

Abstract
Objective
Circulating cardiac troponin has been associated with adverse prognosis in the acute respiratory distress syndrome (ARDS) in small and single center studies, however comprehensive studies of myocardial injury in ARDS using modern high-sensitivity troponin (hsTn) assays, which can detect troponin at much lower circulating concentrations, have not been performed.
Design
We performed a prospective cohort study.
Setting
We included patients enrolled in previously completed trials of ARDS.
Patients
1,057 ARDS patients were included.
Interventions
To determine the association of circulating hsTnI (Abbott ARCHITECT), with ARDS outcomes, we measured hsTnI within 24 h of intubation. The primary outcome was 60-day mortality.
Measurements and Main Results
Detectable hsTnI was present in 94% of patients; 38% of patients had detectable levels below the 99th percentile of a healthy reference population (26 ng/L), while 56% of patients had levels above the 99th percentile cut point. After multivariable adjustment, age, cause of ARDS, temperature, heart rate, vasopressor use, SOFA score, creatinine, and pCO2 were associated with higher hsTnI concentration. After adjustment for age, sex, and randomized trial assignment, the hazard ratio for 60-day mortality comparing the 5th to the 1st quintiles of hsTnI was 1.61 (95% CI 1.11 – 2.32; P-trend = 0.003). Adjusting for SOFA score suggested that this association was not independent of disease severity (hazard ratio 0.95 (95% CI 0.64 – 1.39; P = 0.93).
Conclusions
Circulating troponin is detectable in over 90% of patients with ARDS and is associated with degree of critical illness. The magnitude of myocardial injury correlated with mortality.
Keywords: myocardial injury, cardiac, respiratory failure
Introduction
The acute respiratory distress syndrome (ARDS), a common cause of acute respiratory failure, is associated with high rates of morbidity and mortality (1, 2). Cardiac involvement, manifested by hemodynamic (3) and echocardiographic (4) evidence of right heart dysfunction, has emerged as an important predictor of outcome in ARDS which may be amenable to targeted therapy (5). High-sensitivity assays for cardiac troponin (hsTn) detect myocardial injury with very high analytical sensitivity (6), and myocardial injury even at these low levels has prognostic significance (7). While elevated troponin levels have been associated with adverse prognosis in ARDS, the evidence is limited to small or single center studies using conventional rather than hsTn assays (8, 9). The prognostic relevance of very low concentrations of circulating troponin assessed by hsTn assays in ARDS has not been established (7, 10, 11), however use of these assays could identify many more ARDS patients manifesting myocardial injury and facilitate investigation into mechanism of myocardial injury in ARDS. Consequently, we performed a multicenter cohort study to determine the association of hsTn with mortality, ICU free days, and ventilator free days in ARDS. We hypothesized that higher hsTn values would be associated with worse ARDS outcomes. We also investigated which clinical factors, such as co-morbidities and treatments, were associated with hsTn concentration in ARDS.
Methods
Patient population
Our study population included patients from two NHLBI-sponsored ARDS Network trials, the ALVEOLI trial (12) of higher versus lower positive end-expiratory pressure (PEEP) and the Fluid and Catheter Treatment Trial (FACTT) (13, 14) of fluid management and hemodynamic monitoring strategies. The inclusion criteria and ARDS definition for both trials were identical and included a ratio of partial pressure of oxygen (PaO2) to fraction of inspired oxygen (fiO2) less than 300, bilateral pulmonary infiltrates, and no clinical evidence of left atrial hypertension. Both trials excluded patients with acute myocardial infarction and those patients with severe chronic pulmonary or neuromuscular disease. There were very few patients with chronic heart failure enrolled in FACTT- 27 of 1000 patients were noted to have a history of prior heart failure. The ALVEOLI trial data set did not report information regarding prior episodes of heart failure. Patients on hemodialysis were excluded from the FACTT trial, and no comment is made regarding hemodialysis status in the ALVEOLI trial dataset. The ALVEOLI trial, conducted in 23 ICUs, randomized 549 subjects with ARDS already receiving low tidal volume ventilation to receive treatment with a lower or a higher positive end-expiratory pressure (PEEP) strategy. FACTT randomized 1000 subjects with ARDS from 20 ICUs in a 2×2 design to a liberal or conservative fluid management strategy and to fluid management strategies guided by a pulmonary artery catheter or central venous catheter placement (13, 14).
We obtained plasma samples via the NIH Biologic Specimen and Data Repository Information Coordinating Center (BioLINCC) (15). Samples were drawn within the first 24 hours of study randomization (day 0). Our study cohort consisted of all patients in both trials who had plasma for analysis and who were intubated within the 24 hours prior to trial enrollment. We excluded patients intubated 24 hours or more before trial enrollment and one patient from the ALVEOLI trial with missing outcome information (Supplemental Figure). The final sample size was 1,057 participants (417 from ALVEOLI and 640 from FACTT.
The FACTT and ALVEOLI studies were approved by all participating Institutional Review Boards (12-14). The Johns Hopkins Hospital Institutional Review Board exempted our study, given that all study data was publicly available from the BIOLINCC program (15), and that the data sets contained no protected health information.
High sensitivity troponin I (hsTnI) measurements
We measured cardiac troponin at Johns Hopkins Hospital in EDTA-anticoagulated plasma using the ARCHITECT STAT hsTnI assay (Abbott Laboratories). The applied limit of detection (LoD) was 2 ng/L. For subjects with undetectable levels (hsTnI <LoD; n = 64), the value was set at 1.3 ng/L for analysis. The upper reference limit, corresponding to the 99th percentile value of a healthy reference population, was 26 ng/L (11, 16). The number of patients with troponin levels greater than 99th percentile reference using sex-specific cutpoints of 16 ng/L for women and 34 ng/L (17) is also reported.
