

Abstract
The discovery of the endogenous melanocortin agonists in the 1950s have resulted in sixty years of melanocortin ligand research. Early efforts involved truncations or select modifications of the naturally occurring agonists leading to the development of many potent and selective ligands. With the identification and cloning of the five known melanocortin receptors, many ligands were improved upon through bench-top in vitro assays. Optimization of select properties resulted in ligands adopted as clinical candidates. A summary of every melanocortin ligand is outside the scope of this review. Instead, this review will focus on the following topics: classic melanocortin ligands, selective ligands, small molecule (non-peptide) ligands, ligands with sex-specific effects, bivalent and multivalent ligands, and ligands advanced to clinical trials. Each topic area will be summarized with current references to update the melanocortin field on recent progress.
Keywords: selective ligands, small molecules, sexual dimorphism, bivalent/multivalent ligands, clinical trials
1. Introduction
The melanocortin system consists of five receptor subtypes, discovered to date, that are involved in numerous biological pathways. The melanocortin-1-receptor (MC1R), expressed in the skin, is primarily involved in pigmentation [1, 2]. The melanocortin-2 receptor (MC2R) is involved in steroidogenesis and is expressed in the adrenal cortex [2]. The centrally expressed melanocortin-3 and melanocortin-4 receptors (MC3R and MC4R) are linked to energy homeostasis [3-9]. Additionally, the MC4R has a role in sexual function in humans [10, 11]. While the exact role of the melanocortin-5 receptor (MC5R) has not been elucidated [12, 13], it has been linked to exocrine function [14].
A variety of endogenous ligands interact with the melanocortin receptors (MCRs). The naturally occurring agonists, derived from the proopiomelanocortin (POMC) gene transcript [15], stimulate the receptors to increase intracellular cAMP levels. Unique to this GPCR family is the presence of endogenous antagonists, agouti (ASP) and agouti-related protein (AGRP) [16-18]. Additionally, AGRP has been demonstrated to possess inverse agonist activity (directly decreasing cAMP levels within a cell) at the MC4R in mice and humans [19, 20], while agouti has been shown to be an inverse agonist in cells expressing the grey squirrel MC1R [21].
Since changes in pigmentation can be readily visualized, early work on melanocortin ligands focused on the MC1R. The first reports of altered pigmentation dates back to 1916 [22, 23]. Significant advances were achieved with the identification, sequencing, and cloning of the MCRs from 1992 to 1994 [1, 2, 6, 7, 9, 12, 13], coupled with the development of 96-well plate cAMP assays [24]. The genetic information combined with assay platforms generated an experimental paradigm that allowed for the design, synthesis, and investigation of potent, selective compounds for the different receptor subtypes. Many pharmaceutical companies initiated melanocortin ligand programs following the discoveries that the MC4R was linked to food intake, energy homeostasis, obesity, and sexual function in humans [5]. Reports of cardiovascular side effects associated with MC4R ligands [25] coupled with an increase in mergers within the pharmaceutical industry led to diminished industrial interest in melanocortin ligands. However, melanocortin ligands have continued to be advanced to clinical trials.
Over a century of work has been published on the melanocortin receptors, and 60 years of reports focused on melanocortin ligands have resulted in numerous discoveries. As there are too many ligands to summarize succinctly, the scope of this review will focus on the following topics. The first section will review select classic peptide melanocortin ligands followed by a summary of recent advancements in selective ligands. Next, a discussion of small molecule (non-peptide) ligands will focus primarily on the MC4R. Ligands resulting in sex-specific effects will be summarized, followed by an update of bivalent and multivalent ligands. A final section will highlight melanocortin ligands advanced to clinical trials, emphasizing compounds described between 2011 and 2016.
2. Classic Peptide Melanocortin Ligands
Since the first reports of the sequences of adrenocorticotropic hormone (ACTH), β-melanocyte stimulating hormone (β-MSH), and α-MSH in the 1950s [26-28], numerous peptides and small molecule ligands have been developed for the MCRs. This section will focus on some author-perceived classic ligands. In particular, the naturally occurring ligands derived from the POMC gene transcript, the endogenous antagonists ASP and AGRP, and select synthetic derivatives of α-MSH (NDP-MSH, MTII, and SHU9119) will be highlighted (Figure 1).
Figure 1.

Structures of classical melanocortin ligands. (A) POMC-derived naturally occurring agonists (the common His-Phe-Arg-Trp tetrapeptide is highlighted in red). (B) Sequences of the endogenous antagonists AGRP and ASP (the active Arg-Phe-Phe tripeptide is highlighted in blue). (C) NDP-MSH, MTII and SHU9119 (hypothesized pharmacophore region highlighted in red).
2.1 Proopiomelanocortin (POMC) Gene Transcript
The endogenous agonists for the melanocortin receptors are all derived from the POMC gene transcript [15]. Cleavage of the pre-proopiomelanocortin polypeptide sequence by prohormone convertases (PC) generates the melanocortin agonist ligands α-MSH, β-MSH, γ-MSH, and ACTH, as well as other peptides including β-endorphin and β-lipotropin [29-31]. Common to the endogenous melanocortin agonists is a His-Phe-Arg-Trp tetrapeptide sequence which is hypothesized to be the molecular recognition sequence for these ligands (Figure 1A). This sequence is the minimally active truncation product that possesses agonist activity in the classic frog and lizard skin bioassays [32, 33].
Since the endogenous agonists are derived from the POMC gene, the absence of the agonists in POMC-null individual has many effects on pigmentation (MC1R), steroidogenesis (MC2R), and weight gain (MC4R) [34-39]. Following the initial report of POMC-null humans, POMC knock-out (KO) mice were generated by removing the coding region for POMC derived peptides [40, 41]. Similar to the phenotype observed in POMC-null humans, POMC KO mice experienced hyperphagia (MC4R), altered pigmentation (MC1R), and hypocortisolism (MC2R). While it was initially reported that adrenal glands were absent in POMC KO mice [40], it was subsequently observed that POMC mice possess adrenal glands that are significantly smaller than adrenal glands found in wildtype mice [41-43]. An intraperitoneal injection of an exogenous synthetic melanocortin ligand was able to alter the weight gain and pigmentation changes observed in these mice [40]. Untreated, the absence of the POMC gene is fatal in humans [36], underscoring the many critical functions these endogenous ligands perform in vivo.
2.1.1 α-MSH
The α-MSH peptide is derived from the N-terminal 13 residues of ACTH (Figure 1A) and is highly conserved across mammalian species. Both termini of α-MSH are modified, with the N-terminal acetylated and the C-terminal carboxyamidated (Figure 1A). Acetylation of the N-terminal has been demonstrated to increase the stability of α-MSH compared to des-acetylated α-MSH [44, 45]. The full length peptide possesses nonselective sub-nanomolar to nanomolar potencies at the MC1R, MC3R, MC4R, and MC5R [46, 47]. Alanine scans of α-MSH have also indicated the importance of the Met4, Phe7, Arg8, and Trp9 positions for binding and functional activity at the mouse MC1R and rat MC3R [48, 49]. A 2016 report examining the cloned mouse receptors indicated that in addition to positions Met4, Phe7, Arg8, and Trp9, the Glu5 and His6 positions also affect functional activity [47]. Expression of α-MSH in the central nervous system is predominantly in the hypothalamus [50]. Expression of α-MSH is dispersed throughout the arcuate nucleus, however it is found more densely in the lateral regions where it is synthesized [50, 51]. Other locations of α-MSH expression include the dorsomedial nucleus of the hypothalamus (DMH), fibers in the medial preoptic, and the paraventricular, periventricular, and anterior hypothalamic nuclei [50, 52]. The ability of α-MSH to decrease food intake in rodents following intracerebroventricular (icv) administration and alter the skin/hair coloration of humans and small mammals when dosed peripherally demonstrate the importance of this ligand in the regulation of several important pathways [53-59].
2.1.2 β-MSH
The peptide β-MSH consists of 22 amino acids from the POMC gene transcript and is expressed in the hypothalamus [60]. Unlike α-MSH discussed above, β-MSH does not have modifications to either terminal position (Figure 1A). Rodents cannot produce mature β-MSH due to the lack of a di-basic cleavage site [31], although β-MSH has higher affinity at both the human MC4R and the rat MC4R when compared to α-MSH (30- and 4-fold, respectively) [61]. At the human melanocortin receptors, β-MSH has reported to possess single digit nanomolar binding affinity at the MC1R, approximately 10-, 300-, and 12,000-fold higher affinity when compared to the MC3R, MC4R, and MC5R [62, 63]. It was reported that icv administration of β-MSH decreased spontaneous food intake in rats, and was as potent as α-MSH in rats that were fasted for 24 h [64]. However, a separate study showed that β-MSH had no significant inhibition of food intake after fasting for 48 h [57], perhaps due to the increased fasting time overwhelming the pharmacological response. In humans, β-MSH has also been shown to be important in the regulation of energy homeostasis. A missense mutation of the POMC gene transcript encoding region (Tyr221Cys) producing a nonfunctional β-MSH has been reported [65]. Obese children with the Tyr221Cys mutation experience hyperphagia and increased linear growth, similar phenotypes to those observed in MC4R-deficient individuals [65]. Another mutation in the POMC gene (Arg236Gly) generates a β-MSH/β-endorphin fusion protein which cannot activate the MC4R and results in a similar phenotype [66], underscoring the importance of β-MSH as a physiologically relevant melanocortin ligand.
2.1.3 γ-MSH
The N-terminal domain of POMC encodes for the three γ-MSH peptides, consisting of γ1-MSH, γ2-MSH, and γ3-MSH (Figure 1A). The 23-residue N-glycosylated γ3-MSH [67, 68] can be further cleaved to γ2-MSH (N-terminal 12 amino acids of γ3-MSH) and γ1-MSH (N-terminal 11 amino acids of γ3-MSH with a C-terminal carboxyamidate). An alanine positional scan of γ2-MSH indicated residues Met3, His5, Phe6, Arg7, and Trp8 were all functionally important for stimulation of the MC3-5R, similar to residues in α-MSH important for activity [69]. Differences in functional receptor selectivity have been observed between species, where γ2-MSH possessed a 100-fold selectivity for the human (h)MC3R over the hMC5R [69, 70], whereas there was no potency difference between the mouse (m)MC3R and mMC5R [71]. When the activity of γ2-MSH was compared in parallel between the MC1R, MC3R, MC4R, and MC5R, both the mouse and human MC1R and MC4R possessed similar sub-micromolar potencies [71, 72], although the potency of γ2-MSH at the hMC3R [72] was approximately 100-fold lower than previous reports [9, 69, 70]. Expression of γ-MSH in the brain is predominantly in the pituitary and hypothalamic arcuate [73-76], but has also been reported in the adrenal medulla [77]. The greater than 10-fold selectivity of γ2-MSH for the MC3R over the MC4R led to several investigations of the role the MC3R may play on food intake in vivo by administering γ-MSH ligands, with mixed results. While icv administration of γ1-MSH did not inhibit food intake in rats after a 48 h fast [64], icv administration of γ2-MSH in rats fasted for 48 h caused a significant, yet delayed inhibition of food intake [57]. In another report, icv administration of γ2-MSH in rats fasted for 24 h yielded no effect on food intake [78], confounding the role γ-MSH peptides may play in regulating food intake. Additional studies of γ-MSH ligands have examined the role of this ligand on cardiovascular, sodium, and blood pressure regulation [79-81].
2.1.4 ACTH
Pro-ACTH is cleaved by PC1 in the anterior pituitary corticotrophs to produce ACTH(1-39) [82, 83], which can be further processed through PC2 to produce ACTH(1-13)NH2 and α-MSH primarily in the pars intermedia of the anterior lobe of the pituitary [83]. As the only endogenous ligand known to stimulate the MC2R, ACTH is the only endogenous agonist that can stimulate all five melanocortin receptor subtypes [84]. While full length ACTH is 39 residues long, ACTH(1-24) is believed to be the molecular recognition domain while ACTH(25-39) is hypothesized to protect ACTH(1-24) from enzymatic degradation [85]. The N-terminal ACTH(1-24) possessed activity at the central melanocortin receptors, as a 4 μg dose of ACTH(1-24) injected via the lateral ventricle results in 70-80% decreased food intake in rats after a 24 h fast [86]. Direct injection of the same dose into the ventromedial hypothalamus also decreased food intake in free feeding rats during the nocturnal phase [86]. An anorectic effect was reported 4 hours after icv administration of ACTH(1-24) for doses as low as 0.05 μg/animal in mice and 10 μg/animal in rabbits [87]. Exogenous ACTH-mediated feeding inhibition is believed to be controlled solely by the central nervous system and not through peripheral feeding-regulatory pathways [88]. Doses up to 200 μg/kg of ACTH(1-24) administered subcutaneously in rats has no effect on feeding behaviors [88].
2.2 Agouti and AGRP
Unique to the melanocortin system is the presence of two endogenous antagonists, ASP and AGRP, the only naturally occurring GPCR antagonists discovered to date (Figure 1B). Full-length ASP consists of 132 amino acids and expression in the skin has been shown to affect pigmentation through MC1R antagonism [89, 90]. The active form of ASP has been hypothesized to be ASP(23-132), following cleavage of the N-terminal 22 residue signal peptide [91, 92]. The C-terminal domain possesses five disulfide bonds and was found to be equipotent to the full-length peptide [90], with an Arg-Phe-Phe tripeptide sequence shown to be critical for binding to the MC1R [93]. Ectopic expression of agouti due to a mutation at the agouti locus results in the lethal yellow strain of mice (Ay) [89, 94]. The constant antagonism of the MC1R is characterized by overexpression of the skin pigment pheomelanin, resulting in the observed yellow coat color [90, 94]. These mice are also characterized by increased weight gain and increased linear growth [95], characteristic of altered MC4R signaling. Indeed, ASP was found to be a competitive antagonist at the MC4R, but did not interact with the MC3R or MC5R [96]. In a subsequent publication, a synthetic C-terminal agouti fragment with two amino acid substitutions (Q115Y and S124Y, corresponding to the homologous residues in AGRP) was reported to antagonize the MC3R, as well as the MC1R and MC4R [97]. These substitutions to form agouti-YY were required for proper protein folding to generate sufficient quantities of the ligand for in vitro assays and NMR characterization [97]. In addition to the skin, ASP has also been found to be expressed in testis, ovary, and adipose tissue [98].
Similar to ASP, AGRP possesses a C-terminal domain with five disulfide bonds that is as active as the full length protein (Figure 1B), as well as an Arg-Phe-Phe tripeptide sequence shown to be critical for antagonist function [18, 99]. The C-terminal domains of agouti-YY, AGRP, and a truncated “mini-AGRP” have all been shown to possess similar structures by solution NMR [97, 100-102]. Despite these structural similarities, these antagonists possess different pharmacological profiles at the melanocortin receptor subtypes. While ASP has been shown to antagonize the MC1R, MC3R and MC4R, AGRP is not an antagonist at the MC1R but does interact with the centrally expressed MC3R and MC4R [17, 18]. Truncated and chimeric ASP-AGRP ligands indicated that the C-terminal loop of ASP was responsible for MC1R selectivity [103]. An additional difference between the antagonists is AGRP has been demonstrated to be further processed into AGRP(83-132), the proposed functional form in vivo [104]. Expression of AGRP is primarily in the arcuate nucleus, the adrenal cortex, posterior hypothalamus, paraventricular nucleus regions of the brain [50, 98, 105]. Similar to the Ay strain in mice, ectopic expression of AGRP results in mice displaying hyperphagia and increased linear growth, purportedly to be due to MC4R antagonism [18, 106]. The orexigenic effect of AGRP(83-132) has been demonstrated to last up to 7 days [107, 108], indicating a long-term mechanism for inducing increased food intake.
2.3 Synthetic Ligands
Since the sequence of α-MSH was reported in 1957 [27], this peptide has been subjected to numerous structure-activity relationship studies, including the classic truncation and alanine-positional scanning experiments [32, 33, 47-49]. Although α-MSH possesses nanomolar to sub-nanomolar potencies at the MCRs, key discoveries led to synthetic compounds with increased potency, length of activity, and altered receptor pharmacological profiles.
2.3.1 NDP-MSH (Melanotan-I)
The [Nle4,DPhe7]α-MSH (NDP-MSH) ligand was reported to have enhanced potency, increased resistant to proteolysis, and increased duration of action relative to α-MSH in 1980 [109]. Two amino acid residues, Nle4 and DPhe7, differ between NDP-MSH and α-MSH (Figure 1C). The methionine to norleucine substitution in position 4 was selected since the methionine amino acid was reported to be prone to oxidation when attempting to radiolabel α- or β-MSH [110, 111]. This modification was shown to increase potency relative to α-MSH [111, 112]. The Phe7 to DPhe7 substitution was explored due to the observation that heat-alkali treatment of α-MSH enhanced activity [113], and the Phe7 position was a major site of racemization [109]. Incorporation of the two modifications resulted in the NDP-MSH ligand, a sub-nanomolar, nonselective melanocortin receptor agonist. Truncation studies of NDP-MSH indicated an Ac-DPhe-Arg-Trp-NH2 tripeptide sequence to be the minimally active fragment in both the frog skin bioassay and at the cloned MCRs [46, 114]. An alanine-positional scan of NDP-MSH reported decreased potencies when either the DPhe7 or Trp9 positions were substituted, indicating the importance of these two aromatic residues for the high potency of NDP-MSH [47]. Thirty-four years after its discovery, NDP-MSH was approved in the European Union as a treatment for adult erythropoietic protoporphria in 2014 [115].
2.3.2 MTII (Melanotan-II)
In 1989, a series of lactam cyclized α-MSH/NDP-MSH analogs were synthesized in order to develop more potent and prolonged-acting melanocortin ligands [116, 117]. Due to a hypothesized salt bridge between the Glu5 and Lys11 of α-MSH/NDP-MSH based upon NMR and computer modeling, truncated ligands were cyclized through a lactam bridge between positions 5 and 10, maintaining the His-DPhe-Arg-Trp active tetrapeptide sequence of NDP-MSH. Truncation of three residues from both the N- and C-termini, in addition to Glu5 to Asp5 and Gly10 to Lys10 substitutions and subsequent lactam bridge formation resulted in MTII, a potent, non-selective melanocortin ligand with agonist activity at the MC1R, MC3R, MC4R, and MC5R [116, 117]. Since its discovery, MTII has been used as an in vitro and in vivo probe, with central icv administration of MTII inhibiting food intake in mice [5].
2.3.3 SHU9119
The MTII scaffold has been utilized in many structure-activity relationship studies to develop new melanocortin ligands with different selectivity and potency profiles. An early report substituted the DPhe of MTII with a DNal(2′) residue to generate SHU9119 (Figure 1C) [118]. The alteration of one residue changed the pharmacology of the resulting ligand: while SHU9119 maintained potent agonist activity at the MC1R and MC5R, it possessed sub-nanomolar antagonist potency at the MC4R and antagonist/partial agonist activity at the MC3R [118]. As the first peptide ligand discovered with potent antagonist activity at the MC3R and MC4R, icv administration of SHU9119 was shown to significantly increase food intake in mice [5, 108].
3. Selective Compounds
There have been a number of purported selective ligands published for the melanocortin receptor subtypes, with varying degrees of selectivity depending on the definition of the authors. A search of “selective melanocortin ligands” in PubMed (as of October 2016) yields 41 results from 2006 to 2016 alone, with many of these papers reporting a number of ligands. For these reasons, the current review is not meant to provide a comprehensive review of every selective melanocortin ligand, but to highlight selective compounds developed since 2006. For the purposes of this review, a selective compound must possess a 100-fold potency difference between at least two melanocortin receptors. This review will also only discuss ligands with potencies determined at the MC1R, MC3R, MC4R, and MC5R; the MC2R will not be discussed as it is only stimulated by ACTH.
3.1 “Selective” Melanocortin Agonists
In this section, agonists were selected for discussion when functional data (EC50 values) at the MC1R, MC3R, MC4R, and MC5R were determined. Ligands for which activity was determined at three or fewer receptors were not included. The search terms “melanocortin” and “melanocortin agonist” were used to search PubMed databases for papers containing agonist melanocortin ligands, focusing on ligands reported since 2006. Selective ligands (ligands with 100-fold differences in potency between at least two receptors) are listed in Table 1 and Table 2. Compounds were separated based upon whether they were assayed at the human (Table 1) or mouse (Table 2) melanocortin receptors. Ligands were additionally divided by selectivity for a particular receptor subtype. The same ligand may be selective for multiple receptors and are listed multiple times in the tables.
Table 1.
Table of selective melanocortin agonists for the human MCRs. Selectivity was defined to be at least 100-fold more potent between at least two receptor subtypes. Ligands are grouped by receptor selectivity. In some publications, numerous ligands were reported with the same selectivity profiles. In these instances, only the most potent ligands were chosen.
| Selective For: | First Author | Year | Ref | Compound ID | Compound EC50 (nM) | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| hMC1R | hMC3R | hMC4R | hMC5R | |||||
| hMC1R | Bednarek, Maria A | 2007 | [119] | 11 | 1.4 (59%) | 0.54 (43%) | 4% @ 2.5μM | 0.11 |
| Cai, Minying | 2015 | [121] | 1 | 15 | >10000 | >10000 | 7 | |
| Bednarek, Maria A | 2007 | [119] | 5 | 0.23 (67%) | 3% @ 2.5μM | 0% @ 2.5μM | 0.088 | |
| Bednarek, Maria A | 2007 | [120] | 2 | 90 | 0% @ 10μM | 0% @ 10μM | 50 | |
| Conde-Frieboes, K | 2011 | [122] | 6a | 12 | >10000 | 700 | >10000 | |
| Conde-Frieboes, K | 2011 | [122] | 7 | 16 | >10000 | 470 | >10000 | |
| Cai, Minying | 2015 | [121] | 15 | 3.7 | >10000 | >10000 | >10000 | |
| Cai, Minying | 2015 | [121] | 8 | 2.1 | >10000 | >10000 | 450 | |
| Bednarek, Maria A | 2007 | [123] | 2 | 2.4 | 34% @ 1μM | 22% @ 2.5μM | 26% @ 1.5μM | |
|
| ||||||||
| hMC3R | Bednarek, Maria A | 2007 | [119] | 11 | 1.4 (59%) | 0.54 (43%) | 4% @ 2.5μM | 0.11 |
| Carotenuto, Alfonso | 2015 | [127] | 2 | 940 | 1.9 | >1000 | 10 | |
|
| ||||||||
| hMC4R | Hong, Qingmei | 2010 | [128] | 21A | 1200 | 90 | 5.4 | 490 |
| Hong, Qingmei | 2010 | [128] | 23A | 520 | 79 | 6.4 | 1000 | |
| Hong, Qingmei | 2010 | [128] | 20B | 540 | 1800 | 13 | >5000 | |
| Hong, Qingmei | 2010 | [128] | 25B | >5000 | 2500 | 19 | >5000 | |
| Cai, Minying | 2015 | [121] | 17 | >10000 | >10000 | 5 (67%) | >10000 | |
|
| ||||||||
| hMC5R | Bednarek, Maria A | 2007 | [119] | 11 | 1.4 (59%) | 0.54 (43%) | 4% @ 2.5μM | 0.11 |
| Cai, Minying | 2015 | [121] | 1 | 15 | >10000 | >10000 | 7 | |
| Bednarek, Maria A | 2007 | [119] | 5 | 0.23 (67%) | 3% @ 2.5μM | 0% @ 2.5μM | 0.088 | |
| Bednarek, Maria A | 2007 | [120] | 23 | 3.8 (46%) | 8% @ 5μM | 3% @ 5μm | 0.15 | |
| Bednarek, Maria A | 2007 | [119] | 7 | 20% @ 5μM | 54% @ 5μM | 36% @ 5μM | 0.41 | |
Table 2.
