

Nature repeatedly repurposes, in that molecules that serve as metabolites, energy depots, or polymer subunits are at the same time used to deliver signals within and between cells. The preeminent example of this repurposing is adenosine triphosphate, which functions as a building block for nucleic acids, an energy source for enzymatic reactions, a phosphate donor to regulate intracellular signaling, and a neurotransmitter to control the activity of neurons. A series of recent studies now consolidate the view that phosphatidylserine (PtdSer), a common phospholipid constituent of membrane bilayers, is similarly repurposed for use as a signal between cells, and that the ligands and receptors of the TAM family of receptor tyrosine kinases (RTKs) are prominent transducers of this signal.
PtdSer asymmetry
PtdSer is, at first glance, a boring molecule: it is an everyday phospholipid that serves as a structural component of the plasma and secretory membranes of all eukaryotic cells [1]. (You can even buy it as a nutritional supplement at your local drugstore – caveat emptor [2].) Like phosphatidylethanolamine, phosphatidylinositol, and phosphatidylcholine, however, PtdSer has the property of being asymmetrically distributed between the inner and outer leaflets of bilayered membranes [3]. This asymmetry is especially striking for PtdSer: nearly all PtdSer is normally confined to the inner, cytoplasm-facing leaflet of the plasma membrane, and to the cytoplasm-facing leaflets of endosomal and Golgi membranes [1]. As such, its ability to act as a signaling molecule relies not on changes in its expression level, modification, or release, but rather on a regulated change in its membrane localization [4, 5]. In addition, its confinement to the membrane means that the signaling events that PtdSer mediates (see below) all involve direct cell-cell or cell-vesicle contact.
PtdSer asymmetry is established and maintained by the action of 10-transmembrane domain aminophospholipid translocases, a family of P4-type ATPases commonly referred to as ‘flippases’ [6]. There are 14 of these enzymes encoded in the human genome [6, 7], which are expressed in different tissues and in different membrane compartments (e.g., plasma membrane, Golgi, lysosomes) within the cell [7]. A subset of flippases, including ATP11C, are inactivated by caspase cleavage during apoptosis [8, 9].
Scramblases and PtdSer externalization
PtdSer can only signal between cells if it displayed on the outer leaflet of the plasma membrane, but established PtdSer asymmetry does not collapse spontaneously. Inner-outer transmembrane exchange of any phospholipid has a high energy barrier (15–50 kcal/mol) because the hydrophilic headgroup of the phospholipid must dehydrate during transit across the hydrophobic core of the bilayer, and so the spontaneous rate of such exchange is very slow [10, 11]. The enzymes that facilitate the trans-bilayer exchange of PtdSer and other phospholipids have been designated phospholipid ‘scramblases’ [4, 7, 12]. For several years, the molecular identity of these enzymes was debated, but an emerging consensus has coalesced on proteins of the TMEM16 and XKR families [4, 13–15]. Also referred to as Anoctamins (they were originally thought to be anion channels with 8 transmembrane helices), the 10-transmembrane domain TMEM16 proteins are especially interesting with respect to the biology discussed below. Although two of these proteins – Ano1 and 2 – appear to function exclusively as Ca2+-activated ion channels, there is now good evidence that Ano3, 4, 5, 7, 9, and especially Ano6 (TMEM16F), function as Ca2+-dependent phospholipid scramblases [11, 13]. Ano6 and a fungal homolog externalize PtdSer [13, 16, 17], and so the activation of these enzymes by Ca2+ is almost certainly key to the deployment of PtdSer as an intercellular signal. One XKR protein – XKR8 – is activated by caspase cleavage, and appears to be particularly important for the externalization of PtdSer on apoptotic cells [4, 15, 18].
PtdSer recognition by Gla domains
There are multiple families of proteins that bind to PtdSer specifically and with high affinity [19]. These include Brain angiogenesis inhibitor 1 (BAI1) [20], T-cell immunoglobulin and mucin receptors 1, 3, and 4 (TIM-1/3/4) [21, 22], and Stabilin 1 and 2 [23, 24]. In addition, PtdSer binding by members of the large Annexin family [25–27], notably Annexin A5 and B12, has been exploited to generate fluorescent and radiolabeled derivatives of these proteins for imaging PtdSer in cells and tissues [28, 29].
The most-studied PtdSer recognition proteins, however, are those that employ a ‘Gla domain’ for binding. These domains are relatively short (~45 amino acid) polypeptide segments that are rich in glutamic residues whose gamma carbons have been carboxylated in a vitamin K-dependent post-translational modification [30–32]. (Vitamin K is an essential co-factor for γ-glutamyl carboxylase [33].) Gamma-carboxylation of glutamic acid in proteins is ancient, and is found in Drosophila, marine snails (Conus), and pre-vertebrate chordates (Ciona) [30]. Gla domains appear in approximately two dozen human proteins, including osteocalcin and periostin, but are best known for the critical role they play in blood coagulation [34, 35]. The coagulation factors Prothrombin, Factors VII, IX, and X, and Proteins C and Z all contain Gla domains that allow them to assemble onto the PtdSer-rich surface of activated platelets. Warfarin and related blood thinners are effective because they antagonize γ-carboxylation of these coagulation factor Gla domains [36]. Importantly, the PtdSer externalization required for Gla domain binding to the surface of platelets and endothelial cells during blood coagulation is driven by Ca2+-dependent scramblases [37], and can be induced by Ca2+ ionophores [37].
TAM receptors, TAM ligands, and PtdSer
Two additional Gla-domain-containing proteins are growth arrest specific-6 (Gas6) and Protein S (Pros1), both of which have a Gla domain positioned at their amino terminus (Figure 1). These proteins are the ligands for the TAM family of cell surface receptor tyrosine kinases (RTKs) [38–40]. (Pros1 also plays an essential, TAM-independent role as an anti-coagulant in the blood coagulation cascade [41].) The Gla domains of Gas6 and Pros1 are not involved in their binding to TAM receptors (Figure 1), but are nonetheless critical to TAM receptor activation.
Figure 1.
