng/ml) or PP242 (1 μM) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-S6K1 (Thr389), p-4E-BP1 (Thr70), and survivin were semi-quantified. (C) The cell proliferation was evaluated by cell counting. (D) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP<0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group.
mTORC1-mediated S6K1 pathway is essential for rapamycin’s suppression of hsBAFF-induced B-cell proliferation and viability
S6K1 is one of the best-characterized downstream effector molecules of mTORC1 (Cornu et al., 2013; Laplante and Sabatini, 2012). To understand the role of S6K1 in rapamycin’s inhibition of hsBAFF-induced B-cell proliferation/viability, Raji cells were firstly infected with recombinant adenoviruses expressing HA-tagged wild-type (wt) S6K1 (Ad-S6K1-wt), constitutively active S6K1 (Ad-S6K1-ca) and control virus encoding GFP alone. Western blot results showed high levels of recombinant S6K1-wt or S6K1-ca in the cells infected with Ad-S6K1-wt or Ad-S6K1-ca, but not in the cells infected with Ad-GFP (Fig. 4A). Of note, cells expressing S6K1-wt or S6K1-ca, but not GFP, had a robust phosphorylation of S6K1 and S6 (Fig. 4A and B). Addition of hsBAFF failed to further enhance the phosphorylation of these proteins (Fig. 4A and B). Nevertheless, cells expressing S6K1-ca, but not S6K1-wt or GFP, were greatly resistant to rapamycin inhibition of the basal and hsBAFF-induced phospho-S6K1, phospho-S6, survivin, and cell proliferation/viability (Fig. 4A–D), suggesting that inhibition of mTORC1 by rapamycin mediates the blockage of hsBAFF-induced S6K1 pathway contributing to proliferation/viability in B cells.
Fig. 4.
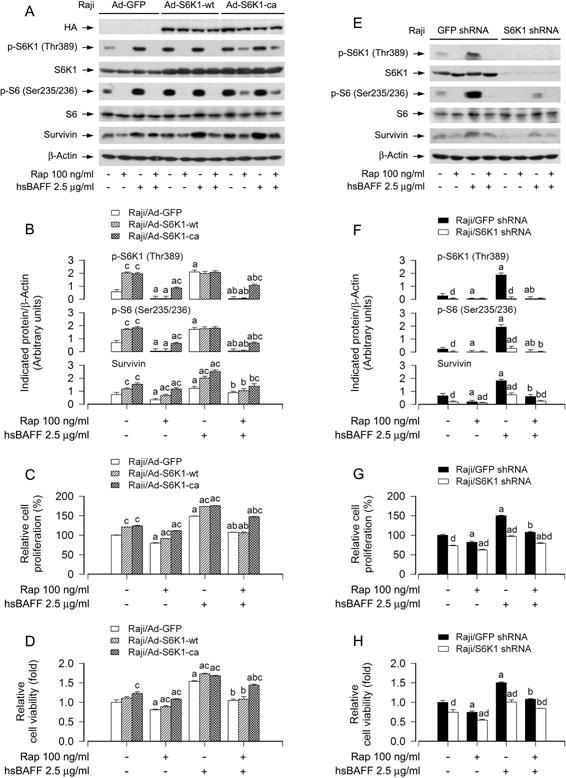
Effects of ectopic expression of wild-type or constitutively active S6K1 or down-regulation of S6K1 in Raji cells on rapamycin’s inhibition of hsBAFF-stimulated cell proliferation/viability. Raji cells, infected with Ad-S6K1-wt, Ad-S6K1-ca, or Ad-GFP (for control), were pretreated with/without rapamycin (Rap, 100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A and E) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and F) The blots for p-S6K1 (Thr389), p-S6 (Ser235/236), and survivin were semi-quantified. (C and G) The cell proliferation was evaluated by cell counting. (D and H) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP<0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group; cP<0.05, Ad-S6K1-wt group or Ad-S6K1-ca group vs Ad-GFP group; dP<0.05, S6K1 shRNA group vs GFP shRNA group.
To substantiate the role of S6K1 in rapamycin’s suppression of hsBAFF-induced B-cell proliferation/viability, lentiviral shRNA to S6K1 was used to silence the expression of S6K1 in Raji cells. As shown in Fig. 4E, expression of S6K1 protein was downregulated by ~ 90% in the cells infected with lentiviral shRNA to S6K1. Silencing S6K1 obviously attenuated the basal and hsBAFF-induced phosphorylation of S6K1/S6 and survivin (Fig. 4E and F). Rapamycin was able to almost completely block the basal and hsBAFF-induced phosphorylation of S6K1 or S6 in Raji cells, regardless of infection with lentiviral shRNA to S6K1 or not (Fig. 4E and F). However, silencing S6K1 reinforced the inhibitory effect of rapamycin on the basal and hsBAFF-induced surviving expression compared to the control (GFP shRNA) (Fig. 4E and F). Consistently, by cell counting and the MTS assay, we observed that silencing S6K1 alone partially prevented the basal and hsBAFF-induced proliferation/viability in the cells (Fig. 4G and H). Addition of rapamycin more significantly reduced cell proliferation/viability induced by haBAFF (Fig. 4G and H). Taken together, the findings clearly indicate that rapamycin inhibits excessive hsBAFF-induced B-cell proliferation/viability in part by suppressing mTORC1-mediated S6K1 pathway.
