

Abstract
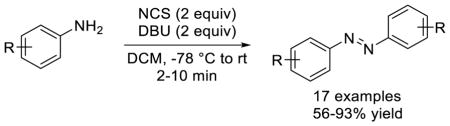
A convenient method for the synthesis of symmetrical azobenzenes is reported. This one-step procedure involves treatment of anilines with N-chlorosuccinimide (NCS) and organic base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). A wide range of commercially available substituted anilines readily participate in this reaction to produce the corresponding azobenzenes in moderate-to-excellent yields in minutes.
Graphical abstract
Azobenzenes have long attracted interests from organic materials community due to their rapid, reversible photoswitching properties upon irradiation with UV or visible light. Because the cis and trans isomers exhibit distinct structural and photophysical properties,1 azobenzenes have been extensively employed in the design of photoresponsive materials including polymers,2 crystals,3,4 and bioactive ligands.5,6 From a synthetic point of view, azobenzenes can be readily accessed using a number of methods.7 A classic method involves oxidation of anilines by potassium permanganate and copper sulfate or ferrous sulfate in refluxing methylene chloride for two days, which limits the type of functional groups that can be attached to the benzene ring 8 (Eq. 1 in Chart 1). To avoid this problem, a number of milder reaction conditions have been developed recently. For example, Jiao and coworker9 reported the copper-catalyzed aerobic oxidative dehydrogenative coupling of anilines to generate the azobenzenes (Eq. 2 in Chart 1). Minakata and coworkers10,11 reported oxidative dimerization of anilines using organic oxidant, tert-butylhypoiodite (tBuOI), at room temperature to generate a variety of aromatic azo compounds with excellent tolerance of the functional groups10,11 (Eq. 3 in Chart 1).
Chart 1.

Oxidative coupling to form azobenzenes
In our pursuit of harnessing the photoswitching properties of azobenzenes for photoregulation of protein function in live cells, we were interested in synthetic methods that allow facile synthesis of red-shifted, highly functionalized azobenzenes carrying functional groups that can be readily incorporated into biological systems. To this end, we previously reported the synthesis of a series of red-shifted, amino acid-functionalized azobenzenes by reacting the appropriately substituted phenylhydrazines with the amino acid spirolactone in the presence of catalytic amount of ceric ammonium nitrate.12 During the preparation of some of the substrates, we serendipitously discovered a simple method for direct synthesis of symmetric azobenzenes. Herein, we report the rapid synthesis of symmetric azobenzenes through oxidative coupling of anilines using N-chlorosuccinimide (NCS) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (Eq. 4 in Chart 1), and propose a mechanism to explain this unprecedented transformation.
Our initial observation was made during the preparation of 4-bromo-2,6-difluoroaniline using NBS and pyridine, where we detected trace amount of the corresponding symmetric azobenzene. We surmised it should be possible to use NBS as an organic oxidant for oxidative homo-coupling of anilines to form the azobenzenes. Interestingly, Takeda and coworkers have examined NBS as an organic oxidant for homo-dimerization of toluidine without much success.11 In our own hands, when toluidine was treated with NBS and pyridine in methylene chloride at room temperature, the brominated aniline product, instead of the azobenzene, was generated exclusively. To distinguish the aryl bromination reactivity from the nitrogen oxidation reactivity of NBS, we were encouraged by an earlier literature report in which combination of NBS with a base at low temperature gave rise to a predominant oxidative bromine species.13 Later work by Keillor and coworkers showed that treatment of aryl carboxamides with NBS/DBU produced the Hofmann rearrangement products in excellent yields, suggesting that the combination of NBS with DBU serves principally as an oxidant.14
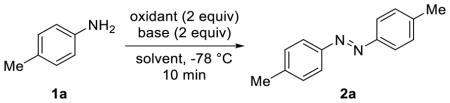
With these results in mind, we screened several organic and inorganic bases in combination with NBS and found DBU gave the highest yield in azobenzene formation (entries 1–4, Table 1). We then examined various solvents including polar, nonpolar, protic, and aprotic solvents and found dichloromethane gave the highest yield (entries 4–8, Table 1). Subsequently, we investigated other electrophilic halogen sources, including N-bromophthalimide (NBP), N-iodosuccinimide (NIS), and N-chlorosuccinimide (NCS). Compared to NBS, NCS gave a higher yield while NBP and NIS gave lower yields (compare entries 9–11 to entry 4, Table 1). The reactivity trend of NCS > NBS > NIS suggests that complete dissociation of the electrophilic halogen cation is not conducive to oxidative coupling of toluidine. We also assessed the reactivity of two other electrophilic N-chloroimide reagents, trichlorocyanuric acid (TCCA) and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH); both gave slightly lower yields than NCS (compare entries 12–13 to 11, Table 1), presumably due to their more rapid ionization to form a free Cl+ that participates competing aromatic chlorination reactions. Yield remains the same when the amidine base DBU was changed to 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) (entry 14, Table 1). However, the use of a stronger organic base such as KOtBu led to a lower yield (entry 15, Table 1), suggesting that the higher yields afforded by the amidine bases are not a result of their higher basicity.