Study outcomes and covariates
Our primary outcome was in hospital death prior to 60 days. Secondary outcomes included ventilator-free days, defined as the number of days within 30 days of trial enrollment that the patient was not receiving mechanical ventilation, and ICU-free days, defined as the number of days within the first 30 days of trial enrollment that the patient was not in the ICU. The methods for covariate assessment including demographic and clinical and laboratory data have been described in detail elsewhere (12-14). The Sequential Organ Failure Assessment (SOFA) score was calculated as a marker of global illness severity (18), and the Murray Lung Injury Score as a marker of ARDS severity (19).
Statistical analysis
While information for the main exposure variable- hsTnI levels- and for all study outcomes was complete, information on some covariates was missing in some participants (Supplemental Table 1). We thus used multiple imputation to obtain unbiased estimates of the association between hsTnI and study outcomes taking into account missing data. Multiple imputation was performed using chained equations with 50 imputed data sets. The results with and without multiple imputation were similar. All results in the paper used multiple imputation methods.
For the main analysis, hsTnI was categorized in quintiles based on the overall sample distribution. We estimated hazard ratios (HR) with 95% confidence intervals (CI) for 60-day mortality (primary outcome) comparing the 4 highest quintiles of hsTnI to the lowest quintile using Cox proportional hazards models. The proportional hazards assumption was met in all cases. For the secondary outcomes, we estimated average differences with 95% CI in ventilator-free days and ICU-free days comparing the 4 highest quintiles of hsTnI to the lowest quintile using multiple linear regression. For analyses of the primary and secondary outcomes, we used 3 models with progressive degrees of adjustment. Model 1 included age, sex, and randomized trial assignment. Model 2 further adjusted for SOFA score. Model 3 further adjusted for use of vasopressors and heart rate.
As secondary analyses, we used hsTnI as a loge-transformed continuous variable and calculated the HR for mortality and the average difference in ventilator-free days and ICU-free days comparing participants at the 90th vs. the 10th percentiles of the hsTnI distribution (804.7 and 2.8 ng/L, respectively). For sensitivity analyses, we also grouped hsTnI into assay defined clinical categories as <2 ng/L (undetectable), 2 – <26 ng/L (hsTn present but less than 99% reference value), 26 – <130 ng/L (“positive” hsTn), and ≥130 ng/L (>5 times the 99% cut-point), and the findings were similar to the quintile based analysis (not shown).
To evaluate determinants of hsTnI at day 0, we used multiple linear regression with loge-transformed hsTnI as dependent variable. The final model included variables with p<0.10 in univariate analyses. Non-linear associations were evaluated using restricted cubic splines with 3 knots at the 5th, 50th, and 95th percentiles of the predictor variables.
A two-tailed p value <0.05 was considered statistically significant. Statistical analyses were performed using Stata version 14.0 (StataCorp Inc, College Station, TX).
Results
Clinical Correlates of hsTn
HsTnI was detected in 94% of the cohort (Figure 1); 38% of the cohort had detectable hsTnI levels but below the 99th percentile of a healthy reference population, while 56% of patients had levels above the 99th percentile upper reference limit (URL). Using sex-specific cutoffs, 59.4% of the cohort had levels above the 99th percentile cut point. ARDS patients with higher hsTnI levels were older, had higher average heart rate, temperature, tidal volume, serum creatinine, and SOFA scores, and were more likely be non-White and have been treated with vasopressors. The primary cause of ARDS was more likely to be sepsis, transfusion, or trauma than aspiration or pneumonia (Table 1). Lower pH and lower pCO2 were also associated with higher hsTnI levels. Serum creatinine concentration, heart rate, and SOFA score had the strongest univariate associations with higher hsTnI levels (Table 2). In contrast, mean arterial pressure and ventilator variables such as PEEP, plateau pressure, and tidal volume were not associated with hsTnI levels after adjustment. After multivariable adjustment, age, cause of ARDS, temperature, heart rate, vasopressor use, SOFA score, creatinine, and pCO2 remained associated with higher hsTnI concentration.
Figure 1.
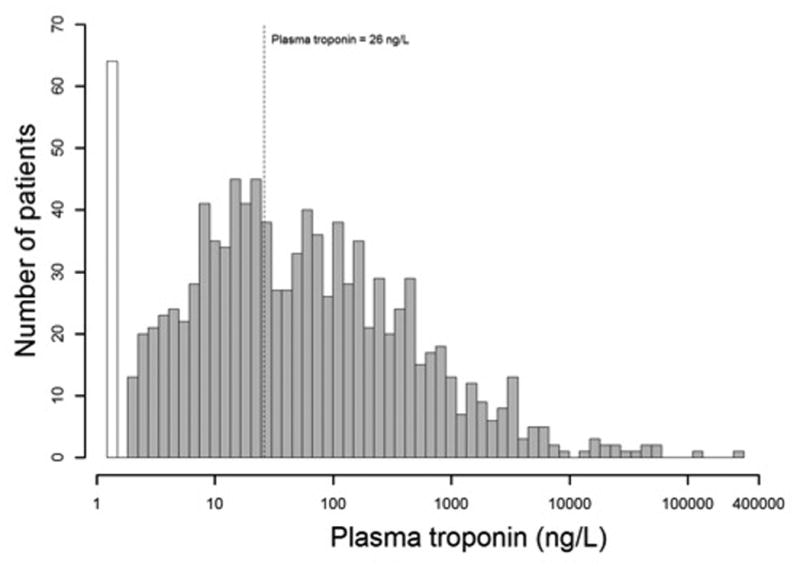
Distribution of high sensitivity troponin I levels in study population.The white bar represents 6% of the cohort with no detectable high sensitivity troponin I levels at the day of intubation (day 0, baseline). The dotted line represents a high sensitivity troponin I level of 26 ng/L, corresponding to the 99th percentile reference value of a healthy population.