mTORC1-mediated 4E-BP1 pathway is necessary for rapamycin’s repression of hsBAFF-induced B-cell proliferation and viability
4E-BP1/eIF4E is another best-known downstream signaling pathway of mTORC1 (Cornu et al., 2013; Laplante and Sabatini, 2012). To define whether rapamycin inhibits hsBAFF-induced B-cell proliferation/viability by targeting 4E-BP1/eIF4E pathway, we employed recombinant adenovirus encoding HA-tagged 4E-BP1 mutants where Thr36, Thr45, Ser64, Thr69 andSer82 are replaced by Ala residues (Ad-4EBP1-5A) mimicking hypophosphorylated residues, thus tightly binding to and sequester eIF4E in cells (Mothe-Satney et al., 2000). As shown in Fig. 5A, HA-tagged 4E-BP1 and higher levels of 4E-BP1 were detected in Ad-4EBP1-5A-infected Raji cells. Administration of rapamycin failed to alter the mobility of 4E-BP1-5A (Fig. 5A). Interestingly, expression of 4E-BP1-5A obviously strengthened the inhibitory effects of rapamycin on the basal and hsBAFF-induced survivin and proliferation/viability in the cells (Fig. 5B–D), suggesting that rapamycin suppresses hsBAFF-induced B-cell proliferation/viability also at least in part by targeting mTORC1-mediated 4E-BP1/eIF4E pathway.
Fig. 5.
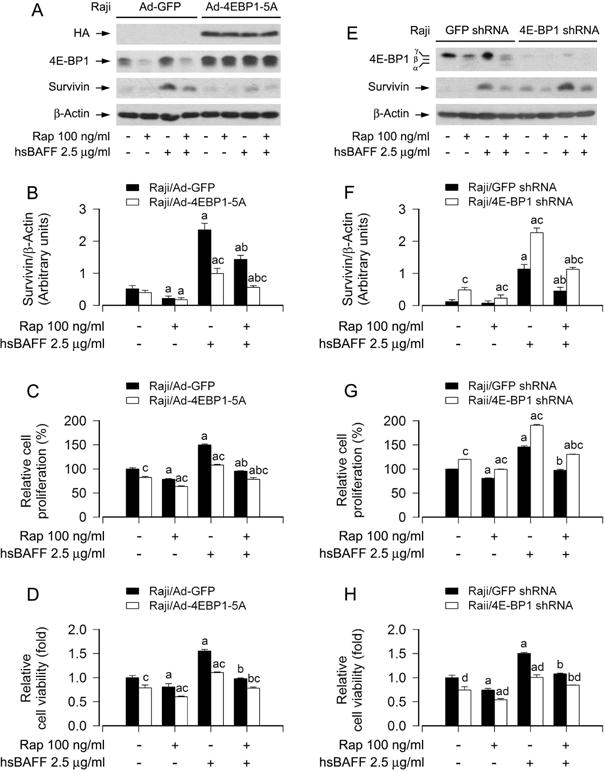
Effects of ectopic expression of 4E-BP1-5A or down-regulation of 4E-BP1 in Raji cells on rapamycin’s hindering from hsBAFF-stimulated cell proliferation/viability. Raji cells, infected with lentiviral shRNAs to 4E-BP1 or GFP (for control), or with Ad-4EBP1-5A and Ad-GFP were pretreated with/without rapamycin (Rap, 100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A and E) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B and F) The blots for survivin were semi-quantified. (C and G) The cell proliferation was evaluated by cell counting. (D and H) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP<0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group; cP<0.05, Ad-4EBP1-5A group vs Ad-GFP group or 4E-BP1 shRNA group vs GFP shRNA group.
To further confirm the above findings, genetic silence for 4E-BP1 was carried out. As demonstrated in Fig. 5E, infection of Raji cells with lentiviral shRNA to 4E-BP1 caused an obvious downregulation of 4E-BP1 protein expression compared to control cells infected with lentiviral shRNA to GFP, as determined by Western blotting with antibodies to 4E-BP1. Consequently, the survivin expression and cell proliferation/viability in 4E-BP1-knockdown Raji cells treated with/without hsBAFF ± rapamycin were dramatically elevated (Fig. 5F–H). The findings verify that mTORC1-mediated 4E-BP1 pathway is as vital as S6K1 pathway for rapamycin’s repression of hsBAFF-induced B-cell proliferation and viability.
mTORC2-mediated Akt pathway contributes to rapamycin’s attenuation of hsBAFF-induced B-cell proliferation and viability
We have observed that mTORC2 modulates rapamycin’s inhibition of hsBAFF-induced B-cell proliferation/viability (Fig. 2–3). Akt is a well characterized substrate of mTORC2, and mTORC2 directly phosphorylates Akt on Ser473 (Cornu et al., 2013; Sarbassov et al., 2005). Therefore, we continued to test whether mTORC2-mediated Akt pathway plays an important role in rapamycin’s inhibition of hsBAFF-induced B-cell proliferation/viability. To this end, Akt inhibitor X, a selective Akt inhibitor, was used. We found that pretreatment with Akt inhibitor X or rapamycin obviously suppressed the basal and hsBAFF-induced phosphorylation of Akt (Ser473 and Thr308) and its substrate GSK3β (Ser9) in Raji cells and primary B cells (Fig. 6A and B). In addition, similar effects for the basal or hsBAFF-induced phosphorylation of S6K1 (Thr389) and 4E-BP1 (Thr70), as well as expression of survivin were also observed in the cells treated with Akt inhibitor X or rapamycin (Fig. 6A and B). In particular, co-treatment with rapamycin/Akt inhibitor X exhibited a stronger inhibitory effect on hsBAFF-induced events in the cells (Fig. 6A and B). Consistently, co-treatment with rapamycin/Akt inhibitor X also inhibited the basal and hsBAFF-stimulated B-cell proliferation/viability more potently than rapamycin or Akt inhibitor X alone (Fig. 6C and D), suggesting that mTORC2-mediated Akt pathway is involved in rapamycin’s inhibition of hsBAFF-induced B-cell proliferation and viability as well.
