

Abstract
The Escobar variant of multiple pterygium syndrome (OMIM #265000) is a rare, autosomal recessive disorder associated with mutations in the γ-subunit of the nicotinic acetylcholine receptor (CHRNG). CHRNG is expressed in fetal muscle during motor development and contributes to the formation of neuromuscular junctions. Anomalies in neuromuscular junction structure and function have not been investigated in patients with Escobar syndrome. We report five patients identified as having Escobar syndrome, from four families. In three families, the same mutation (c.459dupA) was identified in CHRNG. A biopsy from brachioradialis muscle was collected from a patient from one of these families and analyzed for neuromuscular junction organization using fluorescence microscopy. Compared to spinalis muscle from control patients with idiopathic scoliosis or cerebral palsy, the patient with Escobar syndrome had a significantly higher degree of acetylcholine receptor present outside acetylcholinesterase; and significantly less acetylcholinesterase outside acetylcholine receptor. Given the role of the acetylcholine receotor γ-subunit in fetal neuromuscular signal transduction and in establishing the primary encounter of muscle and motor nerve terminal, the CHRNG mutation described in Escobar syndrome may cause a broader disruption of postsynaptic proteins and result in aberrant development of the NMJ due to impaired prenatal neuromuscular transmission and/or abnormal neuromuscular synaptogenesis.
Keywords: Escobar syndrome, multiple pterygium syndrome, CHRNG, acetylcholine receptor, acetylcholinesterase, neuromuscular junction
INTRODUCTION
The Escobar variant of multiple pterygium syndrome (OMIM #265000) is a rare, autosomal recessive disorder characterized by pterygia, cutaneous webbing associated with joint contractures, and scoliosis. Variable features include growth retardation, congenital respiratory distress, craniofacial dysmorphism with ptosis and low-set ears, cleft palate, and cryptorchidism [Hoffmann, et al., 2006]. The disorder is associated with mutations in the gamma-subunit of the nicotinic acetylcholine receptor (CHRNG), which is expressed in muscle during motor development and contributes to the formation of neuromuscular junctions [Chen, 2012; Hesselmans, et al., 1993; Hoffmann, et al., 2006; Monnier, et al., 2009; Morgan, et al., 2006]; however, a lack of CHRNG mutation has been reported in several families with Escobar-like symptoms, suggesting that mutations in another gene may be responsible for some cases [Kodaganur, et al., 2013; Vogt, et al., 2012]. Mutations in CHRNA1, CHRND, RAPSN and DOK7 have been described in lethal multiple pterygium syndrome (OMIM #253290) and fetal akinesia deformation sequence (FADS, OMIM #208150) without pterygia [Michalk, et al., 2008; Vogt, et al., 2008; Vogt, et al., 2009; Vogt, et al., 2012]. It is unknown if mutations in these genes can result in a phenotype resembling Escobar syndrome with CHRNG mutations.
CHRNG dysfunction leads to fetal myasthenia and akinesia, which are major contributors to the disease presentation; however, anomalies in neuromuscular junction structure and function have not been investigated. The current study was undertaken to elucidate whether a patient with Escobar syndrome had disrupted motor synapse microanatomy relative to unaffected controls.
MATERIAL AND METHODS
Four individuals with a clinical diagnosis of Escobar syndrome were enrolled in a research study approved by the local Institutional Review Board (#203398). If not previously completed, CHRNG mutations were identified by Sanger sequencing in the Nemours Biomolecular Core Laboratory. Only individuals positive for CHRNG mutations were considered affected.
One additional patient (Patient 2) was not enrolled into the research study, but she and her parents consented to share clinical and molecular information and patient photographs.
