

Abstract
Trapoxin A is a microbial cyclic tetrapeptide that is an essentially irreversible inhibitor of class I histone deacetylases (HDACs). The inhibitory warhead is the α,β-epoxyketone side-chain of (2S,9S)-2-amino-8-oxo-9,10-epoxydecanoic acid (L-Aoe), which mimics the side-chain of the HDAC substrate acetyl-L-lysine. We now report the crystal structure of the HDAC8–trapoxin A complex at 1.24 Å resolution, revealing that the ketone moiety of L-Aoe undergoes nucleophilic attack to form a zinc-bound tetrahedral gem-diolate that mimics the tetrahedral intermediate and its flanking transition states in catalysis. Mass spectrometry, activity measurements, and isothermal titration calorimetry confirm that trapoxin A binds tightly (Kd = 3 ± 1 nM) and does not covalently modify the enzyme, so the epoxide moiety of L-Aoe remains intact. Comparison of the HDAC8–trapoxin A complex with the HDAC6-HC toxin complex provides new insight regarding the inhibitory potency of L-Aoe-containing natural products against class I and class II HDACs.
Ever since the discovery of histone acetylation in transcriptional regulation more than 50 years ago,1 thousands of histone and non-histone proteins have been identified as lysine acetylation targets in the mammalian acetylome.2-4 The reversible acetylation of L-lysine side chains is a critical molecular strategy for the regulation of protein function in vivo and rivals phosphorylation in the regulation of diverse biological processes, including the cell cycle and central carbon metabolism.5,6 The acetylation of a specific L-lysine residue on a target protein is catalyzed by a histone acetyl transferase (HAT) using acetyl CoA as a co-substrate;7,8 the hydrolysis of acetyl-L-lysine to form free L-lysine and acetate is catalyzed by a histone deacetylase (HDAC).9,10
Upregulated HDAC activity is associated with tumorigenesis, neurodegenerative diseases, and immune disorders, so this enzyme family represents an attractive target for therapeutic intervention.11,12 Phylogenetic analysis indicates four classes of deacetylases: class I HDACs (1, 2, 3, and 8), class IIa (4, 5, 7, and 9) and class IIb (6 and 10) HDACs, class III HDACs (sirtuins 1-7), and the single class IV enzyme HDAC11.13 Class I, II, and IV HDACs require Zn2+ or Fe2+ for optimal catalytic activity.14 The first crystal structure of a metal-dependent deacetylase15 revealed an α/β fold identical to that first observed in arginase.16,17 This fold is distinct from that adopted by sirtuins, which employ a different catalytic mechanism that requires the cofactor NAD+.18 Among the metal-dependent HDACs, HDAC8 was the first to yield a crystal structure.19,20 Important catalytic residues in the HDAC8 active site include tandem histidines (electrostatic catalyst H142 and general base-general acid H143)21,22 and a conformationally-flexible23 tyrosine residue that assists the Zn2+ ion in the polarization of the substrate carbonyl group.24,25
The tightest-binding HDAC inhibitors are those capable of coordinating to the active site Zn2+ ion and also forming hydrogen bonds with catalytically-important active site residues. Currently, four HDAC inhibitors are approved for clinical use in cancer chemotherapy, one of which is the cyclic depsipeptide romidepsin.26,27 Macrocyclic HDAC inhibitors such as romidepsin or the related depsipeptide largazole28,29 comprise rigid scaffolds to which a Zn2+-coordinating functional group is attached through a linker that is the approximate length of a lysine side chain.