Fig. 6.
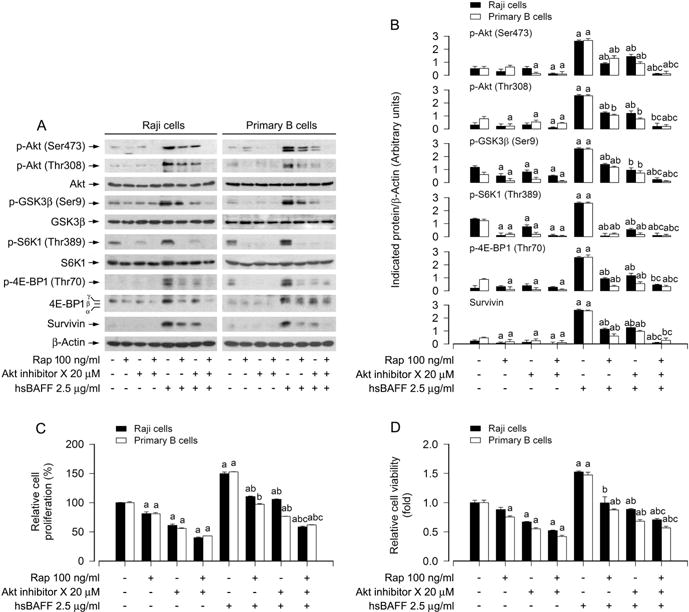
Preventive effects of administered rapamycin and/or Akt inhibitor X in Raji cells and primary B lymphocytes on hsBAFF-induced activation of Akt/mTOR pathway and cell proliferation/viability. Raji cells and purified mouse splenic B lymphocytes were pretreated with/without rapamycin (100 ng/ml) for 2 h and then pretreated with/without Akt inhibitor X (20 μM) for 1 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-Akt (Thr308), p-GSK3β (Ser9), p-S6K1 (Thr389), p-4E-BP1 (Thr70), and survivin were semi-quantified. (C) The cell proliferation was evaluated by cell counting. (D) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP< 0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group; cP<0.05, difference with hsBAFF/rapamycin group or hsBAFF/Akt inhibitor X group.
To corroborate the functional significance of mTORC2-mediated Akt pathway in rapamycin’s intervention in hsBAFF-induced B-cell proliferation/viability, Raji cells, infected with recombinant adenoviruses encoding HA-tagged constitutively active Akt (Ad-myr-Akt), dominant negative Akt (Ad-dn-Akt) or GFP (Ad-GFP) (as control), were pretreated with/without rapamycin (100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h or 48 h. Western blotting results showed that the infection with Ad-myr-Akt and Ad-dn-Akt, but not Ad-GFP, resulted in expression of high levels of HA-tagged Akt mutants (Fig. 7A). The basal phosphorylation level of Akt was obviously raised and reduced by the infection with Ad-myr-Akt and Ad-dn-Akt, respectively, compared to the control infection with Ad-GFP (Fig. 7A and B). Importantly, ectopic expression of myr-Akt apparently elevated the basal and hsBAFF-induced phospho-Akt, survivin and proliferation/viability in the cells, and conferred resistance to the inhibitory effect of rapamycin on the events (Fig. 7A–D), whereas overexpression of dn-Akt attenuated the basal and hsBAFF-induced phospho-Akt, survivin and proliferation/viability in the cells, and potentiated the inhibitory activity of rapamycin (Fig. 7A–D). Taken together, these findings indicate that rapamycin inhibits hsBAFF-induced B-cell proliferation and survival also in part by inhibiting mTORC2-mediated Akt pathway.
Fig. 7.