Patients
Patient 1
The patient was born to a 27-year-old father and a 24-year-old mother who reported decreased fetal movement. At 24 weeks gestation, an ultrasound revealed abnormal bilateral hand position suggesting arthrogryposis, and a follow-up evaluation showed slightly elevated amniotic fluid levels. The patient was delivered at 37 weeks by cesarian secondary to breech presentation. His birth weight was 2.24 kg (10th–25th centile for gestational age), length 39.4 cm (<10th centile, resulting from contractures), and head circumference 33 cm (50th centile), with Apgar scores of 7 at one minute and 9 at five minutes. Examination at birth revealed camptodactyly of the hands and feet, contractures of his wrists, elbows, knees, ankles, and hips, low-set ears, a webbed neck, hypospadias, a sacral dimple, and rocker bottom feet (Figure 1). A high-resolution banding karyotype (550 bands) showed a normal 46,XY result. Echocardiography revealed a structurally normal heart with a patent foramen ovale (PFO) and a small patent ductus arteriosus (PDA); an echocardiogram performed at 13 months showed closure of the PFO and PDA. An ultrasound study of the brain completed in the neonate had a normal result. A renal ultrasound revealed moderate hydronephrosis of the left kidney that improved by age 1 week. At age 1 month, a genetics evaluation for Escobar syndrome and CHRNG mutation analysis were performed. Exons 1 to 5 of CHRNG were analyzed by PCR and bi-directional sequencing. A homozygous duplication c.459dupA (Table I) was identified, which results in a frame-shift and a premature termination at codon 177. The parents were subsequently found to be heterozygous for this duplication.
Figure 1. Photographic images of Patient 1 as neonate and at age 4 years.

A) At nine days of age, note flexion contractures of elbows, wrists, hips and knees, and foot positional abnormality. B) At age 4 years, note short neck and low set ear, webbing of shoulders and elbows and wrist position. C) Lower extremities at age 4 years. Note low muscle mass, abnormal foot position and scars over lower legs.
Table I.
Molecular information and a summary of phenotypic findings in five patients with Escobar syndrome.
| Patient | CHRNG mutation | Parental results | Current age (years) | Palate | Contractures | Other |
|---|---|---|---|---|---|---|
| 1 | c.459dupA (p.Val154Ser fs*24) | heterozygous | 5 | Cleft | Wrists, elbows, knees, ankles, hips | Low-set ears, webbed neck, hypospadias, sacral dimple, and vertical tali |
| 2 | c.459dupA (p.Val154Ser fs*24) | heterozygous | 13 | High, narrow | “arthrogryposis multiplex congenital”; upper and lower extremities, hands | Vertical tali; hip dysplasia; surgical removal of “excess tissue” for stridor |
| 3 | c.459dupA (p.Val154Ser fs*24) | 4 | Normal | Hips, knees | Bilateral vertical tali; cryptorchidism; dislocated hips; scoliosis | |
| 4 (sibling to 3) | (sibling to Patient 3) | 6 | Normal | Sternocleidomastoid muscle on the left, right knee | Paralyzed right hemidiaphragm; bilateral dislocated hips; bilateral vertical tali; flexible thoracic scoliosis; left torticollus; captodactyly of fingers; micrognathia; prominent forehead | |
| 5 | c.753_754delCT (p.Val253Alafs*44) | heterozygous | 11 | No cleft | “arthrogryposis multiplex congenita”; upper and lower extremities | Vertical tali; unilateral hip dysplasia; eventration right diaphragm; tracheostomy; gastrostomy; ASD resolved |
In addition to the symptoms noted at birth, Patient 1 had radioulnar synostosis, mild scoliosis, short stature, cleft palate, vertical tali, inguinal hernias, cryptorchidism, ptosis, and a small chest cavity. Surgical procedures included correction of vertical tali with anterior tibial and Achilles tendon lengthenings at age 4 months and again at 36 months; hypospadias, cryptorchidism, and bilateral inguinal hernia repair at 13 months; ptosis correction at 14 months; cleft palate repair at 14 months; bilateral knee flexion contracture release at 16 months; wrist flexion contracture release and tendon transfer to thumb at 33 months; and three sets of myringotomy tubes.