Table 1.
Characteristics of study participants by quintile of high sensitivity troponin I levels at day of intubation (day 0, baseline).
| Characteristic | Overall | Quintile of HS-troponin (range, ng/L) | P value | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| First (1.3 – 7.2) | Second (7.2 – 20.9) | Third (20.9 – 71.9) | Fourth (71.9 – 296.5) | Fifth (>296.5) | |||
| Number | 1,057 | 211 | 211 | 212 | 211 | 212 | |
| HS-troponin, ng/L | 1073.5 | 3.3 | 13.1 | 41.3 | 152.7 | 5142.6 | |
| Age, years | 50.4 | 46.6 | 51.4 | 51.3 | 51.0 | 51.6 | <0.001 |
| Ethnicity, % | 0.01 | ||||||
| White | 68.9 | 74.4 | 69.7 | 66.5 | 74.4 | 59.4 | |
| Black | 18.8 | 11.9 | 19.0 | 20.8 | 17.5 | 25.0 | |
| Other | 12.3 | 13.7 | 11.4 | 12.7 | 8.1 | 15.6 | |
| Male sex, % | 51.9 | 53.6 | 48.8 | 52.8 | 47.9 | 56.6 | 0.36 |
| Primary cause of ARDS, % | 0.001 | ||||||
| Aspiration | 16.0 | 14.2 | 15.6 | 20.3 | 12.3 | 17.0 | |
| Pneumonia | 47.8 | 56.4 | 55.0 | 44.3 | 45.5 | 38.2 | |
| Sepsis | 22.2 | 15.2 | 17.1 | 25.0 | 22.8 | 31.1 | |
| Transfusion | 1.8 | 1.4 | 1.4 | 0.0 | 3.3 | 2.8 | |
| Trauma | 4.2 | 1.9 | 2.4 | 4.2 | 8.1 | 4.2 | |
| Other | 8.0 | 10.9 | 8.5 | 6.1 | 8.1 | 6.6 | |
| Temperature, °C | 37.5 | 37.3 | 37.6 | 37.6 | 37.6 | 37.7 | 0.002 |
| Heart rate, beats per min | 102.0 | 96.8 | 100.6 | 102.0 | 102.7 | 107.8 | <0.001 |
| Systolic BP, mmHg | 112.6 | 113.4 | 114.1 | 113.6 | 111.6 | 110.2 | 0.28 |
| Diastolic BP, mmHg | 59.0 | 58.5 | 58.4 | 60.0 | 59.2 | 58.8 | 0.66 |
| Vasopressor use, % | 32.2 | 19.4 | 28.0 | 34.4 | 35.0 | 44.3 | <0.001 |
| Height, cm | 169.5 | 168.7 | 169.1 | 169.8 | 169.8 | 170.3 | 0.59 |
| Weight, kg | 79.9 | 78.9 | 78.9 | 81.6 | 80.2 | 79.3 | 0.68 |
| SOFA score | 7.7 | 6.6 | 7.3 | 7.7 | 7.9 | 8.4 | <0.001 |
| Tidal volume, mL | 477.5 | 466.2 | 480.0 | 474.0 | 473.0 | 491.4 | 0.21 |
| PEEP, cm H2O | 9.47 | 9.16 | 9.16 | 9.52 | 9.78 | 9.67 | 0.37 |
| Plateau pressure, cm H2O | 26.2 | 25.5 | 26.9 | 25.9 | 25.9 | 26.7 | 0.28 |
| FiO2, | 0.65 | 0.64 | 0.66 | 0.65 | 0.65 | 0.67 | 0.64 |
| PaO2/FiO2 ratio, | 149.0 | 151.3 | 146.5 | 142.8 | 152.3 | 155.3 | 0.37 |
| Lung compliance, mL/cmH2 | 31.0 | 32.8 | 30.6 | 34.1 | 33.8 | 31.9 | 0.33 |
| Murray Lung Injury score | 2.87 | 2.90 | 2.98 | 2.78 | 2.79 | 2.84 | 0.11 |
| pH | 7.36 | 7.39 | 7.36 | 7.36 | 7.36 | 7.35 | 0.003 |
| pCO2, mmHg | 39.6 | 40.9 | 40.6 | 39.0 | 39.2 | 38.3 | 0.04 |
| pO2, mmHg | 90.3 | 91.0 | 86.9 | 86.2 | 90.1 | 96.0 | 0.13 |
| Creatinine, mg/dL | 1.52 | 1.08 | 1.46 | 1.54 | 1.54 | 1.97 | <0.001 |
Values in the Table were calculated using multiple imputation to take missing data into account (see Supplemental Table 1 for description of data available for each variable).
Table 2.