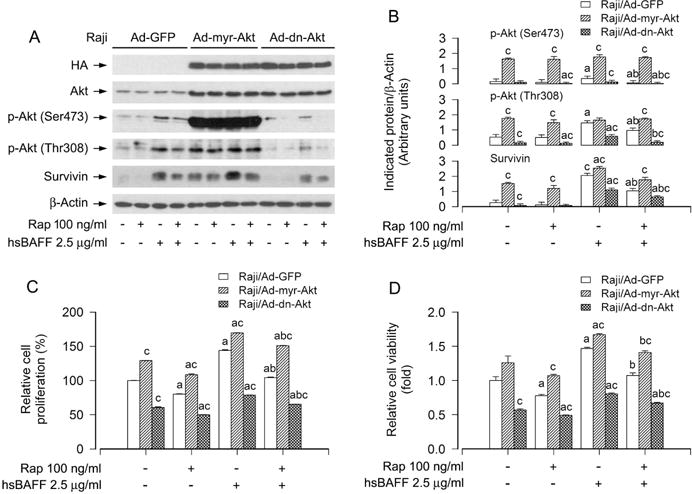
Effects of ectopic expression of constitutively active or dominant negative Akt in Raji cells on rapamycin’s prevention from hsBAFF-stimulated cell proliferation/viability. Raji cells infected with Ad-myr-Akt, Ad-dn-Akt, or Ad-GFP (for control), respectively, were pretreated with/without rapamycin (Rap, 100 ng/ml) for 2 h, followed by stimulation with/without hsBAFF (2.5 μg/ml) for 12 h (for Western blotting) or 48 h (for cell proliferation and viability assay). (A) Total cell lysates were subjected to Western blotting using indicated antibodies. The blots were probed for β-actin as a loading control. Similar results were observed in at least three independent experiments. (B) The blots for p-Akt (Ser473), p-Akt (Thr308), and survivin were semi-quantified. (C) The cell proliferation was evaluated by cell counting. (D) The cell viability was determined by the MTS assay. Results are presented as mean ± SEM (n = 3–5). aP< 0.05, difference with control group; bP<0.05, difference with 2.5 μg/ml hsBAFF group; cP<0.05, Ad-myr-Akt group or Ad-dn-Akt group vs Ad-GFP group.
Discussion
BAFF is critical for B cell transformation, maturation and survival, and excessive BAFF is linked to aggressive/neoplastic B-cell disorders (e.g. SLE, RA, SS) (Mackay et al., 2007; Pers et al., 2005; Sanz and Lee, 2010). It has been described that excessive endogenous or transgenic BAFF in mice prolongs the life span and increases the population of peripheral B lymphocytes (Moisini and Davidson, 2009; Sanz and Lee, 2010). The SLE patients suffer from a great quantity of superfluous autoantibodies secreted from aggressive B cells, due to excessive BAFF (Kaneko et al., 2014; Moisini and Davidson, 2009; Sanz and Lee, 2010). These data point to an important role of excess BAFF-evoked aggressive or neoplastic B-cell disorders in the pathophysiology of autoimmune diseases (Cornec et al., 2012; Ramanujam and Davidson, 2008). Unfortunately, so far we have no effective intervention to prevent the development of the BAFF-induced disorders. Therefore, it is of great importance to find a novel therapeutic target and strategy to control BAFF-promoted B-cell expansion. Rapamycin, a macrocyclic lactone, is a well-known specific mTOR inhibitor (Zhou et al., 2010). A series of studies have recently shown that rapamycin can prevent development of murine lupus (Lui et al., 2008) and mitigate symptoms of patients with juvenile rheumatoid arthritis (Foroncewicz et al., 2005). Especially, rapamycin can provide effective therapeutic benefit in patients with certain autoimmune diseases (Blachly and Baiocchi, 2014; Eyre et al., 2014; Mohindra et al., 2014). In view of BAFF’s close relationship with aggressive/neoplastic B-cell disorders associated with autoimmune diseases, in this study, we focused on studying how rapamycin acts as a pharmacological agent for fighting excessive BAFF-evoked aggressive B-cell proliferation and survival.
mTOR is a central modulator of cell growth, proliferation and survival (Cornu et al., 2013; Laplante and Sabatini, 2012). In mammalian cells, mTOR is a catalytic subunit of two structurally and functionally distinct complexes, mTORC1 and mTORC2 (Cornu et al., 2013; Laplante and Sabatini, 2012). mTORC1 and mTORC2 were originally found to be rapamycin-sensitive and rapamycin-insensitive, respectively, on the basis of their differential sensitivity to the inhibitory effects of rapamycin (Jacinto et al., 2004; Sarbassov et al., 2004). However, further research has revealed that prolonged (> 24 h) or high-concentrated rapamycin treatment can also inhibit mTORC2 activity in a cell line dependent manner (Sarbassov et al., 2006). Our recent studies have shown that rapamycin inhibits B-cell proliferation and survival by suppressing mTOR pathway in hsBAFF-stimulated normal and neoplastic B-lymphoid cells (Ke et al., 2013; Zeng et al., 2015). However, it is unclear whether this is through targeting mTORC1 and/or mTORC2. In the current study, we found that pretreatment with 100 ng/ml of rapamycin for 2 h was able to strongly block hsBAFF-induced phosphorylation of S6K1 (Thr389), 4E-BP1 (Thr70) and Akt (Ser473) in Raji cells, Daudi cells and primary B lymphocytes (Fig. 1), suggesting that a short exposure to rapamycin may inhibit both mTORC1 andmTORC2 in hsBAFF-stimulated B cells. Furthermore, we also showed that rapamycin dramatically inhibited the basal or hsBAFF-induced expression of survivin in these B cells (Fig. 1D and E). The results suggest that rapamycin may suppress hsBAFF-stimulated survivin and cell proliferation/survival in normal and neoplastic B-lymphoid cells by blocking mTORC1 and mTORC2 signaling pathways.