Table of selective melanocortin agonists for the mouse MCRs. Selectivity was defined to be at least 100-fold more potent between at least two receptor subtypes. Ligands are grouped by receptor selectivity. In some publications, numerous ligands were reported with the same selectivity profiles. In these instances, only the most potent ligands were chosen.
| Selective For: | First Author | Year | Ref | Compound ID | Compound EC50 (nM) | |||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| mMC1R | mMC3R | mMC4R | mMC5R | |||||
| mMC1R | Todorovic, Aleksandar | 2007 | [124] | 10 | 5.9 | >100000 | 41 | 30 |
| Todorovic, Aleksandar | 2007 | [124] | 11 | 14 | >100000 | >100000 | 25 | |
| Todorovic, Aleksandar | 2007 | [124] | 12 | 5.1 | 10% @ 100μM | 63% @ 100μM | 19 | |
| Singh, Anamika | 2014 | [125] | AMW6103 | 0.81 | 5300 | 440 | 31 | |
| Singh, Anamika | 2013 | [126] | 4 | 1.9 | 4500 | 290 | 415 | |
|
| ||||||||
| mMC3R | Todorovic, Aleksandar | 2016 | [47] | [Ala4]α-MSH | 12000 | 17 | 120 | 60 |
|
| ||||||||
| mMC4R | Todorovic, Aleksandar | 2007 | [124] | 10 | 5.9 | >100000 | 41 | 30 |
| Hess, Shmuel | 2008 | [129] | 4 | 28 | 1300 | 4 | 2 | |
| Proneth, Bettina | 2008 | [130] | 11 | 280 | 240 | 2.8 | 2.9 | |
|
| ||||||||
| mMC5R | Todorovic, Aleksandar | 2007 | [124] | 10 | 5.9 | >100000 | 41 | 30 |
| Hess, Shmuel | 2008 | [129] | 4 | 28 | 1300 | 4 | 2 | |
There have been a number of selective peptide, peptidomimetic, and small molecule ligands reported for the melanocortin receptors. Of the agonist compounds selective for the hMC1R (Table 1), one was selective for the hMC1R over the hMC4R [119], three were selective for the hMC1R over the hMC3R and hMC4R [119-121], two were selective for the hMC1R over the hMC3R and hMC5R [122], and three possessed at least 100-fold selectivity for the hMC1R over the hMC3R, hMC4R, and hMC5R [121, 123]. For agonist compounds selective for the mMC1R (Table 2), one ligand was selective for the mMC1R over the mMC3R [124], three were selective for the mMC1R over the mMC3R and mMC4R [124, 125], and one compound was at least 100-fold selective for the mMC1R over the three remaining receptors [126]. Of the ligands selective for the hMC1R or mMC1R, three were based upon the linear structure of α-MSH [119, 123], four were substitutions of the MTII/SHU9119 scaffold [120, 121], five were small molecules [122, 124], and two were cyclic analogues of AGRP possessing a thioether heterocyclic [125, 126].
Unlike the other melanocortin receptors, selectivity for the MC3R has been more difficult to achieve. Two selective agonists have been reported for the hMC3R (Table 1): a partial agonist at the hMC3R was selective over the hMC4R [119] and one compound has been reported selective for the hMC3R over the hMC1R and hMC4R [127]. At the mMC3R (Table 2), one compound has also shown to be selective for the mMC3R over the mMC1R [47]. Of the MC3R selective compounds, two were derivatives of α-MSH [47, 119] and one based upon the MTII/SHU9119 template [127].
Perhaps due to the correlation between the MC4R and obesity, a number of selective ligands have been reported for this receptor subtype. At the hMC4R (Table 1), one compound has been reported for selectivity for the hMC4R over the hMC1R [128], one compound selective for the hMC4R over the hMC5R [128], one ligand for the hMC4R over the hMC3R and hMC5R [128], and two were at least 100-fold selective for the hMC4R over the remaining three receptor subtypes [121, 128]. At the mMC4R (Table 2), two compounds were selective for the mMC4R over the mMC3R [124, 129] and another ligand was reported 100-fold selective for the mMC4R over the mMC1R [130]. Of the selective ligands reported for the MC4R, five were small molecules [124, 128], two were based upon the His-DPhe-Arg-Trp tetrapeptide sequence [129, 130], and one was a derivative of the MTII/SHU9119 scaffold [121].
Investigations into the MC5R have also resulted in several selective ligands. At the hMC5R (Table 1), one compound was selective for the hMC5R over the hMC4R [119], three were selective for the hMC5R over the hMC3R and hMC4R [119-121], and one was selective for the hMC5R over the remaining three receptor subtypes [119]. At the mMC5R (Table 2), two compounds have been reported to be selective for the mMC5R over the mMC3R [124, 129]. Of compounds selective for the MC5R, three were derivatives of α-MSH [119], two were substitutions within the MTII/SHU9119 scaffold [120, 121], one was based upon the tetrapeptide His-DPhe-Arg-Trp [129], and one was a small molecule [124].
3.2 “Selective” Melanocortin Antagonists
Whereas agonists at the melanocortin receptor stimulate the production of cAMP, antagonists inhibit the ability of an agonist to stimulate cAMP production. Antagonists must therefore be assayed in the presence of an agonist, preferably at multiple concentrations of antagonist to generate a Schild analysis [131]. There are no studies which reported functional antagonist pA2 values at each of the four melanocortin receptors investigated in this review. Therefore, publications were selected when pA2 values were reported at a minimum of two of the four melanocortin receptors, and functional agonist data at the remaining receptors. For melanocortin receptor antagonists, selectivity was defined as a 100-fold difference in potency between the two melanocortin receptors assayed for antagonist activity. Since pA2 values represent a log scale, compounds with pA2 values different by two pA2 units are 100-fold different. Search terms used for the analysis of melanocortin antagonists in the PubMed database were “melanocortin” and “melanocortin antagonist,” focusing on ligands reported 2006 - 2016.
From these search parameters, three studies reported selective pA2 values at two melanocortin receptors (mMC3R and mMC4R) and reported functional agonist EC50 values for the other two receptors (mMC1R and mMC5R, Table 3) [132-134]. For the sake of clarity, antagonists in Table 3 were divided into two sections: ligands with antagonist activity at the mMC4R but no agonist or antagonist activity observed at the mMC3R at concentrations up to 10 μM (pA2 < 5) and antagonists that were active at the mMC3R and mMC4R. The antagonists reported by Doering et al. were derivatives of the tetrapeptide Ac-Trp-DPhe(p-I)-Arg-Trp-NH2 [132]. Scaffolds reported by Ericson et al. were AGRP macrocyclic derivatives [133]. The antagonist reported by Lensing et al. possessed the structure Ac-His-DNal(2′)-Arg-Trp-(PEDG20)-NH2. No ligands within the search parameters were MC1R, MC3R or MC5R selective antagonists. Interestingly, although most reported MC3R antagonists possess partial agonist activity at the MC3R, the peptides developed by Doering et al. and Ericson et al. did not possess partial agonist activity at the MC3R [132, 133].
Table 3.
Table of selective mouse MC4R antagonists. Selectivity was defined to be greater than 100-fold more potent between at least two receptors (two pA2 units). Ligands are grouped by possessing or not possessing agonist or antagonist activity at the mMC3R at concentrations up to 10 μM (pA2 < 5). In some publications, numerous ligands were reported with the same selectivity profiles. In these instances, only the most potent ligands were chosen.
| First Author | Year | Ref | Compound ID | Receptor Activity | |||
|---|---|---|---|---|---|---|---|
|
| |||||||
| mMC1R EC50 (nM) | mMC3R pA2 | mMC4R pA2 | mMC5R EC50 (nM) | ||||
| Ericson, Mark D | 2015 | [133] | 17 | 20000 nM | < 5 | 8.2 | >100000 |
| Doering, Skye R | 2015 | [132] | 3 | 8300 nM | < 5 | 7.1 | 24000 nM |
| Ericson, Mark D | 2015 | [133] | 10 | 1500 nM | < 5 | 7 | >100000 |
| Doering, Skye R | 2015 | [132] | 2 | 21000 nM | < 5 | 6.3 | 7800 nM |
|
| |||||||
| Ericson, Mark D | 2015 | [133] | 22 | 75% @ 100μM | 6.9 | 9.1 | >100000 |
| Lensing, Cody J | 2016 | [134] | 10 | 110 nM | 6.1 | 8.4 | 70% @ 100μM |
| Doering, Skye R | 2015 | [132] | 6 | 5500 nM | 5.9 | 8.3 | 20000 nM |
| Doering, Skye R | 2015 | [132] | 7 | 5800 nM | 5.7 | 8.3 | 20000 nM |
| Doering, Skye R | 2015 | [132] | 1 | 2000 nM | 5.4 | 7.8 | 2800 nM |
| Ericson, Mark D | 2015 | [133] | 19 | 13000 nM | 5.6 | 7.8 | >100000 |
4. Small Molecule Ligands
This section will focus on the development of small molecule melanocortin ligands. This topic has been previously reviewed in 2005 [135] and 2007 [136], so the current review will focus primarily on small molecules published since 2008. Numerous non-peptide small molecule ligands have been developed for the melanocortin receptors. In particular, the MC4R was heavily targeted by the pharmaceutical industry due to correlation of the MC4R with obesity [8]. Since MC4R agonists demonstrated an ability to decrease food intake in rodent models [5], small molecule MC4R agonists were investigated as potential therapeutics to promote weight loss. However, a clinical trial in 2009 indicated potent side effects including an increase in blood pressure with the use of the peptide agonist LY2112688 (Ac-DArg-c[Cys-Glu-His-DPhe-Arg-Trp-Cys]-NH2) [25] and decreased the rate of MC4R-selective small molecule agonist development as anti-obesity treatments. The potential uses of MC4R agonists as pro-erectile agents or MC4R antagonists as weight gain therapeutics for cachexia patients have also been investigated [137-139].
While many different small molecule scaffolds have been reported, they can broadly be divided into two categories. One category originates from the selective small molecule ligand developed by Merck for the MC4R, a tetrahydroisoquinolone ligand THIQ. The other category consists of de novo designs or resulted from library screening of ligands not based upon the THIQ scaffold. This review will discuss both sets of molecules, beginning with the THIQ-based ligands. The focus will be on small molecule ligands that have been disclosed and functionally characterized in peer-reviewed publications. Molecules, including SNT207707, SNT209858, and BL-6020/979, that have been described but not pharmacologically characterized or which structures have not been clearly identified outside of the patent literature are not included [140, 141]. When describing the compounds derived from the THIQ ligand, the compounds are described in the approximate order of their publications.
4.1 Historic Overview of Small Molecule Melanocortin Ligands
The first small molecule ligands reported for the melanocortin receptors in 1999 were heterocycles containing a β-turn motif that possessed micromolar agonist potency at the MC1R, but were inactive at the MC3R and MC4R at concentrations up to 100 μM (Figure 2, EL1 and EL2) [142]. In 2002, two groups reported small molecules possessing activity at the MC4R. Bondebjerg et al. reported a thioether scaffold that generated one ligand possessing sub-micromolar potencies at the MC1R, MC4R, and MC5R (Figure 2, JB1n) [143]. The first small molecule with single-digit nanomolar potency at the MC4R, THIQ, was described by Sebhat et al. (Figure 2, THIQ) [144]. Following these initial ligands, many additional compounds have been reported as described herein and in previous reviews [135, 136].
Figure 2.
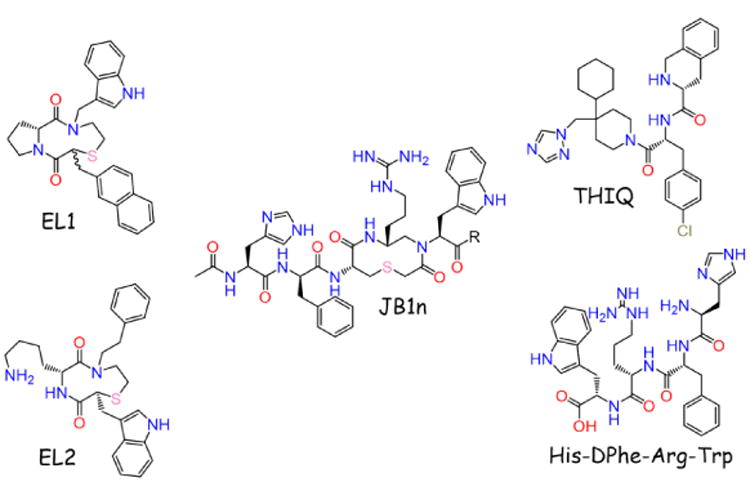
First reported melanocortin small molecule ligands, along with the hypothesized Ac-His-DPhe-Arg-Trp-NH2 melanocortin pharmacophore. The C-terminus of JB1n was reported as a mixture of an amide and carboxylic acid.
The postulated agonist pharmacophore of the endogenous melanocortin ligands is a His-Phe-Arg-Trp tetrapeptide. It has been shown that stereochemical inversion of the Phe to DPhe increased potency, as discussed in section 2.3.1 NDP-MSH (Melanotan-I). In efforts to generate a small molecule melanocortin ligand, a group at Merck noted the similarity between the melanocortin His-Phe-Arg-Trp sequence and the active core of the growth hormone secretagogue peptide GHRP-6 (His-DTrp-Ala-Trp) [144]. A clinical candidate was developed by Merck for GHRP-6 with a spiroindanyl piperidine (Figure 3) functioning as an Ala-Trp mimetic. A search of the Merck sample collection for similar compounds resulted in the optimization of the THIQ melanocortin-4 selective agonist (Figure 3), a nanomolar potent ligand with greater than 100-fold selectivity for the hMC4R over the remaining hMCRs [144]. Comparing the structures of His-DPhe-Arg-Trp and THIQ (Figure 3), it may be observed that the His is replaced with a constrained tetrahydroisoquinoline moiety in THIQ, DPhe by a 4-chlorophenyl ring, and the Trp may be in close proximity to the triazole heterocycle. Later modifications by Merck included replacing the 4-chlorophenyl with a 4-fluorophenyl to improve potency at the hMC4R, and potentially minimizing off-target effects by replacing the triazole with a t-butylamide group and substituting the tetrahydroisoquinoline moiety with a piperazine ring (MB243, Figure 3) [145]. Another key contribution from the Merck group was the discovery of the t-butylpyrrolidine containing MC4R-selective ligands (including MK0493, Figure 3) [146], a scaffold that is evident in many of the molecules presently reviewed. Through the successive generation of ligands, a piperidine core was maintained along with a halogenated phenyl ring (Figure 3). While many additional structure-activity relation studies and developments around these core structures have been reported, this review updates compounds disclosed since 2008 due to comprehensive reviews published from 2007 and earlier [135, 136].
Figure 3.
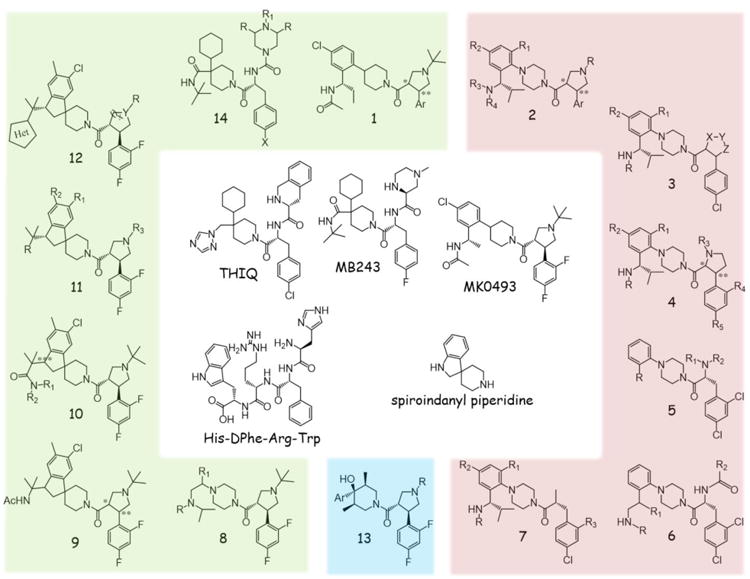
Small-molecule melanocortin scaffolds derived from the Merck compound THIQ. Scaffolds are grouped by company disclosing the structures: Merck (green), Neurocrine Biosciences, Inc. (red), or Pfizer (blue). In the center, THIQ, MB243, and MK0493 are select Merck compounds disclosed before 2007, the His-DPhe-Arg-Trp hypothesized melanocortin pharmacophore, and an illustrative spiroinanyl piperidine core structure.
4.2 THIQ-derived Small Molecule Scaffolds
Guo et al. reported a series of N-tert-butylpyrrolidine analogues (1, Figure 3), maintaining the 4-chloro phenylpiperidine ring of MK0493 with an elongated ethyl group replacing the methyl next to the acetamide [147]. In efforts to improve MC4R binding and functional activity, they investigated aromatic substitutions to replace the 2,4-difluorophenyl group. Both 4-chloro-2-fluorophenyl and 4-bromo-2-fluorophenyl substitutions increased affinity and agonist potency, indicating larger halogens in the 4-position may be beneficial [147]. In examining the affinity and functional effects of the trans diastereomers relative to the pyrrolidine ring (1, stereocenters indicated by * and ** in Figure 3) of select compounds, they reported that the (*,** in Figure 3) S,R isomers acted as full agonists, while the R,S isomers maintained similar affinity but resulted in decreased cAMP production. This work resulted in several nanomolar ligands for the hMC4R [147].
Chen et al. from Neurocrine Biosciences, Inc. described a series of pyrrolidine derivatives possessing a piperazine ring in place of the piperidine ring of MK0493 (2, Figure 3) [148]. This series optimized a previous reported ligand set combining the pyrrolidine moiety of MK0493 with other work utilizing piperazinbenylamines [149]. Substitutions at the aromatic ring attached to the pyrrolidine, the nitrogen of the pyrrolidine, the 2 and 4 positions of the piperazinephenyl ring, and the benzylamine nitrogen were explored using binding affinity and agonist potency at the hMC4R. Off the pyrrolidine ring, a 4-chlorophenyl group possessed the highest affinity and potency [148]. For substitution at the pyrrolidine nitrogen, a tetrahydropyran derivative exhibited the highest affinity and potency. Similar to the work by Guo et al. [147] investigating the two trans pyrrolidine diastereomers indicated the (*,** in Figure 3) S,R compound possessed high agonist activity while the R,S ligand possessed high affinity but was unable to stimulate cAMP production. Relatively small differences in potencies were observed when either a 2-fluoro or 4-methyl phenylpiperazine was used. Further investigation at the benzylamine position indicated that attachment of a N,N-dimethylaminopropionic acid through an amide bond resulted in the highest binding affinity, potency, and efficacy (Ki = 1.0 nM, EC50 = 3.8 nM, 122 % max signal for α-MSH) and possessed 200-fold selective binding for the hMC4R over other receptor subtypes [148]. This compound was shown to dose dependently decrease food intake in diet-induced obese (DIO) mice [148].
Maintaining the 4-chlorophenyl group, the Neurocrine Biosciences group next investigated the pyrrolidine ring by substituting in tetrahydrothiophenes and tetrahydrofurans in efforts to develop more potent hMC4R antagonists for cachexia treatment (3, Figure 3) [138]. Additionally, they explored 4-chloro, 4-trifluoromethyl, 4-methyl, or 2-fluoro phenylpiperazine substitutions. As desired, none of the compounds significantly increased cAMP production. Select compounds possessed nanomolar binding affinity and were able to block α-MSH stimulated cAMP release (with IC50 values of 590 nM or higher) [138], indicating functional antagonism of the MC4R. Notably, none of the compounds significantly increased cAMP production.
A similar study by the Neurocrine Biosciences group modified the location of the pyrrolidine nitrogen, explored various substitutions in the 2 and 4 positions of both phenyl ring systems, varied the benzylamide substitution, and attached different carboxylic acids to the pyrrolidine nitrogen (4, Figure 3) [139]. Similar to the tetrahydrothiophenes and tetrahydrofurans (3) [138], this compound series was unable to stimulate hMC4R induced cAMP production even though several compounds possessed nanomolar binding affinities [139]. The most potent compound in this series possessed a 4-chlorophenyl group, a 4-methyl substitution at the phenylpyrrolinidine ring, and an urea off of the pyrrolidine nitrogen. This compound blocked α-MSH stimulated cAMP production with a IC50 value of 93 nM. Interestingly, the cis-isomers of the pyrrolidine substitution were found to impart higher binding affinity, in contrast to the trans-isomers previously reported when the pyrrolidine nitrogen is in a different location (4 versus 1 and 2, Figure 3) [147, 148].
The group at Neurocrine also investigated a series of piperazinebenzylalcohols and related ketones and amine analogs without the pyrrolidine ring (5, Figure 3) [150]. An isopropyl group off of the benzyl position increased binding affinity, similar to the increased affinity for a benzylamine or cyclohexyl-carboxylate at a similar position [151, 152]. A trend for increased binding affinities for benzylamines over the benzylketones or benzylalcohols was observed. The benzylalcohols were all found to possess significantly more binding affinity for the hMC4R over the other hMCRs, though none of the compounds possessed agonist activity. A pharmacokinetic comparison between the most potent alcohol with the corresponding amine indicated similar trends, although the charged amine had slightly higher brain penetration than the uncharged alcohol [150].
With this same general scaffold, the group from Neurocrine also reported a series of analogues with an amine one methylene further removed from the phenylpiperazine rings (6, Figure 3) [153]. Due to previous enhancements in potency by incorporating a basic nitrogen from the phenylpiperazine ring, the group wanted to further remove the nitrogen further removed from this ring system. Several compounds were shown to possess sub-nanomolar binding affinities at the hMC4R, although their reported functional activity was greater than 100 nM [153].
Another non-pyrrolidine scaffold from the Neurocrine group probed the benzylamine position and substitutions on both aromatic rings (7, Figure 3) [154]. Ligands with 6-fluoro, 6-chloro, or 4-methyl substitutions in the phenylpiperazine ring were synthesized, all of which possessed nanomolar binding affinities. Insertion of 4-H, 4-methoxy, and 4-chloro in the halogenated ring resulted in similar affinities. Substitutions at the benzylamine position were also found to be beneficial. This series possessed minimal affinity at the hMC1R and was >40-fold selective for the hMC4R over the hMC3R. One compound evaluated in a mouse cachexia model was able to increase body weight in treated mice versus vehicle [154].
A group from Merck group also utilized a piperazine scaffold possessing a 2,4-difluorophenyl aromatic ring attached to the N-tert-butyl pyrrolidine (8, Figure 3) [128]. Bulkier aliphatic groups (iso-propyl, tert-butyl, or cyclohexyl) were incorporated off of the piperazine ring in a similar orientation to the cyclohexyl group of MB243, in addition to examining a variety of amine substitutions. Both cyclohexyl and iso-propyl substitutions increased potency relative to a tert-butyl group. Select amide substitutions resulted in nanomolar potent MC4R agonists with 10-fold selectivity for this receptor over other melanocortin receptor subtypes [128].
The Merck group also generated a series of analogs using a spiroindane motif attached to the piperidine ring (9-12, Figure 3) [155-158]. The spiroindanyl piperidine motif had previously been utilized in a growth hormone secretagoue project at Merck. It was developed to mimic an Ala-Trp functionality with improved pharmacokinetics parameters for the growth hormone program [144, 157, 159]. They hypothesized this motif may have similar beneficial results in the MC4R ligand project. This scaffold initially maintained the 2,4 difluorophenyl group and N-tert-butylpyrrolidine elements of MK0493 (9, Figure 3). The stereochemistry of the two pyrrolidine ring substitutions was explored [157]. Similar to the pyrrolidine scaffolds with a piperazine (2) or piperidine (1) core [147, 148], a (*,** in Figure 3) S,R substitution possessed more potent binding and agonist potency at the hMC4R, although the R,S substitution in this scaffold still possessed agonist pharmacology with 10-fold decreased potency [157].
As a follow-up study, the Merck group utilized the spiroindanyl piperinde scaffold, but reversed the amide bond off of spiroindane (10, Figure 3) [158]. This modification allowed the rapid screening of many amines through amide bond formation with the carboxylic acid. The stereochemistry of the carboxylic acid attached to the spiroindanyl ring was demonstrated to be important (*** in Figure 3), as an S-conformation increased agonist potency and binding at the hMC4R. From this series, a number of ligands were generated which possessed sub-nanomolar agonist potencies at the hMC4R and were greater than 100-fold selective for the hMC4R over the hMC1R [158].
The Merck group continued to utilize the spiroindanyl piperidine core and examined positions 3 and 4 off the spiroindane phenyl group with additional substitutions off of the spiroinanyl piperidine, and pyrrolidine amine substitutions (11, Figure 3). The 3-chloro, 4-methyl substitutions were found to possess the highest affinity and agonist potency compared to a 3-fluoro, 4-fluoro or 3-fluoro, 4-chloro substitutions. Primarily a nitrile was used off of the spiroindanyl core, although morpholine or pyrrolinol substitutions further increased agonist potency to sub-nanomolar values. While the N-tert-butyl substitution at the pyrrolidine amine was the most potent and possessed the highest affinity, a 2-fold decrease in binding affinity and agonist potency was observed when the N-tert-butyl group was replaced with a tetrahydrofuran analog. The tetrahydrofuran substitution decreased lipophilicity and addressed an unpublished metabolic issue observed for the N-tert-butyl substitution [155]. Combining these data resulted in the 3-chloro, 4-methyl spiroindanyl substitution pattern with a nitrile group off of the spiroindanyl moiety coupled with a tetrahydropyranal amine substitution ligand that possess nanomolar potency at the hMC4R and was greater than 620-fold selective for the hMC4R over the hMC1R [155]. This ligand decreased food intake and bodyweight at 10 mg/kg orally dosed in DIO wt mice and had no significant effect in MC3R/MC4R KO mice [155].