Determinants of high sensitivity troponin I levels at day of intubation (day 0, baseline).
| Characteristic | Univariable models | Multivariable models* | ||
|---|---|---|---|---|
|
| ||||
| Ratio of geometric means (95% confidence interval) | P-value | Ratio of geometric means (95% confidence interval) | P-value | |
| Age† | 1.85 (1.31 – 2.61) | <0.001 | 2.39 (1.68 – 3.42) | <0.001 |
| Ethnicity | 0.01 | 0.019 | ||
| White | 1.00 (reference) | 1.00 (reference) | ||
| Black | 1.69 (1.20 – 2.39) | 1.59 (1.15 – 2.20) | ||
| Other | 1.20 (0.79 – 1.80) | 1.07 (0.73 – 1.58) | ||
| Male sex | 1.06 (0.81 – 1.38) | 0.66 | ||
| Primary cause of ARDS | <0.001 | 0.008 | ||
| Aspiration | 1.00 (reference) | 1.00 (reference) | ||
| Pneumonia | 0.68 (0.47 – 0.99) | 0.74 (0.51 – 1.05) | ||
| Sepsis | 1.61 (1.05 – 2.47) | 1.08 (0.72 – 1.64) | ||
| Transfusion | 2.56 (0.92 – 7.15) | 1.89 (0.71 – 5.00) | ||
| Trauma | 1.70 (0.83 – 3.49) | 1.83 (0.92 – 3.61) | ||
| Other | 0.62 (0.36 – 1.10) | 0.67 (0.39 – 1.15) | ||
| Temperature | 1.83 (1.31 – 2.56) | <0.001 | 1.60 (1.14 – 2.24) | 0.007 |
| Heart rate | 2.60 (1.86 – 3.64) | <0.001 | 2.34 (1.61 – 3.41) | <0.001 |
| Mean arterial pressure† | 0.88 (0.64 – 1.22) | 0.45 | ||
| Vasopressor use (yes vs. no) | 2.35 (1,78 – 3.11) | <0.001 | 1.39 (1.03 – 1.86) | 0.029 |
| Height | 1.27 (0.92 – 1.76) | 0.15 | ||
| Weight | 1.10 (0.79 – 1.55) | 0.56 | ||
| SOFA score | 3.38 (2.37 – 4.82) | <0.001 | 1.57 (1.01 – 2.45) | 0.047 |
| Tidal volume | 1.48 (1.09 – 2.01) | 0.01 | 0.97 (0.71 – 1.32) | 0.85 |
| PEEP | 1.24 (0.89 – 1.72) | 0.20 | ||
| Plateau pressure | 1.12 (0.78 – 1.61) | 0.54 | ||
| FiO2 | 1.25 (0.85 – 1.83) | 0.26 | ||
| PaO2/FiO2 ratio† | 1.05 (0.74 – 1.49) | 0.08 | 1.15 (0.74 – 1.80) | 0.63 |
| Lung compliance | 1.04 (0.80 – 1.35) | 0.78 | ||
| Murray Lung Injury score | 0.74 (0.51 – 1.09) | 0.12 | ||
| pH† | 0.71 (0.51 – 0.99) | 0.02 | 1.02 (0.68 – 1.54) | 0.54 |
| pCO2† | 0.47 (0.34 – 0.66) | <0.001 | 0.63 (0.45 – 0.88) | 0.007 |
| pO2 | 1.23 (0.99 – 1.54) | 0.06 | 1.24 (0.93 – 1.64) | 0.14 |
| Creatinine† | 5.34 (3.82 – 7.46) | <0.001 | 2.59 (1.73 – 3.89) | <0.001 |
Values in the Table are ratios of geometric means calculated from linear regression models with log-high sensitivity troponin I levels as outcome. For categorical variables, the ratios are compared with respect to the reference category. For continuous variables, the ratios compare the 90th to the 10th percentiles of the predictor variable in the overall study sample. The 90th and 10th percentiles were, for age, 73 and 30 years; for temperature, 38.8 and 36.3 °C; for heart rate, 128 and 75 beats per minute; for mean arteria pressure, 94.3 and 61.7 mmHg; for height, 182.9 and 157.0 cm; for weight, 109.1 and 55.7 kg; for SOFA score, 11 and 4 points; for tidal volume, 600 and 350 cc/kg IBW; for PEEP, 15 and 5 cm H2O; for plateau pressure, 35 and 18 cm H2O; for FiO2, 1.0 and 0.4; for PaO2/FiO2 ratio, 237.3 and 73.5; for lung compliance, 48 and 17.9 mL/cmH2O; for Murray Lung Injury score, 4 and 2 points; for pH, 7.47 and 7.23; for pCO2, 51 and 29 mmHg; for pO2, 128 and 60 mmHg; for creatinine, 2.8 and 0.6 mg/dL.
Values in the Table were calculated using multiple imputation to take missing data into account (see Supplemental Table 1 for description of data available for each variable).
Values in the Table were calculated using multiple linear regression with ventilator free days or ICU free days as outcome variables and multiple imputation to take missing data into account (see Supplemental Table 1 for description of data available for each variable).”
Variables were included in the multivariable model if P <0.10 in the univariable model.
Modeled as restricted cubic splines to accommodate non-linear associations (see Methods for details).
hsTnI and Mortality in ARDS
After 60-days, cumulative mortality in quintiles 1 – 5 of hsTnI at day 0 was 22.2, 26.1, 26.4, 28.4, and 34.9%, respectively (P = 0.02; Figure 2). After adjustment for age, sex, and randomized trial assignment, the HR for 60-day mortality comparing the 5th to the 1st quintiles of hsTnI was 1.61 (95% CI 1.11 – 2.32; P-trend = 0.003) (Table 3). This association, however, was not independent of the severity of underlying illness assessed by SOFA score, heart rate, and vasopressor requirement. Increasing hsTnI concentration was associated with fewer ventilator free days and ICU free days in a model adjusted for age, sex, and randomized trial assignment (Supplemental Table 2). These associations also atttenuated after adjusting for SOFA score, heart rate, and vasopressor use. Those patients with troponin levels between 2 – <26 ng/L (representing those with hsTn present but less than 99% reference value) did not fare worse than those with undetectable troponin (HR for 60 day mortality 1.17 95% CI 0.67-2.04; P = 0.6). Findings were similar among patients without sepsis as a primary cause of ARDS (HRs for 60-day mortality in model 3 for quintiles 2 – 5 of hsTnI compared to quintile 1 restricted to this subgroup were 0.78 (0.49 – 1.23), 0.73 (0.46 – 1.17), 0.74 (0.46 – 1.18), and 1.07 (0.68 – 1.67), respectively (p-trend 0.62)).