In this study, we showed that rapamycin dramatically inhibited the basal and hsBAFF-induced cell proliferation/viability in Raji cells, Daudi cells and primary B lymphocytes (Fig. 1A–C). It is known that cell proliferation is associated with cell cycle progression (Malumbres and Barbacid, 2009). Multiple studies have demonstrated that rapamycin inhibits proliferation/growth of normal and malignant B cells by affecting expression of key regulatory proteins related to the G1/S cell cycle progression (Decker et al., 2003; Vaysberg et al., 2007). For instance, rapamycin induces G1 cell cycle arrest by decreasing the expression of cyclin D2, cyclin D3, and CDK4 in Epstein-Barr virus (EBV)-positive B-cell lymphomas (Vaysberg et al., 2007), and by reducing the expression of cyclin D3, cyclin E, and cyclin A in B-cell chronic lymphocytic leukemia (B-CLL) cells (Decker et al., 2003). Furthermore, it is noteworthy that rapamycin elicits apoptosis or reduces survival by decreasing the expression of Mcl-1, Bcl-2, Bcl-xL, and cFLIP, and by increasing the expression of Bax in JN-DSRCT-1 cells (Tirado et al., 2005), anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma (ALCL) cells (Vega et al., 2006) and Mantle cell lymphoma (MCL) cells (Peponi et al., 2006). Recently, our group has demonstrated that rapamycin represses the expression of CDK2, CDK4, CDK6, cyclin A, cyclin D1, and cyclin E in Raji cells, and unveiled that rapamycin inhibits hsBAFF-simulated cell proliferation and survival through down-regulating the expression of cyclin A, cyclin E, cyclin D1, CDK2, CDK4 and CDK6, and up-regulating the expression of p21 and p27 in the cells (Zeng et al., 2015). In this study, although we did not repeatedly determine whether rapamycin inhibits the expression of anti-apoptotic proteins Bcl-2, Bcl-xL, Mcl-1, and cFLIP, we observed that rapamycin substantially inhibited the basal and hsBAFF-induced expression of survivin, an anti-apoptotic protein, in Raji cells, Daudi cells and primary B lymphocytes (Fig. 1D and E). Together, based on the data from us and others, rapamycin inhibits BAFF-stimulated cell proliferation and survival, at least in part by inhibiting the activities of G1-CDKs and the expression of anti-apoptotic proteins in normal and neoplastic B-lymphoid cells.
A number of studies have described that the functions of mTORC1 and mTORC2 are substantially impacted by the complex integrity, especially their associations with raptor and rictor, respectively (Cornu et al., 2013; Jacinto et al., 2004; Kim et al., 2002; Sarbassov et al., 2004). In the present study, we found that single or double disruption of mTORC1 and mTORC2 by using lentiviral shRNAs to raptor and/or rictor attenuated hsBAFF-induced survivin expression and cell proliferation/viability in Raji cells (Fig. 2). Pretreatment of rapamycin for 2 h effectively strengthened this inhibitory effect of lentiviral shRNA to raptor or rictor on the cell proliferation/viability (Fig. 2). Also, there existed a more potent inhibition for the double raptor/rictor-silenced cells than for the single raptor or rictor-silenced cells (Fig. 2). Furthermore, PP242 (an mTORC1/2 kinase inhibitor) repressed the basal or hsBAFF-stimulated survivin expression and cell proliferation/viability in Raji cells and primary B lymphocytes more potently than rapamycin (Fig. 3). Collectively, our data support the idea that both mTORC1 and mTORC2 signaling pathways play an important role in hsBAFF-induced proliferation and survival in normal and neoplastic B-lymphoid cells.
Recently, it has been described that rapamycin inhibits mSin1 phosphorylation, which is independent of mTORC1 and mTORC2, but is possibly dependent on a new mTOR complex, which at least contains mTOR and mLST8 (Luo et al., 2015). Of interest, in this study, we also noticed that rapamycin was able to further enhance the inhibitory effect of raptor/rictor double knockdown on hsBAFF-induced proliferation and survival in Raji cells (Fig. 2C and D). The results imply that rapamycin might inhibit some BAFF-stimulated functions of mTOR, which are beyond the regulation of mTORC1 and mTORC2. In addition, our group has demonstrated that BAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway (Ke et al., 2013), BAFF activates Erk1/2 promoting cell proliferation and survival by Ca2+-CaMKII-dependent inhibition of PP2A in normal and neoplastic B-lymphoid cells (Liang et al., 2014). Especially, we have found that rapamycin inhibits BAFF-stimulated B-cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway (Zeng et al., 2015), suggesting that Ca2+ signaling may play a crucial role in rapamycin’s regulation of crosstalk among mTORC1, mTORC2, PP2A and Erk1/2. Further research is needed to address these issues.
S6K1 and 4E-BP1 are two best-characterized downstream targets of mTORC1 (Cornu et al., 2013; Laplante and Sabatini, 2012). To discern whether rapamycin inhibits hsBAFF-induced B-cell proliferation and survival via targeting mTORC1-mediated S6K1 and/or 4E-BP1/eIF4E pathways, the levels or activities of S6K1 and 4E-BP1 were individually manipulated genetically. We found that ectopic expression of S6K1-ca or downregulation of 4E-BP1 conferred resistance to rapamycin’s attenuation of hsBAFF-induced survivin expression and B-cell proliferation/viability, whereas overexpression of 4E-BP1-5A or downregulation of S6K1 potentiated the inhibitory effects of rapamycin. The findings highlight that both mTORC1-mediated S6K1 and 4E-BP1pathways are involved in rapamycin’s suppression of hsBAFF-induced B-cell proliferation and survival.