Trapoxin A, first isolated from the microbial parasite Helicoma ambiens, is a macrocyclic tetrapeptide HDAC inhibitor with the amino acid sequence cyclo-[L-Phe–L-Phe–D-hPro–L-Aoe] (hPro = homoproline, also known as pipecolic acid; L-Aoe = (2S,9S)-2-amino-8-oxo-9,10-epoxydecanoic acid).30 The α,β-epoxyketone moiety of the L-Aoe side chain serves as the Zn2+-coordinating group and its ketone carbonyl group is isosteric with the scissile carbonyl of the HDAC substrate acetyl-L-lysine (Figure 1). Trapoxin A is proposed to act as an irreversible inhibitor of class I HDACs31,32 and was used for the first isolation of HDAC1 from the nuclear extracts of human Jurkat T cells.33 The epoxide moiety of the L-Aoe side chain was thought to react with the nucleophilic side chain of an active site residue; however, a covalent enzyme-inhibitor complex was not observable by SDS-PAGE/autoradiography.32 Curiously, although trapoxin A was found to irreversibly inhibit HDAC1, a class I enzyme, it was found to reversibly inhibit the class II enzyme HDAC6.34 The molecular basis of these activity differences has remained unclear in the absence of structural data.
Figure 1.
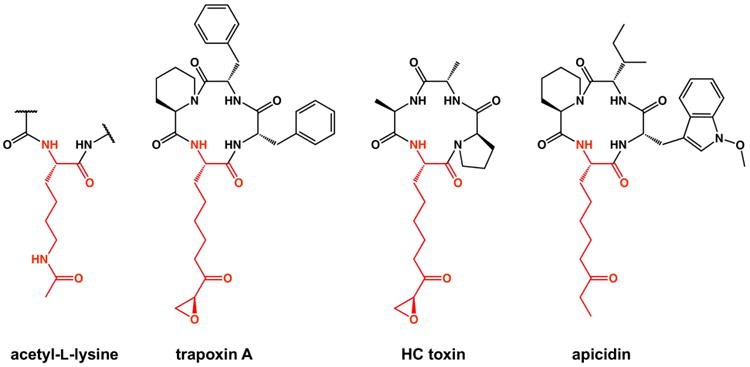
The canonical HDAC substrate peptidyl acetyl-L-lysine and structurally-related cyclic tetrapeptide inhibitors. The ketone carbonyl group of each inhibitor is isosteric with the scissile carbonyl of acetyl-L-lysine.
Here, we report the first X-ray crystal structure of trapoxin A complexed with a class I histone deacetylase, HDAC8, at 1.24 Å resolution (Figure 2a). Experimental procedures, including the preparation of a new HDAC8 construct for crystallographic studies, are outlined in the Supporting Information (data collection and refinement statistics are recorded in Supporting Information Table 1). The structure of the HDAC8–trapoxin A complex reveals that the conformation of the tetrapeptide backbone of trapoxin A is identical to that observed in the crystal structure of the uncomplexed inhibitor,30 with root-mean-square (rms) deviations of 0.21–0.24 Å for 16 main chain atoms of the cyclic peptide backbone; the L-Aoe side chains adopt different conformations (Supporting Information Figure S1). All peptide linkages of trapoxin A adopt the trans configuration except for the Phe-hPro linkage, which forms a cis-peptide. All three backbone NH groups donate hydrogen bonds to the side-chain carboxylate group of D101. The hydrogen bonds between D101 and the backbone NH groups of L-Aoe and the adjacent L-Phe residue are similar to those observed between D101 and linear tetrapeptide substrates bound to inactivated HDAC8 (Supporting Information Figure S2).24,25 Apart from interactions described below for the L-Aoe side chain, no other direct enzyme-inhibitor hydrogen bonds are observed. Although there are no intramolecular hydrogen bonds in trapoxin A, the side chains of its two L-Phe residues make a favorable quadrupole-quadrupole interaction with edge-to-face geometry.
Figure 2.