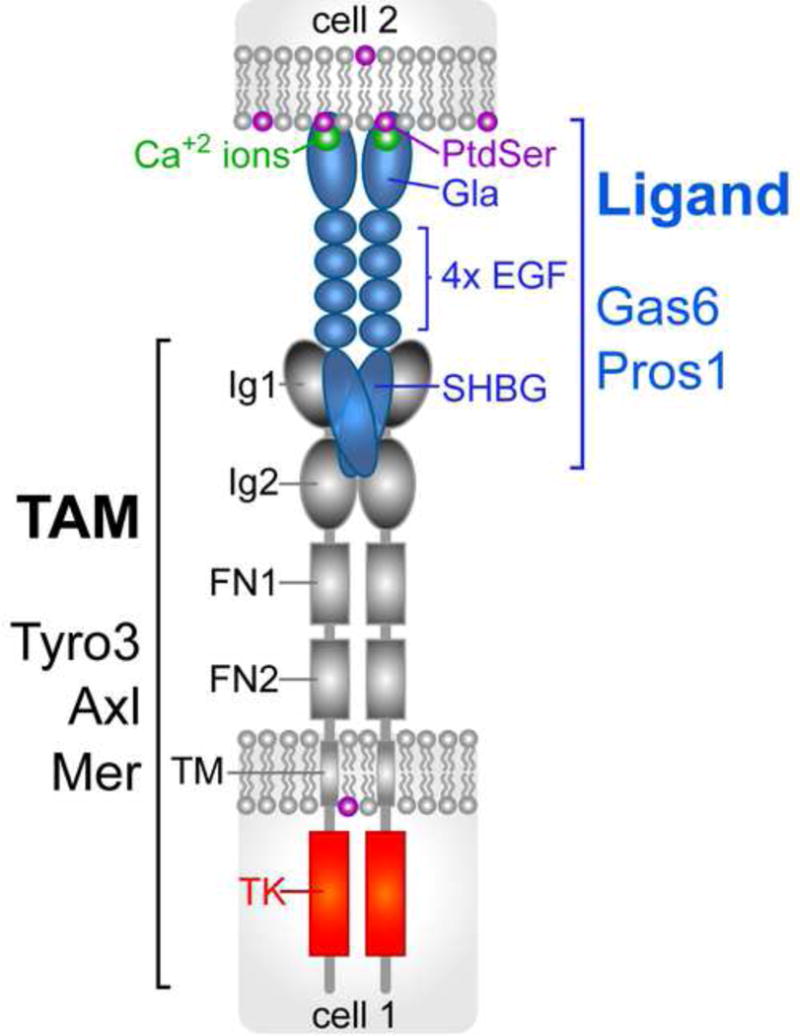
Schematic of the TAM signaling module. Tyro3, Axl, and Mer (gray) display closely-related structures. Gas6 binds and activates all three receptors; Pros1 binds and activates Tyro3 and Mer, but not Axl [39]. Binding of Gas6 and Pros1 (blue) occurs through the two immunoglobulin-related domains (Ig1/Ig2) of the receptors. The two fibronectin type III repeats of the receptors (FN1/FN2) are presumed to facilitate TAM dimerization. A single transmembrane domain (TM) links to a potent tyrosine kinase (TK, red) that is catalytically quiescent unless activated by ligand binding [39]. Binding of Gas6 and Pros1 to the receptors is mediated exclusively by their sex hormone binding globulin (SHBG)-like domain [39, 93, 94], which is composed of two tandem laminin G domains. This domain is separated from the amino-terminal Gla domain by four epidermal growth factor (EGF) domains. The structure of the Gla domain is stabilized by the binding of multiple (~7) [53], essential [39, 54] Ca2+ ions (green), several of which may be liganded to the head group of phosphatidylserine (PtdSer, purple). All known instances of operation of the TAM module involve the expression of a TAM receptor on the surface of one cell, with PtdSer expressed on the surface of an apposed cell (or virus or vesicle); Gas6 or Pros1 are interposed between the PtdSer-expressing cell and the TAM-expressing cell. At present, there is little evidence for heterodimerization between either receptors or ligands.
Comprised of Tyro3, Axl, and Mer (gene name Mertk), the TAM RTKs act as important regulators in adult tissues that are subject to constant challenge and renewal [38]. Uniquely for an RTK family, these receptors as a group play no essential role in embryonic development [42]. Their activities in adults include regulation and feedback inhibition of the innate immune response in dendritic cells and other immune sentinels [43, 44], the engulfment of apoptotic cells and membranes by macrophages and other phagocytes [45, 46], the facilitation of infection of target cells by enveloped viruses (e.g. Zika virus) [47, 48], and prominently, the development and metastasis of many cancers [38, 49–51]. The ‘rules of engagement’ for TAM receptor-ligand interaction have only recently been established definitively [39], and it is now clear that while Gas6 binds to and activates all three receptors, Pros1 is a ligand only for Tyro3 and Mer [39, 52].
Importantly, the ability of Gas6 and Pros1 to activate TAM receptors optimally requires the presence of their Gla domains. This is the case even though these domains are completely irrelevant to receptor binding, which is mediated by the carboxy-terminal ‘SHBG’ (for ‘sex hormone binding globulin’ like) domains of both ligands (Figure 1). For example, a ‘Gla-less’, truncated version of recombinant Gas6 has been shown to bind to Axl with the same sub-nanomolar affinity (KD) as full-length Gas6 in cell-based equilibrium displacement assays, but to be entirely incapable of stimulating Axl activation (monitored by autophosphorylation) in these cells [39]. This same Gla-less Gas6 is also severely weakened as a Tyro3 and Mer agonist, although not completely disabled [39]. In contrast, full-length Gla-containing Gas6 is an exceptionally potent TAM agonist in cell-based assays [39], because there are always PtdSer-containing membranes present in the culture media for these assays [39]. TAM RTK activation by Gas6 and Pros1 also requires that their Gla domains be gamma-carboxylated, since production of recombinant full-length ligands in the presence of warfarin abrogates their activity [52]. Finally, Gla domains, including those of Gas6 and Pros1, require the binding of multiple Ca2+ ions to stabilize their structure and bind PtdSer [53, 54], and Gas6 tightly bound to its receptor in the presence of the Ca2+ chelator EDTA cannot activate the receptor [39]. The TAMs are therefore unusual vis-à-vis other RTKs in that ligand binding does not obligatorily translate into receptor activation. The stoichiometry in the only intact PtdSer-Gla domain complex for which a structure has been determined by X-ray crystallography – that of the Prothrombin Gla domain complexed with an artificial membrane – is 1:1; that is, one molecule of PtdSer to one Gla domain [53].