At age 6 months, a nerve conduction study on the ulnar motor nerve was normal. Repetitive nerve stimulation studies at slow and fast rates in upper and lower limbs showed no abnormality in neuromuscular transmission. Needle electromyography of muscles in the upper and lower limbs showed myopathy with normal spontaneous activity. A trial of pyridostigmine initially resulted in lethargy within a few days of starting therapy. Several months later, pyridostigmine was restarted with modest improvement in muscle strength and overall stamina. To our knowledge, the other four patients were never treated with pyridostigmine or other acetylcholinesterase inhibitors.
Patient 2
This female was the second child born to her non-consanguineous parents of German and mixed North European ancestry. Fetal movements were described as different in quality compared to the prior pregnancy. Delivery at 42 weeks gestation was by cesarian due to breech presentation. Her weight was 3.37 kg (50–75th centile), length and OFC were not recorded. Multiple joint contractures included camptodactyly, limited range of motion in hips and shoulders, and foot positional abnormalities due to vertical tali. Physical therapy and splinting resulted in improved range of motion. Patient 2 required surgical removal of redundant tissue in her airway for severe stridor; bilateral myringotomy tube placement for chronic otitis; surgical correction of the vertical tali; several procedures to correct bilateral congenital hip dysplasia. While her cognitive development progressed age-appropriately, her motor development was delayed with independent walking at age 3.5 years. At age 10 7/12 years, her height was 120 cm (<3rd centile; 50th centile for age 6 ¾ years), weight 19.6 kg (<3rd centile; 50th centile for 6 years) and OFC 51.5 cm (within 2 SD of mean for age). She had a very soft voice. Her face showed mild asymmetry and a faint glabellar nevus flammeus (Figure 2). Her neck was webbed. Apparent myopathy resulted in decreased muscle mass and strength. She had limited shoulder abduction; limited pronation and supination bilaterally with almost complete extension of the right elbow and limited extension of the left. Hands showed webbing of the thumbs, bilateral flexion contractures of her fingers and an absence of palmar flexion creases (Figure 2C,D) Mild hip and knee contractures (Figure 2E) were noted upon standing. Feet showed high arches and hallux valgus. Her labia majora appeared hypoplastic. A clinical diagnosis of Escobar syndrome was confirmed by diagnostic CHRNG sequencing, which revealed a homozygous mutation, c.459dupA (Table I).
Figure 2. Photographic images of Patient 2 at age 10 years.

Note facial asymmetry and short and webbed neck (A,B), low set ears, camptodactyly with absent palmar flexion creases (C, D), and knee contractures (E).
Patients 3–5
Molecular information and a summary of the phenotypic findings are provided in Table I. Limited clinical information is available and skeletal muscle specimens were not available. Patients 3 and 4 are siblings; molecular testing showed a homozygeous CHRNG mutation, c.459dupA, in Patient 3 (Table I). Patient 3 was diagnosed with multiple pterygium syndrome based on ultrasound evaluation at about 18 weeks gestation and the family history. He moved a fair amount during pregnancy. A cesarian was performed at 37 weeks due to breech position and decreasing amniotic fluid. He was born with flexion contractures of elbows, tight fists, flexion of hips, knees, and feet, rocker bottom feet, dislocated hip on the left side, a myopathic facial appearance, and a high right diaphragm. He had fusion of C1 and C2 vertebrae, scoliosis, and a tethered spinal cord that was corrected at 15 months. He had significant lower limb involvement, and his left side was more severely affected than his right. He had feeding problems for the first few months that gradually resolved. He was noted to have webbing of the neck, a small jaw, a large forehead, and undescended testes.
Patient 4 is an older sibling of Patient 3 and was noted to have a less severe phenotype. The pregnancy was complicated by severe hyperemesis. The fetus was noted to move less than an older sibling. Patient 4 was born at 39 weeks gestation to a 29-year-old mother by vaginal delivery after induced labor and breech positioning. Apgar scores were 6 at 1 minute and 7 at 5 minutes. She required blow-by oxygen after delivery and had mild respiratory insufficiency. A paralyzed right hemidiaphragm was surgically repaired at age 5 months. She was evaluated the day after birth for multiple dysmorphic features and arthogryposis. She had bilateral dislocated hips, bilateral vertical tali, flexible thoracic scoliosis, captodactyly of her fingers, and left torticollis. She was noted in the newborn period to have micrognathia and a prominent forehead.