Table 1.
Condition Optimization a
| ||||
|---|---|---|---|---|
| entry | solvent | oxidant | base | yield (%)b |
| 1 | DCM | NBS | pyridine | NR |
| 2 | DCM | NBS | Cs2CO3 | NR |
| 3 | DCM | NBS | Et3N | 24 |
| 4 | DCM | NBS | DBU | 80 |
| 5 | THF | NBS | DBU | NR |
| 6 | EtOAc | NBS | DBU | 15 |
| 7 | EtOH | NBS | DBU | 37 |
| 8 | MEK | NBS | DBU | 12 |
| 9 | DCM | NBP | DBU | 71 |
| 10 | DCM | NIS | DBU | 17 |
| 11 | DCM | NCS | DBU | 87 |
| 12 | DCM | TCCAc | DBU | 85 |
| 13 | DCM | DCDMHd | DBU | 73 |
| 14 | DCM | NCS | DBN | 87 |
| 15 | DCM | NCS | KOtBu | 41 |
The reaction was set up by incubating 1 equiv of aniline and 2 equiv of base in appropriate solvent followed by addition of 2 equiv of oxidant.
Yields are isolated yields and include both cis and trans-2a.
0.67 mmol trichlorocyanuric acid (TCCA) was used to generate 2 equiv of electrophilic Cl+ species.
1.0 mmol 1,3-dichloro-5,5-dimethylhydantoin (DCDMH) was used to generate 2 equiv of electrophilic Cl+ species. NR, no reaction. MEK = methyl ethyl ketone.
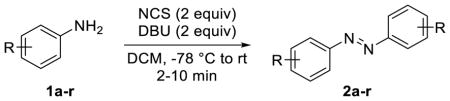
Since NCS is commercially available and inexpensive, we decided to use NCS as organic oxidant in the substrate scope study. Under the optimized conditions, a wide range of mono-substituted anilines carrying alkyl (1a, 1b), ethynyl (1c), electron-withdrawing groups (1d-1g), and halogens (1h-1m) were smoothly converted into their azobenzene products with 56–93% yields (entries 1–12, Table 2). Since tetra-ortho-fluoroazobenzenes have shown robust photoswitching properties under visible light,15 we assessed whether our condition is suitable for making this type of highly substituted azobenzenes. To this end, five poly-substituted fluorine-containing anilines were treated with NCS/DBU in DCM for 2–5 min and the desired tetra-ortho-fluoroazobenzenes were obtained uneventfully in 58–70% yields (entries 13–17, Table 2). Compared to KMnO4/FeSO4·7H2O-mediated oxidative coupling that gave rise to the azobenzenes in 10–25% yield after overnight reflux,15 the NCS/DBU method produces the desired azobenzene products in higher yields and shorter time.
Table 2.
Substrate Scope
| ||
|---|---|---|
| entry | aniline | yield (%) a |
| 1 | p-Me (1a) | 87 |
| 2 | p-iPr (1b) | 66 |
| 3 | p-acetylene (1c) | 88 |
| 4 | p-CF3 (1d) | 93 |
| 5 | p-CN (1e) | 85 |
| 6 | p -CO2Et (1f) | 82 |
| 7 | m-CN (1g) | 56 |
| 8 | p–F (1h) | 83 |
| 9 | p-Cl (1i) | 86 |
| 10 | p-Br (1j) | 59 |
| 11 | p-I (1k) | 76 |
| 12 | m-F (1m) | 75 |
| 13 | 2,4,6-F3 (1n) | 70 |
| 14 | 4-Br-2,6-F2 (1o) | 64 |
| 15 | 4-I-2,6-F2 (1p) | 63 |
| 16 | 4-CO2Et-2,6-F2 (1q) | 62 |
| 17 | 2,3,4,5,6-F5 (1r) | 58 |
Yields are isolated yields and include both cis and trans-azobenzene.