An additional series from the spiroindanyl piperidine scaffold by the Merck group investigated the substitution of a heterocycle ring motif off of the spiroindanyl core, similar to the ring system observed in THIQ (12 and THIQ, Figure 3) [156]. Additionally, they probed substitutions off of the pyrrolidine. Incorporating a cyclopentyl ring with a tertiary nitrogen attached to a methyl group and tetrahydropyran ring resulted in the compound with the highest affinity (0.43 nM) and potency (0.11 nM), and decreased food intake in a rat DIO model when orally dosed at 1 and 3 mg/kg [156].
A series of analogs possessing the N-tert-butylpyrrolidine core with an attached phenylpiperidine ring system were studied by Pfizer for potentially treating male sexual dysfunction. (13, Figure 3) [137]. A previous MC4R agonist developed by this group possessing a 4-alcohol on the phenylpiperidine ring suggested small flanking substitutions on the C3 and C5 position of the piperidine ring had a beneficial effect, and was the basis for their structure-activity relation study. Probing substitutions on the aryl portion of the phenylpiperidine ring and substitution on the pyrrolidine nitrogen led to two compounds, a phenyl/tert-butyl or 4-fluorophenyl/tert-butyl substitution pattern possessing 12 and 18 nM agonist potency at the hMC4R and approximately 100-fold selectivity for this receptor [137]. The phenyl/tert-butyl compound was advanced into a human trial for male erectile dysfunction. A 200 mg dose demonstrated similar efficacy to sildenafil and demonstrated a significant effect compared to the placebo control [137].
As a final small molecule for the MC4R derived from the MB243 scaffold, the Merck group incorporated a substituted piperazine attached through a urea linkage while preserving the halogenated aromatic ring and tert-buyl/cyclohexyl substituted piperidine of MB243 (14, Figure 3).[160] This series was designed to partially activate the receptor with the aims of decreasing food intake while not inducing an erectile response. The 4-chlorophenyl ring substitution did not possess activity at the hMC4R, and so a 4-fluorophenyl motif was used. A cis-methyl substitution at the C3 and C5 positions along with an unsubstituted nitrogen in the piperazine ring resulted in a 4.9 nM binding affinity compound with 22 nM agonist potency at the hMC4R with an efficacy of 59% relative to α-MSH. This compound was able to reduce body weight in an DIO rat model without causing an erectile response, indicating that it was possible to modulate weight without pro-erectile activities in rodents [160].
4.3 De novo designed/library screened small molecule scaffolds
In addition to small molecule melanocortin scaffolds with origins from the THIQ ligand, other novel scaffolds have been described. These de novo scaffolds were the result of serendipitous discoveries, screening GPCR privileged structures, or by adopting peptide structures into small molecule designs (Figure 4)
Figure 4. De novo.
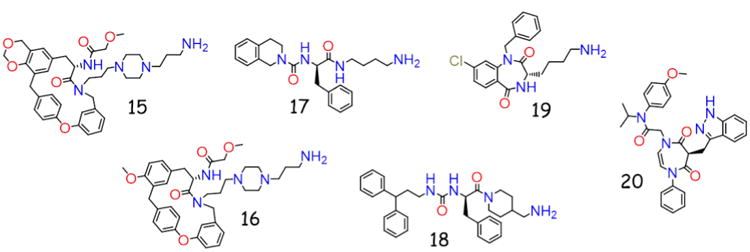
melanocortin ligand small molecule scaffolds.
In a melanocortin program at Novo Nordisk for the treatment of obesity, a scaffold presenting four pharmacophores was synthesized to contain 384 random compounds from 41 building blocks [122]. While possessing a piperazine ring and aromatic substitutions (15 and 16, Figure 4), a cyclophane scaffold was discovered through a random screening approach. The structures of the lead ligands were also initially incorrectly assigned, as the active compounds were side reactions that occurred during syntheses, demonstrating the importance of resynthesizing and screening lead ligands compounds. Small molecules 15 and 16 possessed 12 and 16 nM agonist potency at the hMC1R, 700 and 470 nM agonist potency at the hMC4R, and were inactive at the hMC3R and hMC5R at concentrations up to 10 μM [122]. Compound 16 was also shown to dose-dependently decrease food intake in schedule fed male rats [122].
Another scaffold reported by Singh et al. incorporated an urea motif with an additional aromatic moiety and primary amine (17 and 18 as examples, Figure 4) [161]. This was a follow-up to a previously synthesized library from the same group which reported a micromolar MC3R agonist that was unable to stimulate the MC4R [162]. A series of 27 analogues were synthesized which resulted in five compounds with micromolar agonist potencies at the MCRs [161]. A comparison of 15 and 16 to 17 and 18 shows both possess a primary amine and aromatic moieties in approximately the same orientation, resulting in some similarities between these scaffolds.
Two groups have also reported small molecule melanocortin ligands based upon diazepine scaffolds, a privileged structure for other GPCR systems. Joseph et al. generated a series of 1,4-benzodiazepine-2,5-dione ligands [163], a scaffold previously reviewed to possess activity at the cholecystokinin receptor, opiate receptor, and antitumor properties [164]. The most potent of this compound set (19, Figure 4) resulted in sub-micromolar agonist activity at the MCRs [163]. Another diazepine scaffold was developed from a 1-4-dihydro-[1,4]diazepine-5,7-dione core, reported by Szewczyk et al., after screening compounds from this series against numerous GPCRs [165]. Their analogs (20, Figure 4) possessed partial agonist activity (relative to α-MSH) at the MC1R and MC4R, with sub-nanomolar potencies at these receptors.
5. Sex-Specific Effects of Melanocortin Ligands
Various studies have reported a sexual dimorphism within the melanocortin system. An example is the differences in body weight gained between MC4R knockout male and female mice that are similar to the trends in human genetic epidemiology studies [8, 166-168]. Based on physiological observations of sex differences, it has been hypothesized that melanocortin ligands with sex-specific effects may be developed. This section will review published reports that directly compare the effects of melanocortin ligands in males and females on various physiological properties, summarized in Table 4. Non-pharmacological data is beyond the scope of this review, but has previously been examined [169].
Table 4.
Examples of melanocortin ligands affecting males and females differently.
| Ligand | Effect | Species | Strain | Ref |
|---|---|---|---|---|
| α-MSH | Improved learning tasks in males, but not females. | Rats | Holtzman albino | [175, 176] |
|
| ||||
| MTII | Reduced juvenile play behaviors in males, but not females after neonatal administration | Voles | Prairie Voles | [177] |
|
| ||||
| Ac-Nle-Asp-Trp-DPhe-Nle-Trp-Lys-NH2 | Effected κ-opioid analgesia in females, but not in males | Mice | Crl:CD-1 | [178] |
| C[Gly-Cpg-DNal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 | Effected κ-opioid analgesia in females, but not in males | Mice | Crl:CD-1 | [178] |
| C[(CH2)3CO-Gly-His-DPhe-Arg-DTrp-Cys(S-)]-Asp-Arg-Phe-Gly-NH2 | Reversed morphine hyperalgesia in female mice, but had no effect in males | Mice | C57BL/6J; CD-1 | [179, 180] |
|
| ||||
| AGRP | Reduced energy expenditure (measured by vO2) in female rats more than in male rats. | Rats | Long-Evans | [181] |
| Ac-Trp-DPhe(p-I)-Arg-Trp-NH2 | Different doses necessary to affect energy homeostasis in males and females | Mice | Mixed C57BL/6J and 129/Sv background | [182] |
|
| ||||
| SHU9119 | Increased blood pressure in males, but not in females | Rats | Spontaneously hypertensive rats (SRH) | [183] |
Melanocortin ligands have utilized in basic and clinical research in both males and females for treating sexual function disorders [170]. Although the MTII derivative bremelanotide has shown efficacy in both males and females, bremelanotide is currently only in clinical trials for female sexual desire disorder (For structure, see Figure 6 below) [170-173]. To the authors knowledge, the decision to focus on females for therapeutic development is the first clinical attempt in utilizing sex differences of melanocortin ligands. The differential effects of melanocortin ligands on sexual function in males and females has been extensively reviewed [170, 171, 174], and will not be discussed in further detail herein.
Figure 6.
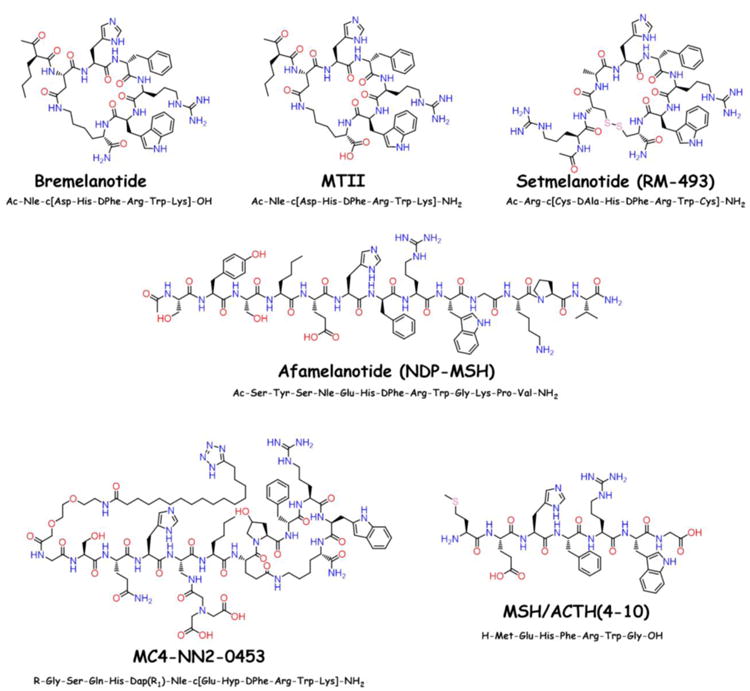
Chemical structures of the melanocortin ligands used in clinical trials from 2011 to 2016. The ligand MTII is included as a comparison to bremelanotide.
In 1977, Beckwith and coworkers reported melanocortin ligands had differential effects on male and female rats [176]. Neonatal administration of α-MSH in male rats resulted in better performance on learning tasks as adults than control animals, an effect not observed in female rats [176]. This group also demonstrated sex differences in response to neonatal administration of α-MSH in an open-field test. Male rats treated with α-MSH demonstrated enhanced effects at 45 days old and 120 days old, but significant effects were observed only at 45 days old in female rats [175]. In more recent studies, neonatal administration of melanocortin agonists to prairie voles affects social behaviors in a sex-dependent manner [177]. Daily neonatal peripheral injection of MTII reduced juvenile play behaviors in males, but not females. In contrast females receiving daily neonatal injection of MTII displayed enhancements in partner preference after non-mated cohabitation with males, which was not observed in males after cohabitation with females. Classical melanocortin agonist responses were observed in both prairie vole sexes after MTII administration, including darkened pigmentation (MC1R activation) and reduced body weight (MC3R/MC4R activation) [177].
Another established line of melanocortin sexual dimorphism has focused on the role of the MC1R in meditating a female-specific mechanism of κ-opioid analgesia [178-180, 184, 185]. Agonist κ-opioid analgesia could be blocked by N-methyl-D-aspartate (NMDA) receptor antagonism in males, but not females [186]. It has been postulated that female mice use a MC1R-mediated pathway instead of the NMDA receptor [178, 180, 184, 187]. It was demonstrated that icv administration of the melanocortin ligands Ac-Nle-Asp-Trp-DPhe-Nle-Trp-Lys-NH2 (pA2=8.4 in the frog skin assay, IC50 = 260, 60, and 910 nM at the hMC3R, hMC4R, and hMC5R) respectively or c[Gly-Cpg-DNal(2′)-Arg-Trp-Glu]-Val-Val-Gly-NH2 (Ki = 53 nM at the hMC1R by Schild analysis, IC50 = 12, 44, and 1300 nM at the hMC3R, hMC4R, and hMC5R, respectively) potentiated κ-opioid analgesia in female but not male mice [178, 188, 189]. The presumed melanocortin antagonism rendered females sensitized to blockage of κ-opioid analgesia by a NMDA receptor antagonist similar to male mice [178]. These melanocortin pharmacological studies were supported by studies in mice and humans that lack functional MC1R, implicating the role of the MC1R over the other melanocortin receptor subtypes in the analgesic response [178, 185, 187]. Similar to rodent studies, women with mutations in the MC1R displayed greater analgesic responses to pentazocine on thermal and ischemic pain stimuli compared to women or men with no variant MC1R [178]. Ovariectomized (OVX) female mice possessed NMDA antagonist sensitivity while treatment with estrogen or progesterone to OVX mice reinstates NMDA resistance, implicating sex hormones for this effect [184, 186, 190, 191].
Further studies on the icv administration of a presumed MC1R antagonist MSG606 (c[(CH2)3CO-Gly-His-DPhe-Arg-DTrp-Cys(S-)]-Asp-Arg-Phe-Gly-NH2) demonstrated this ligand reversed morphine hyperalgesia in female mice, but had no effect in male mice in two different strains (CD-1 and C57BL/6J) [180]. The MSG606 ligand was reported to be have IC50 = 17, 3900, >10000, and 1100 nM, and EC50 = >10000, 59, >10000, and 1300 nM at the hMC1R, hMC3R, hMC4R, and hMC5R, respectively [192]. However, MSG606 was not fully functionally characterized as a competitive MC1R antagonist by a traditional Schild analysis. In 2015, it was observed that both icv and intrathecal (i.t.) administration of MSG606 or a NMDA receptor antagonist in mice followed the same sex-specific pattern as previously observed. Administration of MSG606 reversed morphine-induced hyperalgesia in females, but not males. Antagonism of the NMDA receptor reversed morphine-induced hyperalgesia in males, but not females. Ovariectomized female mice were sensitized to NMDA receptor antagonism, but not to MSG606 administration. Progesterone treatment administered to OVX females re-sensitized them to both icv and i.t. administration of MSG606 to reverse morphine-induced hyperalgesia [179]. Male mice treated with progesterone were sensitized to icv administration of MSG606 to reverse morphine-induced hyperalgesia, but not to i.t. administration [179]. While these studies suggest mMC1R activation may be responsible for the algetic effects, all of the ligands utilized bind or activate multiple melanocortin receptor subtypes. Studies with selective functionally confirmed MC1R antagonists will be necessary to fully understand the pharmacological effects in relation to the other melanocortin receptors, especially as the MC4R has also been implicated in analgesia [193-195]. To date, pharmacological and genetic data support the hypothesis that males primarily use the NDMA receptor pathway and females use a melanocortin pathway, although both sexes can compensate with the other pathway depending on sex hormone levels [184].
Melanocortin ligands have been suggested to differentially affect energy homeostasis in males and females. For example, AGRP was reported to reduce energy expenditure (measured by vO2) in female rats more than in male rats [181]. Removal of the gonads attenuated the observed differences in energy expenditure in females, suggesting sex hormones mediated the effect. Following icv administration, AGRP induced significantly elevated feeding for 5 days in males compared to only 3 days in female mice [181]. Lensing et al. reported a tetrapeptide, Ac-Trp-DPhe(p-I)-Arg-Trp-NH2, that displayed sex-specific metabolic responses in mice [182]. The Ac-Trp-DPhe(p-I)-Arg-Trp-NH2 peptide possesses micromolar antagonist potency at the mMC3R (pA2 = 5.4) and nanomolar antagonist potency at the mMC4R (pA2 = 7.8) [132]. This ligand is also a micromolar agonist at the mMC1R (EC50 = 2000 nM) and mMC5R (EC50 = 2800 nM) [132]. A 7.5 nmol icv administered dose increased food intake, increased respiratory exchange ratio (RER), and trended towards decreasing energy expenditure in male mice, but had minimal effects in female mice. A 2.5 nmol dose significantly increased food intake, RER, and energy expenditure in female mice, but had minimal effects in males at this dose. These data suggest that Ac-Trp-DPhe(p-I)-Arg-Trp-NH2 could be used as a sex-specific probe in vivo to study the underlying mechanisms of the melanocortin sexual dimorphism as it related to energy homeostasis [182]. From these results, it may be hypothesized that melanocortin ligands have different therapeutic windows in males and females, with clinically relevant implications. However, there have been several reports indicating no ligand differences on energy homeostasis between male and female rodents [196-199].
A surprising sex-specific effect was observed in 2014, when icv administration of SHU9119 resulted in no significant effect on blood pressure in young or old female rats, but reduced blood pressure in male rats [183]. Food intake increased in all mice after SHU9119 administration, signifying MC3R/MC4R antagonism in both sexes [183]. This suggests ligands may be designed with ideal pharmacological profiles for therapeutic use in only one sex. For example, melanocortin anti-obesity therapeutics may be developed that decrease food intake but lack cardiovascular side effects in females.
Overall, it appears melanocortin ligands may differentially affect males and females (Table 4). However, more research will be necessary to exploit the sexual dimorphism for sex-specific therapeutics. Establishing whether the sex-specific effects are ligand dependent or present with all ligands with similar in vitro pharmacologies and the underlying causes (including melanocortin receptor, POMC, AGRP or other gene expression levels, changes in neuron connectivity, sex hormones differences, or other system wide changes) of the sexual dimorphisms will aid in the development of sex-specific ligands.
One current difficulty discovering these ligands is that no current in vitro technology is capable of detecting melanocortin sex-specific properties. Therefore, detection of ligands with sex difference relies on in vivo experimental paradigms, presenting no opportunity to optimize ligands prior to more extensive animal studies. The development of in vitro assays that can detect sex differences may represent a major advancement in the design of melanocortin ligands with sex-specific effects. Melanocortin ligands with pre-established sex-specific effects will be necessary to validate that the in vitro assays developed are physiological relevant and correlate with the in vivo pharmacology. Important considerations include: 1) reporting the sex of the cell lines used in vitro [200], 2) studying pharmacological effects in both males and females, and reporting null effects if observed only in a single sex, 3) clearly reporting the sex of animals used in individual experiments, especially when multiple paradigms are performed in a single manuscript, and 4) when combining data that is not significantly different between males and females, reporting the sexes separately in the supplemental materials or the number of females and males used within a data set. These suggestions may allow the development of ligands with sex-specific effects that possess reduced side effects and potential therapeutic applications in one sex.
6. Bivalent and Multivalent Melanocortin Ligands
Bivalent and multivalent ligand design strategies targeting the melanocortin receptors have often been utilized in order to achieve high affinity ligands. These strategies lower the entropic cost of binding by allowing multiple binding interactions per ligand resulting in cooperative binding affinity (Figure 5 A-C) [201-206]. These probes may take advantage of the aggregation or “clumping” of multiple receptors together on the cell membrane (Figure 5 D-E). Recent studies have suggested the presence of melanocortin receptors dimers (or higher-order oligomers) for every known melanocortin subtype [207-214]. Furthermore, radiolabeled ligand binding studies suggest that there are two tandem binding sites with different binding properties on cells expressing melanocortin receptors, indicating targetable dimers [215, 216]. Bivalent and multivalent ligands may therefore preferentially interact with melanocortin receptor dimers or higher-order oligomers.
Figure 5.
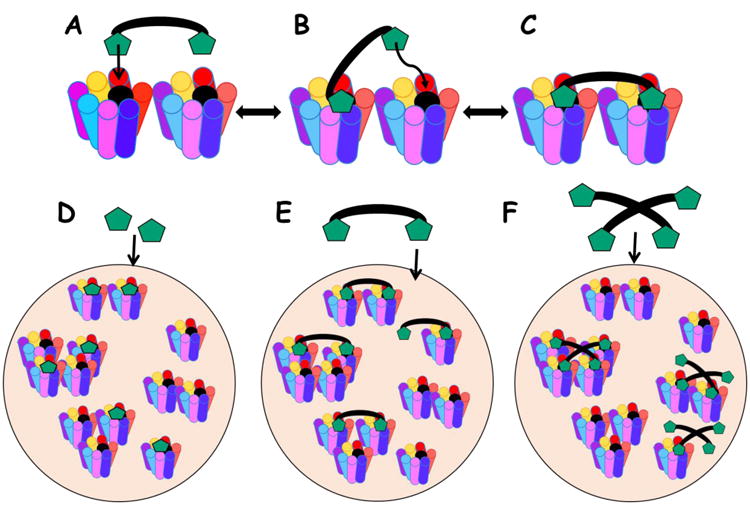
Bivalent and multivalent ligand binding modes. (A) The bivalent ligand first binds a receptor with one pharmacophore in a monovalent fashion. (B) The second pharmacophore is tethered in close proximity to the second binding site. (C) The second pharmacophore can bind the second receptor with reduced entropic cost. Similar binding mode may exist for multivalent ligands with more than two pharmacophores. (D) Monovalent ligands bind monomers, dimers, and higher-order oligomers equally. (E) Bivalent ligands bind dimers and higher-order oligomers in a cooperative synergistic fashion. (F) Multivalent ligands bind GPCR clusters in a cooperative synergistic fashion. Image modified from Lensing, et. al. [134].
One difficulty in understanding the pharmacology of bivalent and multivalent ligands is discerning whether the effects are from a cooperative synergistic binding mode or due to the effect of increasing the pharmacophore concentration without synergy effects. Large increases in binding affinity compared to the monovalent controls (>10-fold) are hypothesized to be due to a cooperative synergistic bivalent or multivalent binding mode (Figure 5). While bivalent ligands have been shown to enhance binding affinity, rarely do additional pharmacophores beyond two result in further potency gains [205, 217].
Melanocortin multivalent ligands were first reported in 1977 [218]. Eberle et al. coupled α-MSH peptides (six to several hundred) to albumin, thyroglobulin, and tobacco mosaic virus which resulted in 1500-fold higher potency than α-MSH alone [218, 219]. Bivalent ligands were first reported in the clinic by Barb et al. as diagnostic tools [220-223]. Since melanoma cells often express elevated levels of the MC1R, it was hypothesized that ligands with high MC1R affinity could be conjugated to dyes or other labels and utilized as imagining or diagnostic tools [224-228]. These high affinity ligands might also be used to deliver therapeutics selectively to melanoma cells. However, this targeting strategy has been criticized because stimulation of the MC1R has been shown to increase melanocyte proliferation, and potentially could lead to melanoma growth [227, 229]. The use of multivalent melanocortin ligands as diagnostic tools has been reviewed previously [224-228]. Therefore, this review will focus on updating the use of bivalent and multivalent ligands as imagining tools. Bivalent ligands containing two pharmacophores will be reviewed first, followed by multivalent ligands possessing more than two pharmacophores.
6.1 Melanocortin Bivalent Ligands
Bivalent ligands are the simplest form of multivalent ligands featuring two pharmacophores separated by a linker or spacer. The two pharmacophores are intended to target two different binding sites. A subclass of bivalent ligands is bitopic ligands, which target both an orthosteric and an allosteric binding site on the same receptor. To the authors' knowledge, there are no known bitopic melanocortin ligands. Therefore, in this review the term bivalent ligand will be used exclusively for ligands which target two orthosteric binding sites on two different receptors.
Bivalent ligands for various GPCRs have been demonstrated to have unique effects compared to their monovalent counterparts. In particular, bivalents ligands are uniquely poised to study GPCR dimerization [202, 230-232]. Carrithers and Lerner developed a series of homobivalent (containing two of the same pharmacophores) melanocortin ligands in 1996 to target two separate melanocortin receptors by crosslinking them. They utilized either an agonist pharmacophore based on α-MSH or the pharmacophore Met-Pro-DPhe-Arg-DTrp-Phe-Lys-Pro-Val tethered by a poly-lysine linker [233, 234]. They demonstrated that the agonist bivalent ligand increased functional activity 5- to 7-fold. In the frog melanocyte dispersion assay, the Met-Pro-DPhe-Arg-DTrp-Phe-Lys-Pro-Val based monovalent ligand was reported to antagonize α-MSH mediated dispersion in a dose-dependent manner. At high concentrations, the bivalent based on Met-Pro-DPhe-Arg-DTrp-Phe-Lys-Pro Val resulted in an agonist functional response [233]. This was the first report demonstrating improvement in functional activity with melanocortin bivalent ligands targeting putative dimers.
After the above report, homobivalent ligand development primarily focused on increasing the binding affinity at the hMC4R through various bivalent ligand design strategies [134, 201, 205, 217, 235-242], with some reports focusing on the MC1R and in vivo imaging [134, 243-245]. While high affinity ligands are desirable for biological responses, the use of low affinity pharmacophores in bivalent ligand design allow the greatest detection of synergistic binding [201, 204, 206, 217, 235, 236, 239, 243]. Therefore, analogs with lower initial monovalent binding affinities result in the greatest observable fold enhancements via a bivalent design strategy. Research programs primarily utilized different analogs of the agonist NDP-MSH. These included the tetrapeptide His-DPhe-Arg-Trp [134, 201, 205, 217, 235, 236, 238, 240-242], six residue analogs[235, 244, 245], seven residue analogs [237, 239, 243], and full length NDP-MSH [201, 236, 238]. Antagonist analogs in which the DPhe was replaced with DNal(2′) have been utilized to produce antagonist analogs with increased binding affinity [134, 239]. There has also been one report in which a melanocortin agonist pharmacophore was attached to an antagonist pharmacophore via a linker that also resulted in increased binding affinity [239].