As Akt is a well characterized substrate of mTORC2 (Laplante and Sabatini, 2012), we also speculated that Akt pathway may play a critical role in rapamycin’s inhibition of hsBAFF-induced B cell proliferation and survival. For this, Akt inhibitor and genetic inhibition/activation of Akt were utilized. We found that pharmacological inhibition of Akt with Akt inhibitor X or ectopic expression of dn-Akt enhanced rapamycin’s prevention of hsBAFF-induced phosphorylation of S6K1/4E-BP1, expression of survivin and cell proliferation/viability, whereas ectopic expression of myr-Akt rendered resistance to rapamycin’s inhibition of hsBAFF-induced events in B cells (Fig. 6–7). The results confirm that rapamycin inhibits hsBAFF-induced B-cell proliferation and survival also in part by inhibiting mTORC2-mediated Akt pathway. Since mTORC2 not only regulates the phosphorylation of Akt, but also regulates the phosphorylation or activity of SGK1, PKCα, small GTPases, and focal adhesion proteins (Cornu et al., 2013; Laplante and Sabatini, 2012), more research is required to address whether any of other mTORC2-mediated signaling molecules is involved in the inhibitory effects of rapamycin on hsBAFF-induced B-cell proliferation and survival.
In conclusion, we have identified that both mTORC1 and mTORC2 participate in hsBAFF-stimulated cell proliferation and survival in normal and neoplastic B-lymphoid cells. Rapamycin inhibits both mTORC1-mediated S6K1/4E-BP1 pathways and mTORC2-mediated Akt pathway, thereby attenuating hsBAFF-induced proliferation and survival in the cells (Fig. 8). Our data underscore that rapamycin may be a potential agent for preventing excessive BAFF-evoked aggressive B-cell malignancies and autoimmune diseases.
Fig. 8.

Schematic model of the preventive effect of rapamycin on hsBAFF-evoked B-cell proliferation and survival. Rapamycin prevents B cells from hsBAFF-promoted proliferation/survival, not only by targeting mTORC1-mediated S6K1/4E-BP1 pathways, but also via targeting mTORC2-mediated Akt pathway.
Acknowledgments
This work was supported in part by the grants from National Natural Science Foundation of China (No.31172083; LC), NIH (CA115414; SH), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010; LC), American Cancer Society (RSG-08-135-01-CNE; SH), Louisiana Board of Regents (NSF-2009-PFUND-144; SH), and Innovative Research Program of Jiangsu College Graduate of China (No. KYLX_0714; QZ).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
Literature Cited
- Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1:27–36. doi: 10.1007/s12154-008-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batten M, Groom J, Cachero TG, Qian F, Schneider P, Tschopp J, Browning JL, Mackay F. BAFF mediates survival of peripheral immature B lymphocytes. J Exp Med. 2000;192:1453–1466. doi: 10.1084/jem.192.10.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blachly JS, Baiocchi RA. Targeting PI3-kinase (PI3K), AKT and mTOR axis in lymphoma. Br J Haematol. 2014;167:19–32. doi: 10.1111/bjh.13065. [DOI] [PubMed] [Google Scholar]
- Bosello S, Youinou P, Daridon C, Tolusso B, Bendaoud B, Pietrapertosa D, Morelli A, Ferraccioli G. Concentrations of BAFF correlate with autoantibody levels, clinical disease activity, and response to treatment in early rheumatoid arthritis. J Rheumatol. 2008;35:1256–1264. [PubMed] [Google Scholar]
- Cao P, Mei JJ, Diao ZY, Zhang S. Expression, refolding, and characterization of human soluble BAFF synthesized in Escherichia coli. Protein Expr Purif. 2005;41:199–206. doi: 10.1016/j.pep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S. Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells. Lab Invest. 2010;90:762–773. doi: 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornec D, Devauchelle-Pensec V, Tobon GJ, Pers JO, Jousse-Joulin S, Saraux A. B cells in Sjogren’s syndrome: from pathophysiology to diagnosis and treatment. J Autoimmun. 2012;39:161–167. doi: 10.1016/j.jaut.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev. 2013;23:53–62. doi: 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Decker T, Hipp S, Ringshausen I, Bogner C, Oelsner M, Schneller F, Peschel C. Rapamycin-induced G1 arrest in cycling B-CLL cells is associated with reduced expression of cyclin D3, cyclin E, cyclin A, and survivin. Blood. 2003;101:278–285. doi: 10.1182/blood-2002-01-0189. [DOI] [PubMed] [Google Scholar]
- Eyre TA, Collins GP, Goldstone AH, Cwynarski K. Time now to TORC the TORC? New developments in mTOR pathway inhibition in lymphoid malignancies. Br J Haematol. 2014;166:336–351. doi: 10.1111/bjh.12945. [DOI] [PubMed] [Google Scholar]
- Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discov Med. 2010;9:173–178. [PMC free article] [PubMed] [Google Scholar]
- Foroncewicz B, Mucha K, Paczek L, Chmura A, Rowinski W. Efficacy of rapamycin in patient with juvenile rheumatoid arthritis. Transpl Int. 2005;18:366–368. doi: 10.1111/j.1432-2277.2004.00070.x. [DOI] [PubMed] [Google Scholar]