The above findings now suggest that a TAM RTK-ligand pair should in fact be viewed as a bi-molecular receptor for PtdSer, and that the real job of this RTK-ligand hybrid is to detect PtdSer upon its externalization. Two additional observations are consistent with this hypothesis. First, the genomes of pre-vertebrate ascidians such as Ciona and Halocynthia, which lack blood coagulation, encode both a Gas6-like, Gla-domain-containing protein (a TAM ligand), but also a TAM-like transmembrane RTK whose extracellular domain consists only of a Gas6-related Gla domain [55, 56]. These ascidian RTKs effectively fuse the vertebrate TAM ligands and receptors into a single protein, and could in principle act as a direct PtdSer detector. Second, it has recently been discovered that most or all of the Gas6 in cells and tissues in vivo is, remarkably, already pre-bound specifically to Axl – without substantial steady-state activation of the receptor [46, 57]. That is, Axl and Gas6 are already pre-assembled to function as a hybrid PtdSer receptor in vivo. Moreover, Gas6 expression in cells and tissues is specifically dependent on Axl, as it is lost in Axl−/− but not Mertk−/− or Tyro3−/− mice [46, 57]. Although cis interactions between PtdSer, Gas6/Pros1, and a TAM receptor on the same cell membrane surface have not been ruled out, all biological interactions that have been studied to date (see below) involve the display of PtdSer on the surface of one cell (or vesicle or virus particle), and the expression of a TAM-receptor-ligand complex on the surface of a distinct, apposed, interacting cell (Figure 1).
Biological phenomena driven by PtdSer/TAM signaling
Apoptotic cell engulfment
The regulated display of externalized PtdSer appears to be the trigger for TAM receptor/ligand activation in many settings. The best known of these is the phagocytosis of apoptotic cells by macrophages and other phagocytes (Figure 2A). Indeed, externalized PtdSer is the most common and among the most potent of the so-called ‘eat-me signals’ by which apoptotic cells are recognized by phagocytes [5, 19, 58]; and TAM receptors, expressed by phagocytes, have been shown to be critical to the PtdSer-triggered clearance of apoptotic cells in multiple tissues [45, 46, 50, 59]. Much of this phagocytosis is continuous and routine: it has been estimated that in the human body ~2 million cells undergo apoptosis each and every second [60]. In most examples of ‘immunologically silent’ phagocytosis (or ‘efferocytosis’), TAM-mediated recognition and engulfment of continuously-generated PtdSer-positive apoptotic cells is mediated by Mer and Gas6/Pros1, although in some instances, including infection, inflammation, and tissue injury, this job is also handled by Axl and Gas6 [46]. The largest routine apoptotic burden in the body is the corpus of senescent and injured erythrocytes, which are cleared by red pulp macrophages in the spleen [61]. These red pulp macrophages express the highest known levels of both Mertk and Axl mRNA [62]. The second most significant apoptotic burden in the body is spent neutrophils, which are cleared by tissue macrophages that are also strongly Mer-positive [62].
Figure 2.
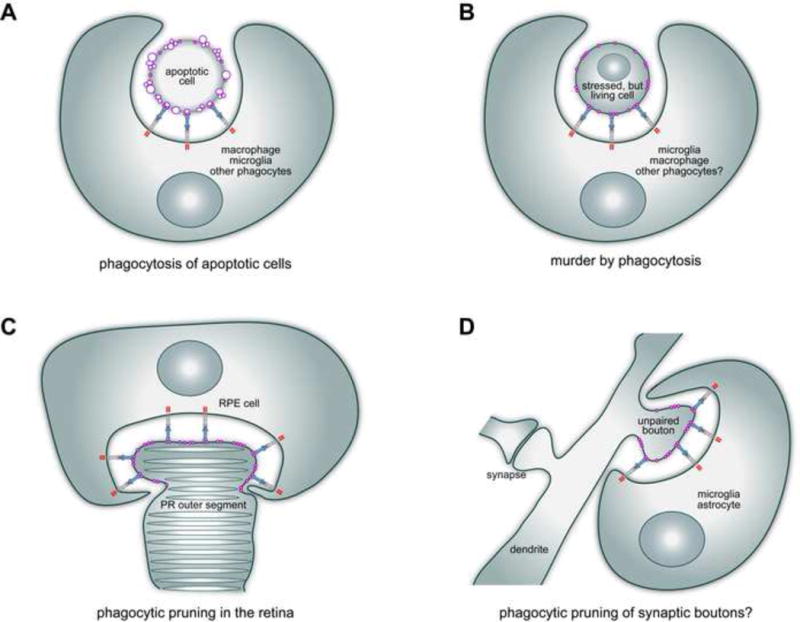
Four examples of the operation of PtdSer-TAM signaling in biology. A. TAM receptors and ligands mediate the phagocytic clearance of apoptotic cells, by macrophages, Sertoli cells, and other phagocytes, in many tissues [38, 45, 46, 50]. This phagocytosis is always driven by externalized PtdSer (purple) as an ‘eat-me’ signal, which is often present on membrane blebs (purple circles). B. TAM receptors also facilitate the phagocytic killing of live cells. When stressed by trauma, metabolic insult, or infection, live cells may externalize PtdSer on their plasma membrane surface and thereby be ‘eaten alive’ by TAM-dependent engulfment [65–67, 70]. Although only recently discovered, this ‘murder by phagocytosis’ may be a very general phenomenon. C. On a smaller scale, PtdSer-TAM signaling also drives the phagocytic eating of only part of a cell. In the retina, the retinal pigment epithelial (RPE) cell is a phagocyte that clips off the distal ends of rhodopsin-containing photoreceptor (PR) outer segments, at or around subjective dawn, every day of life [71–73]. This phagocytic pruning is TAM (Mer)-dependent, and is driven and delimited by the highly localized externalization of PtdSer at only the tips of the outer segments [75]. D. On an even finer scale, TAM signaling has been implicated in the phagocytic pruning of synaptic boutons on neuronal dendrites in the postnatal brain [79]. The neuron-intrinsic signal that initiates and delimits this pruning, which is carried out by microglia and astrocytes, remains to be established. However, it is reasonable to propose that this signal, by analogy with the outer segments of photoreceptor neurons, is PtdSer.