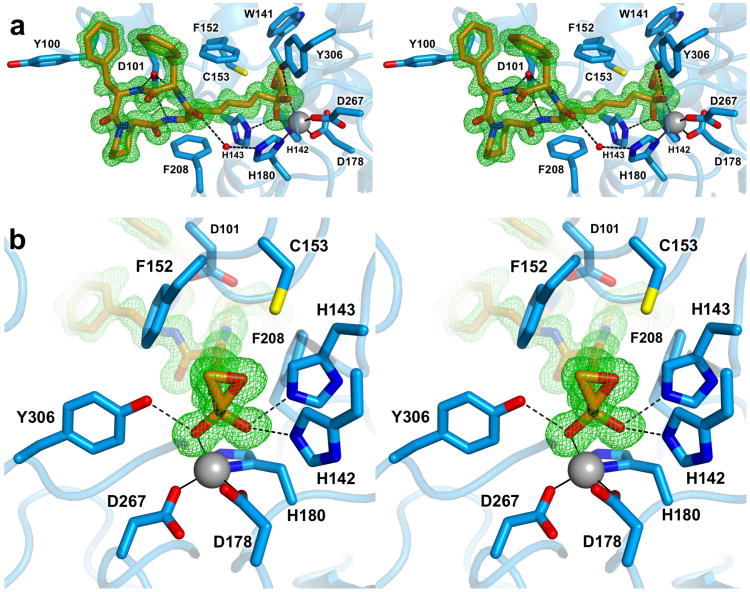
1.24 Å-resolution structure of the HDAC8–trapoxin A complex. (a) Stereoview of trapoxin A (C = orange, N = blue, O = red) bound in the active site of HDAC8 (C = light blue, N = blue, O = red). The simulated annealing omit map of trapoxin A (green) is contoured at 3.0σ. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Close-up stereoview showing the zinc-bound gem-diolate. Atomic color coding is identical to that in (a).
Conformational changes are required in the enzyme active site to accommodate the steric bulk of the rigid peptide macrocycle in comparison with substrate binding, primarily in the L2 loop (residues G86-I108, of which D87-I94 are disordered). Relative to the structure of H143A HDAC8 complexed with a tetrapeptide substrate,25 the greatest change is observed for Y100, which undergoes a 116° change in side chain torsion angle χ1 (Supporting Information Figure S2). Similar conformational flexibility of Y100 accommodates the binding of the macrocyclic depsipeptide inhibitor largazole.29 In the HDAC8–trapoxin A complex, the conformational change of Y100 is triggered by one of the inhibitor L-Phe residues, with which one of two observed conformers makes a favorable edge-to-face interaction.
Surprisingly, the ketone carbonyl of the L-Aoe side chain undergoes nucleophilic attack by water upon binding to HDAC8, such that the inhibitory species is a gem-diolate (or perhaps a gem-diol) stabilized by Zn2+ coordination and three hydrogen bonds. Thus, trapoxin A mimics the binding of the tetrahedral intermediate and its flanking transition states in catalysis; the origins of high affinity are undoubtedly rooted in the fact that trapoxin A binds as an analogue of the postulated transition state. The Zn2+–O1 and Zn2+–O2 distances for the gem-diolate are 2.5 Å and 1.9 Å, respectively. The O1 hydroxyl group also forms hydrogen bonds with H142 and H143 (O–O separations = 2.7 Å each), and the O2 oxyanion accepts a hydrogen bond from Y306 (O–O separation = 2.6 Å). While it is unusual to see an unactivated ketone binding as a tetrahedral gem-diolate, which exists to less than 0.2% in aqueous solution (based on the hydration of the unactivated ketone carbonyl of acetone in aqueous solution35), it is notable that the L-Aoe side chain of the cyclic tetrapeptide inhibitor HC toxin (Figure 1) similarly undergoes nucleophilic attack in the recently-determined structure of its complex with catalytic domain 2 of HDAC6.36 This behavior is also reminiscent of the binding of unactivated aldehyde and ketone substrate analogues to carboxypeptidase A, which similarly bind as tetrahedral gem-diolate transition state analogues.37,38
In the HDAC8–trapoxin A complex, the epoxide ring of the L-Aoe side chain is clearly intact and makes no hydrogen bond interactions with any enzyme residues or water molecules (Figure 2b). The closest side chains to the epoxide moiety are those of W141, H142, C153, and Y306 with interatomic separations of 3.3–3.7 Å. The epoxide moiety is believed to be required for essentially irreversible inhibitory activity against class I HDACs, based on the lack of irreversible inhibitory activity for the cyclic tetrapeptide inhibitor apicidin (Figure 1), which lacks an epoxide moiety.30 Thus, it is curious that the epoxide moiety of trapoxin A does not react with the enzyme. However, the crystal structure reveals that although the epoxide moiety binds in the vicinity of catalytic general base H143 and highly conserved C153, neither of these potential nucleophiles is positioned or oriented for nucleophilic attack at the epoxide (Figure 2b).