Figure 2.
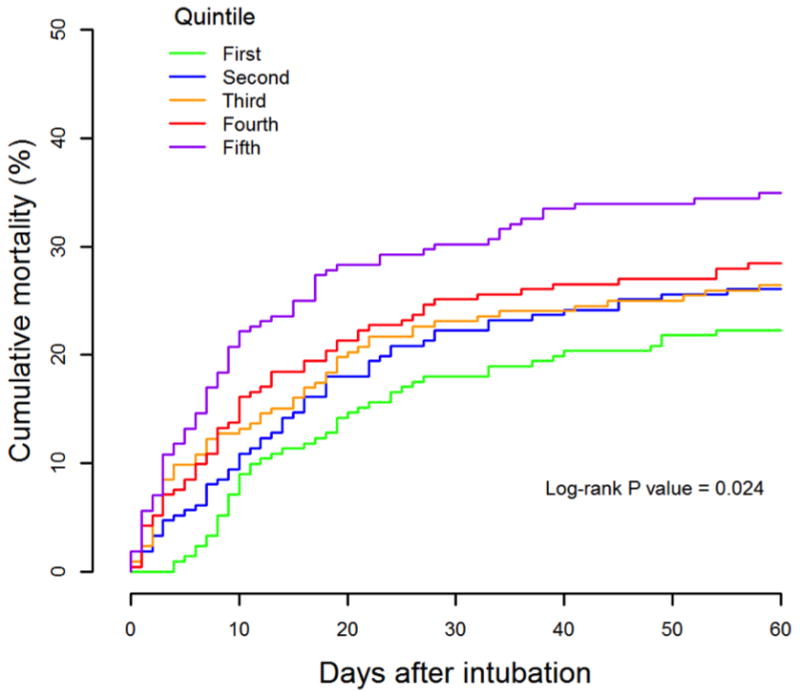
Cumulative mortality by quintile of high sensitivity troponin I levels at day the day of intubation (day 0, baseline).
Table 3.
Hazard ratios for 60-day mortality by quintile of high sensitivity troponin I levels at day of intubation (day 0, baseline).
| Model | Quintile of HS-troponin (range, ng/L) | 90th vs. 10th percentile | ||||
|---|---|---|---|---|---|---|
|
| ||||||
| First (1.3 – 7.2) | Second (7.2 – 20.9) | Third (20.9 – 71.9) | Fourth (71.9 – 296.5) | Fifth (>296.5) | ||
| Number of participants | 211 | 211 | 212 | 211 | 212 | |
| Mean HS-troponin | 3.3 | 13.1 | 41.3 | 152.7 | 5142.6 | |
| Number of deaths (%) | 47 (22.2) | 55 (26.1) | 56 (26.4) | 60 (28.4) | 74 (34.9) | |
| Model 1 | ||||||
| Hazard ratio | 1.00 | 1.07 | 1.12 | 1.24 | 1.60 | 1.57 |
| (95% confidence interval) | (reference) | (0.73 – 1.60) | (0.76 – 1.65) | (0.85 – 1.83) | (1.11 – 2.32) | (1.17 – 2.11) |
| P-trend | 0.003 | |||||
| Model 2 | ||||||
| Hazard ratio | 1.00 | 1.00 | 0.95 | 0.99 | 1.18 | 1.21 |
| (95% confidence interval) | (reference) | (0.67 – 1.49) | (0.64 – 1.40) | (0.66 – 1.46) | (0.81 – 1.73) | (0.88 – 1.65) |
| P-trend | 0.24 | |||||
| Model 3 | ||||||
| Hazard ratio | 1.00 | 0.92 | 0.80 | 0.88 | 0.94 | 1.01 |
| (95% confidence interval) | (reference) | (0.62 – 1.37) | (0.53 – 1.19) | (0.59 – 1.30) | (0.64 – 1.39) | (0.73 - 1.39) |
| P-trend | 0.94 | |||||
Values in the Table were calculated using multiple imputation to take missing data into account (see Supplemental Table 1 for description of data available for each variable).
Model 1: Adjusted for age, sex, and randomized trial assignment.
Model 2: Further adjusted for SOFA score.
Model 3: Further adjusted for use of vasopressors and heart rate.
Discussion
We present the largest multicenter cohort study assessing the prevalence and prognostic impact of circulating troponin in ARDS. We report three major findings. First, circulating hsTnI levels can be detected in almost all patients with ARDS (94% of our study population) and 56% of the population had hsTnI levels >99th percentile of a healthy reference population, suggesting that myocardial injury is highly prevalent in ARDS. Second, elevated hsTnI levels were associated with higher mortality and lower ventilator free and ICU free days, indicating worse clinical prognosis. Third, indicators of the severity of underlining critical illness such as SOFA score, heart rate, and vasopressor use, were associated with higher hsTnI levels, and adjusting for critical illness severity. After adjusting for these clinical factors, hsTn was not independently associated with outcomes in ARDS beyond these potential mechanisms of myocardial injury. Our findings provide strong evidence that myocardial damage is common in ARDS, and is associated with outcome as a function of underlying critical illness.