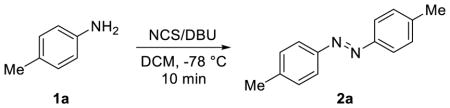
Based on the previous report of using the halogen-containing oxidants for oxidative dimerization of aromatic amines,10,11 a plausible mechanism for NCS/DBU-mediated homo-coupling of anilines is put forward in Scheme 1. The combination of NCS and DBU would allow formation of the chlorinated DBU (DBU-Cl+), an organic oxidant whose balanced reactivity and selectivity may be responsible for the two key chlorination/oxidation steps: with aniline in step (i) to generate N-chloro-aniline 3 after proton transfer in step (ii); and with hydrazine 4 in step (v) to generate N-chloro-hydrazine 5 after proton transfer in step (vi). The use of DBU in facilitating chloronium ion transfer is catalytic as DBU is regenerated after N-chlorination (steps i and v). However, two equivalents of DBU (relative to one-half of the azobenzene structure) are consumed as a base during the proton transfer in step (iv) after nucleophilic substitution and during the final elimination step (vii). Therefore, for symmetric azobenzenes only one equivalent of NCS and DBU (relative to aniline starting materials) are needed to complete the overall transformation. To confirm this, we varied the amount of the NCS in the reaction from 0.5 equiv to 3.0 equiv and obtained 73% isolated yield when 1.0 equiv of NCS was used (Table 3), consistent with our mechanistic hypothesis. Furthermore, when hydrazine 4, which was prepared separately using a literature procedure,16 was exposed to NCS and DBU, quantitative conversion to azobenzene 2a was observed, indicating that hydrazine 4 is a competent intermediate and likely involved in the reaction. This mechanism is reminiscent of the use of NBS/DBU for generation of more electrophilic bromine species for allylic amination reaction17 as well as tandem bromoamination/debromination reaction18 reported in the literature.
Scheme 1.

Plausible reaction mechanism.
Table 3.
Effect of NCS Equivalency a
| |
|---|---|
| NCS equiv. | yield (%) |
| 0.5 | 42 |
| 1.0 | 73 |
| 2.0 | 87 |
| 3.0 | 84 |
The amount of DBU was kept constant at 2 equivalent relative to toluidine.
In summary, we have discovered a new oxidative homo-coupling method for one-step synthesis of the symmetric azobenzenes from the substituted anilines. This method employs stable and inexpensive NCS and DBU reagents, which may produce an electrophilic chloro-DBU species likely responsible for selective N-chlorination/oxidation of the anilines. The substrate scope study revealed that the tetra-ortho-substituted azobenzene analogs can be rapidly accessed in good yields using this method.
Experimental Section
General methods
All reactions were performed under ambient atmosphere. The commercially available reagents were used without further purification. The NMR spectra were recorded in CDCl3 or DMSO-d6 on a Varian Mercury-300 or Inova-400 MHz instrument. NMR data are presented as follows: chemical shift (ppm); multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet); coupling constants in hertz (Hz). Flash column chromatography was performed with SiliCycle P60 silica gel (40–63 μm, 60 Å).
Synthetic procedure
To a solution of aniline (1 mmol) dissolved in 15 mL DCM was added DBU (2 mmol). The solution was stirred at room temperature for 5 min before cooled down to a −78°C. The oxidant (2 mmol) was added as a solid to the reaction mixture. The orange solution was stirred for 10 min at −78 °C before quenching by addition of saturated bicarbonate solution. The organic layer was separated, washed sequentially with 50 mL water and 50 mL 1 N HCl, dried over anhydrous sodium sulfate, and concentrated to dryness in vacuo. Compounds with compatible functional groups were treated with 5 mL TFA at room temperature for 20 min to maximize the conversion to the trans-azobenzene, and TFA was removed in vacuo. The residue was purified by silica gel flash chromatography using DCM-hexanes as eluent.
1,2-Di-p-tolyldiazene (2a).9
Orange solid; 91 mg, 87% yield: 1H NMR (300 MHz, CDCl3) δ 7.81 (d, J = 8.3 Hz, 4H), 7.30 (d, J = 8.5 Hz, 4H), 2.43 (s, 6H).
1,2-Bis(4-isopropylphenyl)diazene (2b).19
Orange solid; 88 mg, 66% yield: 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.4 Hz, 4H), 7.41 – 7.32 (m, 4H), 3.10 – 2.92 (m, 2H), 1.31 (d, J = 6.9 Hz, 12H).