We confirmed the irreversibility of trapoxin A inhibition by assaying HDAC8 activity following multiple rounds of dialysis of the enzyme-inhibitor complex. Regeneration of activity was not observed for HDAC8 preincubated with a 10-fold molar excess of trapoxin A, but was observed under the same conditions using apicidin (Figure 3). This result confirms that the epoxide moiety is required for essentially irreversible inhibition. However, this result does not prove that a covalent enzyme-inhibitor complex is formed.
Figure 3.
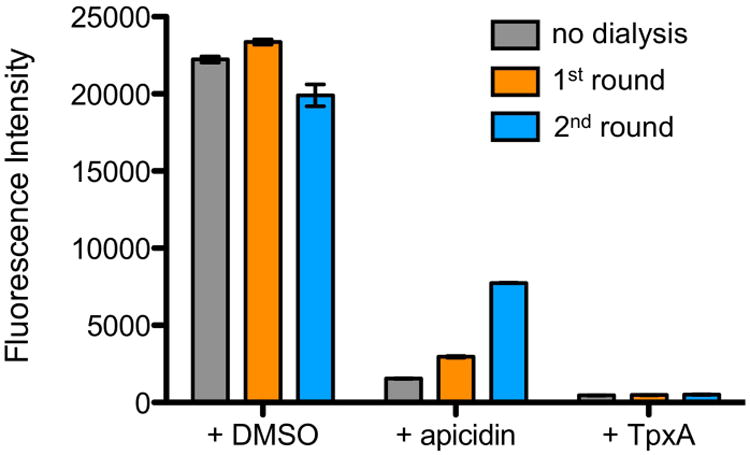
HDAC activity assays. Recovered HDAC8 activity from samples incubated with DMSO, apicidin, and trapoxin A (TpxA) after no (gray), one (orange) or two (blue) rounds of dialysis against 104-fold excess fresh buffer.
To study the covalent modification of HDAC8 by trapoxin A in solution, we employed liquid chromatography-tandem mass spectrometry (LC-MS/MS) on HDAC8 preincubated for 18 hours with 10 molar equivalents of trapoxin A and subsequently digested with trypsin. Prior to digestion, mass peaks corresponding to HDAC8 covalently modified with one or two trapoxin A molecules were observed for this sample by MALDI mass spectrometry (Supporting Information Figure S3). Following trypsin digestion, LC-MS/MS analysis, and sequence analysis for residue modifications, a mass shift corresponding to the molecular weight of trapoxin A was sporadically observed for various cysteine and histidine residues located on the surface of HDAC8 (Supporting Information Figure S4 and Table S2). Only nonspecific labeling of the enzyme was observed in the presence of excess inhibitor, with no particular preference for covalent modification in the active site. Additionally, incubation of trapoxin A for 1 hour in the presence and absence of HDAC8 followed by LC-MS analysis indicated the presence of only intact trapoxin A (i.e., with an intact epoxide ring; Supporting Information Figure S5). These results strongly suggest that trapoxin A is simply an exceptionally tight-binding, noncovalent transition state analogue inhibitor of HDAC8. As noted by Schramm and colleagues,39 transition state analogues with essentially irreversible binding behavior are feasible for enzymes that exhibit catalytic rate enhancements of 1010 or greater, which is likely the case for an amide hydrolase such as HDAC8 based on uncatalyzed amide bond hydrolysis half-lives measured in centuries by Radzicka and Wolfenden.40
Isothermal titration calorimetry (ITC) measurements yield HDAC8 dissociation constants Kd = 3 ± 1 nM for trapoxin A and Kd = 250 ± 70 nM for apicidin (Supporting Information Figure S6), indicating 83-fold enhanced binding affinity for trapoxin A relative to apicidin. Given the structural similarity between the two cyclic tetrapeptides, and the fact that the macrocycle makes very few intermolecular interactions in the HDAC8–trapoxin A complex, it is clear that the epoxide moiety of trapoxin A is indeed responsible for tight binding to HDAC8.