Prevalence of myocardial injury
With an hsTn assay, we could identify some level of myocardial injury in over 90% of ARDS patients, and hsTnI levels above the 99th percentile of a healthy reference population in 56% of participants. In two smaller single-center studies of 177 and 305 ARDS patients using conventional (non-high sensitivity) troponin assays, 67% of patients had detectable troponin levels (9) and 55% had levels >99% cut point of the conventional assay (8). Overall mortality in both these studies was greater than 40%, higher than our reported overall mortality of 22%. This may reflect a discrepancy between the illness of patients enrolled in clinical trials versus those seen in clinical practice. Using a high sensitivity assay thus seems to result in a substantial increase in the number of ARDS patients identified with myonecrosis. Using an hsTn assay, 85% of 995 patients with sepsis (of whom 70% received mechanical ventilation) had detectable troponin levels (20), which is consistent with our findings. As hsTn assays continue to gain widespread use in regions around the world, clinicians should be mindful that these assays will detect circulating troponin in nearly all patients with ARDS.
Causes of elevated troponin
Circulating troponin is associated with several syndromes including acute myocardial infarction due to intracoronary thrombus (type I myocardial infarction), oxygen supply-demand mismatch at the cardiac myocyte level with signs and symptoms of cardiac ischemia (type II myocardial infarction), and acute or chronic myocardial injury not meeting criteria for myocardial infarction (21). In the FACTT and ALVEOLI trials, patients were excluded if they had signs or symptoms of acute cardiac ischemia (12-14), thus most of the myocardial injury in our cohort most likely represents acute myocardial injury without infarction. Myocardial infarction was not an adjudicated endpoint in either trial. Indeed, increased serum troponin concentrations have been observed after rapid atrial pacing without infarction (22, 23), in the proinflammatory milieu of sepsis (20), and after increased cardiac work in stress testing (24). Potential mechanisms for circulating troponins levels in ARDS patients include myocyte necrosis in the setting of critical illness (25), as well as cellular changes without myocyte necrosis (26) including increased myocyte permeability, membrane blebs or cell release of troponin degradation products (22).
Factors associated with increased troponin in our study included markers of other organ dysfunction such as creatinine and SOFA score, indices of ventilation such as pH and pCO2, as well as heart rate and body temperature. Elevated troponin in ARDS was also associated with more tricuspid regurgitation and regional wall motion abnormalities on echocardiography (9) in other studies. Given that hemodynamic and echocardiographic right ventricular function is associated with outcome in ARDS (3, 4, 27), the cause-effect relationship between myocardial injury and right ventricular function in ARDS should be further studied.
Troponin and outcomes
In our study, circulating hsTnI levels were associated with ARDS outcome in minimally adjusted models and the association was no longer present after adjusting for underlying critical illness. This contrasts with a prior prospective single center study of 177 ARDS patients in which troponin was independently associated with mortality adjusting for APACHE score (9). In another small single center study, troponin levels were independently associated with mortality in ARDS, but the association was not present among subgroups of patients with higher APACHE scores and among those with higher age (8). Several factors could account for the observed differences between our results and studies reporting an independent association of troponin level with ARDS outcomes, including single center versus multi-center study design, different enrollment criteria or patient profile in ARDS Network studies (12-14) compared to ARDS patients in clinical practice (1), and possible selection bias in prior trials. Finally, our results in ARDS were similar to a study of elevated troponin levels in patients with severe sepsis (20), in which troponin levels were no longer predictive of outcomes after adjustment for measures of illness severity. It is possible that troponin elevation manifests as a result of critical illness through several potential mechanisms. Whether targeting treatment to reduce troponin elevation could improve clinical outcomes requires further study.
Therapeutic implications
Whether treatment aimed at reducing myocardial injury would improve outcomes is not known- myocardial injury could be a treatable manifestation of underlying critical illness. Esmolol to control heart rate in sepsis was associated with reduced mortality (28), but it is unclear if this effect was mediated through reduced myocardial injury. Propranolol in severely burned children reduced heart rate and improved catabolism (29). In animal models, β-agonism was associated with increased lipopolysaccharide-induced myocyte necrosis (30) and β-blockade improved cardiac function (28), but the clinical implications of these experimental findings are unclear. Treatments targeted at alleviating right ventricular afterload and reducing right ventricular strain in ARDS are also appealing (27).
Limitations of our study include observational design, although the parent trials and endpoints were determined prospectively. Not every enrolled patient in FACTT and ALVEOLI had available plasma at the time of intubation. Furthermore, there was no information on electrocardiography or assessment of cardiac structure or function, and the association of these factors with troponin in ARDS would shed important light on the mechanism of injury including possible occult myocardial infarction. Serial measurements of troponin within the first 24 hours were not available, and such measurements could provide clarity on the mechanism of myocardial injury. Data on concomitant medical therapy such as with beta blockers, aspirin, and anticoagulants was not available for our cohort. Finally, the mechanism of death in ARDS was unavailable, and it is still possible that troponin levels could be predictive of deaths with a major cardiovascular component. The mode of death among ARDS patients with increased troponin levels should be assessed in future studies.
In conclusion, circulating cardiac troponin detected by a high-sensitivity assay is present in over 90% of patients. Elevated hsTnI was associated with markers of severity of critical illness and did not confer incremental information regarding survival, ventilator free days or ICU free days. Thus, the extent of myocardial injury correlates with underlying critical illness in ARDS and is another indicator of end-organ dysfunction in this syndrome Future studies should further assess the mechanisms of myocardial injury in ARDS, which could identify pathophysiologic targets to improve outcomes.