Patient 5 was born after a pregnancy complicated by polyhydramnios and was suspected to have a myopathy as an infant. Subsequently, joint contractures became more noticeable, and a suspected diagnosis of Escobar syndrome was confirmed by diagnostic CHRNG sequencing (Table I).
Muscle sample acquisition and analysis
At age 3 years, Patient 1 underwent a tendon transfer on his thumb and a flexion contracture release on his wrist. A brachioradialis muscle biopsy from the right forearm obtained at Scottish Rite Hospital in Atlanta was shipped on wet ice to the A.I. duPont Hospital for Children in Wilmington, Delaware. The sample was snap-frozen in liquid nitrogen-chilled isopentane, stored at −80°C, and sectioned for Hematoxylin and Eosin (H&E) and fluorescence staining analyses. H&E staining revealed areas of normal-appearing muscle adjacent to areas that appeared to have been damaged during acquisition or transport (Figure 3). Fluorescence staining methods were employed to assess the organization of the neuromotor synapse. Sections of frozen tissue (8–10 microns thick) were fixed with 2% paraformaldehyde (Electron Microscopy Sciences) for 5 min, permeabilized with 0.1% Triton X-100 (Sigma) for 15 min, blocked with 3% bovine serum albumin (Sigma) for 30 min. All samples were stained with Alexa Fluor 488-conjugated alpha-bungarotoxin (Life Technologies) at 0.16 μg/mL to label acetylcholine receptors (AChR) and Alexa Fluor 594-conjugated (Life Technologies) fasciculin-2 (Alomone Labs) at 1 μg/mL to label acetylcholine esterase (AChE). In addition, samples were stained with an antibody against laminin β2 (C4 hybridoma medium, Developmental Studies Hybridoma Bank (DSHB)) diluted 1:10 followed by an Alexa Fluor 647-conugated secondary antibody (Life Technologies). Samples were digitally imaged on an Olympus BX-60 fluorescence microscope equipped with an Evolution QEi monochrome 12-bit (1360 x 1036 pixels) digital camera (Media Cybernetics) controlled by Image Pro Plus software (version 6.3; Media Cybernetics). The relative distributions of AChR, AChE, and laminin β2 were compared pair-wise in a two-step process that used the Image Pro Plus thresholding algorithm. The algorithm provided an objective value for the threshold level based on an iterative analysis of an image’s histogram that yielded the best split between foreground and background pixels, followed by a customized software macro to calculate an appositional score indicating the degree of non-colocalization.
Figure 3. Hematoxylin and eosin stain of a brachioradialis muscle biopsy from a patient with Escobar syndrome.
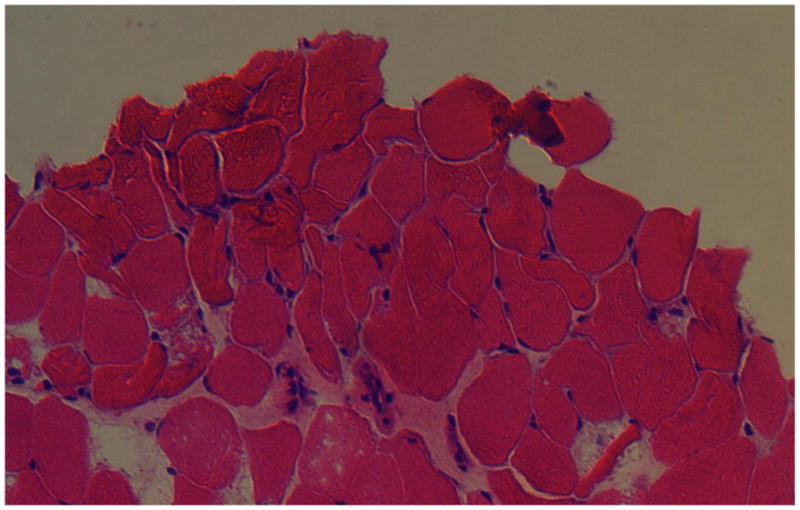
H&E staining revealed areas of normal-appearing muscle adjacent to areas that appeared to have been damaged during acquisition and transport.