ITC measurements of HDAC8 active site variants indicate that H142 is critical for trapoxin A binding just as it is important for catalysis, since the H142A mutation results in significantly weaker affinity with Kd = 17 ± 5 μM – H142A HDAC8 exhibits a 233-fold reduced kcat/KM value relative to wild-type HDAC8.22 This residue functions in catalysis with a positively charged imidazolium group that serves as an electrostatic catalyst.22 Electron density is observed for the Nε-H proton of H142 in the 1.24 Å electron density map (Supporting Information Figure S7), so the H142 imidazolium group must similarly stabilize the zinc-bound gem-diolate.
Although the L-Aoe side chains of trapoxin A and HC toxin bind to HDAC8 and HDAC6, respectively, as the gem-diolate, there are notable differences between the structures of each enzyme-inhibitor complex that may explain the tighter binding of these inhibitors to class I HDACs.34 In the class IIb HDAC6-HC toxin complex (Ki = 350 nM),36 the C–O bond of the epoxide adopts an energetically unfavorable eclipsed conformation with respect to the C–O bond of the hydroxyl group of the zinc-bound gem-diolate (Figure 4a); in the class I HDAC8–trapoxin A complex (Kd = 3 nM), the C–O bond of the epoxide adopts an energetically favorable staggered conformation (Figure 4b). The energetically unfavorable eclipsed conformation in the HDAC6-HC toxin complex appears to be caused by the bulky P571 residue in the L3 loop, conserved in all class II HDACs – if the epoxide adopted an energetically favorable staggered conformation in the HDAC6 active site, the epoxide methylene group would clash with P571. In class I HDACs, P571 is not conserved and the active site is more open. Thus, a more favorable binding conformation is accessible to the epoxyketone moiety only in the active site of a class I HDAC.
Figure 4.
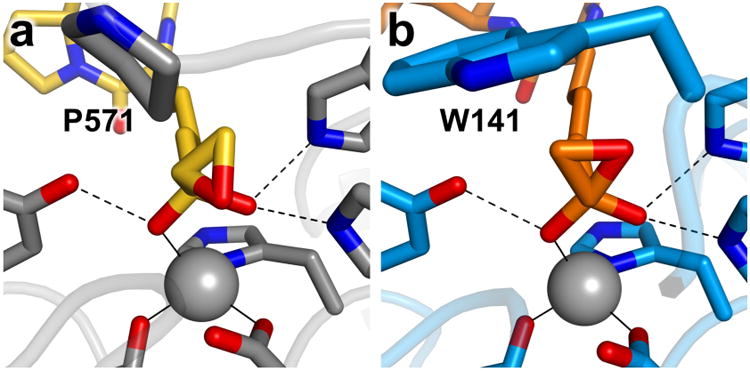
Epoxyketone binding modes. (a) HC toxin (C = yellow, N = blue, O = red) bound to HDAC6 (C = gray, N = blue, O = red). (b) Trapoxin A (C = orange, N = blue, O = red) bound to HDAC8 (C = light blue, N = blue, O = red). Residues affecting epoxide conformations are labeled. Zinc ions are shown as gray spheres.