Supplementary Material
Acknowledgments
Disclosures: Abbott Laboratories provided reagents and financial support for the study; the study was designed and executed solely by the study investigators without industry involvement. Dr. Sokoll received further research funding from Abbott Laboratories. Dr. Morrow reports grants to the TIMI Study Group from Abbott Laboratories, Amgen, AstraZeneca, Daiichi Sankyo, Eisai, GlaxoSmithKline, Merck, Novartis, Roche Diagnostics, Singulex and consultant fees from Abbott Laboratories, AstraZeneca, diaDexus, GlaxoSmithKline, Merck, Peloton, Roche Diagnostics, and Verseon. Dr. Metkus performs consulting unrelated to this subject matter for BestDoctors Inc and Oakstone/EBIX. Dr. Metkus received royalties for a textbook publication for McGraw-Hill publishing, unrelated to this subject matter. From 2014-2016, Dr. Metkus received salary support from NIH-NHLBI grant number T32-HL007227-40.
Copyright form disclosure: Dr. Metkus's institution received funding from Abbott Laboratories and the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (salary support from grant number T32-HL007227-40, 2014-2016); he received funding from BestDoctors Inc (consulting), Oakstone/EBIX (consulting), and McGraw-Hill Publishing (royalties, unrelated to this subject matter); he disclosed that Abbott Laboratories provided reagents and financial support for the study, but the study was designed and executed solely by the study investigators without industry involvement; he; received support for article research from the NIH; and he disclosed off-label product use of high sensitivity troponin assays, which are not yet approved or have just recently been approved for clinical use in the United States (these assays are in routine clinical use in Europe). Dr. Sokoll's institution received funding from Abbott Laboratories, and disclosed reagent support from Abbott Laboratories. Dr. Morrow received funding from consulting for Abbott Laboratories, Roche Diagnostics, diaDexus; he reports grants to the TIMI Study Group (for studies other than the one in this article) from Abbott Laboratories, Amgen, AstraZeneca, Daiichi Sankyo, Eisai, GlaxoSmithKline, Merck, Novartis, Roche Diagnostics, and Singulex; and he received consultant fees from Abbott Laboratories, AstraZeneca, diaDexus, GlaxoSmithKline, Merck, Peloton, Roche Diagnostics, and Verseon. Dr. Tomaselli received support for article research from the NIH. Dr. Brower received funding from Applied Clinical Intelligence and Global Therapeutics. Dr. Korley's institution received funding from Abbott Laboratories, and he disclosed funding from prior consulting for Abbott Laboratories and Roche Diagnostics.
Footnotes
Author contributions: Conception and design: TM, EG, DM, RB, SS, FK; Analysis and interpretation: TM, EG, LS, DM, GT, RB, SS, FK; Drafting the manuscript for important intellectual content: TM, EG, LS, DM, GT, RB, SS, FK.
The remaining authors have disclosed that they do not have any potential conflicts of interest.
References
- 1.Bellani G, Laffey JG, Pham T, et al. Epidemiology, Patterns of Care, and Mortality for Patients With Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. Jama. 2016;315(8):788–800. doi: 10.1001/jama.2016.0291. [DOI] [PubMed] [Google Scholar]
- 2.Herridge MS, Tansey CM, Matte A, et al. Functional disability 5 years after acute respiratory distress syndrome. The New England journal of medicine. 2011;364(14):1293–1304. doi: 10.1056/NEJMoa1011802. [DOI] [PubMed] [Google Scholar]
- 3.Bull TM, Clark B, McFann K, et al. Pulmonary vascular dysfunction is associated with poor outcomes in patients with acute lung injury. American journal of respiratory and critical care medicine. 2010;182(9):1123–1128. doi: 10.1164/rccm.201002-0250OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mekontso Dessap A, Boissier F, Charron C, et al. Acute cor pulmonale during protective ventilation for acute respiratory distress syndrome: prevalence, predictors, and clinical impact. Intensive care medicine. 2016;42(5):862–870. doi: 10.1007/s00134-015-4141-2. [DOI] [PubMed] [Google Scholar]
- 5.Repesse X, Charron C, Vieillard-Baron A. Acute cor pulmonale in ARDS: rationale for protecting the right ventricle. Chest. 2015;147(1):259–265. doi: 10.1378/chest.14-0877. [DOI] [PubMed] [Google Scholar]
- 6.Sandoval Y, Smith SW, Apple FS. Present and Future of Cardiac Troponin in Clinical Practice: A Paradigm Shift to High-Sensitivity Assays. The American journal of medicine. 2016;129(4):354–365. doi: 10.1016/j.amjmed.2015.12.005. [DOI] [PubMed] [Google Scholar]
- 7.Grinstein J, Bonaca MP, Jarolim P, et al. Prognostic implications of low level cardiac troponin elevation using high-sensitivity cardiac troponin T. Clinical cardiology. 2015;38(4):230–235. doi: 10.1002/clc.22379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bajwa EK, Boyce PD, Januzzi JL, et al. Biomarker evidence of myocardial cell injury is associated with mortality in acute respiratory distress syndrome. Critical care medicine. 2007;35(11):2484–2490. doi: 10.1097/01.ccm.0000281852.36573.22. [DOI] [PubMed] [Google Scholar]
- 9.Rivara MB, Bajwa EK, Januzzi JL, et al. Prognostic significance of elevated cardiac troponin-T levels in acute respiratory distress syndrome patients. PloS one. 2012;7(7):e40515. doi: 10.1371/journal.pone.0040515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korley FK, Jaffe AS. Preparing the United States for high-sensitivity cardiac troponin assays. Journal of the American College of Cardiology. 2013;61(17):1753–1758. doi: 10.1016/j.jacc.2012.09.069. [DOI] [PubMed] [Google Scholar]