Cyclic apoptosis and phagocytic clearance are evident at other sites in the body where new cells are continuously generated throughout adult life, and PtdSer triggered TAM signaling operates at these sites as well. Millions of apoptotic cells are generated at each cycle of mammalian spermatogenesis, for example, and the trigger for the engulfment of these corpses by Sertoli cells, the somatic phagocytes of the testes, is externalized PtdSer [63]. Sertoli cell phagocytosis is TAM-dependent, as Tyro3−/−Axl−/−Mertk−/− triple mutant male mice display a massive accumulation of apoptotic cells in the seminiferous tubules of the testes, and are sterile [39, 42]. Similarly, most of the cells newly generated at sites of continuing neurogenesis in the adult central nervous system (CNS), including those in the subventricular zone (SVZ) adjacent to the lateral ventricle and the dentate gyrus of the hippocampus, undergo apoptosis before they can differentiate into neurons; and these apoptotic neural cells are cleared by microglia, the resident tissue macrophages of the CNS [64]. Recent work has demonstrated this phagocytic clearance is once again TAM dependent, as apoptotic cells accumulate in the CNS neurogenic regions of Mertk−/− and Axl−/−Mertk−/− mice [65].
Murder by phagocytosis
Remarkably, several recent analyses have further indicated that distressed but nonetheless living cells may also externalize PtdSer over much of their surface, leading them to be ‘eaten alive’ via TAM-dependent phagocytosis (Figure 2B). For example, when neurogenic cells in the adult SVZ are BrdU pulse-labeled during cell division, many more healthy BrdU-labeled neurons are present in the olfactory bulb (to which newborn SVZ cells migrate) one month after labeling in Axl−/−Mertk−/− mice than in wild-type [65]. More definitively, adenovirus-transduced astrocytes have been found to transiently externalize PtdSer, and over the course of several days after the transduction, to be phagocytically cleared by activated microglia [66]. This phagocytosis is both TAM- and PtdSer-dependent, since clearance does not occur in either Axl−/−Mertk−/− mutants or when PtdSer externalization is genetically inhibited [66]. Moreover, in these mutant settings, the non-eaten transduced cells persist as healthy functioning astrocytes for many months [66]. These studies are consistent with: (a) earlier analyses of tissue recovery following focal brain ischemia, in which areas (brain volume) of neuronal atrophy are reduced, and recovery is enhanced, in Mertk−/− mice relative to wild-type [67]; and (b) even earlier work in C. elegans embryos, in which genetic inactivation of engulfment (phagocytosis) genes results in the long-term survival of a subset of cells that would otherwise die [68, 69]. It is therefore possible that phagocytic ‘culling’ of cell populations (to ensure ‘survival of the fittest’?) is a very general phenomenon. Together, these data indicate that microglia, and by extension other vertebrate phagocytes, can eat live cells – a process that has been christened ‘phagoptosis’ [70]. They demonstrate that TAM-dependent activities include the killing of cells by phagocytosis (Figure 2B). These findings have important implications for the contribution of microglia, macrophages, and other phagocytes to degenerative disease.
Local membrane excision and synaptic pruning
Externalized PtdSer also drives routine TAM-mediated phagocytic events in normal healthy cells. In the eye, retinal pigment epithelial (RPE) cells perform a surgical phagocytic pruning of just the distal tips of photoreceptor outer segments, at or around subjective dawn, every day of life. This pruning removes toxic oxidized proteins, generated during phototransduction, that would otherwise kill the photoreceptors [71–74]. It is a Mer-dependent process that involves the phagocytosis of only a small part of a neuron, as opposed to an entire apoptotic cell (Figure 2C). Importantly, imaging of live photoreceptors with a fluorescent Annexin B12 derivative has shown that RPE phagocytosis is both triggered and delimited by the localized externalization of PtdSer at just the distal tips of the photoreceptors [75]. It remains a mystery how such localized externalization of PtdSer, which is also seen in other cells [76], is achieved. The daily pruning of photoreceptor outer segments is very important: loss-of-function Mertk mutations severely compromise this phagocytosis, and result in inherited forms of retinitis pigmentosa, photoreceptor death, and blindness [73, 77].
A similar but even more restricted and much smaller segment of neuronal membrane – that of dendritic boutons in the maturing, postnatal CNS – is selectively engulfed in a process referred to as ‘synaptic pruning’ [78]. This engulfment (Figure 2D), which is critical to synaptic maturation and plasticity, is mediated by microglia [78] and perhaps by astrocytes. Mer expression by macroglial and microglial populations has been implicated in synaptic pruning [79], although delimited externalization of PtdSer on dendritic boutons that are to be eliminated has not been demonstrated. It is nonetheless reasonable to posit that this might be the case, since C1q and other complement proteins that have been implicated in the ‘decoration’ of boutons targeted for pruning [80, 81] are known PtdSer-binding proteins [82, 83]. Moreover, while complement proteins are soluble agents extrinsic to the neuron, PtdSer is intrinsic to the cell, and can in principle be localized to the membrane segment (bouton) that is to be eliminated through the delimited activation of Ca2+-dependent scramblases.
Infection by enveloped viruses
Finally, there is considerable current foment in the literature with respect to the role that TAM receptors play in the infection of cells by enveloped viruses. When HIV, vaccinia, West Nile, Zika, and other viruses bud from virus-producing cells, their membrane envelopes can display significant levels of externalized PtdSer [84]. If these virus particles are exposed to environments containing TAM ligands – for example, if they pass through the blood where Pros1 is present at ~300 nM – they will effectively be opsonized with these ligands, to a greater or lesser extent as a function of the fraction of the virus surface that is exposed PtdSer. If these TAM-ligand-coated virus particles then encounter a TAM-receptor-expressing cell, they can use the TAM receptor to dock to the cell [47]. This docking will also activate the TAM receptor, which both promotes endocytic engulfment of the particle and at the same time inhibits the innate immune response of the cell to the virus [47]. In this scenario, virus particles are effectively ‘nano versions’ of PtdSer-displaying apoptotic cells, and the process has been termed viral infection by ‘apoptotic mimicry’ [85]. The introduction of a TAM receptor into an infection-resistant cell in culture (Axl and Tyro3 have been most commonly used) has repeatedly been observed to greatly potentiate infection by HIV, Dengue, vaccinia, Ebola and other enveloped viruses [47, 86–89], and Axl has been the subject of intense recent study as a candidate PtdSer-dependent receptor for the Zika virus [48, 90, 91]. The significance of these in vitro observations to virus infection in vivo remains to be established [92].