- Fu L, Lin-Lee YC, Pham LV, Tamayo AT, Yoshimura LC, Ford RJ. BAFF-R promotes cell proliferation and survival through interaction with IKKbeta and NF-kappaB/c-Rel in the nucleus of normal and neoplastic B-lymphoid cells. Blood. 2009;113:4627–4636. doi: 10.1182/blood-2008-10-183467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guertin DA, Stevens DM, Saitoh M, Kinkel S, Crosby K, Sheen JH, Mullholland DJ, Magnuson MA, Wu H, Sabatini DM. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell. 2009;15:148–159. doi: 10.1016/j.ccr.2008.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley T, Kovesdi D, Turner M. B-cell responses to B-cell activation factor of the TNF family (BAFF) are impaired in the absence of PI3K delta. Eur J Immunol. 2008;38:3543–3548. doi: 10.1002/eji.200838618. [DOI] [PubMed] [Google Scholar]
- Hietakangas V, Cohen SM. TOR complex 2 is needed for cell cycle progression and anchorage-independent growth of MCF7 and PC3 tumor cells. BMC Cancer. 2008;8:282. doi: 10.1186/1471-2407-8-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Amano H, Kawano S, Minowa K, Ando S, Watanabe T, Nakano S, Suzuki J, Morimoto S, Tokano Y, Takasaki Y. Increased serum concentration of BAFF/APRIL and IgA2 subclass in patients with mixed connective tissue disease complicated by interstitial lung disease. Modern rheumatology/the Japan Rheumatism Association. 2014;24:310–315. doi: 10.3109/14397595.2013.843748. [DOI] [PubMed] [Google Scholar]
- Ke Z, Liang D, Zeng Q, Ren Q, Ma H, Gui L, Chen S, Guo M, Xu Y, Gao W, Zhang S, Chen L. hsBAFF promotes proliferation and survival in cultured B lymphocytes via calcium signaling activation of mTOR pathway. Cytokine. 2013;62:310–321. doi: 10.1016/j.cyto.2013.03.011. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laragione T, Gulko PS. mTOR regulates the invasive properties of synovial fibroblasts in rheumatoid arthritis. Mol Med. 2010;16:352–358. doi: 10.2119/molmed.2010.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Zeng Q, Xu Z, Zhang H, Gui L, Xu C, Chen S, Zhang S, Huang S, Chen L. BAFF activates Erk1/2 promoting cell proliferation and survival by Ca2+-CaMKII-dependent inhibition of PP2A in normal and neoplastic B-lymphoid cells. Biochem Pharmacol. 2014;87:332–343. doi: 10.1016/j.bcp.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Chen L, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27:4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Liu L, Luo Y, Chen L, Shen T, Xu B, Chen W, Zhou H, Han X, Huang S. Rapamycin inhibits cytoskeleton reorganization and cell motility by suppressing RhoA expression and activity. J Biol Chem. 2010;285:38362–38373. doi: 10.1074/jbc.M110.141168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui SL, Yung S, Tsang R, Zhang F, Chan KW, Tam S, Chan TM. Rapamycin prevents the development of nephritis in lupus-prone NZB/W F1 mice. Lupus. 2008;17:305–313. doi: 10.1177/0961203307088289. [DOI] [PubMed] [Google Scholar]
- Luo Y, Liu L, Wu Y, Singh K, Su B, Zhang N, Liu X, Shen Y, Huang S. Rapamycin inhibits mSin1 phosphorylation independently of mTORC1 and mTORC2. Oncotarget. 2015;6:4286–4298. doi: 10.18632/oncotarget.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay F, Ambrose C. The TNF family members BAFF and APRIL: the growing complexity. Cytokine Growth Factor Rev. 2003;14:311–324. doi: 10.1016/s1359-6101(03)00023-6. [DOI] [PubMed] [Google Scholar]
- Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- Mackay F, Silveira PA, Brink R. B cells and the BAFF/APRIL axis: fast-forward on autoimmunity and signaling. Curr Opin Immunol. 2007;19:327–336. doi: 10.1016/j.coi.2007.04.008. [DOI] [PubMed] [Google Scholar]
- Malemud CJ. Intracellular Signaling Pathways in Rheumatoid Arthritis. J Clin Cell Immunol. 2013;4:160. doi: 10.4172/2155-9899.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M. Cell cycle, CDKs and cancer: a changing paradigm. Nature reviews. 2009;9:153–166. doi: 10.1038/nrc2602. [DOI] [PubMed] [Google Scholar]
- Mohindra NA, Giles FJ, Platanias LC. Use of mTOR inhibitors in the treatment of malignancies. Expert Opin Pharmacother. 2014;15:979–990. doi: 10.1517/14656566.2014.899582. [DOI] [PubMed] [Google Scholar]
- Moisini I, Davidson A. BAFF: a local and systemic target in autoimmune diseases. Clin Exp Immunol. 2009;158:155–163. doi: 10.1111/j.1365-2249.2009.04007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore PA, Belvedere O, Orr A, Pieri K, LaFleur DW, Feng P, Soppet D, Charters M, Gentz R, Parmelee D, Li Y, Galperina O, Giri J, Roschke V, Nardelli B, Carrell J, Sosnovtseva S, Greenfield W, Ruben SM, Olsen HS, Fikes J, Hilbert DM. BLyS: member of the tumor necrosis factor family and B lymphocyte stimulator. Science. 1999;285:260–263. doi: 10.1126/science.285.5425.260. [DOI] [PubMed] [Google Scholar]