RESULTS
Based on our ascertainment procedures, all patients survived the infant period and had CHRNG mutations. Mutations were homozygous, despite a reported absence of consanguinity in each family. All families were of Northern European, Caucasian ancestry. Interestingly, in three apparently unrelated families the same mutation (c.459dupA) was identified. Patient 5 was homozygous for a different mutation (Table I).
Representative patterns of muscle staining from Patient 1 are shown in Figure 4. Co-localization of neuromuscular junction (NMJ) components was determined by calculating appositional scores using previously described methods (PLoS One, manuscript in revision). Compared to spinalis muscle from control patients with idiopathic scoliosis or cerebral palsy (CP), Patient 1 had a significantly higher degree of AChR present outside AChE and significantly less AChE outside AChR (p < 0.05 by Mann-Whitney; Figure 5, Table II). The patient was not significantly different from control or CP in regard to laminin β2 relative to AChR or AChE.
Figure 4. Typical staining patterns for an idiopathic scoliosis patient and the Escobar patient with the corresponding threshold images.

Skeletal muscle samples were triple stained for AChR (BTX; yellow), AChE (Fas2; magenta), and laminin β2 (cyan). Representative staining patterns of the three stains are shown. Threshold images for each stain were determined by the Image Pro software based on the number of pixels at each gray level (PLoS One, manuscript in revision). In the idiopathic scoliosis patient (A,B), the stains were highly colocalized as the formation of tightly apposed pre- and post-synaptic specializations at NMJs are hallmarks of neuromotor maturation. Compared to the idiopathic scoliosis patients, the Escobar patient (C,D) had more AChR present outside AChE (arrow) and less AChE present outside AChR.
Figure 5. Comparison of non-colocalization scores at the NMJs of spinalis from idiopathic scoliosis patients and brachioradialis from an Escobar patient.

Threshold images for each stain (BTX = AChR, FAS = AChE, LAMB2 = laminin β2) were determined by the Image Pro software based on the number of pixels at each gray level. After the software set the threshold levels, the pixels above those levels were categorized and counted. The software used this method to count the pixels for the entire NMJ in two dimensions and the stains were compared pair-wise to determine the proportion of pixels from one stain that resided outside the other. The Escobar patient had a significantly higher score for AChR outside AChE and a significantly lower score for AChE outside AChR (*** = p<0.05 by Mann-Whitney).
Table II.
Degree of non-colocalization of NMJ components in muscle from Patient 1 compared to back muscle from control patients [Theroux, et al., 2005], back muscle from spastic CP patients (PLoS One, manuscript in revision), leg muscle from ambulatory and non-ambulatory spastic CP patients [Theroux, et al., 2002;Theroux, et al., 2005], and Duchenne Muscular Dystrophy patients [Theroux, et al., 2008].
| Control Patients | Escobar Patient | Back of Spastic CP Patients | Leg of Ambulatory Spastic CP Patients | Leg of Non-Ambulatory Spastic CP Patients | DMD Patients | |
|---|---|---|---|---|---|---|
| AChR > AChE | 0.07 ± 0.04 | 0.32 ± 0.16* | 0.07 ± 0.08 | 0.16 ± 0.08* | 0.23 ± 0.14* | 0.11 ± 0.10 |
| AChE > AChR | 0.32 ± 0.13 | 0.18 ± 0.15* | 0.33 ± 0.14* | - | - | - |
| Laminin β2 > AChE | 0.40 ± 0.16 | 0.51 ± 0.16 | 0.37 ± 0.16* | - | - | - |
| AChE > Laminin β2 | 0.09 ± 0.10 | 0.08 ± 0.10 | 0.14 ± 0.13* | - | - | - |
p < 0.05 (by Mann-Whitney/Kruskal Wallis) compared to control, - = not examined.