Emerging PtdSer/TAM questions
It is not yet clear how PtdSer controls the signaling competence of Gas6 and Pros1 at a molecular level. Crystal structures of a Gla-less segment of Gas6, primarily the SHBG domain alone bound to the two Ig domains of Axl (see Figure 1), have been independently determined by two groups [93, 94]. As noted above, these structures, which are the same, depict a complex that may not be signaling competent [39]; and they also have several unusual features. The stoichiometry of the complex is 2:2, with two molecules of Gas6 (SHBG) and two molecules of Axl (Ig1+Ig2), but no contacts between the individual Gas6 or Axl polypeptides themselves. Instead, the two Gas6 molecules hold the complex together by bridging the Ig1 domain of one Axl protein to the Ig2 domain of the other. This has the effect of placing the two Axl Ig2 domains, which are proximal to the plasma membrane of the Axl-expressing cell, far apart and pointed away from one another in the crystal [93, 94]. These Ig2 domains are linked to the two FNIII repeats of the TAMs, which in other RTKs facilitate dimerization, and it is not clear how well these FNIII domains might find one another when attached to the distant Ig2 domains of the published structures. It is possible that PtdSer binding to a TAM ligand Gla domain, which is separated from the SHBG domain by four EGF-like repeats (Figure 1), completely re-configures the Gas6-Axl interaction (Figure 3A). Alternatively, Gla-PtdSer interaction might instead result in ligand multimerization that greatly increases TAM ligand-receptor avidity (Figure 3B). There is precedent for this second possibility in the Eph-ephrin RTK-ligand family, where the ligands are either membrane associated GPI-anchored ephrin-As or transmembrane ephrin-Bs, both of which enhance signaling by promoting receptor clustering [95]. Definitive answers to these questions may require the determination of high resolution TAM structures composed of full-length receptor ectodomains and full-length ligands, with and without PtdSer.
Figure 3.
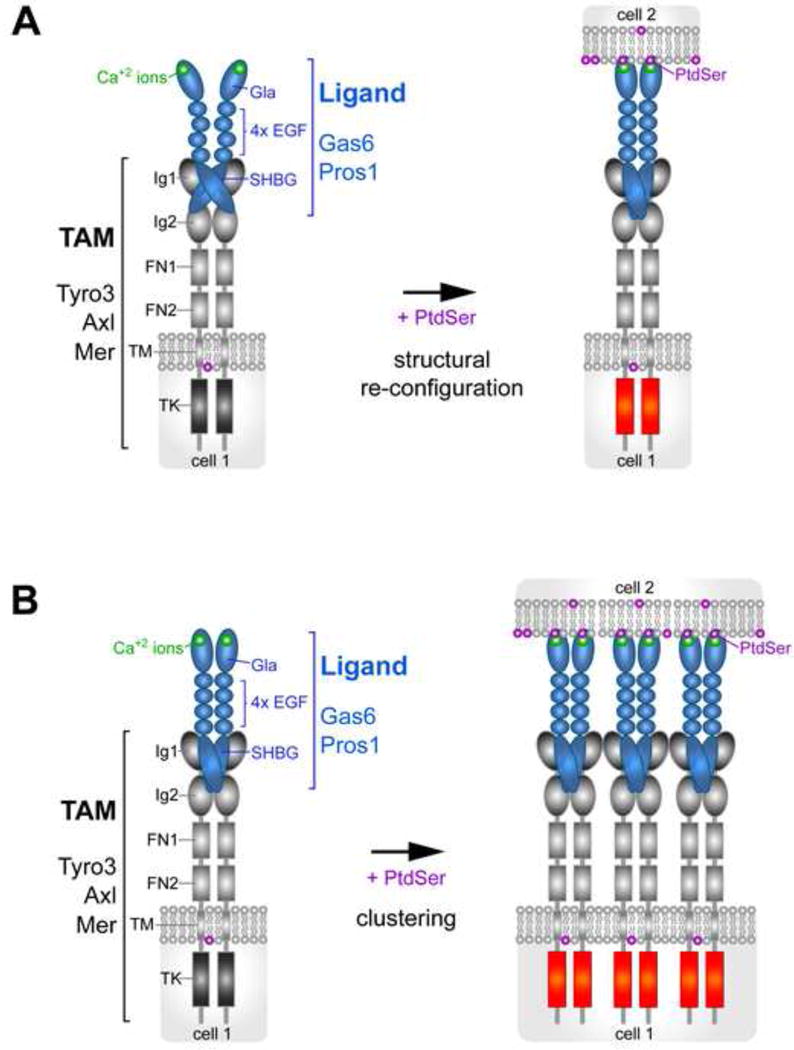
Two alternative models for how PtdSer may regulate TAM receptor-ligand interaction. A. Structural re-configuration. The binding of PtdSer to the Gla domain of TAM ligands significantly re-orients the binding of the ligand’s SHBG domain to the Ig1 and Ig2 domains of the TAM receptor. B. Multimerization. The presence of an array of PtdSer on a surface membrane provides a platform for the clustering of TAM ligands and the consequent clustering of TAM receptors on the surface of an apposed cell, greatly increasing the apparent avidity of TAM ligand-receptor complexes.
We also have little or no understanding as to how PtdSer might be locally exposed only within restricted domains on a cell surface, as is seen with photoreceptor outer segments and synaptic T cells [75, 76]. It is possible that localized activation of TMEM16F or other Ca2+-dependent scramblases by the localized expression or opening of Ca2+ channels in the plasma membrane might account for this. PtdSer has been shown to be externalized at delimited foci on the surface of neuroendocrine (chromaffin) cells when these cells are depolarized, and this externalization is Ca2+-dependent [96, 97]. However, the extent to which PtdSer is in fact externalized specifically at synaptic boutons that are to be phagocytically eliminated from neurons in the neonatal brain is unknown, and so the PtdSer-driven pruning model of Figure 2D is at this point only a hypothesis.