- Mothe-Satney I, Yang D, Fadden P, Haystead TA, Lawrence JC., Jr Multiple mechanisms control phosphorylation of PHAS-I in five (S/T)P sites that govern translational repression. Mol Cell Biol. 2000;20:3558–3567. doi: 10.1128/mcb.20.10.3558-3567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller CG, Boix C, Kwan WH, Daussy C, Fournier E, Fridman WH, Molina TJ. Critical role of monocytes to support normal B cell and diffuse large B cell lymphoma survival and proliferation. J Leukoc Biol. 2007;82:567–575. doi: 10.1189/jlb.0706481. [DOI] [PubMed] [Google Scholar]
- Peponi E, Drakos E, Reyes G, Leventaki V, Rassidakis GZ, Medeiros LJ. Activation of mammalian target of rapamycin signaling promotes cell cycle progression and protects cells from apoptosis in mantle cell lymphoma. The American journal of pathology. 2006;169:2171–2180. doi: 10.2353/ajpath.2006.051078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pers JO, Daridon C, Devauchelle V, Jousse S, Saraux A, Jamin C, Youinou P. BAFF overexpression is associated with autoantibody production in autoimmune diseases. Ann N Y Acad Sci. 2005;1050:34–39. doi: 10.1196/annals.1313.004. [DOI] [PubMed] [Google Scholar]
- Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell. 2009;137:873–886. doi: 10.1016/j.cell.2009.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanujam M, Davidson A. BAFF blockade for systemic lupus erythematosus: will the promise be fulfilled? Immunol Rev. 2008;223:156–174. doi: 10.1111/j.1600-065X.2008.00625.x. [DOI] [PubMed] [Google Scholar]
- Sanz I, Lee FE. B cells as therapeutic targets in SLE. Nat Rev Rheumatol. 2010;6:326–337. doi: 10.1038/nrrheum.2010.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Derudder E, Hobeika E, Pelanda R, Reth M, Rajewsky K, Schmidt-Supprian M. Canonical NF-kappaB activity, dispensable for B cell development, replaces BAFF-receptor signals and promotes B cell proliferation upon activation. Immunity. 2006;24:729–739. doi: 10.1016/j.immuni.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189(11):1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider P, Tschopp J. BAFF and the regulation of B cell survival. Immunol Lett. 2003;88:57–62. doi: 10.1016/s0165-2478(03)00050-6. [DOI] [PubMed] [Google Scholar]
- Schweighoffer E, Vanes L, Nys J, Cantrell D, McCleary S, Smithers N, Tybulewicz VL. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity. 2013;38:475–488. doi: 10.1016/j.immuni.2012.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado OM, Mateo-Lozano S, Notario V. Rapamycin induces apoptosis of JN-DSRCT-1 cells by increasing the Bax : Bcl-xL ratio through concurrent mechanisms dependent and independent of its mTOR inhibitory activity. Oncogene. 2005;24:3348–3357. doi: 10.1038/sj.onc.1208471. [DOI] [PubMed] [Google Scholar]
- Vaysberg M, Balatoni CE, Nepomuceno RR, Krams SM, Martinez OM. Rapamycin inhibits proliferation of Epstein-Barr virus-positive B-cell lymphomas through modulation of cell-cycle protein expression. Transplantation. 2007;83:1114–1121. doi: 10.1097/01.tp.0000260142.38619.9c. [DOI] [PubMed] [Google Scholar]
- Vega F, Medeiros LJ, Leventaki V, Atwell C, Cho-Vega JH, Tian L, Claret FX, Rassidakis GZ. Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer research. 2006;66:6589–6597. doi: 10.1158/0008-5472.CAN-05-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weichhart T, Saemann MD. The multiple facets of mTOR in immunity. Trends Immunol. 2009;30:218–226. doi: 10.1016/j.it.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S, Vincelette ND, Knorr KL, Almada LL, Schneider PA, Peterson KL, Flatten KS, Dai H, Pratz KW, Hess AD, Smith BD, Karp JE, Hendrickson AE, Fernandez-Zapico ME, Kaufmann SH. 4EBP1/c-MYC/PUMA and NF-kappaB/EGR1/BIM pathways underlie cytotoxicity of mTOR dual inhibitors in malignant lymphoid cells. Blood. 2016;127:2711–2722. doi: 10.1182/blood-2015-02-629485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H, Chi H. mTOR and lymphocyte metabolism. Curr Opin Immunol. 2013;25:347–355. doi: 10.1016/j.coi.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Q, Zhang H, Qin J, Xu Z, Gui L, Liu B, Liu C, Xu C, Liu W, Zhang S, Huang S, Chen L. Rapamycin inhibits BAFF-stimulated cell proliferation and survival by suppressing mTOR-mediated PP2A-Erk1/2 signaling pathway in normal and neoplastic B-lymphoid cells. Cell Mol Life Sci. 2015;72:4867–4884. doi: 10.1007/s00018-015-1976-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Roschke V, Baker KP, Wang Z, Alarcon GS, Fessler BJ, Bastian H, Kimberly RP, Zhou T. Cutting edge: a role for B lymphocyte stimulator in systemic lupus erythematosus. J Immunol. 2001;166:6–10. doi: 10.4049/jimmunol.166.1.6. [DOI] [PubMed] [Google Scholar]
- Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem. 2010;10:571–581. doi: 10.2174/187152010793498663. [DOI] [PMC free article] [PubMed] [Google Scholar]