Linker optimization has been a primary goal of melanocortin homobivalent ligand design. The optimal linker length must be long enough to bridge or crosslink two receptors, but not too long to eliminate entropic gains. Various linker systems have been incorporated, including poly-lysine [233, 237], polyethylene glycol [134, 235, 237-239, 241], Ala-Gly [235], Pro-Gly [134, 235, 237, 239, 240], rigid amino acids [236], squalene [201], glycerol [241], D-mannitol [241], phloroglucinol [242], tripropargylamine [242], 1,4,7-triazacyclononane [242], others [205, 217, 243], and mixtures of these different linker systems together. Improper linker design may result in some increased binding affinity (<10-fold) that can be attributed to simply doubling the pharmacophore concentration [235-237]. Greater fold enhancements (>10-fold) are observed with linkers that appear to bridge two receptors resulting in cooperative synergistic binding (Figure 5A-C). An optimal linker length of approximately 23 ± 5 Å has been suggested by multiple studies at the hMC4R [217, 237, 239, 241, 242].
An assumption in the field was that a bivalent ligand optimized for the MC4R would be effective as a diagnostic tool for melanoma (that highly expresses the MC1R), as long as a non-selective pharmacophore was incorporated. As a result, the majority of studies only investigated in vitro pharmacology using HEK293 cells overexpressing the hMC4R, which resulted in ligands optimized for the hMC4R. In contrast, Lensing et al. reported the linker systems, independent of the binding pharmacophore, displayed preferential patterns for different melanocortin receptor subtypes [134]. A 36 atom (Pro-Gly)6 linker system was optimal for mMC1R binding (14-fold enhancement compared to monovalent counterpart), suggesting a cooperative bivalent binding mode (Figure 1 A-C). However, a 20 atom polyethylene glycol-based linker was less effective at the mMC1R. The lower-fold enhancement was mostly likely an effect of increasing the pharmacophore concentration and is not indicative of a synergistic bivalent binding mode. The 20 atom polyethylene glycol-based linker was optimal at the mMC4R (22-fold enhancement compared to monovalent counterpart). The 36 atom (Pro-Gly)6 linker system resulted in only a 6-fold enhancement at the mMC4R. Both linker systems had binding affinity increases consistent with bivalent binding at the mMC3R (23- to 25-fold enhancement). Currently, this is the only parallel structure activity relationship (SAR) study of various linker systems between different melanocortin receptor subtypes [134]. Similar trends for other GPCR systems have been reported [230, 246, 247], highlighting the importance of optimizing the linker and pharmacophore for a specific receptor subtype if high affinity is desired.
Binding affinity is usually the optimized parameter in melanocortin bivalent designs, but there are a few reports discussing bivalent ligand effects on functional potency. As described above, Carrithers and Lerner observed a 5- to 7-fold enhancement relative to the monovalent ligand in a functional frog-melanocyte dispersion assay (presumably through the MC1R) [233]. Another study by Brabez et al. compared the effects of a monovalent, bivalent, and trivalent ligands on cAMP signaling in HEK293 cells expressing the hMC4R and observed increased cAMP signaling corresponding to increased valency [217]. Lensing et al. reported that although bivalent ligands increased binding affinity (14- to 25-fold), more moderate increases in cAMP signaling potency were observed (3- to 5-fold). Considering that at least a doubling in functional potency would be expected due to doubling the pharmacophore concentration, these increases suggested minimal synergy in function due to bivalent ligand design [134]. Several hypotheses have been proposed for the possible divergences between binding affinity and functional potency, including unique βγ subunit signaling, potency masking, auxiliary binding sites, and asymmetric dimer signaling [134, 217, 233]. Given the limited reports of bivalent functional effects, further studies may be warranted to investigate the mechanism of bivalent ligands' functional signaling and how they may differ from monovalent ligands.
In vivo studies on melanocortin bivalent ligands have focused on their utility as diagnostic tools [220, 221, 224, 225, 243-245]. As noted above, the use of high affinity ligands for the MC1R coupled to dyes or radiolabeled have been identified as possible imagining tools, diagnostic tools, or targeting molecules for melanoma. In 1992, a bivalent chelating derivative of α-MSH was examined in humans, but showed high non-specific uptake in the liver and other organs [220-223, 245]. In 2012, Morais and coworkers demonstrated that 99mTc(I)-labeled bivalent analogs of NDP-MSH resulted in approximately 20-fold enhancements in binding affinity to B16F1 murine melanoma cells (presumably expressing the MC1R) compared to a monovalent counterpart [243]. These ligands displayed negligible degradation when incubated in vitro with human blood serum [243]. The biodistribution of 99mTc(I)-labeled bivalent analogs were studied in melanoma-bearing mice. The lead bivalent analog displayed increased tumor retention in which 98% of the signal from a tumor at 1 h was still detected at 4 h compared to 72% with the monovalent analog [243]. The enhanced tumor retention, hypothesized to be due to increased kinetics of binding, partially accounted for the better tumor-to-blood and tumor-to-muscle ratios 4 h after injection compared to the monovalent ligand. However, despite improved in vitro MC1R binding affinity and high cellular internalization/retention, the bivalent ligand did not increase tumor uptake or improve the pharmacokinetic profiles relative to the monovalent counterpart [243]. Previous reports demonstrated a similar result that bivalent ligands increased in vitro binding affinities at the MC1R but not enhanced in vivo properties relative to monovalent ligands, possibly due to reduced tissue penetration [220-223, 244, 245].
The first report of in vivo functional effects of a melanocortin homobivalent ligand was of CJL-1-87 (Ac-His-DPhe-Arg-Trp-PEDG20-His-DPhe-Arg-Trp-NH2) [134]. This ligand consists of two His-DPhe-Arg-Trp pharmacophores connected by a 20 atom polyethylene glycol-based linker. Administration of CJL-1-87 icv resulted in dose-dependent decreased food intake [134]. Comparison to the monovalent ligand Ac-His-DPhe-Arg-Trp-NH2 suggested little improvement in a nocturnal feeding paradigm [108, 134, 214]. However, a direct comparison study utilizing a fast-refeeding paradigm showed significant differences between CJL-1-87 and Ac-His-DPhe-Arg-Trp-NH2 after ICV administration [214]. Administration of the bivalent CJL-1-87 icv resulted in 50% less food intake than the monovalent ligand 2 to 8 h post-treatment [214]. Treatment also resulted in significantly lowered respiratory exchange ratio (RER) as well as significantly decreased insulin, C-peptide, leptin, and resistin plasma levels compared to the monovalent ligand Ac-His-DPhe-Arg-Trp-NH2 [214].
6.2 Heterobivalent Ligands
Another promising tool for targeting melanoma or other cancers has been the utilization of heterobivalent ligands featuring pharmacophores for two different GPCR types [248-254]. These ligands feature a pharmacophore for the melanocortin receptors and a pharmacophore for a different receptor system (including opioid receptors or cholecystokinin). Monovalent binding occurs to cells expressing one receptor subtype. However, synergistic bivalent binding occurs only on cells expressing both receptor types [248-254]. If appropriate receptors pairs are selected that are co-expressed in cancer cells but not in normal cells, the synergistic bivalent binding will selectively occur on cancer cells. A fluorescent label can be conjugated in the inert linker region, resulting in high affinity ligands that can be used for cancer imagining and diagnostics. Replacement of the fluorescent label with a chemotherapeutic can result in a highly selective drug targeting strategy [248-254]. This heterobivalent ligand targeting strategy has been validated both in vitro and in vivo [248-253]. In 2012, Xu et al. synthesized heterobivalent ligands containing a melanocortin pharmacophore and a cholecystokinin pharmacophore separated by a fluorescently-labeled synthetic linker. They observed the ligands had up to a 12-fold higher specificity for tumors co-expressing both of these receptors than for tumors expressing one receptor, providing a proof-of-principle for future studies [253].
6.3 Melanocortin Multivalent Ligands
Multivalent ligands are defined as ligands that feature more than two pharmacophores for the melanocortin receptors, ranging from three to several hundred. Conjugates of multiple copies α-MSH derivatives to larger biomolecules were reported starting in 1977 [218, 255]. Eberle et al. reported conjugating α-MSH to human serum albumin at a ratio of four and six α-MSH hormones to one molecule of albumin and showed approximately equal activity to non-conjugated α-MSH analogs in testing on Rana pipiens. Schwyzer et al. reported loading the tobacco mosaic virus (TMV) with approximately 400-600 molecules of α-MSH analogs, resulting in enhanced potency, affinity, resistance towards enzymatic degradation, and prolonged activity at target cells [219, 256-260]. Sharma et al. developed a class of multivalent fluorescent melanotropin-macromolecule conjugates [261, 262]. They used a polyvinyl alcohol (PVA) scaffold that had an approximate molecular weight of 110,000 and 2500 hydroxyl groups available for derivation. The hydroxyl groups were conjugated to introduce 10-16 molecules of a melanocortin pharmacophore (based on NDP-MSH) and 10-16 molecules of a fluorophore (fluorescein isothiocyanate or FTIC) to create macromolecular conjugates (MSH-PVA-FITC). These conjugates possessed increased binding affinity and increased levels of melanocortin receptor detection in labeling experiments comparing different cells that did or did not express the melanocortin receptors [261, 262]. Sharma et al. also developed both latex bead and polyamide bead conjugates to NDP-MSH analogs and achieved similar results as Schwyzer et al. [263-265]. The latex beads were considered microspheres (∼1 μm in diameter) and the polyamide beads were classified as macrospheres (40 to 100 μm in diameter). Electron and light microscopy imaging indicated that mutiple latex microspheres were bound to B16/F10 mouse melanoma cells (∼10-15 μm in diameter). The larger polyamide macrospheres were bound to multiple cells, a difference presumed to be due to the relative sizes of the conjugated beads and the cells.
In 2007, Newton et al. engineered and fused α-MSH analogs to phages and used these multivalent phage constructs to image B16-F1 mouse melanoma in vitro and in vivo [266]. In 2011 and 2013, Barkey et al. attached hMC1R selective α-MSH analogs to stabilized triblock polymer micelles through Cu-catalyzed click chemistry [267, 268]. Though the ligand decreased binding affinity of the polymer micelles after attachment, it increased specificity for the hMC1R over the hMC4R and hMC5R [268]. Further cross-linking the targeted polymer micelles generated constructs that were used as delivery systems for contrast-enhancing gadolinium complexes of texaphyrin (Gd-Tx) [267]. These agents were efficacious at penetrating and delivering the contrast agent into xenografted tumors in mice with minimal accumulation in healthy tissues, including the kidney and liver [267].
Besides conjugated multivalent ligands, additional research has focused on the design of smaller, synthetic ligands. Trivalent ligands have been synthesized by incorporating a lysine in the linker of bivalent ligands, providing an additional chemical handle to add another pharmacophore. Trivalent ligands possessed increased binding affinity over bivalent and monovalent ligands at the hMC4R, albeit the result was not indicative of trivalent binding [235].
Additional trivalent ligands were shown to increase binding affinity and supported a cooperative binding mode. In 2011, Brabez et al. reported a series of trivalent melanocortin ligands with increased binding affinity. They found the optimal distance between His-DPhe-Arg-Trp tetrapeptide pharmacophores to be 24 ± 5 Å when targeting the hMC4R [217]. In competitive binding experiments with HEK293 cells expressing the hMC4R, the monovalent, bivalent, and trivalent analogs had IC50 values of 4900, 310, and 14 nM, respectively. The increased affinity with each valency suggested that three receptors were involved in a trivalent cooperative binding mechanism with the trivalent ligand [205, 217]. The authors noted that although cAMP signaling potency increased with each valency, the levels of cAMP signaling corresponded to receptor occupancy independent of valency. This suggested that the ligands activated cAMP signaling in a monovalent fashion and no allosterism or synergy in function was detected [217]. In a follow-up study, the authors combined the trivalent ligands onto additional scaffolds, resulting in ligands with 6 and 9 pharmacophores [269]. The 6- and 9-valent compounds decreased binding affinity 3-fold at the hMC4R compared to the trivalent analog, but were approximately 100-fold more potent than the monovalent compound. These data suggest the 6- and 9- pharmacophore ligands achieved cooperative binding but three or fewer receptors were involved. The cAMP signaling was also independent of the number of pharmacophores present, as previously described. They reported that all compounds were internalized within 90 minutes, suggesting these constructs could potentially be used for drug delivery purposes [269].
Solanesol-derived and sucrose-derived scaffolds were utilized to make both bivalent and tetravalent ligands attached to the His-DPhe-Arg-Trp tetrapeptide sequence [270, 271]. Moderate improvement in binding affinity at the hMC4R was observed, likely due to proximity effects and increasing the moles of pharmacophore present, but not indicative of cooperative or multivalent binding. The authors hypothesized their ligands may not possess the correct linker length or improperly presented the pharmacophores for cooperative binding [270, 271].
An unique strategy to synthesize multivalent ligands featuring the His-DPhe-Arg-Trp tetrapeptide was reported by Dehigaspitiya et al. in 2015. Linear ligands that had up to eight His-DPhe-Arg-Trp units were synthesized and separated by a (Pro-Gly)3 linker. The binding affinities for the hMC4R were slightly enhanced in competitive binding experiments when adjusted for pharmacophore concentrations, suggesting the observed enhancement were not from cooperativity or multivalent binding [240].
In 2015, Elshan et al. presented trigonal scaffolds and compared monovalent, bivalent and trivalent ligands featuring the His-DPhe-Arg-Trp tetrapeptide pharmacophore. The bivalent ligands increased binding affinity 10- to 30-fold, indicating synergistic bivalent binding affinity at the hMC4R. The trivalent ligands were reported to be marginally better binders (∼2-fold) than the bivalent ligands, supporting a model where trivalent ligands bind in a synergistic bivalent binding mode, but not a trivalent mode [242]. Their results suggested the optimal linker length to bridge two receptors is between 17 and 23 Å. However, considering a third receptor did not appear to be utilized, it is likely that the third pharmacophore linker is not optimized to achieve trivalent binding [242]. Dehigaspitiya et al. reported similar results when comparing monovalent, bivalent, trivalent and tetravalent ligands featuring the tetrapeptide His-DPhe-Arg-Trp in different scaffolds [241]. All multivalent compounds possessed 30- to 40-fold higher binding affinities at the hMC4R compared to monovalent controls, although valencies beyond two did not result in further affinity gains. This finding is consistent with bivalent binding to putative melanocortin receptor dimers, without evidence for trivalent or tetravalent binding. They also reported the optimal distance between pharmacophores was between 17 and 23 Å, as previously reported [241, 242].
In summarizing the reported bivalent and multivalent ligand designs for the melanocortin receptors, two key observations can be made. First, in order to observe synergistic effects, the proper pharmacophore must be used. In almost all cases, the tetrapeptide His-DPhe-Arg-Trp was observed to result in the greatest-fold affinity enhancements, presumably due to synergistic binding. This is likely due to the lower initial binding affinity of the tetrapeptide compared to longer analogs that allows easier detection and observation of the synergistic binding mode. The second key is the design of proper linker length to bridge putative melanocortin dimers. The greatest-fold enhancements were with linkers of approximately 23 ± 5 Å. One difficulty in estimating the exact length is that the linkers are flexible and therefore nearly impossible to measure precisely. The estimated range for the optimal melanocortin linker is similar to that of other GPCR systems including the oxytocin (∼25 Å) [272], opioid (∼22 Å) [246, 273, 274], and dopamine receptors (∼25 Å) [247]. This provides strong evidence for a common design of bivalent ligands targeting GPCR systems and suggests this length may be the result of a common GPCR phenomenon (dimerization or high-order oligomerization).
Although the increases in binding affinity support the use of bivalent and multivalent melanocortin ligands as diagnostic tools, imaging probes, and drug delivery vehicles for melanoma, their functional effects require further investigation. In the limited studies evaluating functional effects, some increases in potency were observed. However, these enhanced in vitro potencies observed could be due to increased concentration and increased binding affinity, and not due to allosterism or synergistic functional effects [134, 217]. Future studies on bivalent and multivalent ligands may need to focus on evaluating the functional effects of this ligand class both in vitro and in vivo, on developing SAR at different receptor subtypes (and heterodimers), and optimizing the linker length to a third pharmacophore to extend beyond bivalent binding.
7. Clinical Candidates
There has been a concerted effort to translate melanocortin ligands into clinical therapies. Both α-MSH and β-MSH were injected in humans in 1961 [55]. However, many unexpected challenges such as receptor-mediated pressor effects limit the translation of these pharmacologically active compounds into viable therapies [25]. The clinical study involving Eli-Lilly compound LY2112688, which had unexpected cardiovascular effects, is the archetype of unexpected challenges targeting the melanocortin receptors [25].
This section will focus on clinical studies from 2011 to 2016, in peer-reviewed publications on melanocortin compounds in preclinical and clinical studies to complement previous reviews [171, 275]. A survey was conducted on the databases ClinicalTrials.gov, PubMed, and the Cochrane Central Register of Controlled Trials (CENTRAL). Search criteria included “melanocortin” and “pharmacotherapy OR drug therapy OR pharmaceutic OR drug.” Studies were excluded if they were not peer-reviewed or did not possess patient data. In addition, studies focusing on polymorphisms of the melanocortin receptors and the resulting effects on different pathways were excluded. These criteria generated more than 70 articles that were evaluated. Compounds currently in clinical trial (Figure 6) include bremelanotide, afamelanotide, setmelanotide, MC4-NN2-0453, and MSH/ACTH(4-10).
7.1 Bremelanotide
The cyclic heptapeptide, sequence Ac-Nle-c[Asp-His-DPhe-Arg-Trp-Lys]-OH (bremelanotide, Figure 6), is a derivative of the potent melanotan-II (MTII, Figure 6) with a C-terminal carboxylic acid [116, 117]. Bremelanotide is in clinical development by Palatin Technologies for the treatment of hypoactive sexual desire disorder in women. The initial development of these cyclic α-MSH analogues targeted sunless tanning. It was discovered by Dr. Hadley et al. that a single 10 mg injection of MTII was able to induce an instantaneous and unrelenting erection lasting approximately 8 hours in duration [174], indicating sexual effects in males with other sexual effects also observed in females.
A phase 2 clinical study consisting of 394 women experiencing sexual dysfunction reported clinical efficacy was achieved with 1.25 and 1.75 mg subcutaneous doses. These doses induced a maximal 3.0 mm Hg pressure increase in both systolic and diastolic blood pressures between 0 and 4 h post injection. Importantly, the increases in blood pressure were transient, approximately 15 minutes in duration, and quickly reduced back to pretreatment levels [276].
Bremelanotide was taken into a phase 2 trial in postmenopausal women with self-reported sexual dysfunction [277]. A total of 327 women with female sexual arousal disorder and/or hypoactive sexual desire disorder were randomized in a double-blind placebo-controlled study. Treatment consisted of placebo, 0.75, 1.25, or 1.75 mg subcutaneous doses of bremelanotide. Two week base-line measurements were followed by a 12 week at-home study where the compound was administered as desired once daily (up to 16 doses in a 4-week period) 45 minutes prior to anticipated sexual activity. Efficacy was determined as changes in score using the self-reported female sexual distress scale-desire/arousal/orgasm via an electronic diary for all sexual encounters for the duration of the study. Results indicated a significant increase in the number of sexual encounters per month (+0.75 for the 1.25/1.75 mg pooled data) in addition to a decrease in the distressed scores produced from the self-reported questionnaire (indicating greater satisfaction) [277]. Positive results were also reported from a phase 3 clinical study in August 2016 in premenopausal women, and may perhaps be the first US approved melanocortin ligand drug.
7.2 Afamelanotide
The linear 13-residue afamelanotide is another name for the synthetic melanocortin analog NDP-MSH (Figure 6) that was initially reported in 1980 [109, 278]. Afamelanotide has been used in phase 2 trials for the treatment of skin conditions associated with inflammation. These studies have focused on rare skin conditions such as Hailey-Hailey disease in addition to diseases with a broader scope such as acne vulgaris, although these studies are limited due to small patient size [279, 280].
In clinical development and evaluation sponsored partially by Clinuvel Pharmaceuticals, afamelanotide has produced successful results in phase 3 clinical trials for erythropoietic protoporphyria, a photosensitivity skin condition (ClinicalTrials.gov identifiers NCT01605136 and NCT00979745), summarized in 2015 [281]. Presumably signaling through the MC1R, afamelanotide induces the production of eumelanin in the skin, serving as a chemical tanning agent that would allow increased exposure to sunlight for individuals afflicted with erythropoietic protoporphyria. These studies were conducted in both the United States (94 patients) and the European Union (74 patients) using similar methodology. In a randomized, placebo-controlled, double-blind study, patients were treated with 16 mg injections of afamelanotide (proprietary name Scenesse) formulated in a biodegradable, implantable polymer matrix [282]. In the European study, the patients received five doses over 9-months, while in the United States study patients received three doses over 6-months. The results from both locations indicated less-severe phototoxic reactions with shorter recovery times. In a self-reporting questionnaire, patients indicated the treatment produced a positive impact on their daily lives [281]. Patients also experienced a significant improvement to light tolerance [281]. Afamelanotide was approved by European regulators to treat erythropoietic protoporphyria in 2014 [115] and is the first approved melanocortin ligand for therapeutic use.
7.3 Setmelanotide
The disulfide cyclized octapeptide setmelanotide (RM-493, formerly BIM-22493, IRC-022493, Figure 6), Ac-Arg-c[Cys-DAla-His-DPhe-Arg-Trp-Cys]-NH2, is in clinical evaluation for weight-loss by Rhythm Pharmaceuticals. Preclinical studies in obese rhesus macaques indicated chronic subcutaneous administration of setmelanotide reduced overall food intake, decreased body weight, improved glucose tolerance, and did not induce negative cardiac effects [283]. Phase 1 and 2 studies have successfully evaluated the safety, efficacy, tolerability, pharmacokinetics, and pharmacodynamics of the octapeptide in obese volunteers (ClinicalTrials.gov identifiers NCT02431442 and NCT02041195). A phase 2 clinical trial (ClinicalTrials.gov identifier NCT01867437) evaluated the efficacy and safety of setmelanotide, and consisted of 12 obese, mean body mass index (BMI) 35.7 kg/m2, otherwise healthy individuals. In a randomized crossover experimental paradigm, individuals received a 1 mg/24 hour subcutaneous infusion of drug or placebo for 72 hours. A significant increase in resting energy expenditure was observed, without corresponding increases in blood pressure or heart rate [284]. An active study of volunteers with Prader-Willi syndrome to evaluate the compound efficacy is ongoing in 2016 (ClinicalTrials.gov identifier NCT02311673). In addition, there are results from an investigator initiated study consisting of two obese patients with proopiomelanocortin deficiencies [285]. Daily subcutaneous administration of setmelanotide in these patients resulted in substantial weight loss (patient 1 = 51.0 kg in 42 weeks, patient 2 = 20.5 kg in 12 weeks) and significantly decreased blood pressure in 1 patient [285].
7.4 MC4-NN2-0453
In development by Novo Nordisk, MC4-NN2-0453 is a α-MSH analog with an N-terminal fatty acid extension [286]. The modified peptide, 16-(tetrazol-5-yl)hexadecanoyl-Oeg-Gly-Ser-Gln-His-Dap[bis(carboxymethyl)amino]acetyl-Nle-c[Glu-Hyp-DPhe-Arg-Trp-Lys]-NH2 (Figure 6), has Ki of 2700, 71, 0.58, and 13 nM at the hMC1R, hMC3R, hMC4R, hMC5R, respectively [286]. These in vitro results did not translate into clinically relevant effects on body weight in obese, otherwise healthy, patients [287]. A four day multi-dose, randomized, double-blind, placebo-controlled trial tested multiple concentrations administered subcutaneously up to 3.0 mg/day [287]. Analysis of the pharmacokinetic parameters indicated the compound possessed a long (>200 hour) t1/2 half-life, but did not alter body weight. Side effects of this compound included non-serious skin adverse events, primarily hyperpigmentation. Further development of this compound as an anti-obesity therapy appears to be halted, perhaps due to lack of efficacy.
7.5 MSH/ACTH(4-10)
The linear heptapeptide H-Met-Glu-His-Phe-Arg-Trp-Gly-OH (Figure 6), also referred to as MSH/ACTH(4-10), has been subject to at least one human study between 2011 and 2016. In this study, 10 healthy, BMI between 20 and 25 kg/m2, male volunteers participated in a double-blind randomized crossover study. The participants received a 10 mg dose via intranasal administration. Interstitial glycerol, an indicator of lipid hydrolysis, was monitored via microdialysis in abdominal white adipose tissue and in skeletal muscle of the forearm. Results indicated a significant increase in glycerol in the white adipose tissue 45 minutes after dosing compared to no change in the glycerol levels in the skeletal muscle tissue. These results indicate the peptide alters lipid metabolism in humans [288].