The TAM RTKs and their ligands display an unusual signaling biochemistry that arises from their dependence on the phospholipid phosphatidylserine. This signaling biochemistry in turn underlies the diverse biological phenomena that the TAMs control in cells and tissues that are subject to continuous challenge and renewal. As such, this signaling ensemble promises to supply many additional surprises in the years to come.
Trends Box.
The Tyro3/Axl/Mer (TAM) family of receptor tyrosine kinases (RTKs) are bound by two protein ligands – Gas6 and Protein S (Pros1) – but activation of these RTKs requires the coincident binding of the phospholipid phosphatidylserine (PtdSer) to the amino termini of the ligands.
PtdSer is normally confined to the inner leaflet of the plasma membrane bilayer, but can be externalized to the outer leaflet via the action of one of several Ca2+-activated or caspase-activated PtdSer ‘scramblases’.
When externalized, PtdSer is a potent signal. It may indicate that a cell has died by apoptosis, or that it is metabolically stressed by oxidative trauma or infection, or that delimited segments of its surface are to be phagocytically ‘pruned’, or that an enveloped virus is primed to infect.
In many settings, the PtdSer signal is transduced by a TAM RTK-ligand pair acting as a hybrid detector for the phospholipid.
Outstanding Questions Box.
What is the biochemical consequence of PtdSer binding for TAM ligand-receptor interaction? Does PtdSer binding re-order this interaction? Or does PtdSer promote ligand-receptor clustering on the cell surface?
What is the biochemical nature of TAM RTK-ligand interaction with other ligand-receptor systems that mediate recognition of PtdSer?
Most of the Gas6 seen in tissues in vivo appears to be constitutively pre-bound to Axl, and furthermore, to depend on Axl for its stable expression. How does Gas6 then reach the Mer and Tyro3 receptors that it is known to activate?
How is the localized externalization of PtdSer within a delimited membrane domain – as opposed to the entire cell surface – achieved? What are the scramblases that act to externalize PtdSer, both globally and locally, in diverse cell types? How is the activity of these scramblases regulated by calcium?
Does PtdSer-TAM signaling mediate the elimination of synaptic boutons in the neonatal brain? Is PtdSer the neuron-intrinsic signal that targets boutons for phagocytic removal?
Acknowledgments
I thank Paqui González, Kaisa Happonen, Erin Lew, and Sheng Miao for helpful comments on and corrections to the manuscript. Work in my laboratory is supported by grants from the NIH (R01 NS085296 and R01 AI101400), the Leona M. and Harry B. Helmsley Charitable Trust (#2017-PG-MED001 to the Salk Institute), and the Nomis, H. N. and Frances C. Berger, Fritz B. Burns, and HKT Foundations.
References
- 1.Leventis PA, Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu Rev Biophys. 2010;39:407–27. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- 2.Jorissen BL, et al. The influence of soy-derived phosphatidylserine on cognition in age-associated memory impairment. Nutr Neurosci. 2001;4(2):121–34. doi: 10.1080/1028415x.2001.11747356. [DOI] [PubMed] [Google Scholar]
- 3.van Meer G. Dynamic transbilayer lipid asymmetry. Cold Spring Harb Perspect Biol. 2011;3(5) doi: 10.1101/cshperspect.a004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bevers EM, Williamson PL. Getting to the Outer Leaflet: Physiology of Phosphatidylserine Exposure at the Plasma Membrane. Physiol Rev. 2016;96(2):605–45. doi: 10.1152/physrev.00020.2015. [DOI] [PubMed] [Google Scholar]
- 5.Birge RB, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23(6):962–78. doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Andersen JP, et al. P4-ATPases as Phospholipid Flippases-Structure, Function, and Enigmas. Front Physiol. 2016;7:275. doi: 10.3389/fphys.2016.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nagata S, et al. Exposure of phosphatidylserine on the cell surface. Cell Death Differ. 2016;23(6):952–61. doi: 10.1038/cdd.2016.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segawa K, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344(6188):1164–8. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 9.Segawa K, et al. Human Type IV P-type ATPases That Work as Plasma Membrane Phospholipid Flippases and Their Regulation by Caspase and Calcium. J Biol Chem. 2016;291(2):762–72. doi: 10.1074/jbc.M115.690727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kornberg RD, McConnell HM. Inside-outside transitions of phospholipids in vesicle membranes. Biochemistry. 1971;10(7):1111–20. doi: 10.1021/bi00783a003. [DOI] [PubMed] [Google Scholar]
- 11.Whitlock JM, Hartzell HC. Anoctamins/TMEM16 Proteins: Chloride Channels Flirting with Lipids and Extracellular Vesicles. Annu Rev Physiol. 2017;79:119–143. doi: 10.1146/annurev-physiol-022516-034031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kodigepalli KM, et al. Roles and regulation of phospholipid scramblases. FEBS Lett. 2015;589(1):3–14. doi: 10.1016/j.febslet.2014.11.036. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki J, et al. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468(7325):834–8. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki J, et al. Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J Biol Chem. 2013;288(19):13305–16. doi: 10.1074/jbc.M113.457937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suzuki J, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341(6144):403–6. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 16.Brunner JD, et al. X-ray structure of a calcium-activated TMEM16 lipid scramblase. Nature. 2014;516(7530):207–12. doi: 10.1038/nature13984. [DOI] [PubMed] [Google Scholar]
- 17.Malvezzi M, et al. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun. 2013;4:2367. doi: 10.1038/ncomms3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suzuki J, et al. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2016;113(34):9509–14. doi: 10.1073/pnas.1610403113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev. 2016;269(1):44–59. doi: 10.1111/imr.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450(7168):430–4. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- 21.Freeman GJ, et al. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–89. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuchroo VK, et al. New roles for TIM family members in immune regulation. Nat Rev Immunol. 2008;8(8):577–80. doi: 10.1038/nri2366. [DOI] [PubMed] [Google Scholar]
- 23.Park SY, et al. Stabilin-1 mediates phosphatidylserine-dependent clearance of cell corpses in alternatively activated macrophages. J Cell Sci. 2009;122(Pt 18):3365–73. doi: 10.1242/jcs.049569. [DOI] [PubMed] [Google Scholar]