- 11.Korley FK, Schulman SP, Sokoll LJ, et al. Troponin elevations only detected with a high-sensitivity assay: clinical correlations and prognostic significance. Academic emergency medicine : official journal of the Society for Academic Emergency Medicine. 2014;21(7):727–735. doi: 10.1111/acem.12417. [DOI] [PubMed] [Google Scholar]
- 12.Brower RG, Lanken PN, MacIntyre N, et al. Higher versus lower positive end-expiratory pressures in patients with the acute respiratory distress syndrome. The New England journal of medicine. 2004;351(4):327–336. doi: 10.1056/NEJMoa032193. [DOI] [PubMed] [Google Scholar]
- 13.National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Wheeler AP, et al. Pulmonary-artery versus central venous catheter to guide treatment of acute lung injury. The New England journal of medicine. 2006;354(21):2213–2224. doi: 10.1056/NEJMoa061895. [DOI] [PubMed] [Google Scholar]
- 14.National Heart L, Blood Institute Acute Respiratory Distress Syndrome Clinical Trials N. Wiedemann HP, et al. Comparison of two fluid-management strategies in acute lung injury. The New England journal of medicine. 2006;354(24):2564–2575. doi: 10.1056/NEJMoa062200. [DOI] [PubMed] [Google Scholar]
- 15.Giffen CA, Carroll LE, Adams JT, et al. Providing Contemporary Access to Historical Biospecimen Collections: Development of the NHLBI Biologic Specimen and Data Repository Information Coordinating Center (BioLINCC) Biopreservation and biobanking. 2015;13(4):271–279. doi: 10.1089/bio.2014.0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Apple FS, Ler R, Murakami MM. Determination of 19 cardiac troponin I and T assay 99th percentile values from a common presumably healthy population. Clin Chem. 2012;58(11):1574–1581. doi: 10.1373/clinchem.2012.192716. [DOI] [PubMed] [Google Scholar]
- 17.Bohula May EA, Bonaca MP, Jarolim P, et al. Prognostic performance of a high-sensitivity cardiac troponin I assay in patients with non-ST-elevation acute coronary syndrome. Clin Chem. 2014;60(1):158–164. doi: 10.1373/clinchem.2013.206441. [DOI] [PubMed] [Google Scholar]
- 18.Vincent JL, Moreno R, Takala J, et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive care medicine. 1996;22(7):707–710. doi: 10.1007/BF01709751. [DOI] [PubMed] [Google Scholar]
- 19.Murray JF, Matthay MA, Luce JM, et al. An expanded definition of the adult respiratory distress syndrome. Am Rev Respir Dis. 1988;138(3):720–723. doi: 10.1164/ajrccm/138.3.720. [DOI] [PubMed] [Google Scholar]
- 20.Masson S, Caironi P, Fanizza C, et al. Sequential N-Terminal Pro-B-Type Natriuretic Peptide and High-Sensitivity Cardiac Troponin Measurements During Albumin Replacement in Patients With Severe Sepsis or Septic Shock. Critical care medicine. 2016;44(4):707–716. doi: 10.1097/CCM.0000000000001473. [DOI] [PubMed] [Google Scholar]
- 21.Thygesen K, Alpert JS, Jaffe AS, et al. Third universal definition of myocardial infarction. Journal of the American College of Cardiology. 2012;60(16):1581–1598. doi: 10.1016/j.jacc.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 22.White HD. Pathobiology of troponin elevations: do elevations occur with myocardial ischemia as well as necrosis? Journal of the American College of Cardiology. 2011;57(24):2406–2408. doi: 10.1016/j.jacc.2011.01.029. [DOI] [PubMed] [Google Scholar]
- 23.Turer AT, Addo TA, Martin JL, et al. Myocardial ischemia induced by rapid atrial pacing causes troponin T release detectable by a highly sensitive assay: insights from a coronary sinus sampling study. Journal of the American College of Cardiology. 2011;57(24):2398–2405. doi: 10.1016/j.jacc.2010.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabatine MS, Morrow DA, de Lemos JA, et al. Detection of acute changes in circulating troponin in the setting of transient stress test-induced myocardial ischaemia using an ultrasensitive assay: results from TIMI 35. Eur Heart J. 2009;30(2):162–169. doi: 10.1093/eurheartj/ehn504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapman AR, Adamson PD, Mills NL. Assessment and classification of patients with myocardial injury and infarction in clinical practice. Heart. 2017;103(1):10–18. doi: 10.1136/heartjnl-2016-309530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang W, Schulze CJ, Suarez-Pinzon WL, et al. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106(12):1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 27.Metkus TS, Tampakakis E, Mullin CJ, et al. Pulmonary Arterial Compliance in Acute Respiratory Distress Syndrome: Clinical Determinants and Association With Outcome From the Fluid and Catheter Treatment Trial Cohort. Crit Care Med. 2017;45(3):422–429. doi: 10.1097/CCM.0000000000002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morelli A, Ertmer C, Westphal M, et al. Effect of heart rate control with esmolol on hemodynamic and clinical outcomes in patients with septic shock: a randomized clinical trial. Jama. 2013;310(16):1683–1691. doi: 10.1001/jama.2013.278477. [DOI] [PubMed] [Google Scholar]
- 29.Herndon DN, Hart DW, Wolf SE, et al. Reversal of catabolism by beta-blockade after severe burns. The New England journal of medicine. 2001;345(17):1223–1229. doi: 10.1056/NEJMoa010342. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Wang Y, Yang D, et al. beta(1)-adrenoceptor stimulation promotes LPS-induced cardiomyocyte apoptosis through activating PKA and enhancing CaMKII and IkappaBalpha phosphorylation. Crit Care. 2015;19:76. doi: 10.1186/s13054-015-0820-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.