1,2-Bis(4-ethynylphenyl)diazene (2c).20
Orange solid; 101 mg, 88% yield: 1H NMR (300 MHz, CDCl3) δ 7.89 (s, 4H), 7.64 (s, 4H), 3.24 (s, 2H).
1,2-Bis(4-(trifluoromethyl)phenyl)diazene (2d).21
Yellow solid; 147 mg, 93% yield: 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 8.1 Hz, 4H), 7.81 (d, J = 8.1 Hz, 4H).
4,4′-(Diazene-1,2-diyl)dibenzonitrile (2e).11
Orange-yellow solid; 98 mg, 85% yield: 1H NMR (300 MHz, CDCl3) δ 8.04 (d, J = 8.3 Hz, 4H), 7.85 (d, J = 8.5 Hz, 4H).
Diethyl 4,4′-(diazene-1,2-diyl)-dibenzoate (2f).11
Red-orange solid; 134 mg, 82% yield: 1H NMR (400 MHz, CDCl3) δ 8.21 (d, J = 8.4 Hz, 4H), 7.98 (d, J = 8.4 Hz, 4H), 4.42 (q, 4H), 1.43 (t, 6H).
3,3′-(Diazene-1,2-diyl)dibenzonitrile (2g).22
Yellow solid; 65 mg, 56% yield: 1H NMR (400 MHz, CDCl3) δ 8.49 – 7.96 (m, 4H), 7.81 (dd, J = 7.7, 1.1 Hz, 2H), 7.69 (t, J = 7.9 Hz, 2H).
1,2-Bis(4-fluorophenyl)diazene (2h).11
Orange-yellow solid; 90 mg, 83% yield: 1H NMR (400 MHz, CDCl3) δ 7.94-7.91 (m, 4H), 7.22-7.18 (m, 4H).
1,2-Bis(4-chlorophenyl)diazene (2i).11
Orange solid; 108 mg, 86% yield: 1H NMR (400 MHz, CDCl3) δ 7.86 (d, J = 8.7 Hz, 4H), 7.49 (d, J = 8.7 Hz, 4H).
1,2-Bis(4-bromophenyl)diazene (2j).11
Orange solid; 101 mg, 59% yield: 1H NMR (300 MHz, CDCl3) δ 7.79 (d, J = 8.4 Hz, 4H), 7.65 (d, J = 8.3 Hz, 4H).
1,2-Bis(4-iodophenyl)diazene (2k).11
Orange-red solid; 164 mg, 76% yield: 1H NMR (400 MHz, CDCl3) δ 7.87 (d, J = 8.4 Hz, 4H), 7.64 (d, J = 8.4 Hz, 4H).
1,2-Bis(3-fluorophenyl)diazene (2m).23
Orange solid; 82 mg, 75% yield: 1H NMR (400 MHz, DMSO-d6) δ 7.79 (d, J = 7.9 Hz, 2H), 7.69-7.61 (m, 4H), 7.55 – 7.35 (m, 2H).
1,2-Bis(2,4,6-trifluorophenyl)diazene (2n).24
The reaction was stirred at −78°C for 5 min. The solvent was removed in vacuo and the residue was loaded directly onto silica gel for flash chromatography. Orange-yellow solid; 102 mg, 70% yield: 1H NMR (300 MHz, CDCl3) δ 6.84 (s, 4H).
1,2-Bis(4-bromo-2,6-difluorophenyl)diazene (2o)
Red solid; 132 mg, 64% yield: 1H NMR (400 MHz, DMSO-d6) δ 7.77 (d, J = 9.1 Hz, 4H).
1,2-Bis(2,6-difluoro-4-iodophenyl)diazene (2p)
Red solid; 160 mg, 63% yield: 1H NMR (400 MHz, DMSO-d6) δ 7.81 (d, J = 8.9 Hz, 4H).
Diethyl 4,4′-(diazene-1,2-diyl)-bis(3,5-difluorobenzoate) (2q)
Red solid; 124 mg, 62% yield: 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 9.0 Hz, 2H), 4.43 (q, J = 7.1 Hz, 2H), 1.43 (t, J = 7.1 Hz, 3H).
1,2-Bis(perfluorophenyl)diazene (2r)
The reaction was stirred at −78°C for 5 min. The solvent was removed in vacuo and the residue was loaded directly onto silica gel for flash chromatography. Orange-yellow solid; 104 mg, 58% yield: 19 F NMR (282 MHz, CDCl3) δ −147.46 – −149.35 (m, 6F), −160.72 – −162.12 (m, 4F).