8. Conclusions
Numerous melanocortin ligands have been developed in the 60 years since the sequences of the first endogenous ligands were elucidated. While much of the early focus was on the development of compounds that alter pigmentation, the cloning of the receptors and identification of other biological pathways controlled by these receptors (including obesity and sexual function) led to the development of potent and selective ligands. Many classical medicinal chemistry structure-activity relationship study techniques have led to a greater understanding of ligand/receptor interactions. Although many unique ligands have been reported, the expanding signaling pathways associated with the melanocortin receptors provides opportunities for continued investigations. With new biological functions and pathways associated with the melanocortin receptors, it may be unwise to “believe that we have reached the limits of insight that can be reasonably provided by structure-activity studies” for melanocortin ligands [85].
Highlights.
An updated melanocortin ligand review.
Classic, selective, small-molecule, sex-specific, bivalent, and clinical ligands.
Focuses on melanocortin ligand publications from 2006-2016.
Acknowledgments
Funding Sources: This work has been supported by NIH Grants R01DK108893, R01DK091906 and R01DK097838 (CHL). Mark D. Ericson is a recipient of an NIH Postdoctoral Fellowship (F32DK108402). Cody J. Lensing is a recipient of an Olsteins Graduate Fellowship and a Doctoral Dissertation Fellowship from the University of Minnesota.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chhajlani V, Wikberg JE. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Letters. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 2.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 3.Butler AA, Kesterson RA, Khong K, Cullen MJ, Pelleymounter MA, Dekoning J, Baetscher M, Cone RD. A unique metabolic syndrome causes obesity in the melanocortin-3 receptor-deficient mouse. Endocrinology. 2000;141:3518–3521. doi: 10.1210/endo.141.9.7791. [DOI] [PubMed] [Google Scholar]
- 4.Chen AS, Marsh DJ, Trumbauer ME, Frazier EG, Guan XM, Yu H, Rosenblum CI, Vongs A, Feng Y, Cao LH, Metzger JM, Strack AM, Camacho RE, Mellin TN, Nunes CN, Min W, Fisher J, Gopal-Truter S, MacIntyre DE, Chen HY, Van der Ploeg LHT. Inactivation of the mouse melanocortin-3 receptor results in increased fat mass and reduced lean body mass. Nature Genetics. 2000;26:97–102. doi: 10.1038/79254. [DOI] [PubMed] [Google Scholar]
- 5.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 6.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. The Journal of Biological Chemistry. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 7.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. The Journal of Biological Chemistry. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 8.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 9.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for γ melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proceedings of the National Academy of Sciences. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorr RT, Lines R, Levine N, Brooks C, Xiang L, Hruby VJ, Hadley ME. Evaluation of Melanotan-II, a superpotent cyclic melanotropic peptide in a pilot phase-I clinical study. Life Sciences. 1996;58:1777–1784. doi: 10.1016/0024-3205(96)00160-9. [DOI] [PubMed] [Google Scholar]
- 11.Van der Ploeg LHT, Martin WJ, Howard AD, Nargund RP, Austin CP, Guan XM, Drisko J, Cashen D, Sebhat I, Patchett AA, Figueroa DJ, DiLella AG, Connolly BM, Weinberg DH, Tan CP, Palyha OC, Pong SS, MacNeil T, Rosenblum C, Vongs A, Tang R, Yu H, Sailer AW, Fong TM, Huang C, Tota MR, Chang RS, Stearns R, Tamvakopoulos C, Christ G, Drazen DL, Spar BD, Nelson RJ, MacIntyre DE. A role for the melanocortin 4 receptor in sexual function. Proceedings of the National Academy of Sciences. 2002;99:11381–11386. doi: 10.1073/pnas.172378699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochemical and Biophysical Research Communications. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 13.Griffon N, Mignon V, Facchinetti P, Diaz J, Schwartz JC, Sokoloff P. Molecular cloning and characterization of the rat fifth melanocortin receptor. Biochemical and Biophysical Research Communications. 1994;200:1007–1014. doi: 10.1006/bbrc.1994.1550. [DOI] [PubMed] [Google Scholar]
- 14.Chen W, Kelly MA, Opitz-Araya X, Thomas RE, Low MJ, Cone RD. Exocrine gland dysfunction in MC5-R-deficient mice: evidence for coordinated regulation of exocrine gland function by melanocortin peptides. Cell. 1997;91:789–798. doi: 10.1016/s0092-8674(00)80467-5. [DOI] [PubMed] [Google Scholar]
- 15.Nakanishi S, Inoue A, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-β-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 16.Blanchard SG, Harris CO, Ittoop OR, Nichols JS, Parks DJ, Truesdale AT, Wilkison WO. Agouti antagonism of melanocortin binding and action in the B16F10 murine melanoma cell line. Biochemistry. 1995;34:10406–10411. doi: 10.1021/bi00033a012. [DOI] [PubMed] [Google Scholar]
- 17.Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, Van der Ploeg LHT. ART (protein product of agouti-related transcript) as an antagonist of MC-3 and MC-4 receptors. Biochemical and Biophysical Research Communications. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- 18.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen YR, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 19.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regulatory Peptides. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 20.Nijenhuis WAJ, Oosterom J, Adan RAH. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Molecular Endocrinology. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 21.McRobie HR, King LM, Fanutti C, Symmons MF, Coussons PJ. Agouti signalling protein is an inverse agonist to the wildtype and agonist to the melanic variant of the melanocortin-1 receptor in the grey squirrel (Sciurus carolinensis) FEBS Letters. 2014;588:2335–2343. doi: 10.1016/j.febslet.2014.05.032. [DOI] [PubMed] [Google Scholar]
- 22.Allen BM. The results of extirpation of the anterior lobe of the hypophysis and of the thyroid of rana pipiens larvae. Science. 1916;44:755–758. doi: 10.1126/science.44.1143.755. [DOI] [PubMed] [Google Scholar]
- 23.Smith PE. Experimental ablation of the hypophysis in the frog embryo. Science. 1916;44:280–282. doi: 10.1126/science.44.1130.280. [DOI] [PubMed] [Google Scholar]
- 24.Chen WB, Shields TS, Stork PJS, Cone RD. A colorimetric assay for measuring activation of Gs-and Gq-coupled signaling pathways. Analytical Biochemistry. 1995;226:349–354. doi: 10.1006/abio.1995.1235. [DOI] [PubMed] [Google Scholar]
- 25.Greenfield JR, Miller JW, Keogh JM, Henning E, Satterwhite JH, Cameron GS, Astruc B, Mayer JP, Brage S, See TC, Lomas DJ, O'Rahilly S, Farooqi IS. Modulation of blood pressure by central melanocortinergic pathways. New England Journal of Medicine. 2009;360:44–52. doi: 10.1056/NEJMoa0803085. [DOI] [PubMed] [Google Scholar]
- 26.Bell PH. Purification and structure of β-corticotropin. Journal of the American Chemical Society. 1954;76:5565–5567. [Google Scholar]
- 27.Harris JI, Lerner AB. Amino-acid sequence of the α-melanocyte-stimulating hormone. Nature. 1957;179:1346–1347. doi: 10.1038/1791346a0. [DOI] [PubMed] [Google Scholar]
- 28.Harris JI, Roos P. Amino-acid sequence of a melanophore-stimulating peptide. Nature. 1956;178:90. doi: 10.1038/178090a0. [DOI] [PubMed] [Google Scholar]
- 29.Cawley NX, Li ZJ, Loh YP. 60 Years of POMC: Biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. Journal of Molecular Endocrinology. 2016;56:T77–T97. doi: 10.1530/JME-15-0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irani BG, Holder JR, Todorovic A, Wilczynski AM, Joseph CG, Wilson KR, Haskell-Luevano C. Progress in the development of melanocortin receptor selective ligands. Current Pharmaceutical Design. 2004;10:3443–3479. doi: 10.2174/1381612043382891. [DOI] [PubMed] [Google Scholar]
- 31.Pritchard LE, Turnbull AV, White A. Pro-opiomelanocortin processing in the hypothalamus: impact on melanocortin signalling and obesity. Journal of Endocrinology. 2002;172:411–421. doi: 10.1677/joe.0.1720411. [DOI] [PubMed] [Google Scholar]
- 32.Castrucci AM, Hadley ME, Sawyer TK, Wilkes BC, al-Obeidi F, Staples DJ, de Vaux AE, Dym O, Hintz MF, Riehm JP, Rao KR, Hruby VJ. α-Melanotropin: The minimal active sequence in the lizard skin bioassay. General and Comparative Endocrinology. 1989;73:157–163. doi: 10.1016/0016-6480(89)90066-x. [DOI] [PubMed] [Google Scholar]
- 33.Hruby VJ, Wilkes BC, Hadley ME, Al-Obeidi F, Sawyer TK, Staples DJ, de Vaux AE, Dym O, Castrucci AM, Hintz MF, Riehm JP, Rao KR. α-Melanotropin: The minimal active sequence in the frog skin bioassay. Journal of Medicinal Chemistry. 1987;30:2126–2130. doi: 10.1021/jm00394a033. [DOI] [PubMed] [Google Scholar]
- 34.Clement K, Dubern B, Mencarelli M, Czernichow P, Ito S, Wakamatsu K, Barsh GS, Vaisse C, Leger J. Unexpected endocrine features and normal pigmentation in a young adult patient carrying a novel homozygous mutation in the POMC gene. Journal of Clinical Endocrinology & Metabolism. 2008;93:4955–4962. doi: 10.1210/jc.2008-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farooqi IS, Drop S, Clements A, Keogh JM, Biernacka J, Lowenbein S, Challis BG, O'Rahilly S. Heterozygosity for a POMC-null mutation and increased obesity risk in humans. Diabetes. 2006;55:2549–2553. doi: 10.2337/db06-0214. [DOI] [PubMed] [Google Scholar]
- 36.Krude H, Biebermann H, Luck W, Horn R, Brabant G, Gruters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nature Genetics. 1998;19:155–157. doi: 10.1038/509. [DOI] [PubMed] [Google Scholar]
- 37.Krude H, Biebermann H, Schnabel D, Tansek MZ, Theunissen P, Mullis PE, Grüters A. Obesity due to proopiomelanocortin deficiency: Three new cases and treatment trials with thyroid hormone and ACTH4–10. Journal of Clinical Endocrinology & Metabolism. 2003;88:4633–4640. doi: 10.1210/jc.2003-030502. [DOI] [PubMed] [Google Scholar]
- 38.Krude H, Grüters A. Implications of proopiomelanocortin (POMC) mutations in humans: The POMC deficiency syndrome. Trends in Endocrinology & Metabolism. 2000;11:15–22. doi: 10.1016/s1043-2760(99)00213-1. [DOI] [PubMed] [Google Scholar]
- 39.Mendiratta MS, Yang Y, Balazs AE, Willis AS, Eng CM, Karaviti LP, Potocki L. Early onset obesity and adrenal insufficiency associated with a homozygous POMC mutation. International Journal of Pediatric Endocrinology. 2011;2011:5–11. doi: 10.1186/1687-9856-2011-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yaswen L, Diehl N, Brennan MB, Hochgeschwender U. Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nature Medicine. 1999;5:1066–1070. doi: 10.1038/12506. [DOI] [PubMed] [Google Scholar]
- 41.Challis BG, Coll AP, Yeo GS, Pinnock SB, Dickson SL, Thresher RR, Dixon J, Zahn D, Rochford JJ, White A, Oliver RL, Millington G, Aparicio SA, Colledge WH, Russ AP, Carlton MB, O'Rahilly S. Mice lacking pro-opiomelanocortin are sensitive to high-fat feeding but respond normally to the acute anorectic effects of peptide-YY(3-36) Proceedings of the National Academy of Sciences. 2004;101:4695–4700. doi: 10.1073/pnas.0306931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coll AP, Challis BG, Yeo GS, Snell K, Piper SJ, Halsall D, Thresher RR, O'Rahilly S. The effects of proopiomelanocortin deficiency on murine adrenal development and responsiveness to adrenocorticotropin. Endocrinology. 2004;145:4721–4727. doi: 10.1210/en.2004-0491. [DOI] [PubMed] [Google Scholar]
- 43.Smart JL, Low MJ. Lack of proopiomelanocortin peptides results in obesity and defective adrenal function but normal melanocyte pigmentation in the murine C57BL/6 genetic background. Annals of the New York Academy of Sciences. 2003;994:202–210. doi: 10.1111/j.1749-6632.2003.tb03181.x. [DOI] [PubMed] [Google Scholar]
- 44.Guo L, Munzberg H, Stuart RC, Nillni EA, Bjorbaek C. N-acetylation of hypothalamic α-melanocyte-stimulating hormone and regulation by leptin. Proceedings of the National Academy of Sciences. 2004;101:11797–11802. doi: 10.1073/pnas.0403165101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Donohue TL, Handelmann GE, Chaconas T, Miller RL, Jacobowitz DM. Evidence that N-acetylation regulates the behavioral activity of α-MSH in the rat and human central nervous system. Peptides. 1981;2:333–344. doi: 10.1016/s0196-9781(81)80126-x. [DOI] [PubMed] [Google Scholar]
- 46.Haskell-Luevano C, Holder JR, Monck EK, Bauzo RM. Characterization of melanocortin NDP-MSH agonist peptide fragments at the mouse central and peripheral melanocortin receptors. Journal of Medicinal Chemistry. 2001;44:2247–2252. doi: 10.1021/jm010061n. [DOI] [PubMed] [Google Scholar]
- 47.Todorovic A, Ericson MD, Palusak RD, Sorensen NB, Wood MS, Xiang Z, Haskell-Luevano C. Comparative functional alanine positional scanning of the α-melanocyte stimulating hormone and NDP-melanocyte stimulating hormone demonstrates differential structure-activity relationships at the mouse melanocortin receptors. ACS Chemical Neuroscience. 2016;7:984–994. doi: 10.1021/acschemneuro.6b00098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sahm UG, Olivier GW, Branch SK, Moss SH, Pouton CW. Synthesis and biological evaluation of α-MSH analogues substituted with alanine. Peptides. 1994;15:1297–1302. doi: 10.1016/0196-9781(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 49.Sahm UG, Qarawi MA, Olivier GWJ, Ahmed ARH, Branch SK, Moss SH, Pouton CW. The melanocortin (MC3) receptor from rat hypothalamus - Photoaffinity labeling and binding of alanine-substituted α-MSH analogs. FEBS Letters. 1994;350:29–32. doi: 10.1016/0014-5793(94)00725-x. [DOI] [PubMed] [Google Scholar]
- 50.Elias CF, Saper CB, Maratos-Flier E, Tritos NA, Lee C, Kelly J, Tatro JB, Hoffman GE, Ollmann MM, Barsh GS, Sakurai T, Yanagisawa M, Elmquist JK. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. Journal of Comparative Neurology. 1998;402:442–459. [PubMed] [Google Scholar]
- 51.Watson SJ, Akil H. The presence of two α-MSH positive cell groups in rat hypothalamus. European Journal of Pharmacology. 1979;58:101–103. doi: 10.1016/0014-2999(79)90351-0. [DOI] [PubMed] [Google Scholar]
- 52.Jacobowitz DM, O'Donohue TL. α-Melanocyte stimulating hormone: Immunohistochemical identification and mapping in neurons of rat brain. Proceedings of the National Academy of Sciences. 1978;75:6300–6304. doi: 10.1073/pnas.75.12.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hofmann K, Yajima H, Schwartz ET. Studies on polypeptides. XVII. The synthesis of three acyltridecapeptide amides possessing a high level of melanocyte-expanding sctivity in vitro. Journal of the American Chemical Society. 1960;82:3732–3737. [Google Scholar]
- 54.Clive D, Snell R. Effect of the alpha melanocyte stimulating hormone on mammalian hair colors. Journal of Investigative Dermatology. 1967;49:314–321. [Google Scholar]
- 55.Lerner AB, McGuire JS. Effect of alpha- and beta-melanocyte stimulating hormones on the skin colour of man. Nature. 1961;189:176–179. doi: 10.1038/189176a0. [DOI] [PubMed] [Google Scholar]
- 56.Thody AJ, Ridley K, Carter RJ, Lucas AM, Shuster S. α-MSH and coat color changes in the mouse. Peptides. 1984;5:1031–1036. doi: 10.1016/0196-9781(84)90166-9. [DOI] [PubMed] [Google Scholar]
- 57.Millington GW, Tung YC, Hewson AK, O'Rahilly S, Dickson SL. Differential effects of α-, β- and γ2-melanocyte-stimulating hormones on hypothalamic neuronal activation and feeding in the fasted rat. Neuroscience. 2001;108:437–445. doi: 10.1016/s0306-4522(01)00428-6. [DOI] [PubMed] [Google Scholar]
- 58.Poggioli R, Vergoni AV, Bertolini A. ACTH-(1-24) and α-MSH antagonize feeding behavior stimulated by kappa opiate agonists. Peptides. 1986;7:843–848. doi: 10.1016/0196-9781(86)90104-x. [DOI] [PubMed] [Google Scholar]
- 59.Wirth MM, Olszewski PK, Yu C, Levine AS, Giraudo SQ. Paraventricular hypothalamic α-melanocyte-stimulating hormone and MTII reduce feeding without causing aversive effects. Peptides. 2001;22:129–134. doi: 10.1016/s0196-9781(00)00367-3. [DOI] [PubMed] [Google Scholar]
- 60.Bertagna X, Lenne F, Comar D, Massias JF, Wajcman H, Baudin V, Luton JP, Girard F. Human β-melanocyte-stimulating hormone revisited. Proceedings of the National Academy of Sciences. 1986;83:9719–9723. doi: 10.1073/pnas.83.24.9719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harrold JA, Widdowson PS, Williams G. β-MSH: A functional ligand that regulated energy homeostasis via hypothalamic MC4-R? Peptides. 2003;24:397–405. doi: 10.1016/s0196-9781(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 62.Schioth HB, Muceniece R, Wikberg JE. Characterisation of the melanocortin 4 receptor by radioligand binding. Pharmacol Toxicol. 1996;79:161–165. doi: 10.1111/j.1600-0773.1996.tb00261.x. [DOI] [PubMed] [Google Scholar]
- 63.Schioth HB, Muceniece R, Wikberg JE, Chhajlani V. Characterisation of melanocortin receptor subtypes by radioligand binding analysis. Eur J Pharmacol. 1995;288:311–317. doi: 10.1016/0922-4106(95)90043-8. [DOI] [PubMed] [Google Scholar]
- 64.Kask A, Rago L, Wikberg JE, Schioth HB. Differential effects of melanocortin peptides on ingestive behaviour in rats: Evidence against the involvement of MC3 receptor in the regulation of food intake. Neuroscience Letters. 2000;283:1–4. doi: 10.1016/s0304-3940(00)00837-5. [DOI] [PubMed] [Google Scholar]
- 65.Lee YS, Challis BG, Thompson DA, Yeo GS, Keogh JM, Madonna ME, Wraight V, Sims M, Vatin V, Meyre D, Shield J, Burren C, Ibrahim Z, Cheetham T, Swift P, Blackwood A, Hung CC, Wareham NJ, Froguel P, Millhauser GL, O'Rahilly S, Farooqi IS. A POMC variant implicates β-melanocyte-stimulating hormone in the control of human energy balance. Cell Metabolism. 2006;3:135–140. doi: 10.1016/j.cmet.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 66.Challis BG, Pritchard LE, Creemers JW, Delplanque J, Keogh JM, Luan J, Wareham NJ, Yeo GS, Bhattacharyya S, Froguel P, White A, Farooqi IS, O'Rahilly S. A missense mutation disrupting a dibasic prohormone processing site in pro-opiomelanocortin (POMC) increases susceptibility to early-onset obesity through a novel molecular mechanism. Human Molecular Genetics. 2002;11:1997–2004. doi: 10.1093/hmg/11.17.1997. [DOI] [PubMed] [Google Scholar]
- 67.Oki S, Nakao K, Tanaka I, Kinoshita F, Naki Y, Imura H. Characterization of γ-melanotropin-like immunoreactivity and its secretion in an adrenocorticotropin-producing mouse pituitary tumor cell line. Endocrinology. 1982;111:418–424. doi: 10.1210/endo-111-2-418. [DOI] [PubMed] [Google Scholar]
- 68.van Strien FJ, Devreese B, Van Beeumen J, Roubos EW, Jenks BG. Biosynthesis and processing of the N-terminal part of proopiomelanocortin in Xenopus laevis: Characterization of γ-MSH peptides. Journal of Neuroendocrinology. 1995;7:807–815. doi: 10.1111/j.1365-2826.1995.tb00718.x. [DOI] [PubMed] [Google Scholar]
- 69.Grieco P, Balse-Srinivasan P, Han G, Weinberg D, MacNeil T, Van der Ploeg LHT, Hruby VJ. Synthesis and biological evaluation on hMC(3), hMC(4) and hMC(5) receptors of γ-MSH analogs substituted with L-alanine. Journal of Peptide Research. 2002;59:203–210. doi: 10.1034/j.1399-3011.2002.01966.x. [DOI] [PubMed] [Google Scholar]
- 70.Grieco P, Balse PM, Weinberg D, MacNeil T, Hruby VJ. D-amino acid scan of gamma-melanocyte-stimulating hormone: Importance of Trp8 on human MC3 receptor selectivity. Journal of Medicinal Chemistry. 2000;43:4998–5002. doi: 10.1021/jm000211e. [DOI] [PubMed] [Google Scholar]
- 71.Joseph CG, Yao H, Scott JW, Sorensen NB, Marnane RN, Mountjoy KG, Haskell-Luevano C. γ2-Melanocyte stimulation hormone (γ2-MSH) truncation studies results in the cautionary note that γ2-MSH is not selective for the mouse MC3R over the mouse MC5R. Peptides. 2010;31:2304–2313. doi: 10.1016/j.peptides.2010.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cai M, Mayorov AV, Cabello C, Stankova M, Trivedi D, Hruby VJ. Novel 3D pharmacophore of α-MSH/γ-MSH hybrids leads to selective human MC1R and MC3R analogues. J Med Chem. 2005;48:1839–1848. doi: 10.1021/jm049579s. [DOI] [PubMed] [Google Scholar]
- 73.Bloom FE, Battenberg ELF, Shibasaki T, Benoit R, Ling N, Guillemin R. Localization of γ-melanocyte stimulating hormone (γ-MSH) immunoreactivity in rat brain and pituitary. Regulatory Peptides. 1980;1:205–222. doi: 10.1016/0167-0115(80)90272-4. [DOI] [PubMed] [Google Scholar]
- 74.Denef C, Lu J, Swinnen E. γ-MSH peptides in the pituitary. Annals of the New York Academy of Sciences. 2003;994:123–132. doi: 10.1111/j.1749-6632.2003.tb03171.x. [DOI] [PubMed] [Google Scholar]
- 75.Osamura RY, Komatsu N, Watanabe K, Nakai Y, Tanaka I, Imura H. Immunohistochemical and immunocytochemical localization of γ -melanocyte stimulating hormone (γ-MSH)-like immunoreactivity in human and rat hypothalamus. Peptides. 1982;3:781–787. doi: 10.1016/0196-9781(82)90015-8. [DOI] [PubMed] [Google Scholar]
- 76.Shibasaki T, Ling N, Guillemin R. Pituitary immunoreactive γ-melanotropins are glycosylated oligopeptides. Nature. 1980;285:416–417. doi: 10.1038/285416a0. [DOI] [PubMed] [Google Scholar]
- 77.Fodor M, Sluiter A, Frankhuijzen-Sierevogel A, Wiegant VM, Hoogerhout P, De Wildt DJ, Versteeg DHG. Distribution of Lys-γ2-melanocyte-stimulating hormone-(Lys-γ2-MSH)-like immunoreactivity in neuronal elements in the brain and peripheral tissues of the rat. Brain Research. 1996;731:182–189. doi: 10.1016/0006-8993(96)00464-7. [DOI] [PubMed] [Google Scholar]
- 78.Abbott CR, Rossi M, Kim MS, Al Ahmed SH, Taylor GM, Ghatei MA, Smith DM, Bloom SR. Investigation of the melanocyte stimulating hormones on food intake: Lack of evidence to support a role for the melanocortin-3-receptor. Brain Research. 2000;869:203–210. doi: 10.1016/s0006-8993(00)02386-6. [DOI] [PubMed] [Google Scholar]