Although the α,β-epoxyketone epoxide moiety of trapoxin A remains intact in the crystal structure of its complex with HDAC8, it is notable that this novel functionality is chemically reactive in the binding of inhibitors to other enzymes. For example, the proteasome inhibitor carfilzomib contains an α,β-epoxyketone that forms a covalent adduct to block proteasome function.41 The crystal structure of a similar natural product, epoxomicin, complexed with the yeast 20S proteasome indicates a multistep cyclization sequence leading to the formation of a morpholino ring between the former α,β-epoxyketone of the inhibitor and the reactive N-terminal threonine residue of the proteasome subunit.42 A two-step mechanism for inhibitor binding is initiated by nucleophilic attack of the threonine hydroxyl group at the epoxyketone carbonyl followed by a 6 Exo-Tet ring closure reaction between the α-amino group of threonine and the epoxide to generate the morpholino product.43 The chemistry of inhibitor binding in this system indicates that the carbonyl group appears to be more reactive than the epoxide of the α,β-epoxyketone moiety. This is consistent with reactivity trends observed in organic synthesis for various α,β-epoxycarbonyl derivatives, where the carbonyl group preferentially undergoes nucleophilic addition while leaving the epoxide moiety intact.44-46 With respect to trapoxin A, it is notable that an additional peak is observed in mass spectra consistent with gem-diol formation even in the absence of enzyme (Supporting Information Figure S5).
Finally, it is interesting to compare the structures of zinc coordination polyhedra in different HDAC8-inhibitor complexes (Supporting Information Figure S8). Only one oxygen of the gem-diolate of trapoxin A is sufficiently close for inner-sphere coordination, so the overall zinc coordination geometry is best described as 4-coordinate distorted tetrahedral. In this regard, zinc coordination geometry approaches that observed in the HDAC8–largazole complex, which exhibits nearly perfect tetrahedral coordination geometry through the binding of the largazole thiolate group.29 In contrast with these examples, the hydroxamate group of trichostatin A coordinates to zinc in bidentate fashion, so that the overall coordination geometry is 5-coordinate square pyramidal.20
In conclusion, the current work demonstrates that trapoxin A is an essentially irreversible noncovalent inhibitor of HDAC8: the α,β-epoxyketone side chain of the inhibitor undergoes nucleophilic attack by zinc-bound water to bind as a tetrahedral gem-diolate transition state analogue. Along with a favorable staggered conformation of the intact epoxide moiety relative to the zinc-bound gem-diolate, these structural features contribute to an exceptionally tight enzyme-inhibitor complex effectively locked into the enzyme active site.
Supplementary Material
Acknowledgments
N.J.P. thanks the Department of Chemistry at the University of Pennsylvania for the award of a graduate research fellowship. Additionally, we thank C. Smith at beamline 9-2 of the Stanford Synchrotron Radiation Lightsource (SSRL) for assistance with data collection. The SSRL, SLAC National Accelerator Laboratory, is supported by the DOE Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the NIH, National Institute of General Medical Sciences (including P41GM103393).
Funding: We thank the National Institutes of Health for grant GM49758 in support of this research.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: Detailed methods, figures, and tables
Accession Code: The atomic coordinates and crystallographic structure factors of the HDAC8-trapoxin A complex have been deposited in the Protein Data Bank (www.rcsb.org) with accession code 5VI6.
ORCHID: David W. Christianson: 0000-0002-0194-5212
Notes: The authors declare no competing financial interest.