Supplementary Material
Acknowledgments
We gratefully acknowledge the NIH (GM085092) for financial support.
Footnotes
NMR spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bandara HM, Burdette SC. Chem Soc Rev. 2012;41:1809. doi: 10.1039/c1cs15179g. [DOI] [PubMed] [Google Scholar]
- 2.Zhou Q, Fursule I, Berron BJ, Beck MJ. J Phys Chem A. 2016;120:7101. doi: 10.1021/acs.jpca.6b05807. [DOI] [PubMed] [Google Scholar]
- 3.Bushuyev OS, Tomberg A, Friscic T, Barrett CJ. J Am Chem Soc. 2013;135:12556. doi: 10.1021/ja4063019. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Lo CW, Zhu D, Li C, Liu Y, Jiang H. Macromol Rapid Commun. 2009;30:1928. doi: 10.1002/marc.200900421. [DOI] [PubMed] [Google Scholar]
- 5.Beharry AA, Woolley GA. Chem Soc Rev. 2011;40:4422. doi: 10.1039/c1cs15023e. [DOI] [PubMed] [Google Scholar]
- 6.Banghart M, Borges K, Isacoff E, Trauner D, Kramer RH. Nat Neurosci. 2004;7:1381. doi: 10.1038/nn1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merino E. Chem Soc Rev. 2011;40:3835. doi: 10.1039/c0cs00183j. [DOI] [PubMed] [Google Scholar]
- 8.Noureldin NA, Bellegarde JW. Synthesis. 1999:939. [Google Scholar]
- 9.Zhang C, Jiao N. Angew Chem Int Ed Engl. 2010;49:6174. doi: 10.1002/anie.201001651. [DOI] [PubMed] [Google Scholar]
- 10.Takeda Y, Okumura S, Minakata S. Angew Chem Int Ed Engl. 2012;51:7804. doi: 10.1002/anie.201202786. [DOI] [PubMed] [Google Scholar]
- 11.Okumura S, Lin CH, Takeda Y, Minakata S. J Org Chem. 2013;78:12090. doi: 10.1021/jo402120w. [DOI] [PubMed] [Google Scholar]
- 12.John AA, Ramil CP, Tian Y, Cheng G, Lin Q. Org Lett. 2015;17:6258. doi: 10.1021/acs.orglett.5b03268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senanayake CH, Fredenburgh LE, Reamer RA, Larsen RD, Verhoeven TR, Reider PJ. J Am Chem Soc. 1994;116:7947. [Google Scholar]
- 14.Huang X, Seid M, Keillor JW. J Org Chem. 1997;62:7495. doi: 10.1021/jo9708553. [DOI] [PubMed] [Google Scholar]
- 15.Blεger D, Schwarz J, Brouwer AM, Hecht S. J Am Chem Soc. 2012;134:20597. doi: 10.1021/ja310323y. [DOI] [PubMed] [Google Scholar]
- 16.Sridhara MB, Srinivasa GR, Gowda DC. J Chem Res. 2004:74. [Google Scholar]
- 17.Wei Y, Liang F, Zhang X. Org Lett. 2013;15:5186. doi: 10.1021/ol402287n. [DOI] [PubMed] [Google Scholar]
- 18.Wei Y, Lin S, Liang F, Zhang J. Org Lett. 2013;15:852. doi: 10.1021/ol303539u. [DOI] [PubMed] [Google Scholar]
- 19.Zhang LJ, Xia J, Li QH, Li XH, Wang SW. Organometallics. 2011;30:375. [Google Scholar]
- 20.Zeitouny J, Aurisicchio C, Bonifazi D, De Zorzi R, Geremia S, Bonini M, Palma CA, Samori P, Listorti A, Belbakra A, Armaroli N. J Mater Chem. 2009;19:4715. [Google Scholar]
- 21.Wang L, Ishida A, Hashidoko Y, Hashimoto M. Angew Chem Int Ed Engl. 2017;56:870. doi: 10.1002/anie.201610371. [DOI] [PubMed] [Google Scholar]
- 22.Piantek M, Miguel J, Kruger A, Navio C, Bernien M, Ball DK, Hermann K, Kuch W. J Phys Chem C. 2009;113:20307. [Google Scholar]
- 23.Leyva E, Medina C, Moctezuma E, Leyva S. Can J Chem. 2004;82:1712. [Google Scholar]
- 24.Nallaiah C, Strickson JA. Tetrahedron. 1986;42:4083. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.