- 24.Park SY, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15(1):192–201. doi: 10.1038/sj.cdd.4402242. [DOI] [PubMed] [Google Scholar]
- 25.Futter CE, White IJ. Annexins and endocytosis. Traffic. 2007;8(8):951–8. doi: 10.1111/j.1600-0854.2007.00590.x. [DOI] [PubMed] [Google Scholar]
- 26.Gerke V, Moss SE. Annexins: from structure to function. Physiol Rev. 2002;82(2):331–71. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 27.Gerke V, et al. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6(6):449–61. doi: 10.1038/nrm1661. [DOI] [PubMed] [Google Scholar]
- 28.Vermes I, et al. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184(1):39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 29.Kim YE, et al. Monitoring apoptosis and neuronal degeneration by real-time detection of phosphatidylserine externalization using a polarity-sensitive indicator of viability and apoptosis. Nat Protoc. 2010;5(8):1396–405. doi: 10.1038/nprot.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bandyopadhyay PK. Vitamin K-dependent gamma-glutamylcarboxylation: an ancient posttranslational modification. Vitam Horm. 2008;78:157–84. doi: 10.1016/S0083-6729(07)00008-8. [DOI] [PubMed] [Google Scholar]
- 31.Stafford DW. The vitamin K cycle. J Thromb Haemost. 2005;3(8):1873–8. doi: 10.1111/j.1538-7836.2005.01419.x. [DOI] [PubMed] [Google Scholar]
- 32.Berkner KL. Vitamin K-dependent carboxylation. Vitam Horm. 2008;78:131–56. doi: 10.1016/S0083-6729(07)00007-6. [DOI] [PubMed] [Google Scholar]
- 33.Furie B, et al. Vitamin K-dependent biosynthesis of gamma-carboxyglutamic acid. Blood. 1999;93(6):1798–808. [PubMed] [Google Scholar]
- 34.Stenflo J. Contributions of Gla and EGF-like domains to the function of vitamin K-dependent coagulation factors. Crit Rev Eukaryot Gene Expr. 1999;9(1):59–88. [PubMed] [Google Scholar]
- 35.Kalafatis M, et al. Regulation and regulatory role of gamma-carboxyglutamic acid containing clotting factors. Crit Rev Eukaryot Gene Expr. 1996;6(1):87–101. doi: 10.1615/critreveukargeneexpr.v6.i1.60. [DOI] [PubMed] [Google Scholar]
- 36.Van Horn WD. Structural and functional insights into human vitamin K epoxide reductase and vitamin K epoxide reductase-like1. Crit Rev Biochem Mol Biol. 2013;48(4):357–72. doi: 10.3109/10409238.2013.791659. [DOI] [PubMed] [Google Scholar]
- 37.Fujii T, et al. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A. 2015;112(41):12800–5. doi: 10.1073/pnas.1516594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lemke G. Biology of the TAM receptors. Cold Spring Harbor Perspectives. 2013;5(11) doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lew ED, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife. 2014;3:e03385. doi: 10.7554/eLife.03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stitt TN, et al. The anticoagulation factor protein S and its relative, Gas6, are ligands for the Tyro 3/Axl family of receptor tyrosine kinases. Cell. 1995;80(4):661–670. doi: 10.1016/0092-8674(95)90520-0. [DOI] [PubMed] [Google Scholar]
- 41.Burstyn-Cohen T, et al. Lack of protein S in mice causes embryonic lethal coagulopathy and vascular dysgenesis. J Clin Invest. 2009;119(10):2942–53. doi: 10.1172/JCI39325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lu Q, et al. Tyro-3 family receptors are essential regulators of mammalian spermatogenesis. Nature. 1999;398(6729):723–8. doi: 10.1038/19554. [DOI] [PubMed] [Google Scholar]
- 43.Rothlin CV, et al. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124–36. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 44.Carrera Silva EA, et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity. 2013;39(1):160–70. doi: 10.1016/j.immuni.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scott RS, et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature. 2001;411(6834):207–11. doi: 10.1038/35075603. [DOI] [PubMed] [Google Scholar]
- 46.Zagórska A, et al. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15(10):920–928. doi: 10.1038/ni.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bhattacharyya S, et al. Enveloped Viruses Disable Innate Immune Responses in Dendritic Cells by Direct Activation of TAM Receptors. Cell Host Microbe. 2013;14(2):136–47. doi: 10.1016/j.chom.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meertens L, et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017;18(2):324–333. doi: 10.1016/j.celrep.2016.12.045. [DOI] [PubMed] [Google Scholar]
- 49.Lemke G, Rothlin CV. Immunobiology of the TAM receptors. Nat Rev Immunol. 2008;8(5):327–36. doi: 10.1038/nri2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rothlin CV, et al. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355–91. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Graham DK, et al. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14(12):769–85. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 52.Tsou WI, et al. Receptor Tyrosine Kinases, TYRO3, AXL and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-Induced Activation. J Biol Chem. 2014;289:25750–25763. doi: 10.1074/jbc.M114.569020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang M, et al. Structural basis of membrane binding by Gla domains of vitamin K-dependent proteins. Nat Struct Biol. 2003;10(9):751–6. doi: 10.1038/nsb971. [DOI] [PubMed] [Google Scholar]
- 54.Dransfield I, et al. Mer receptor tyrosine kinase mediates both tethering and phagocytosis of apoptotic cells. Cell Death Dis. 2015;6:e1646. doi: 10.1038/cddis.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kulman JD, et al. Vitamin K-dependent proteins in Ciona intestinalis, a basal chordate lacking a blood coagulation cascade. Proc Natl Acad Sci U S A. 2006;103(43):15794–9. doi: 10.1073/pnas.0607543103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang CP, et al. Identification of a gene encoding a typical gamma-carboxyglutamic acid domain in the tunicate Halocynthia roretzi. J Thromb Haemost. 2003;1(1):118–23. doi: 10.1046/j.1538-7836.2003.00069.x. [DOI] [PubMed] [Google Scholar]
- 57.Fujimori T, et al. The Axl receptor tyrosine kinase is a discriminator of macrophage function in the inflamed lung. Mucosal Immunol. 2015;8(5):1021–30. doi: 10.1038/mi.2014.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35(4):445–55. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lemke G, Burstyn-Cohen T. TAM receptors and the clearance of apoptotic cells. Ann N Y Acad Sci. 2010;1209:23–9. doi: 10.1111/j.1749-6632.2010.05744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elliott MR, Ravichandran KS. The Dynamics of Apoptotic Cell Clearance. Dev Cell. 2016;38(2):147–60. doi: 10.1016/j.devcel.2016.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kurotaki D, et al. Functions and development of red pulp macrophages. Microbiol Immunol. 2015;59(2):55–62. doi: 10.1111/1348-0421.12228. [DOI] [PubMed] [Google Scholar]