CP = cerebral palsy, DMD = Duchenne Muscular Dystrophy, AChR = acetylcholine receptor, AChE = acetylcholinesterase
DISCUSSION
We identified the same homozygeous mutation, c.459dupA , in three unrelated families of Caucasian ancestry. As none of the families reported parental consanguinity, one might conclude that this suggests a founder effect. However, haplotype analysis performed by Vogt et al. [2012] for this particular recurrent mutation was not consistent with a founder effect. The same mutation identified in three families in the current study was reported by Vogt et al. [2012] in three kindreds with Escobar syndrome and three with lethal multiple pterygium syndrome, as well as by Morgan et al. [2006].
Morgan et al. [2006] reported a 37-week gestation female fetus found to be hydropic, with bilateral moderate pleural effusions, skin edema, and hydronephrotic right kidney. An emergency cesarian was performed for fetal distress, but the baby died at age 1 day from lung hypoplasia [Morgan, et al., 2006]. An apparent lack of correlation between the specific mutations and the clinical presentation, either lethal or Escobar type, has been noted previously [Morgan, et al., 2006; Vogt, et al., 2012]. While our ascertainment procedure resulted in our patients having Escobar syndrome, the absence of deceased affected siblings in all three families is noteworthy. Despite the variable phenotypic severity at presentation, the provided medical care is likely to have a significant impact upon potential survival. Thus, survival may be possible regardless of the specific underlying CHRNG mutations. Realizing this potential, and considering the expression of CHRNG ends at around 33-weeks-gestation [Hoffmann et al., 2006], early diagnosis may allow for effective treatment or prevention of long term sequelae. Understanding the biological consequences of the abnormal gene product is a necessary step in assessing the potential for drug therapies.
Both Patient 1 and previously-analyzed CP patients, displayed non-colocalization of NMJ proteins; however, the most affected components differed. Patient 1 had significantly more AChR present outside AChE and significantly less AChE outside AChR compared to controls, while the most significant abnormality in CP patients was a significantly higher degree of AChE outside laminin β2 (PLoS One, manuscript in revision). Interestingly, while Escobar syndrome and CP are both associated with immobility, CP is caused by a brain lesion during motor development, whereas Escobar syndrome is caused by CHRNG dysfunction, impacting NMJ formation during development. These different etiologies are likely associated with the resulting differences in the NMJ micro-anatomy.
Given CHRNG’s specific role in neuromuscular synaptogenesis, abnormalities at the mature NMJ are not unexpected. CHRNG encodes the γ-subunit of the nicotinic AChR and is expressed until approximately 33 weeks gestation, when it is replaced by the ε-subunit [Chen, 2012; Hesselmans, et al., 1993; Hoffmann, et al., 2006]. The pterygia and bony abnormalities in Escobar syndrome result from fetal akinesia that is a consequence of abnormal fetal neuromuscular transmission [Vogt, et al., 2012]. Since the γ–subunit is replaced by the ε-subunit in late fetal development, Escobar patients have a normal composition of AChR subunits postnatally and exhibit almost no progression or signs of myasthenia after birth. In addition to the γ-subunit’s role in fetal neuromuscular signal transduction, CHRNG expression is also important for establishing the primary encounter of muscle and motor nerve terminal [Hoffmann, et al., 2006]. Animal studies have shown that Chrng dysfunction affects neuromotor synapse formation and localization. Targeted deletion of the γ–subunit in mice leads to an absence of spontaneous fetal muscle action potentials and premature death [Takahashi, et al., 2002]. In addition, γ −/− mice were devoid of AChR clusters on the surface of muscles until E16.5, when ε-subunit expression first became detectable [Liu, et al., 2008]. The absence of AChR clusters was associated with excessive nerve branching, increased motoneuron survival, and aberrant distribution of the NMJ components AChE and rapsyn [Liu, et al., 2008]. At E18.5, after the γ- to ε-subunit switch occurred, the size of the endplate was larger in the mutant than wild type, but the fluorescence intensity of AChR staining was diminished, suggesting a decrease in the density of AChRs [Liu, et al., 2010]. These results demonstrate that the γ–subunit is required for the assembly of AChR clusters, which in turn play a role in the assembly of the postsynaptic apparatus at the NMJ.