References
- 1.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 3.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan KL. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, Ning ZB, Zeng R, Xiong Y, Guan KL, Zhao S, Zhao GP. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kouzarides T. Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000;19:1176–1179. doi: 10.1093/emboj/19.6.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norvell A, McMahon SB. Rise of the rival. Science. 2010;327:964–965. doi: 10.1126/science.1187159. [DOI] [PubMed] [Google Scholar]
- 7.Sterner DE, Berger SL. Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev. 2000;64:435–459. doi: 10.1128/mmbr.64.2.435-459.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCullough CE, Marmorstein R. Molecular basis for histone acetyltransferase regulation by binding partners, associated domains, and autoacetylation. ACS Chem Biol. 2016;11:632–642. doi: 10.1021/acschembio.5b00841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lombardi PM, Cole KE, Dowling DP, Christianson DW. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr Opin Struct Biol. 2011;21:735–743. doi: 10.1016/j.sbi.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 12.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 13.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 14.Gantt SL, Gattis SG, Fierke CA. Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry. 2006;45:6170–6178. doi: 10.1021/bi060212u. [DOI] [PubMed] [Google Scholar]
- 15.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structure of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 16.Kanyo ZF, Scolnick LR, Ash DE, Christianson DW. Structure of a unique binuclear manganese cluster in arginase. Nature. 1996;383:554–557. doi: 10.1038/383554a0. [DOI] [PubMed] [Google Scholar]
- 17.Ash DE, Cox JD, Christianson DW. Arginase: a binuclear manganese metalloenzyme. Metal Ions Biol Syst. 2000;37:407–428. [PubMed] [Google Scholar]
- 18.Bheda P, Jing H, Wolberger C, Lin H. The substrate specificity of sirtuins. Annu Rev Biochem. 2016;85:405–429. doi: 10.1146/annurev-biochem-060815-014537. [DOI] [PubMed] [Google Scholar]
- 19.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkühler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Somoza JR, Skene RJ, Katz BA, Mol C, Ho JD, Jennings AJ, Luong C, Arvai A, Buggy JJ, Chi E, Tang J, Sang BC, Verner E, Wynands R, Leahy EM, Dougan DR, Snell G, Navre M, Knuth MW, Swanson RV, McRee DE, Tari LW. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 21.Gantt SL, Joseph CG, Fierke CA. Activation and inhibition of histone deacetylase 8 by monovalent cations. J Biol Chem. 2010;285:6036–6043. doi: 10.1074/jbc.M109.033399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gantt SL, Decroos C, Lee MS, Gullett LE, Bowman CM, Christianson DW, Fierke CA. General base-general acid catalysis in human histone deacetylase 8. Biochemistry. 2016;55:820–832. doi: 10.1021/acs.biochem.5b01327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter NJ, Christianson NH, Decroos C, Christianson DW. Structural and functional influence of the glycine-rich loop G302GGGY on the catalytic tyrosine of histone deacetylase 8. Biochemistry. 2016;55:6718–6729. doi: 10.1021/acs.biochem.6b01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vannini A, Volpari C, Galinari P, Jones P, Mattu M, Carfí A, De Francesco R, Steinkühler C, Di Marco S. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. EMBO Rep. 2007;8:879–884. doi: 10.1038/sj.embor.7401047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Furumai R, Matsuyama A, Kobashi N, Lee KH, Nishiyama M, Nakajima H, Tanaka A, Komatsu Y, Nishino N, Yoshida M, Horinouchi S. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–4921. [PubMed] [Google Scholar]
- 27.Guan P, Fang H. Clinical development of histone deacetylase inhibitor romidepsin. Drug Discovery Ther. 2010;4:388–391. [PubMed] [Google Scholar]