- 79.Kathpalia PP, Charlton C, Rajagopal M, Pao AC. The natriuretic mechanism of gamma-melanocyte-stimulating hormone. Peptides. 2011;32:1068–1072. doi: 10.1016/j.peptides.2011.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Bergen P, Kleijne JA, De Wildt DJ, Versteeg DH. Different cardiovascular profiles of three melanocortins in conscious rats; evidence for antagonism between γ2-MSH and ACTH-(1-24) British Journal of Pharmacology. 1997;120:1561–1567. doi: 10.1038/sj.bjp.0701065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Versteeg DHG, Van Bergen P, Adan RAH, De Wildt DJ. Melanocortins and cardiovascular regulation. European Journal of Pharmacology. 1998;360:1–14. doi: 10.1016/s0014-2999(98)00615-3. [DOI] [PubMed] [Google Scholar]
- 82.Bloomquist BT, Eipper BA, Mains RE. Prohormone-converting enzymes: Regulation and evaluation of function using antisense RNA. Molecular Endocrinology. 1991;5:2014–2024. doi: 10.1210/mend-5-12-2014. [DOI] [PubMed] [Google Scholar]
- 83.Zhou A, Bloomquist BT, Mains RE. The prohormone convertases PC1 and PC2 mediate distinct endoproteolytic cleavages in a strict temporal order during proopiomelanocortin biosynthetic processing. Journal of Biological Chemistry. 1993;268:1763–1769. [PubMed] [Google Scholar]
- 84.Schioth HB, Chhajlani V, Muceniece R, Klusa V, Wikberg JE. Major pharmacological distinction of the ACTH receptor from other melanocortin receptors. Life Sciences. 1996;59:797–801. doi: 10.1016/0024-3205(96)00370-0. [DOI] [PubMed] [Google Scholar]
- 85.Schwyzer R. ACTH: A short introductory review. Annals of the New York Academy of Sciences. 1977;297:3–26. doi: 10.1111/j.1749-6632.1977.tb41843.x. [DOI] [PubMed] [Google Scholar]
- 86.Vergoni AV, Poggioli R, Bertolini A. Corticotropin inhibits food intake in rats. Neuropeptides. 1986;7:153–158. doi: 10.1016/0143-4179(86)90091-0. [DOI] [PubMed] [Google Scholar]
- 87.Bertolini A, Poggioli R, Vergoni AV. Cross-species comparison of the ACTH-induced behavioral syndrome. Annals of the New York Academy of Sciences. 1988;525:114–129. doi: 10.1111/j.1749-6632.1988.tb38600.x. [DOI] [PubMed] [Google Scholar]
- 88.Vergoni AV, Poggioli R, Marrama D, Bertolini A. Inhibition of feeding by ACTH-(1-24): Behavioral and pharmacological aspects. European Journal of Pharmacology. 1990;179:347–355. doi: 10.1016/0014-2999(90)90175-6. [DOI] [PubMed] [Google Scholar]
- 89.Miller MW, Duhl DM, Vrieling H, Cordes SP, Ollmann MM, Winkes BM, Barsh GS. Cloning of the mouse agouti gene predicts a secreted protein ubiquitously expressed in mice carrying the lethal yellow mutation. Genes & Development. 1993;7:454–467. doi: 10.1101/gad.7.3.454. [DOI] [PubMed] [Google Scholar]
- 90.Willard DH, Bodnar W, Harris C, Kiefer L, Nichols JS, Blanchard S, Hoffman C, Moyer M, Burkhart W, Weiel J, et al. Agouti structure and function: Characterization of a potent α-melanocyte stimulating hormone receptor antagonist. Biochemistry. 1995;34:12341–12346. doi: 10.1021/bi00038a030. [DOI] [PubMed] [Google Scholar]
- 91.He L, Gunn TM, Bouley DM, Lu XY, Watson SJ, Schlossman SF, Duke-Cohan JS, Barsh GS. A biochemical function for attractin in agouti-induced pigmentation and obesity. Nature Genetics. 2001;27:40–47. doi: 10.1038/83741. [DOI] [PubMed] [Google Scholar]
- 92.Ollmann MM, Lamoreux ML, Wilson BD, Barsh GS. Interaction of Agouti protein with the melanocortin 1 receptor in vitro and in vivo. Genes & Development. 1998;12:316–330. doi: 10.1101/gad.12.3.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kiefer LL, Veal JM, Mountjoy KG, Wilkinson WO. Melanocortin receptor binding determinants in the agouti protein. Biochemistry. 1998;37:991–997. doi: 10.1021/bi971913h. [DOI] [PubMed] [Google Scholar]
- 94.Bultman SJ, Michaud EJ, Woychik RP. Molecular characterization of the mouse agouti locus. Cell. 1992;71:1195–1204. doi: 10.1016/s0092-8674(05)80067-4. [DOI] [PubMed] [Google Scholar]
- 95.Duhl DM, Vrieling H, Miller KA, Wolff GL, Barsh GS. Neomorphic agouti mutations in obese yellow mice. Nature Genetics. 1994;8:59–65. doi: 10.1038/ng0994-59. [DOI] [PubMed] [Google Scholar]
- 96.Lu D, Willard D, Patel IR, Kadwell S, Overton L, Kost T, Luther M, Chen W, Woychik RP, Wilkison WO, et al. Agouti protein is an antagonist of the melanocyte-stimulating-hormone receptor. Nature. 1994;371:799–802. doi: 10.1038/371799a0. [DOI] [PubMed] [Google Scholar]
- 97.McNulty JC, Jackson PJ, Thompson DA, Chai B, Gantz I, Barsh GS, Dawson PE, Millhauser GL. Structures of the agouti signaling protein. Journal of Molecular Biology. 2005;346:1059–1070. doi: 10.1016/j.jmb.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 98.Shutter JR, Graham M, Kinsey AC, Scully S, Luthy R, Stark KL. Hypothalamic expression of ART, a novel gene related to agouti, is up-regulated in obese and diabetic mutant mice. Genes & Development. 1997;11:593–602. doi: 10.1101/gad.11.5.593. [DOI] [PubMed] [Google Scholar]
- 99.Tota MR, Smith TS, Mao C, MacNeil T, Mosley RT, Van der Ploeg LHT, Fong TM. Molecular interaction of agouti protein and agouti-related protein with human melanocortin receptors. Biochemistry. 1999;38:897–904. doi: 10.1021/bi9815602. [DOI] [PubMed] [Google Scholar]
- 100.Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SBH, Gantz I, Millhauser GL. NMR structure of a minimized human agouti related protein prepared by total chemical synthesis. FEBS Letters. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- 101.Jackson PJ, McNulty JC, Yang YK, Thompson DA, Chai B, Gantz I, Barsh GS, Millhauser GL. Design, pharmacology, and NMR structure of a minimized cystine knot with agouti-related protein activity. Biochemistry. 2002;41:7565–7572. doi: 10.1021/bi012000x. [DOI] [PubMed] [Google Scholar]
- 102.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High-resolution NMR structure of the chemically-synthesized melanocortin receptor binding domain AGRP(87-132) of the agouti-related protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 103.Patel MP, Cribb Fabersunne CS, Yang YK, Kaelin CB, Barsh GS, Millhauser GL. Loop-swapped chimeras of the agouti-related protein and the agouti signaling protein identify contacts required for melanocortin 1 receptor selectivity and antagonism. Journal of Molecular Biology. 2010;404:45–55. doi: 10.1016/j.jmb.2010.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Creemers JW, Pritchard LE, Gyte A, Le Rouzic P, Meulemans S, Wardlaw SL, Zhu X, Steiner DF, Davies N, Armstrong D, Lawrence CB, Luckman SM, Schmitz CA, Davies RA, Brennand JC, White A. Agouti-related protein is posttranslationally cleaved by proprotein convertase 1 to generate agouti-related protein (AGRP)83-132: interaction between AGRP83-132 and melanocortin receptors cannot be influenced by syndecan-3. Endocrinology. 2006;147:1621–1631. doi: 10.1210/en.2005-1373. [DOI] [PubMed] [Google Scholar]
- 105.Haskell-Luevano C, Chen P, Li C, Chang K, Smith MS, Cameron JL, Cone RD. Characterization of the neuroanatomical distribution of agouti-related protein immunoreactivity in the rhesus monkey and the rat. Endocrinology. 1999;140:1408–1415. doi: 10.1210/endo.140.3.6544. [DOI] [PubMed] [Google Scholar]
- 106.Mizuno TM, Makimura H, Silverstein J, Roberts JL, Lopingco T, Mobbs CV. Fasting regulates hypothalamic neuropeptide Y, agouti-related peptide, and proopiomelanocortin in diabetic mice independent of changes in leptin or insulin. Endocrinology. 1999;140:4551–4557. doi: 10.1210/endo.140.10.6966. [DOI] [PubMed] [Google Scholar]
- 107.Hagan MM, Rushing PA, Pritchard LM, Schwartz MW, Strack AM, Van Der Ploeg LH, Woods SC, Seeley RJ. Long-term orexigenic effects of AgRP-(83-132) involve mechanisms other than melanocortin receptor blockade. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2000;279:R47–52. doi: 10.1152/ajpregu.2000.279.1.R47. [DOI] [PubMed] [Google Scholar]
- 108.Irani BG, Xiang ZM, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. European Journal of Pharmacology. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sawyer TK, Sanfilippo PJ, Hruby VJ, Engel MH, Heward CB, Burnett JB, Hadley ME. 4-Norleucine, 7-D-phenylalanine-α-melanocyte-stimulating hormone: A highly potent α-melanotropin with ultralong biological activity. Proceedings of the National Academy of Sciences. 1980;77:5754–5758. doi: 10.1073/pnas.77.10.5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heward CB, Yang YCS, Sawyer TK, Bregman MD, Fuller BB, Hruby VJ, Hadley ME. Iodination associated inactivation of β-melanocyte stimulating hormone. Biochemical and Biophysical Research Communications. 1979;88:266–273. doi: 10.1016/0006-291x(79)91725-x. [DOI] [PubMed] [Google Scholar]
- 111.Sawyer TK, Yang YCS, Bregman MD, Hruby VJ, Heward CB, Fuller BB, Hadley ME. Structure-function studies of melanophore stimulating hormones (α-MSH and β-MSH) and their analogs on melanoma plasma membrane adenylate cyclase: comparison with frog skin melanophores. In: Gross E, Meienhofer J, editors. Sixth American Peptide Symposium, Pierce Chemical Company. Georgetown University; Washington D.C: 1979. pp. 1017–1020. [Google Scholar]
- 112.Medzihradszky K. Synthesis and biological activity of adrenocorticotropic and melanotropic hormones. In: Bognar R, Bruckner V, Szantay C, editors. Recent developments in the chemistry of nautral carbon compounds, Akademiai Kiado, Budapest, Hungary. 1976. pp. 119–250. [Google Scholar]
- 113.Smith PE, Graeser JB. A differential response of the melanophore stimulant and oxytocic autocoid of the posterior hyophysis. The Anatomical Record. 1924;27:187. [Google Scholar]
- 114.Haskell-Luevano C, Sawyer TK, Hendrata S, North C, Panahinia L, Stum M, Staples DJ, Castrucci AM, Hadley MF, Hruby VJ. Truncation studies of α-melanotropin peptides identify tripeptide analogues exhibiting prolonged agonist bioactivity. Peptides. 1996;17:995–1002. doi: 10.1016/0196-9781(96)00141-6. [DOI] [PubMed] [Google Scholar]
- 115.Luger TA, Bohm M. An α-MSH analog in erythropoietic protoporphyria. Journal of Investigative Dermatology. 2015;135:929–931. doi: 10.1038/jid.2015.16. [DOI] [PubMed] [Google Scholar]
- 116.Al-Obeidi F, Castrucci AM, Hadley ME, Hruby VJ. Potent and prolonged acting cyclic lactam analogues of α-melanotropin: Design based on molecular dynamics. Journal of Medicinal Chemistry. 1989;32:2555–2561. doi: 10.1021/jm00132a010. [DOI] [PubMed] [Google Scholar]
- 117.Al-Obeidi F, Hadley ME, Pettitt BM, Hruby VJ. Design of a new class of superpotent cyclic α-melanotropins based on quenched dynamic simulations. Journal of the American Chemical Society. 1989;111:3413–3416. [Google Scholar]
- 118.Hruby VJ, Lu DS, Sharma SD, Castrucci AD, Kesterson RA, Alobeidi FA, Hadley ME, Cone RD. Cyclic lactam α-melanotropin analogs of Ac-Nle4-[Asp5,D-Phe7,Lys10] α-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic amino acids at position 7 show high antagonist potency and selectivity at specific melanocortin receptors. Journal of Medicinal Chemistry. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 119.Bednarek MA, MacNeil T, Tang R, Fong TM, Cabello MA, Maroto M, Teran A. Potent and selective agonists of α-melanotropin (α-MSH) action at human melanocortin receptor 5; linear analogs of α-melanotropin. Peptides. 2007;28:1020–1028. doi: 10.1016/j.peptides.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 120.Bednarek MA, Mac Neil T, Tang R, Fong TM, Angeles Cabello M, Maroto M, Teran A. Potent and selective peptide agonists of α-melanocyte stimulating hormone (α-MSH) action at human melanocortin receptor 5; their synthesis and biological evaluation in vitro. Chemical Biology & Drug Design. 2007;69:350–355. doi: 10.1111/j.1747-0285.2007.00513.x. [DOI] [PubMed] [Google Scholar]
- 121.Cai M, Marelli UK, Bao J, Beck JG, Opperer F, Rechenmacher F, McLeod KR, Zingsheim MR, Doedens L, Kessler H, Hruby VJ. Systematic backbone conformational constraints on a cyclic melanotropin ligand leads to highly selective ligands for multiple melanocortin receptors. Journal of Medicinal Chemistry. 2015;58:6359–6367. doi: 10.1021/acs.jmedchem.5b00102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Conde-Frieboes K, Ankersen M, Breinholt J, Hansen BS, Raun K, Thogersen H, Wulff BS. Serendipitous discovery of a new class of agonists for the melanocortin 1 and 4 receptors and a new class of cyclophanes. Bioorganic & Medicinal Chemistry Letters. 2011;21:1459–1463. doi: 10.1016/j.bmcl.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 123.Bednarek MA, MacNeil T, Tang R, Fong TM, Cabello MA, Maroto M, Teran A. Potent and selective peptide agonists for human melanocortin receptors 1b and 5. Journal of Peptide Science. 2008;14:401–408. [Google Scholar]
- 124.Todorovic A, Joseph CG, Sorensen NB, Wood MS, Haskell-Luevano C. Structure-activity relationships of melanocortin agonists containing the benzimidazole scaffold. Chemical Biology & Drug Design. 2007;69:338–349. doi: 10.1111/j.1747-0285.2007.00511.x. [DOI] [PubMed] [Google Scholar]
- 125.Singh A, Dirain ML, Wilczynski A, Chen C, Gosnell BA, Levine AS, Edison AS, Haskell-Luevano C. Synthesis, biophysical, and pharmacological evaluation of the melanocortin agonist AST3-88: Modifications of peptide backbone at Trp 7 position lead to a potent, selective, and stable ligand of the melanocortin-4 receptor (MC4R) ACS Chemical Neuroscience. 2014;5:1020–1031. doi: 10.1021/cn5000953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Singh A, Dirain M, Witek R, Rocca JR, Edison AS, Haskell-Luevano C. Structure–activity relationships of peptides incorporating a bioactive reverse-turn heterocycle at the melanocortin receptors: Identification of a 5800-fold mouse melanocortin-3 receptor (mMC3R), selective antagonist/partial agonist versus the mouse melanocortin-4 receptor (mMC4R) Journal of Medicinal Chemistry. 2013;56:2747–2763. doi: 10.1021/jm301253y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carotenuto A, Merlino F, Cai M, Brancaccio D, Yousif AM, Novellino E, Hruby VJ, Grieco P. Discovery of novel potent and selective agonists at the melanocortin-3 receptor. Journal of Medicinal Chemistry. 2015;58:9773–9778. doi: 10.1021/acs.jmedchem.5b01285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hong QM, Bakshi RK, Dellureficio J, He SW, Ye ZX, Dobbelaar PH, Sebhat IK, Guo LQ, Liu JA, Jian TY, Tang R, Kalyani RN, MacNeil T, Vongs A, Rosenblum CI, Weinberg DH, Peng QP, Tamvakopoulos C, Miller RR, Stearns RA, Cashen D, Martin WJ, Chen AS, Metzger JM, Chen HY, Strack AM, Fong TM, Maclntyre E, Van der Ploeg LHT, Wyvratt MJ, Nargund RP. Optimization of privileged structures for selective and potent melanocortin subtype-4 receptor ligands. Bioorganic & Medicinal Chemistry Letters. 2010;20:4483–4486. doi: 10.1016/j.bmcl.2010.06.038. [DOI] [PubMed] [Google Scholar]
- 129.Hess S, Linde Y, Ovadia O, Safrai E, Shalev DE, Swed A, Halbfinger E, Lapidot T, Winkler I, Gabinet Y, Faier A, Yarden D, Xiang Z, Portillo FP, Haskell-Luevano C, Gilon C, Hoffman A. Backbone cyclic peptidomimetic melanocortin-4 receptor agonist as a novel orally administrated drug lead for treating obesity. Journal of Medicinal Chemistry. 2008;51:1026–1034. doi: 10.1021/jm701093y. [DOI] [PubMed] [Google Scholar]
- 130.Proneth B, Pogozheva ID, Portillo FP, Mosberg HI, Haskell-Luevano C. Melanocortin tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 modified at the para position of the benzyl side chain (DPhe): Importance for mouse melanocortin-3 receptor agonist versus antagonist activity. Journal of Medicinal Chemistry. 2008;51:5585–5593. doi: 10.1021/jm800291b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schild HO. pA, a new scale for the measurement of drug antagonism. British Journal of Pharmacology and Chemotherapy. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Doering SR, Todorovic A, Haskell-Luevano C. Melanocortin antagonist tetrapeptides with minimal agonist activity at the mouse melanocortin-3 receptor. ACS Medicinal Chemistry Letters. 2015;6:123–127. doi: 10.1021/ml500340z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ericson MD, Wilczynski A, Sorensen NB, Xiang ZM, Haskell-Luevano C. Discovery of a β-hairpin octapeptide, c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro], mimetic of agouti-related protein(87-132) [AGRP(87-132)] with equipotent mouse melanocortin-4 receptor (mMC4R) antagonist pharmacology. Journal of Medicinal Chemistry. 2015;58:4638–4647. doi: 10.1021/acs.jmedchem.5b00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lensing CJ, Freeman KT, Schnell SM, Adank DN, Speth RC, Haskell-Luevano C. An in vitro and in vivo investigation of bivalent ligands that display preferential binding and functional activity for different melanocortin receptor homodimers. Journal of Medicinal Chemistry. 2016;59:3112–3128. doi: 10.1021/acs.jmedchem.5b01894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Todorovic A, Haskell-Luevano C. A review of melanocortin receptor small molecule ligands. Peptides. 2005;26:2026–2036. doi: 10.1016/j.peptides.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 136.Ujjainwalla F, Sebhat IK. Small molecule ligands of the human melanocortin-4 receptor. Current Topics in Medicinal Chemistry. 2007;7:1068–1084. doi: 10.2174/156802607780906609. [DOI] [PubMed] [Google Scholar]
- 137.Lansdell MI, Hepworth D, Calabrese A, Brown AD, Blagg J, Burring DJ, Wilson P, Fradet D, Brown TB, Quinton F, Mistry N, Tang K, Mount N, Stacey P, Edmunds N, Adams C, Gaboardi S, Neal-Morgan S, Wayman C, Cole S, Phipps J, Lewis M, Verrier H, Gillon V, Feeder N, Heatherington A, Sultana S, Haughie S, Martin SW, Sudworth M, Tweedy S. Discovery of a selective small-molecule melanocortin-4 receptor agonist with efficacy in a pilot study of sexual dysfunction in humans. Journal of Medicinal Chemistry. 2010;53:3183–3197. doi: 10.1021/jm9017866. [DOI] [PubMed] [Google Scholar]
- 138.Tran JA, Chen CW, Tucci FC, Jiang W, Fleck BA, Chen C. Syntheses of tetrahydrothiophenes and tetrahydrofurans and studies of their derivatives as melanocortin-4 receptor ligands. Bioorganic & Medicinal Chemistry Letters. 2008;18:1124–1130. doi: 10.1016/j.bmcl.2007.11.128. [DOI] [PubMed] [Google Scholar]
- 139.Tran JA, Tucci FC, Arellano M, Jiang WL, Chen CW, Marinkovic D, Fleck BA, Wen J, Foster AC, Chen C. Design and synthesis of 3-arylpyrrolidine-2-carboxamide derivatives as melanocortin-4 receptor ligands. Bioorganic & Medicinal Chemistry Letters. 2008;18:1931–1938. doi: 10.1016/j.bmcl.2008.01.125. [DOI] [PubMed] [Google Scholar]
- 140.Dallmann R, Weyermann P, Anklin C, Boroff M, Bray-French K, Cardel B, Courdier-Fruh I, Deppe H, Dubach-Powell J, Erb M, Haefeli RH, Hennebohle M, Herzner H, Hufschmid M, Marks DL, Nordhoff S, Papp M, Rummey C, Santos G, Scharer F, Siendt H, Soeberdt M, Sumanovski LT, Terinek M, Mondadori C, Guven N, Feurer A. The orally active melanocortin-4 receptor antagonist BL-6020/979: A promising candidate for the treatment of cancer cachexia. Journal of Cachexia Sarcopenia and Muscle. 2011;2:163–174. doi: 10.1007/s13539-011-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Weyermann P, Dallmann R, Magyar J, Anklin C, Hufschmid M, Dubach-Powell J, Courdier-Fruh I, Hennebohle M, Nordhoff S, Mondadori C. Orally available selective melanocortin-4 receptor antagonists stimulate food intake and reduce cancer-induced cachexia in mice. PLOS One. 2009;4 doi: 10.1371/journal.pone.0004774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Haskell-Luevano C, Rosenquist A, Souers A, Khong KC, Ellman JA, Cone RD. Compounds that activate the mouse melanocortin-1 receptor identified by screening a small molecule library based upon the beta-turn. Journal of Medicinal Chemistry. 1999;42:4380–4387. doi: 10.1021/jm990190s. [DOI] [PubMed] [Google Scholar]
- 143.Bondebjerg J, Xiang Z, Bauzo RM, Haskell-Luevano C, Meldal M. A solid-phase approach to mouse melanocortin receptor agonists derived from a novel thioether cyclized peptidomimetic scaffold. Journal of the American Chemical Society. 2002;124:11046–11055. doi: 10.1021/ja0123913. [DOI] [PubMed] [Google Scholar]
- 144.Sebhat IK, Martin WJ, Ye ZX, Barakat K, Mosley RT, Johnston DBR, Bakshi R, Palucki B, Weinberg DH, MacNeil T, Kalyani RN, Tang R, Stearns RA, Miller RR, Tamvakopoulos C, Strack AM, McGowan E, Cashen DE, Drisko JE, Hom GJ, Howard AD, MacIntyre DE, van der Ploeg LHT, Patchett AA, Nargund RP. Design and pharmacology of N-[(3R)-1,2,3,4-tetrahydroisoquinolinium-3-ylcarbonyl]-(1R)-1-(4-chlorobenzyl)2-[4-cyclohexyl-4-(1H-1,2,4-triazol-1-ylmethyl)piperidin-1-yl]-2-oxoethylamine (1), a potent, selective, melanocortin subtype-4 receptor agonist. Journal of Medicinal Chemistry. 2002;45:4589–4593. doi: 10.1021/jm025539h. [DOI] [PubMed] [Google Scholar]
- 145.Palucki BL, Park MK, Nargund RP, Ye ZX, Sebhat IK, Pollard PG, Kalyani RN, Tang R, MacNeil T, Weinberg DH, Vongs A, Rosenblum CI, Doss GA, Miller RR, Stearns RA, Peng QP, Tamvakopoulos C, McGowan E, Martin WJ, Metzger JM, Shepherd CA, Strack AM, MacIntyre DE, Van der Ploeg LHT, Patchett AA. Discovery of (2S)-N-[(1R)-2-[4-cyclohexyl-4-[[(1,1-dimethylethyl)amino]carbonyl]-1-piperidinyl]-1-[(4-fluorophenyl)methyl]-2-oxoethyl]-4-methyl-2-piperazinecarboxamide (MB243), a potent and selective melanocortin subtype-4 receptor agonist. Bioorganic & Medicinal Chemistry Letters. 2005;15:171–175. doi: 10.1016/j.bmcl.2004.10.020. [DOI] [PubMed] [Google Scholar]
- 146.Ujjainwalla F. Design and syntheses of human melanocortin subtype-4 receptor (hMC4R) agonists: Discovery of the tert-butylpyrrolidine archetype. Abstracts of Papers of the American Chemical Society. 2005;230:U2659–U2660. [Google Scholar]