- 62.Gautier EL, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–28. doi: 10.1038/ni.2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shiratsuchi A, et al. Recognition of phosphatidylserine on the surface of apoptotic spermatogenic cells and subsequent phagocytosis by Sertoli cells of the rat. J Biol Chem. 1997;272(4):2354–8. doi: 10.1074/jbc.272.4.2354. [DOI] [PubMed] [Google Scholar]
- 64.Sierra A, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–95. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fourgeaud L, et al. TAM receptors regulate multiple features of microglial physiology. Nature. 2016;532(7598):240–4. doi: 10.1038/nature17630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tufail Y, et al. Phosphatidylserine Exposure Controls Viral Innate Immune Responses by Microglia. Neuron. 2017;93(3):574–586. doi: 10.1016/j.neuron.2016.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Neher JJ, et al. Phagocytosis executes delayed neuronal death after focal brain ischemia. Proc Natl Acad Sci U S A. 2013;110(43):E4098–107. doi: 10.1073/pnas.1308679110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reddien PW, et al. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412(6843):198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- 69.Hoeppner DJ, et al. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412(6843):202–6. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- 70.Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. 2014;15(4):209–16. doi: 10.1038/nrn3710. [DOI] [PubMed] [Google Scholar]
- 71.Burstyn-Cohen T, et al. Genetic dissection of TAM receptor-ligand interaction in retinal pigment epithelial cell phagocytosis. Neuron. 2012;76(6):1123–32. doi: 10.1016/j.neuron.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.D’Cruz PM, et al. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Genet. 2000;9(4):645–51. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- 73.Gal A, et al. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Genet. 2000;26(3):270–1. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- 74.Prasad D, et al. TAM receptor function in the retinal pigment epithelium. Mol Cell Neurosci. 2006;33(1):96–108. doi: 10.1016/j.mcn.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 75.Ruggiero L, et al. Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5−/− or Mfge8−/− mouse retina. Proc Natl Acad Sci U S A. 2012;109(21):8145–8. doi: 10.1073/pnas.1121101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fischer K, et al. Antigen recognition induces phosphatidylserine exposure on the cell surface of human CD8+ T cells. Blood. 2006;108(13):4094–101. doi: 10.1182/blood-2006-03-011742. [DOI] [PubMed] [Google Scholar]
- 77.Parinot C, Nandrot EF. A Comprehensive Review of Mutations in the MERTK Proto-Oncogene. Adv Exp Med Biol. 2016;854:259–65. doi: 10.1007/978-3-319-17121-0_35. [DOI] [PubMed] [Google Scholar]
- 78.Hong S, et al. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016;36:128–34. doi: 10.1016/j.conb.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chung WS, et al. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature. 2013;504(7480):394–400. doi: 10.1038/nature12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vasek MJ, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–43. doi: 10.1038/nature18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mastellos DC. Complement emerges as a masterful regulator of CNS homeostasis, neural synaptic plasticity and cognitive function. Exp Neurol. 2014;261:469–74. doi: 10.1016/j.expneurol.2014.06.019. [DOI] [PubMed] [Google Scholar]
- 82.Paidassi H, et al. C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol. 2008;180(4):2329–38. doi: 10.4049/jimmunol.180.4.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogden CA, Elkon KB. Role of complement and other innate immune mechanisms in the removal of apoptotic cells. Curr Dir Autoimmun. 2006;9:120–42. doi: 10.1159/000090776. [DOI] [PubMed] [Google Scholar]
- 84.Altan-Bonnet N. Lipid Tales of Viral Replication and Transmission. Trends Cell Biol. 2017;27(3):201–213. doi: 10.1016/j.tcb.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320(5875):531–5. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- 86.Shimojima M, et al. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J Virol. 2006;80(20):10109–16. doi: 10.1128/JVI.01157-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Meertens L, et al. TIM and TAM receptors mediate dengue virus infection. Cell Host and Microbe. 2012;12:544–557. doi: 10.1016/j.chom.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brindley MA, et al. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology. 2011;415(2):83–94. doi: 10.1016/j.virol.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moller-Tank S, Maury W. Phosphatidylserine receptors: enhancers of enveloped virus entry and infection. Virology. 2014:468–470. doi: 10.1016/j.virol.2014.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Savidis G, et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016;16(1):232–46. doi: 10.1016/j.celrep.2016.06.028. [DOI] [PubMed] [Google Scholar]
- 91.Nowakowski TJ, et al. Expression Analysis Highlights AXL as a Candidate Zika Virus Entry Receptor in Neural Stem Cells. Cell Stem Cell. 2016;18(5):591–6. doi: 10.1016/j.stem.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miner JJ, et al. The TAM receptor Mertk protects against neuroinvasive viral infection by maintaining blood-brain barrier integrity. Nat Med. 2015;21(12):1464–72. doi: 10.1038/nm.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sasaki T, et al. Structural basis for Gas6-Axl signalling. EMBO J. 2006;25(1):80–7. doi: 10.1038/sj.emboj.7600912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kariolis MS, et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10(11):977–83. doi: 10.1038/nchembio.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Janes PW, et al. Concepts and consequences of Eph receptor clustering. Semin Cell Dev Biol. 2012;23(1):43–50. doi: 10.1016/j.semcdb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 96.Ory S, et al. Phospholipid scramblase-1-induced lipid reorganization regulates compensatory endocytosis in neuroendocrine cells. J Neurosci. 2013;33(8):3545–56. doi: 10.1523/JNEUROSCI.3654-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ceridono M, et al. Selective recapture of secretory granule components after full collapse exocytosis in neuroendocrine chromaffin cells. Traffic. 2011;12(1):72–88. doi: 10.1111/j.1600-0854.2010.01125.x. [DOI] [PubMed] [Google Scholar]