- 28.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total synthesis and molecular target of largazole, a histone deacetylase inhibitor. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 29.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Itazaki H, Nagashima K, Sugita K, Yoshida H, Kawamura Y, Yasuda Y, Matsumoto K, Ishi K, Uotani N, Nakai H, Terui A, Yoshimatsu S, Ikenishi Y, Nakagawa Y. Isolation and structural elucidation of new cyclotetrapeptides, trapoxins A and B, having detransformation activities as antitumor agents. J Antibiot. 1990;43:1524–1532. doi: 10.7164/antibiotics.43.1524. [DOI] [PubMed] [Google Scholar]
- 31.Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T. Trapoxin, an antitumor cyclic tetrapeptide, is an irreversible inhibitor of mammalian histone deacetylase. J Biol Chem. 1993;268:22429–22435. [PubMed] [Google Scholar]
- 32.Taunton J, Collins JL, Schreiber SL. Synthesis of natural and modified trapoxins, useful reagents for exploring histone deacetylase function. J Am Chem Soc. 1996;118:10412–10422. [Google Scholar]
- 33.Taunton J, Hassig CA, Schreiber SL. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996;272:408–411. doi: 10.1126/science.272.5260.408. [DOI] [PubMed] [Google Scholar]
- 34.Fumurai R, Komatsu Y, Nishino N, Khochbin S, Yoshida M, Horinuichi S. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc Nat Acad Sci U S A. 2001;98:87–92. doi: 10.1073/pnas.011405598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lewis CA, Wolfenden R. Antiproteolytic aldehydes and ketones: substituent and secondary deuterium isotope effects on equilibrium addition of water and other nucleophiles. Biochemistry. 1997;16:4886–4890. doi: 10.1021/bi00641a022. [DOI] [PubMed] [Google Scholar]
- 36.Hai Y, Christianson DW. Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat Chem Biol. 2016;12:741–747. doi: 10.1038/nchembio.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christianson DW, Lipscomb WN. Binding of a possible transition state analogue to the active site of carboxypeptidase A. Proc Natl Acad Sci U S A. 1985;82:6840–6844. doi: 10.1073/pnas.82.20.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Christianson DW, David PR, Lipscomb WN. Mechanism of carboxypeptidase A: hydration of a ketonic substrate analogue. Proc Natl Acad Sci U S A. 1987;84:1512–1515. doi: 10.1073/pnas.84.6.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh V, Evans GB, Lenz DH, Mason JM, Clinch K, Mee S, Painter GF, Tyler PC, Furneaux RH, Lee JE, Howell PL, Schramm VL. Femtomolar transition state analogue inhibitors of 5′-methylthioadenosine/S-adenosylhomocysteine nucleosidase from Escherichia coli. J Biol Chem. 2005;280:18265–18273. doi: 10.1074/jbc.M414472200. [DOI] [PubMed] [Google Scholar]
- 40.Radzicka A, Wolfenden R. Rates of uncatalyzed peptide bond hydrolysis in neutral solution and the transition state affinities of proteases. J Am Chem Soc. 1996;118:6105–6109. [Google Scholar]
- 41.Spaltenstein A, Leban JJ, Huang JJ, Reinhardt KR, Viveros OH, Sigafoos J, Crouch R. Design and synthesis of novel protease inhibitors Tripeptide α′,β′-epoxyketones as nanomolar inactivators of the proteasome. Tet Lett. 1996;37:1343–1346. [Google Scholar]
- 42.Johnson HWB, Anderl JL, Bradley EK, Bui J, Jones J, Arastu-Kapur S, Kelly LM, Lowe E, Moebius DC, Muchamuel T, Kirk C, Wang Z, McMinn D. Discovery of highly selective inhibitors of the immunoproteasome low molecular mass polypeptide 2 (LMP2) subunit. ACS Med Chem Lett. 2017;8:413–417. doi: 10.1021/acsmedchemlett.6b00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Groll M, Kim KB, Kairies N, Huber R, Crews CM. Crystal structure of epoxomicin:20S proteasome reveals a molecular basis for selectivity of α′,β′-epoxyketone proteasome inhibitors. J Am Chem Soc. 2000;122:1237–1238. [Google Scholar]
- 44.Urabe H, Matsuka T, Sato F. (2S,3S)-2,3-epoxy-3-trimethylsilylpropanal as a new conjunctive reagent. Tet Lett. 1992;33:4179–4182. [Google Scholar]
- 45.Howe GP, Wang S, Procter G. Stereoselective additions to α,β-epoxyaldehydes; the formation of “non-chelation controlled” products. Tet Lett. 1987;28:2629–2632. [Google Scholar]
- 46.Pégorier L, Petit Y, Larchevêque M. A synthetic route to anti aminoalkyl epoxides by stereocontrolled reductive amination of ketoepoxides. J Chem Soc, Chem Commun. 1994;5:633–634. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.