- 147.Guo LQ, Ye ZX, Ujjainwalla F, Sings HL, Sebhat IK, Huber J, Weinberg DH, Tang R, MacNeil T, Tamvakopoulos C, Peng QP, MacIntyre E, van der Ploeg LHT, Goulet MT, Wyvratt MJ, Nargund RP. Synthesis and SAR of potent and orally bioavailable tert-butylpyrrolidine archetype derived melanocortin subtype-4 receptor modulators. Bioorganic & Medicinal Chemistry Letters. 2008;18:3242–3247. doi: 10.1016/j.bmcl.2008.04.049. [DOI] [PubMed] [Google Scholar]
- 148.Chen C, Jiang W, Tran JA, Tucci FC, Fleck BA, Markison S, Wen J, Madan A, Hoare SR, Foster AC, Marinkovic D, Chen CW, Arellano M, Saunders J. Identification and characterization of pyrrolidine diastereoisomers as potent functional agonists and antagonists of the human melanocortin-4 receptor. Bioorganic & Medicinal Chemistry Letters. 2008;18:129–136. doi: 10.1016/j.bmcl.2007.10.115. [DOI] [PubMed] [Google Scholar]
- 149.Tran JA, Chen CW, Jiang W, Tucci FC, Fleck BA, Marinkovic D, Arellano M, Chen C. Pyrrolidines as potent functional agonists of the human melanocortin-4 receptor. Bioorganic & Medicinal Chemistry Letters. 2007;17:5165–5170. doi: 10.1016/j.bmcl.2007.06.088. [DOI] [PubMed] [Google Scholar]
- 150.Marinkovic D, Tucci FC, Tran JA, Fleck BA, Wen J, Chen C. Structure-activity relationship studies on a series of piperazinebenzylalcohols and their ketone and amine analogs as melanocortin-4 receptor ligands. Bioorganic & Medicinal Chemistry Letters. 2008;18:4817–4822. doi: 10.1016/j.bmcl.2008.07.076. [DOI] [PubMed] [Google Scholar]
- 151.Jiang WL, Tucci FC, Chen CW, Arellano M, Tran JA, White NS, Marinkovic D, Pontillo J, Fleck BA, Wen J, Saunders J, Madan A, Foster AC, Chen C. Arylpropionylpiperazines as antagonists of the human melanocortin-4 receptor. Bioorganic & Medicinal Chemistry Letters. 2006;16:4674–4678. doi: 10.1016/j.bmcl.2006.05.088. [DOI] [PubMed] [Google Scholar]
- 152.Tucci FC, White NS, Markison S, Joppa M, Tran JA, Fleck BA, Madan A, Dyck BP, Parker J, Pontillo J, Arellano LM, Marinkovic D, Jiang WL, Chen CW, Gogas KR, Goodfellow VS, Saunders J, Fosters AC, Chen C. Potent and orally active non-peptide antagonists of the human melanocortin-4 receptor based on a series of trans-2-disubstituted cyclohexylpiperazines. Bioorganic & Medicinal Chemistry Letters. 2005;15:4389–4395. doi: 10.1016/j.bmcl.2005.06.071. [DOI] [PubMed] [Google Scholar]
- 153.Tran JA, Arellano M, Fleck BA, Pontillo J, Marinkovic D, Tucci FC, Wen J, Saunders J, Chen C. Studies on the structure-activity relationship of the basic amine of phenylpiperazines as melanocortin-4 receptor antagonists. Medicinal Chemistry. 2008;4:67–74. doi: 10.2174/157340608783331498. [DOI] [PubMed] [Google Scholar]
- 154.Chen C, Tucci FC, Jiang WL, Tran JA, Fleck BA, Hoare SR, Wen J, Chen TK, Johns M, Markison S, Foster AC, Marinkovic D, Chen CW, Arellano M, Harman J, Saunders J, Bozigian H, Marks D. Pharmacological and pharmacokinetic characterization of 2-piperazine-α-isopropyl benzylamine derivatives as melanocortin-4 receptor antagonists. Bioorganic & Medicinal Chemistry. 2008;16:5606–5618. doi: 10.1016/j.bmc.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 155.Guo LQ, Ye ZX, Liu JA, He SW, Bakshi RK, Sebhat IK, Dobbelaar PH, Hong QM, Jian TY, Dellureficio JP, Tsou NN, Ball RG, Weinberg DH, MacNeil T, Tang R, Tamvakopoulos C, Peng QP, Chen HY, Chen ARS, Martin WJ, MacIntyre DE, Strack AM, Fong TM, Wyvratt MJ, Nargund RP. Discovery of potent, selective, and orally bioavailable 3H-spiro[isobenzofuran-1,4'-piperidine] based melanocortin subtype-4 receptor agonists. Bioorganic & Medicinal Chemistry Letters. 2010;20:4895–4900. doi: 10.1016/j.bmcl.2010.06.068. [DOI] [PubMed] [Google Scholar]
- 156.He SW, Ye ZX, Dobbelaar PH, Bakshi RK, Hong QM, Dellureficio JP, Sebhat IK, Guo LQ, Liu JA, Jian TY, Lai YJ, Franklin CL, Reibarkh M, Holmes MA, Weinberg DH, MacNeil T, Tang R, Tamvakopoulos C, Peng QP, Miller RR, Stearns RA, Chen HY, Chen AS, Strack AM, Fong TM, Wyvratt MJ, Nargund RP. Discovery of highly potent and efficacious MC4R agonists with spiroindane N-Me-1,2,4-triazole privileged structures for the treatment of obesity. Bioorganic & Medicinal Chemistry Letters. 2010;20:6524–6532. doi: 10.1016/j.bmcl.2010.09.049. [DOI] [PubMed] [Google Scholar]
- 157.He SW, Ye ZX, Dobbelaar PH, Sebhat IK, Guo LQ, Liu J, Jian TY, Lai YJ, Franklin CL, Bakshi RK, Dellureficio JP, Hong QM, Tsou NN, Ball RG, Cashen DE, Martin WJ, Weinberg DH, MacNeil T, Tang R, Tamvakopoulos C, Peng QP, Miller RR, Stearns RA, Chen HY, Chen AS, Strack AM, Fong TM, MacIntyre DE, Wyvratt MJ, Nargund RP. Discovery of a spiroindane based compound as a potent, selective, orally bioavailable melanocortin subtype-4 receptor agonist. Bioorganic & Medicinal Chemistry Letters. 2010;20:2106–2110. doi: 10.1016/j.bmcl.2010.02.058. [DOI] [PubMed] [Google Scholar]
- 158.He SW, Ye ZX, Dobbelaar PH, Sebhat IK, Guo LQ, Liu JA, Jian TY, Lai YJ, Franklin CL, Bakshi RK, Dellureficio JP, Hong QM, Weinberg DH, MacNeil T, Tang R, Strack AM, Tamvakopoulos C, Peng QP, Miller RR, Stearns RA, Chen HY, Chen ARS, Fong TM, Wyvratt MJ, Nargund RP. Spiroindane based amides as potent and selective MC4R agonists for the treatment of obesity. Bioorganic & Medicinal Chemistry Letters. 2010;20:4399–4405. doi: 10.1016/j.bmcl.2010.06.062. [DOI] [PubMed] [Google Scholar]
- 159.Patchett AA, Nargund RP, Tata JR, Chen MH, Barakat KJ, Johnston DBR, Cheng K, Chan WWS, Butler B, Hickey G, Jacks T, Schleim K, Pong SS, Chaung LYP, Chen HY, Frazier E, Leung KH, Chiu SHL, Smith RG. Design and biological-activities of l-163,191 (MK-0677) - a potent, orally-active growth-hormone secretagogue. Proceedings of the National Academy of Sciences. 1995;92:7001–7005. doi: 10.1073/pnas.92.15.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hong QM, Bakshi RK, Palucki BL, Park MK, Ye ZX, He SW, Pollard PG, Sebhat IK, Liu JA, Guo LQ, Cashen DE, Martin WJ, Weinberg DH, MacNeil T, Tang R, Tamvakopoulos C, Peng QP, Miller RR, Stearns RA, Chen HY, Chen AS, Strack AM, Fong TM, MacIntyre DE, Wyvratt MJ, Nargund RP. Discovery of a piperazine urea based compound as a potent, selective, orally bioavailable melanocortin subtype-4 receptor partial agonist. Bioorganic & Medicinal Chemistry Letters. 2011;21:2330–2334. doi: 10.1016/j.bmcl.2011.02.090. [DOI] [PubMed] [Google Scholar]
- 161.Singh A, Kast J, Dirain MLS, Huang HS, Haskell-Luevano C. Synthesis and structure-activity relationships of substituted urea derivatives on mouse melanocortin receptors. ACS Chemical Neuroscience. 2016;7:196–205. doi: 10.1021/acschemneuro.5b00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Joseph CG, Bauzo RM, Xiang ZM, Haskell-Luevano C. Urea small molecule agonists on mouse melanocortin receptors. Bioorganic & Medicinal Chemistry Letters. 2003;13:2079–2082. doi: 10.1016/s0960-894x(03)00318-4. [DOI] [PubMed] [Google Scholar]
- 163.Joseph CG, Wilson KR, Wood MS, Sorenson NB, Phan DV, Xiang ZM, Witek RM, Haskell-Luevano C. The 1,4-benzodiazepine-2,5-dione small molecule template results in melanocortin receptor agonists with nanomolar potencies. Journal of Medicinal Chemistry. 2008;51:1423–1431. doi: 10.1021/jm701303z. [DOI] [PubMed] [Google Scholar]
- 164.Horton DA, Bourne GT, Smythe ML. The combinatorial synthesis of bicyclic privileged structures or privileged substructures. Chemical Reviews. 2003;103:893–930. doi: 10.1021/cr020033s. [DOI] [PubMed] [Google Scholar]
- 165.Szewczyk JR, Laudeman CP, Sammond DM, Villeneuve M, Minick DJ, Grizzle MK, Daniels AJ, Andrews JL, Ignar DM. A concise synthesis of 1,4-dihydro- 1,4 diazepine-5,7-dione, a novel 7-TM receptor ligand core structure with melanocortin receptor agonist activity. Bioorganic & Medicinal Chemistry. 2010;18:1822–1833. doi: 10.1016/j.bmc.2010.01.049. [DOI] [PubMed] [Google Scholar]
- 166.Dempfle A, Hinney A, Heinzel-Gutenbrunner M, Raab M, Geller F, Gudermann T, Schafer H, Hebebrand J. Large quantitative effect of melanocortin-4 receptor gene mutations on body mass index. Journal of Medical Genetics. 2004;41:795–800. doi: 10.1136/jmg.2004.018614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stutzmann F, Tan K, Vatin V, Dina C, Jouret B, Tichet J, Balkau B, Potoczna N, Horber F, O'Rahilly S, Farooqi IS, Froguel P, Meyre D. Prevalence of melanocortin-4 receptor deficiency in Europeans and their age-dependent penetrance in multigenerational pedigrees. Diabetes. 2008;57:2511–2518. doi: 10.2337/db08-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sutton GM, Trevaskis JL, Hulver MW, McMillan RP, Markward NJ, Babin MJ, Meyer EA, Butler AA. Diet-genotype interactions in the development of the obese, insulin-resistant phenotype of C57BL/6J mice lacking melanocortin-3 or-4 receptors. Endocrinology. 2006;147:2183–2196. doi: 10.1210/en.2005-1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Asarian L, Geary N. Sex differences in the physiology of eating. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2013;305:R1215–R1267. doi: 10.1152/ajpregu.00446.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Uckert S, Bannowsky A, Albrecht K, Kuczyk MA. Melanocortin receptor agonists in the treatment of male and female sexual dysfunctions: Results from basic research and clinical studies. Expert Opinion on Investigational Drugs. 2014;23:1477–1483. doi: 10.1517/13543784.2014.934805. [DOI] [PubMed] [Google Scholar]
- 171.Wikberg JES, Mutulis F. Targeting melanocortin receptors: An approach to treat weight disorders and sexual dysfunction. Nature Reviews Drug Discovery. 2008;7:307–323. doi: 10.1038/nrd2331. [DOI] [PubMed] [Google Scholar]
- 172.Diamond LE, Earle DC, Rosen RC, Willett MS, Molinoff PB. Double-blind, placebo-controlled evaluation of the safety, pharmacokinetic properties and pharmacodynamic effects of intranasal PT-141, a melanocortin receptor agonist, in healthy males and patients with mild-to-moderate erectile dysfunction. International Journal of Impotence Research. 2004;16:51–59. doi: 10.1038/sj.ijir.3901139. [DOI] [PubMed] [Google Scholar]
- 173.Rosen RC, Diamond LE, Earle DC, Shadiack AM, Molinoff PB. Evaluation of the safety, pharmacokinetics and pharmacodynamic effects of subcutaneously administered PT-141, a melanocortin receptor agonist, in healthy male subjects and in patients with an inadequate response to Viagra. International Journal of Impotence Research. 2004;16:135–142. doi: 10.1038/sj.ijir.3901200. [DOI] [PubMed] [Google Scholar]
- 174.Hadley ME. Discovery that a melanocortin regulates sexual functions in male and female humans. Peptides. 2005;26:1687–1689. doi: 10.1016/j.peptides.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 175.Beckwith BE, Oquin RK, Petro MS, Kastin AJ, Sandman CA. Effects of neonatal injections of α-MSH on open-field behavior of juvenile and adult rats. Physiological Psychology. 1977;5:295–299. [Google Scholar]
- 176.Beckwith BE, Sandman CA, Hothersall D, Kastin AJ. Influence of neonatal injections of α-MSH on learning, memory and attention in rats. Physiology & Behavior. 1977;18:63–71. doi: 10.1016/0031-9384(77)90095-6. [DOI] [PubMed] [Google Scholar]
- 177.Barrett CE, Modi ME, Zhang BC, Walum H, Inoue K, Young LJ. Neonatal melanocortin receptor agonist treatment reduces play fighting and promotes adult attachment in prairie voles in a sex-dependent manner. Neuropharmacology. 2014;85:357–366. doi: 10.1016/j.neuropharm.2014.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mogil JS, Wilson SG, Chesler EJ, Rankin AL, Nemmani KVS, Lariviere WR, Groce MK, Wallace MR, Kaplan L, Staud R, Ness TJ, Glover TL, Stankova M, Mayorov A, Hruby VJ, Grisel JE, Fillingim RB. The melanocortin-1 receptor gene mediates female-specific mechanisms of analgesia in mice and humans. Proceedings of the National Academy of Sciences. 2003;100:4867–4872. doi: 10.1073/pnas.0730053100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Arout CA, Caldwell M, Rossi G, Kest B. Spinal and supraspinal N-methyl-D-aspartate and melanocortin-1 receptors contribute to a qualitative sex difference in morphine-induced hyperalgesia. Physiology & Behavior. 2015;147:364–372. doi: 10.1016/j.physbeh.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 180.Juni A, Cai MY, Stankova M, Waxman AR, Arout C, Klein G, Dahan A, Hruby VJ, Mogil JS, Kest B. Sex-specific mediation of opioid-induced hyperalgesia by the melanocortin-1 receptor. Anesthesiology. 2010;112:181–188. doi: 10.1097/ALN.0b013e3181c53849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Goodin SZ, Keichler AR, Smith M, Wendt D, Strader AD. Effect of gonadectomy on AGRP-induced weight gain in rats. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2008;295:R1747–R1753. doi: 10.1152/ajpregu.90345.2008. [DOI] [PubMed] [Google Scholar]
- 182.Lensing CJ, Adank DN, Doering SR, Wilber SL, Andreasen A, Schaub JW, Xiang ZM, Haskell-Luevano C. Ac-Trp-DPhe(p-l)-Arg-Trp-NH2, a 250-fold selective melanocortin-4 receptor (MC4R) antagonist over the melanocortin-3 receptor (MC3R), affects energy homeostasis in male and female mice differently. ACS Chemical Neuroscience. 2016;7:1283–1291. doi: 10.1021/acschemneuro.6b00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Maranon RO, Lima R, Mathbout M, do Carmo JM, Hall JE, Roman RJ, Reckelhoff JF. Postmenopausal hypertension: Role of the sympathetic nervous system in an animal model. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2014;306:R248–R256. doi: 10.1152/ajpregu.00490.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Mogil JS. Sex differences in pain and pain inhibition: multiple explanations of a controversial phenomenon. Nature Reviews Neuroscience. 2012;13:859–866. doi: 10.1038/nrn3360. [DOI] [PubMed] [Google Scholar]
- 185.Mogil JS, Ritchie J, Smith SB, Strasburg K, Kaplan L, Wallace MR, Romberg RR, Bijl H, Sarton EY, Fillingim RB, Dahan A. Melanocortin-1 receptor gene variants affect pain and μ-opioid analgesia in mice and humans. Journal of Medical Genetics. 2005;42:583–587. doi: 10.1136/jmg.2004.027698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Mogil JS, Sternberg WF, Kest B, Marek P, Liebeskind JC. Sex-differences in the antagonism of swim stress-induced analgesia - Effects of gonadectomy and estrogen replacement. Pain. 1993;53:17–25. doi: 10.1016/0304-3959(93)90050-Y. [DOI] [PubMed] [Google Scholar]
- 187.Delaney A, Keighren M, Fleetwood-Walker SM, Jackson IJ. Involvement of the melanocortin-1 receptor in acute pain and pain of inflammatory but not neuropathic origin. PLOS One. 2010;5 doi: 10.1371/journal.pone.0012498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Han G, Quillan JM, Carlson K, Sadee W, Hruby VJ. Design of novel chimeric melanotropin-deltorphin analogues. Discovery of the first potent human melanocortin 1 receptor antagonist. Journal of Medicinal Chemistry. 2003;46:810–819. doi: 10.1021/jm020355o. [DOI] [PubMed] [Google Scholar]
- 189.Al-Obeidi F, Hruby VJ, Hadley ME, Sawyer TK, Castrucci AMD. Design, synthesis, and biological activities of a potent and selective α-melanotropin antagonist. International Journal of Peptide and Protein Research. 1990;35:228–234. doi: 10.1111/j.1399-3011.1990.tb00942.x. [DOI] [PubMed] [Google Scholar]
- 190.Mogil JS, Bailey AL. Sex and gender differences in pain and analgesia. In: Savic I, editor. Sex Differences in the Human Brain, Their Underpinnings and Implications. Elsevier; Amsterdam: 2010. pp. 141–157. [DOI] [PubMed] [Google Scholar]
- 191.Waxman AR, Juni A, Kowalczyk W, Arout C, Sternberg WF, Kest B. Progesterone rapidly recruits female-typical opioid-induced hyperalgesic mechanisms. Physiology & Behavior. 2010;101:759–763. doi: 10.1016/j.physbeh.2010.08.018. [DOI] [PubMed] [Google Scholar]
- 192.Cai MY, Stankova M, Muthu D, Mayorov A, Yang ZH, Trivedi D, Cabello C, Hruby VJ. An unusual conformation of γ-melanocyte-stimulating hormone analogues leads to a selective human melanocortin 1 receptor antagonist for targeting melanoma cells. Biochemistry. 2013;52:752–764. doi: 10.1021/bi300723f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Kalange AS, Kokare DM, Singru PS, Upadhya MA, Chopde CT, Subhedar NK. Central administration of selective melanocortin 4 receptor antagonist HS014 prevents morphine tolerance and withdrawal hyperalgesia. Brain Research. 2007;1181:10–20. doi: 10.1016/j.brainres.2007.08.054. [DOI] [PubMed] [Google Scholar]
- 194.Starowicz K, Sieja A, Bilecki W, Obara I, Przewlocka B. The effect of morphine on MC4 and CRF receptor mRNAs in the rat amygdala and attenuation of tolerance after their blockade. Brain Research. 2003;990:113–119. doi: 10.1016/s0006-8993(03)03444-9. [DOI] [PubMed] [Google Scholar]
- 195.Niu ZJ, Ma JG, Chu HC, Zhao Y, Feng W, Cheng YW. Melanocortin 4 receptor antagonists attenuates morphine antinociceptive tolerance, astroglial activation and cytokines expression in the spinal cord of rat. Neuroscience Letters. 2012;529:112–117. doi: 10.1016/j.neulet.2012.09.034. [DOI] [PubMed] [Google Scholar]
- 196.Rowland NE, Schaub JW, Robertson KL, Andreasen A, Haskell-Luevano C. Effect of MTII on food intake and brain c-Fos in melanocortin-3, melanocortin-4, and double MC3 and MC4 receptor knockout mice. Peptides. 2010;31:2314–2317. doi: 10.1016/j.peptides.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Clegg DJ, Riedy CA, Smith KAB, Benoit SC, Woods SC. Differential sensitivity to central leptin and insulin in male and female rats. Diabetes. 2003;52:682–687. doi: 10.2337/diabetes.52.3.682. [DOI] [PubMed] [Google Scholar]
- 198.Polidori C, Geary N. Estradiol treatment fails to affect the feeding responses to melanocortin-3/4 receptor agonism or antagonism in ovariectomized rats. Peptides. 2002;23:1697–1700. doi: 10.1016/s0196-9781(02)00112-2. [DOI] [PubMed] [Google Scholar]
- 199.Atalayer D, Robertson KL, Haskell-Luevano C, Andreasen A, Rowland NE. Food demand and meal size in mice with single or combined disruption of melanocortin type 3 and 4 receptors. American Journal of Physiology-Regulatory Integrative and Comparative Physiology. 2010;298:R1667–R1674. doi: 10.1152/ajpregu.00562.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Shah K, McCormack CE, Bradbury NA. Do you know the sex of your cells? American Journal of Physiology-Cell Physiology. 2014;306:C3–C18. doi: 10.1152/ajpcell.00281.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jagadish B, Sankaranarayanan R, Xu L, Richards R, Vagner J, Hruby VJ, Gillies RJ, Mash EA. Squalene-derived flexible linkers for bioactive peptides. Bioorganic & Medicinal Chemistry Letters. 2007;17:3310–3313. doi: 10.1016/j.bmcl.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Opioid agonist and antagonist bivalent ligands as receptor probes. Life Sciences. 1982;31:1283–1286. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 203.Kiessling LL, Gestwicki JE, Strong LE. Synthetic multivalent ligands in the exploration of cell-surface interactions. Current Opinion in Chemical Biology. 2000;4:696–703. doi: 10.1016/s1367-5931(00)00153-8. [DOI] [PubMed] [Google Scholar]
- 204.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. Selective tumor cell targeting using low-affinity, multivalent interactions. ACS Chemical Biology. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 205.Alleti R, Vagner J, Dehigaspitiya DC, Moberg VE, Elshan N, Tafreshi NK, Brabez N, Weber CS, Lynch RM, Hruby VJ, Gillies RJ, Morse DL, Mash EA. Synthesis and characterization of time-resolved fluorescence probes for evaluation of competitive binding to melanocortin receptors. Bioorganic & Medicinal Chemistry. 2013;21:5029–5038. doi: 10.1016/j.bmc.2013.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Kiessling LL, Lamanna AC. Multivalency in biological systems. In: Schneider MP, editor. Chemical Probes in Biology: Science at the Interface of Chemistry, Biology and Medicine. Springer; Dordrecht: 2003. pp. 345–357. [Google Scholar]
- 207.Zanna PT, Sanchez-Laorden BL, Perez-Oliva AB, Turpin MC, Herraiz C, Jimenez-Cervantes C, Garcia-Borron JC. Mechanism of dimerization of the human melanocortin 1 receptor. Biochemical and Biophysical Research Communications. 2008;368:211–216. doi: 10.1016/j.bbrc.2008.01.060. [DOI] [PubMed] [Google Scholar]
- 208.Mandrika I, Petrovska R, Wikberg J. Melanocortin receptors form constitutive homo- and heterodimers. Biochemical and Biophysical Research Communications. 2005;326:349–354. doi: 10.1016/j.bbrc.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 209.Sebag JA, Hinkle PM. Opposite effects of the melanocortin-2 (MC2) receptor accessory protein MRAP on MC2 and MC5 receptor dimerization and trafficking. Journal of Biological Chemistry. 2009;284:22641–22648. doi: 10.1074/jbc.M109.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Piechowski CL, Rediger A, Lagemann C, Muhlhaus J, Muller A, Pratzka J, Tarnow P, Gruters A, Krude H, Kleinau G, Biebermann H. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. Journal of Molecular Endocrinology. 2013;51:109–118. doi: 10.1530/JME-13-0061. [DOI] [PubMed] [Google Scholar]
- 211.Rediger A, Piechowski CL, Habegger K, Gruters A, Krude H, Tschop MH, Kleinau G, Biebermann H. MC4R dimerization in the paraventricular nucleus and GHSR/MC3R heterodimerization in the arcuate nucleus: Is there relevance for body weight regulation? Neuroendocrinology. 2012;95:277–288. doi: 10.1159/000334903. [DOI] [PubMed] [Google Scholar]
- 212.Nickolls SA, Maki RA. Dimerization of the melanocortin 4 receptor: A study using bioluminescence resonance energy transfer. Peptides. 2006;27:380–387. doi: 10.1016/j.peptides.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 213.Biebermann H, Krude H, Elsner A, Chubanov V, Gudermann T, Gruters A. Autosomal-dominant mode of inheritance of a melanocortin-4 receptor mutation in a patient with severe early-onset obesity is due to a dominant-negative effect caused by receptor dimerization. Diabetes. 2003;52:2984–2988. doi: 10.2337/diabetes.52.12.2984. [DOI] [PubMed] [Google Scholar]
- 214.Lensing CJ, Adank DN, Wilber SL, Freeman KT, Schnell SM, Speth RC, Zarth AT, Haskell-Luevano C. A direct in vivo comparison of the melanocortin monovalent agonist Ac-His-DPhe-Arg-Trp-NH2 versus the bivalent agonist Ac-His-DPhe-Arg-Trp-PEDG20-His-DPhe-Arg-Trp-NH2: A bivalent advantage. ACS Chemical Neuroscience. 2017 doi: 10.1021/acschemneuro.6b00399. 10.1021/acschemneuro.6b00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kopanchuk S, Veiksina S, Petrovska R, Mutule I, Szardenings M, Rinken A, Wikberg JES. Cooperative regulation of ligand binding to melanocortin receptor subtypes: Evidence for interacting binding sites. European Journal of Pharmacology. 2005;512:85–95. doi: 10.1016/j.ejphar.2005.02.021. [DOI] [PubMed] [Google Scholar]
- 216.Kopanchuk S, Veiksina S, Mutulis F, Mutule I, Yahorava S, Mandrika I, Petrovska R, Rinken A, Wikberg JES. Kinetic evidence for tandemly arranged ligand binding sites in melanocortin 4 receptor complexes. Neurochemistry International. 2006;49:533–542. doi: 10.1016/j.neuint.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 217.Brabez N, Lynch RM, Xu LP, Gillies RJ, Chassaing G, Lavielle S, Hruby VJ. Design, synthesis, and biological studies of efficient multivalent melanotropin ligands: Tools toward melanoma diagnosis and treatment. Journal of Medicinal Chemistry. 2011;54:7375–7384. doi: 10.1021/jm2009937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Eberle A, Kriwaczek VM, Schwyzer R. Hormone-receptor interactions: Melanotropic activities of covalent serum-albumin complexes with α-melanotropin, α-melanotropin fragments, and enkephalin. FEBS Letters. 1977;80:246–250. doi: 10.1016/0014-5793(77)80450-x. [DOI] [PubMed] [Google Scholar]
- 219.Kriwaczek VM, Eberle AN, Muller M, Schwyzer R. Tobacco mosaic-virus as a carrier for small molecules 1. Preparation and characterization of a TMV-α-melanotropin conjugate. Helvetica Chimica Acta. 1978;61:1232–1240. [Google Scholar]
- 220.Bard DR, Knight CG, Pagethomas DP. A chelating derivative of α-melanocyte stimulating hormone as a potential imaging agent for malignant-melanoma. British Journal of Cancer. 1990;62:919–922. doi: 10.1038/bjc.1990.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wraight EP, Bard DR, Maughan TS, Knight CG, Pagethomas DP. The use of a chelating derivative of alpha melanocyte stimulating hormone for the clinical imaging of malignant-melanoma. British Journal of Radiology. 1992;65:112–118. doi: 10.1259/0007-1285-65-770-112. [DOI] [PubMed] [Google Scholar]
- 222.Bard DR, Wraight EP, Knight CG. BisMSH-DTPA: A potential imaging agent for malignant melanoma. Annals of the New York Academy of Sciences. 1993;680:451–453. doi: 10.1111/j.1749-6632.1993.tb19705.x. [DOI] [PubMed] [Google Scholar]
- 223.Bard DR. An improved imaging agent for malignant-melanoma, based on Nle(4), DPhe(7) α-melanocyte stimulating hormone. Nuclear Medicine Communications. 1995;16:860–866. doi: 10.1097/00006231-199510000-00010. [DOI] [PubMed] [Google Scholar]
- 224.Eberle AN, Froidevaux S. Radiolabeled α-melanocyte-stimulating hormone analogs for receptor-mediated targeting of melanoma: From tritium to indium. Journal of Molecular Recognition. 2003;16:248–254. doi: 10.1002/jmr.633. [DOI] [PubMed] [Google Scholar]
- 225.Eberle AN, Bapst JP, Calame M, Tanner H, Froidevaux S. MSH radiopeptides for targeting melanoma metastases. In: Catania A, editor. Melanocortins: Multiple Actions and Therapeutic Potential. Springer; New York: 2010. pp. 133–142. [DOI] [PubMed] [Google Scholar]
- 226.Ren G, Pan Y, Cheng Z. Molecular probes for malignant melanoma imaging. Current Pharmaceutical Biotechnology. 2010;11:590–602. doi: 10.2174/138920110792246465. [DOI] [PubMed] [Google Scholar]
- 227.Rosenkranz AA, Slastnikova TA, Durymanov MO, Sobolev AS. Malignant melanoma and melanocortin 1 receptor. Biochemistry-Moscow. 2013;78:1228–1237. doi: 10.1134/S0006297913110035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Miao YB, Quinn TP. Alpha-melanocyte stimulating hormone peptide-targeted melanoma imaging. Frontiers in Bioscience. 2007;12:4514–4524. doi: 10.2741/2406. [DOI] [PubMed] [Google Scholar]
- 229.Suzuki I, Cone RD, Im S, Nordlund J, Abdel-Malek ZA. Binding of melanotropic hormones to the melanocortin receptor MC1R on human melanocytes stimulates proliferation and melanogenesis. Endocrinology. 1996;137:1627–1633. doi: 10.1210/endo.137.5.8612494. [DOI] [PubMed] [Google Scholar]
- 230.Erez M, Takemori AE, Portoghese PS. Narcotic antagonistic potency of bivalent ligands which contain β-naltrexamine. Evidence for bridging between proximal recognition sites. Journal of Medicinal Chemistry. 1982;25:847–849. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 231.Conn PM, Rogers DC, Stewart JM, Niedel J, Sheffield T. Conversion of a gonadotropin-releasing hormone antagonist to an agonist. Nature. 1982;296:653–655. doi: 10.1038/296653a0. [DOI] [PubMed] [Google Scholar]
- 232.Blum JJ, Conn PM. Gonadotropin-releasing hormone stimulation of luteinizing-hormone release: A ligand-receptor-effector model. Proceedings of the National Academy of Sciences. 1982;79:7307–7311. doi: 10.1073/pnas.79.23.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Carrithers MD, Lerner MR. Synthesis and characterization of bivalent peptide ligands targeted to G protein-coupled receptors. Chemistry & Biology. 1996;3:537–542. doi: 10.1016/s1074-5521(96)90144-1. [DOI] [PubMed] [Google Scholar]
- 234.Jayawickreme CK, Quillan JM, Graminski GF, Lerner MR. Discovery and structure-function analysis of alpha-melanocyte-stimulating hormone antagonists. Journal of Biological Chemistry. 1994;269:29846–29854. [PubMed] [Google Scholar]
- 235.Vagner J, Handl HL, Gillies RJ, Hruby VJ. Novel targeting strategy based on multimeric ligands for drug delivery and molecular imaging: Homooligomers of α-MSH. Bioorganic & Medicinal Chemistry Letters. 2004;14:211–215. doi: 10.1016/j.bmcl.2003.09.079. [DOI] [PubMed] [Google Scholar]
- 236.Vagner J, Handl HL, Monguchi Y, Jana U, Begay LJ, Mash EA, Hruby VJ, Gillies RJ. Rigid linkers for bioactive peptides. Bioconjugate Chemistry. 2006;17:1545–1550. doi: 10.1021/bc060154p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Handl HL, Sankaranarayanan R, Josan JS, Vagner J, Mash EA, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent NDP-α-MSH(7) peptide ligands for binding to the human melanocortin receptor 4 (hMC4R) Bioconjugate Chemistry. 2007;18:1101–1109. doi: 10.1021/bc0603642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Bowen ME, Monguchi Y, Sankaranarayanan R, Vagner J, Begay LJ, Xu LP, Jagadish B, Hruby VJ, Gillies RJ, Mash EA. Design, synthesis, and validation of a branched flexible linker for bioactive peptides. Journal of Organic Chemistry. 2007;72:1675–1680. doi: 10.1021/jo062276g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Fernandes SM, Lee YS, Gillies RJ, Hruby VJ. Synthesis and evaluation of bivalent ligands for binding to the human melanocortin-4 receptor. Bioorganic & Medicinal Chemistry. 2014;22:6360–6365. doi: 10.1016/j.bmc.2014.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Dehigaspitiya DC, Navath S, Weber CS, Lynch RM, Mash EA. Synthesis and bioactivity of MSH4 oligomers prepared by an A2 + B2 strategy. Tetrahedron Letters. 2015;56:3060–3065. doi: 10.1016/j.tetlet.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Dehigaspitiya DC, Anglin BL, Smith KR, Weber CS, Lynch RM, Mash EA. Linear scaffolds for multivalent targeting of melanocortin receptors. Organic & Biomolecular Chemistry. 2015;13:11507–11517. doi: 10.1039/c5ob01779c. [DOI] [PubMed] [Google Scholar]
- 242.Elshan N, Jayasundera T, Anglin BL, Weber CS, Lynch RM, Mash EA. Trigonal scaffolds for multivalent targeting of melanocortin receptors. Organic & Biomolecular Chemistry. 2015;13:1778–1791. doi: 10.1039/c4ob02094d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Morais M, Raposinho PD, Oliveira MC, Correia JDG, Santos I. Evaluation of novel 99mTc(I)-labeled homobivalent α-melanocyte-stimulating hormone analogs for melanocortin-1 receptor targeting. Journal of Biological Inorganic Chemistry. 2012;17:491–505. doi: 10.1007/s00775-011-0871-y. [DOI] [PubMed] [Google Scholar]
- 244.Bapst JP, Froidevaux S, Calame M, Tanner H, Eberle AN. Dimeric DOTA-α-melanocyte-stimulating hormone analogs: Synthesis and in vivo characteristics of radiopeptides with high in vitro activity. Journal of Receptors and Signal Transduction. 2007;27:383–409. doi: 10.1080/10799890701723528. [DOI] [PubMed] [Google Scholar]
- 245.Bagutti C, Stolz B, Albert R, Bruns C, Pless J, Eberle AN. [111In]-DTPA-labeled analogs of α-melanocyte-stimulating hormone for melanoma targeting: Receptor binding in vitro and in vivo. International Journal of Cancer. 1994;58:749–755. doi: 10.1002/ijc.2910580521. [DOI] [PubMed] [Google Scholar]
- 246.Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. Opioid agonist and antagonist bivalent ligands - the relationship between spacer length and selectivity at multiple opioid receptors. Journal of Medicinal Chemistry. 1986;29:1855–1861. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 247.Kuhhorn J, Hubner H, Gmeiner P. Bivalent dopamine D-2 receptor ligands: Synthesis and binding properties. Journal of Medicinal Chemistry. 2011;54:4896–4903. doi: 10.1021/jm2004859. [DOI] [PubMed] [Google Scholar]
- 248.Josan JS, Vagner J, Handl HL, Sankaranarayanan R, Gillies RJ, Hruby VJ. Solid-phase synthesis of heterobivalent ligands targeted to melanocortin and cholecystokinin receptors. International Journal of Peptide Research and Therapeutics. 2008;14:293–300. doi: 10.1007/s10989-008-9150-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Hruby V, Josan J, Vagner J, Fernandes S, Handl H, Xu LP, Lynch R, Mash E, Gillies R. New approaches to the design, synthesis and biochemical and biophysical evaluation of heteromultivalent ligands for detection and treatment of cancer. Journal of Peptide Science. 2008;14:23–23. [Google Scholar]
- 250.Vagner J, Xu LP, Handl HL, Josan JS, Morse DL, Mash EA, Gillies RJ, Hruby VJ. Heterobivalent ligands crosslink multiple cell-surface receptors: The human melanocortin-4 and δ-opioid receptors. Angewandte Chemie-International Edition. 2008;47:1685–1688. doi: 10.1002/anie.200702770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Xu LP, Vagner J, Josan J, Lynch RM, Morse DL, Baggett B, Han HY, Mash EA, Hruby VJ, Gillies RJ. Enhanced targeting with heterobivalent ligands. Molecular Cancer Therapeutics. 2009;8:2356–2365. doi: 10.1158/1535-7163.MCT-08-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Josan JS, Handl HL, Sankaranarayanan R, Xu LP, Lynch RM, Vagner J, Mash EA, Hruby VJ, Gillies RJ. Cell-specific targeting by heterobivalent ligands. Bioconjugate Chemistry. 2011;22:1270–1278. doi: 10.1021/bc1004284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Xu LP, Josan JS, Vagner J, Caplan MR, Hruby VJ, Mash EA, Lynch RM, Morse DL, Gillies RJ. Heterobivalent ligands target cell-surface receptor combinations in vivo. Proceedings of the National Academy of Sciences. 2012;109:21295–21300. doi: 10.1073/pnas.1211762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yang JQ, Guo HX, Gallazzi F, Berwick M, Padilla RS, Miao YB. Evaluation of a novel Arg-Gly-Asp-conjugated α-melanocyte stimulating hormone hybrid peptide for potential melanoma therapy. Bioconjugate Chemistry. 2009;20:1634–1642. doi: 10.1021/bc9001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Eberle A, Hubscher W, Schwyzer R. Synthesis of radioactive α-melanotropin derivatives containing a bromoacetyl or diazoacetyl group for studies of covalent hormone-macromolecule complexes. Helvetica Chimica Acta. 1977;60:2895–2910. [Google Scholar]
- 256.Schwyzer R, Kriwaczek VM. Tobacco mosaic virus as a carrier for small molecules: Artificial receptor antibodies and superhormones. Biopolymers. 1981;20:2011–2020. doi: 10.1002/bip.1981.360200923. [DOI] [PubMed] [Google Scholar]
- 257.Wunderlin R, Minakakis P, Tunkyi A, Sharma SD, Schwyzer R. Melanotropin receptors 1. Synthesis and biological-activity of Nα-(5-bromovaleryl)-Nα-deacetyl-α-melanotropin. Helvetica Chimica Acta. 1985;68:1–11. [Google Scholar]
- 258.Wunderlin R, Sharma SD, Minakakis P, Schwyzer R. Melanotropin receptors 2. Synthesis and biological-activity of α-melanotropin/tobacco mosaic virus disulfide conjugates. Helvetica Chimica Acta. 1985;68:12–22. [Google Scholar]
- 259.Schwyzer R, Kriwaczek VM, Wunderlin R. A method for mapping peptide receptors. Naturwissenschaften. 1981;68:95–96. doi: 10.1007/BF01047233. [DOI] [PubMed] [Google Scholar]
- 260.Kriwaczek VM, Bristow AF, Eberle AN, Gleed C, Schulster D, Schwyzer R. Superpotency and superaffinity phenomena in the stimulation of steroidogenesis in adrenocortical cells by adrenocorticotropin-tobacco mosaic virus conjugates. Molecular and Cellular Biochemistry. 1981;40:49–59. doi: 10.1007/BF00230187. [DOI] [PubMed] [Google Scholar]
- 261.Sharma SD, Granberry ME, Jiang JW, Leong SPL, Hadley ME, Hruby VJ. Multivalent melanotropic peptide and fluorescent macromolecular conjugates: New reagents for characterization of melanotropin receptors. Bioconjugate Chemistry. 1994;5:591–601. doi: 10.1021/bc00030a015. [DOI] [PubMed] [Google Scholar]
- 262.Sharma SD, Hruby V, Hadley ME, Granberry ME, Leong SPL. Multivalent ligands for diagnosis and therapeutics, Peptides: Chemistry and Biology. Proceedings of the Twelfth American Peptide Symposium. 1992:599–600. [Google Scholar]
- 263.Sharma SD, Jiang JW, Hadley ME, Bentley DL, Hruby VJ. Melanotropic peptide-conjugated beads for microscopic visualization and characterization of melanoma melanotropin receptors. Proceedings of the National Academy of Sciences. 1996;93:13715–13720. doi: 10.1073/pnas.93.24.13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Jiang JW, Sharma SD, Hruby VJ, Bentley DL, Fink JL, Hadley ME. Human epidermal melanocyte and keratinocyte melanocortin receptors: Visualization by melanotropic peptide conjugated microspheres (latex beads) Pigment Cell Research. 1996;9:240–247. doi: 10.1111/j.1600-0749.1996.tb00113.x. [DOI] [PubMed] [Google Scholar]
- 265.Jiang J, Sharma SD, Hruby VJ, Fink JL, Hadley ME. Human epidermal melanocyte and keratinocyte melanotropin receptors: Visualization by melanotropic peptide conjugated macrospheres (polyamide beads) Experimental Dermatology. 1997;6:6–12. doi: 10.1111/j.1600-0625.1997.tb00139.x. [DOI] [PubMed] [Google Scholar]
- 266.Newton JR, Miao YB, Deutscher SL, Quinn TP. Melanoma imaging with pretargeted bivalent bacteriophage. Journal of Nuclear Medicine. 2007;48:429–436. [PubMed] [Google Scholar]
- 267.Barkey NM, Preihs C, Cornnell HH, Martinez G, Carie A, Vagner J, Xu LP, Lloyd MC, Lynch VM, Hruby VJ, Sessler JL, Sill KN, Gillies RJ, Morse DL. Development and in vivo quantitative magnetic resonance imaging of polymer micelles targeted to the melanocortin 1 receptor. Journal of Medicinal Chemistry. 2013;56:6330–6338. doi: 10.1021/jm4005576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Barkey NM, Tafreshi NK, Josan JS, De Silva CR, Sill KN, Hruby VJ, Gillies RJ, Morse DL, Vagner J. Development of melanoma-targeted polymer micelles by conjugation of a melanocortin 1 receptor (MC1R) specific ligand. Journal of Medicinal Chemistry. 2011;54:8078–8084. doi: 10.1021/jm201226w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Brabez N, Saunders K, Nguyen KL, Jayasundera T, Weber C, Lynch RM, Chassaing G, Lavielle S, Hruby VJ. Multivalent interactions: Synthesis and evaluation of melanotropin multimers - tools for melanoma targeting. ACS Medicinal Chemistry Letters. 2013;4:98–102. doi: 10.1021/ml300312b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Alleti R, Rao V, Xu LP, Gillies RJ, Mash EA. A solanesol-derived scaffold for multimerization of bioactive peptides. Journal of Organic Chemistry. 2010;75:5895–5903. doi: 10.1021/jo101043m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Rao V, Alleti R, Xu LP, Tafreshi NK, Morse DL, Gillies RJ, Mash EA. A sucrose-derived scaffold for multimerization of bioactive peptides. Bioorganic & Medicinal Chemistry. 2011;19:6474–6482. doi: 10.1016/j.bmc.2011.08.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Busnelli M, Kleinau G, Muttenthaler M, Stoev S, Manning M, Bibic L, Howell LA, McCormick PJ, Di Lascio S, Braida D, Sala M, Rovati GE, Bellini T, Chini B. Design and characterization of superpotent bivalent ligands targeting oxytocin receptor dimers via a channel-like structure. Journal of Medicinal Chemistry. 2016;59:7152–7166. doi: 10.1021/acs.jmedchem.6b00564. [DOI] [PubMed] [Google Scholar]
- 273.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proceedings of the National Academy of Sciences. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Akgun E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS. Inhibition of inflammatory and neuropathic pain by targeting a mu opioid receptor/chemokine receptor 5 heteromer (MOR-CCR5) Journal of Medicinal Chemistry. 2015;58:8647–8657. doi: 10.1021/acs.jmedchem.5b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Fani L, Bak S, Delhanty P, van Rossum EFC, van den Akker ELT. The melanocortin-4 receptor as target for obesity treatment: A systematic review of emerging pharmacological therapeutic options. International Journal of Obesity. 2014;38:163–169. doi: 10.1038/ijo.2013.80. [DOI] [PubMed] [Google Scholar]
- 276.White WB, Myers MG, Weber MA, Edelsen J, Jordan R, Kowey PR. Effects of the novel melanocortin receptor agonist bremelanotide on ambulatory blood pressure and heart rate in women with sexual dysfunction. Journal of the American Society of Hypertension. 2014;8:e29. [Google Scholar]
- 277.Clayton AH, Althof SE, Kingsberg S, DeRogatis LR, Kroll R, Goldstein I, Kaminetsky J, Spana C, Lucas J, Jordan R, Portman DJ. Bremelanotide for female sexual dysfunctions in premenopausal women: a randomized, placebo-controlled dose-finding trial. Womens Health. 2016;12:325–337. doi: 10.2217/whe-2016-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Dorr RT, Dawson BV, Alobeidi F, Hadley ME, Levine N, Hruby VJ. Toxicologic studies of a superpotent α-melanotropin, [Nle4, D-Phe7]α-MSH. Investigational New Drugs. 1988;6:251–258. doi: 10.1007/BF00173642. [DOI] [PubMed] [Google Scholar]
- 279.Biolcati G, Aurizi C, Barbieri L, Cialfi S, Screpanti I, Talora C. Efficacy of the melanocortin analogue Nle4-DPhe7-α-melanocyte-stimulating hormone in the treatment of patients with Hailey-Hailey disease. Clinical and Experimental Dermatology. 2014;39:168–175. doi: 10.1111/ced.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Boehm M, Ehrchen J, Luger TA. Beneficial effects of the melanocortin analogue Nle4-DPhe7-α-MSH in acne vulgaris. Journal of the European Academy of Dermatology and Venereology. 2014;28:108–111. doi: 10.1111/j.1468-3083.2012.04658.x. [DOI] [PubMed] [Google Scholar]
- 281.Langendonk JG, Balwani M, Anderson KE, Bonkovsky HL, Anstey AV, Bissell DM, Bloomer J, Edwards C, Neumann NJ, Parker C, Phillips JD, Lim HW, Hamzavi I, Deybach JC, Kauppinen R, Rhodes LE, Frank J, Murphy GM, Karstens FPJ, Sijbrands EJG, de Rooij FWM, Lebwohl M, Naik H, Goding CR, Wilson JHP, Desnick RJ. Afamelanotide for erythropoietic protoporphyria. New England Journal of Medicine. 2015;373:48–59. doi: 10.1056/NEJMoa1411481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Haylett AK, Nie Z, Brownrigg M, Taylor R, Rhodes LE. Systemic photoprotection in solar urticaria with α-melanocyte-stimulating hormone analogue [Nle4-D-Phe7]-α-MSH. British Journal of Dermatology. 2011;164:407–414. doi: 10.1111/j.1365-2133.2010.10104.x. [DOI] [PubMed] [Google Scholar]
- 283.Kievit P, Halem H, Marks DL, Dong JZ, Glavas MM, Sinnayah P, Pranger L, Cowley MA, Grove KL, Culler MD. Chronic treatment with a melanocortin-4 receptor agonist causes weight loss, reduces insulin resistance, and improves cardiovascular function in diet-induced obese rhesus macaques. Diabetes. 2013;62:490–497. doi: 10.2337/db12-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Chen KY, Muniyappa R, Abel BS, Mullins KP, Staker P, Brychta RJ, Zhao X, Ring M, Psota TL, Cone RD, Panaro BL, Gottesdiener KM, Van der Ploeg LHT, Reitman ML, Skarulis MC. RM-493, a melanocortin-4 receptor (MC4R) agonist, increases resting energy expenditure in obese individuals. Journal of Clinical Endocrinology & Metabolism. 2015;100:1639–1645. doi: 10.1210/jc.2014-4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Kuehnen P, Clement K, Wiegand S, Blankenstein O, Gottesdiener K, Martini LL, Mai K, Blume-Peytavi U, Grueters A, Krude H. Proopiomelanocortin deficiency treated with a melanocortin-4 receptor agonist. New England Journal of Medicine. 2016;375:240–246. doi: 10.1056/NEJMoa1512693. [DOI] [PubMed] [Google Scholar]
- 286.Conde-Frieboes K, Thogersen H, Lau JF, Sensfuss U, Hansen TK, Christensen L, Spetzler J, Olsen HB, Nilsson C, Raun K, Dahl K, Hansen BS, Wulff BS. Identification and in vivo and in vitro characterization of long acting and melanocortin-4 receptor (MC4-R) selective α-melanocyte-stimulating hormone (α-MSH) analogues. Journal of Medicinal Chemistry. 2012;55:1969–1977. doi: 10.1021/jm201489a. [DOI] [PubMed] [Google Scholar]
- 287.Royalty JE, Konradsen G, Eskerod O, Wulff BS, Hansen BS. Investigation of safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple doses of a long-acting α-MSH analog in healthy overweight and obese subjects. Journal of Clinical Pharmacology. 2014;54:394–404. doi: 10.1002/jcph.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Wellhoener P, Hoerster R, Jacobs F, Sayk F, Lehnert H, Dodt C. Intranasal application of the melanocortin 4 receptor agonist MSH/ACTH(4-10) in humans causes lipolysis in white adipose tissue. International Journal of Obesity. 2012;36:703–708. doi: 10.1038/ijo.2011.105. [DOI] [PubMed] [Google Scholar]