In conclusion, this patient with Escobar syndrome had a significantly altered distribution of neuromotor synapse components. The postnatal abnormalities at the NMJ may reflect the developmental effects of impaired prenatal neuromuscular transmission and/or aberrant neuromuscular synaptogenesis. The CHRNG mutations in Escobar syndrome may, therefore, cause a broader disruption of postsynaptic proteins and result in aberrant development of the NMJ.
Acknowledgments
The authors thank Stacey Koletty for coordination of the study, Dr. Allan Peljovich of The Hand and Upper Extremity Center of Georgia for sample acquisition, the Nemours Biomolecular Core Laboratory, and the Nemours Biomedical Research Histotechnology Core Laboratory. This study was supported by funds from a Clinical Genetics Cluster Award from the Nemours Foundation (KWG and REA); a grant from the Swank Foundation (REA); a grant from the NIH-NIGMS COBRE program (P20-GM-103464; REA).The authors declare no conflict of interest.
References
- Chen CP. Prenatal diagnosis and genetic analysis of fetal akinesia deformation sequence and multiple pterygium syndrome associated with neuromuscular junction disorders: A review. Taiwan J Obstet Gynecol. 2012;51:12–17. doi: 10.1016/j.tjog.2012.01.004. [DOI] [PubMed] [Google Scholar]
- Hesselmans LF, Jennekens FG, Van den Oord CJ, Veldman H, Vincent A. Development of innervation of skeletal muscle fibers in man: Relation to acetylcholine receptors. Anat Rec. 1993;236:553–562. doi: 10.1002/ar.1092360315. [DOI] [PubMed] [Google Scholar]
- Hoffmann K, Muller JS, Stricker S, Megarbane A, Rajab A, Lindner TH, Cohen M, Chouery E, Adaimy L, Ghanem I, Delague V, Boltshauser E, Talim B, Horvath R, Robinson PN, Lochmuller H, Hubner C, Mundlos S. Escobar syndrome is a prenatal myasthenia caused by disruption of the acetylcholine receptor fetal gamma subunit. Am J Hum Genet. 2006;79:303–312. doi: 10.1086/506257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodaganur SG, Tontanahal SJ, Sarda A, Shah MH, Bhat V, Kumar A. Clinical phenotype and the lack of mutations in the CHRNG, CHRND, and CHRNA1 genes in two Indian families with Escobar syndrome. Clin Dysmorphol. 2013;22:54–58. doi: 10.1097/MCD.0b013e32835f9ac0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Padgett D, Takahashi M, Li H, Sayeed A, Teichert RW, Olivera BM, McArdle JJ, Green WN, Lin W. Essential roles of the acetylcholine receptor gamma-subunit in neuromuscular synaptic patterning. Development. 2008;135:1957–1967. doi: 10.1242/dev.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Sugiura Y, Padgett D, Lin W. Postsynaptic development of the neuromuscular junction in mice lacking the gamma-subunit of muscle nicotinic acetylcholine receptor. J Mol Neurosci. 2010;40:21–26. doi: 10.1007/s12031-009-9248-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalk A, Stricker S, Becker J, Rupps R, Pantzar T, Miertus J, Botta G, Naretto VG, Janetzki C, Yaqoob N, Ott CE, Seelow D, Wieczorek D, Fiebig B, Wirth B, Hoopmann M, Walther M, Korber F, Blankenburg M, Mundlos S, Heller R, Hoffmann K. Acetylcholine receptor pathway mutations explain various fetal akinesia deformation sequence disorders. Am J Hum Genet. 2008;82:464–476. doi: 10.1016/j.ajhg.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier N, Lunardi J, Marty I, Mezin P, Labarre-Vila A, Dieterich K, Jouk PS. Absence of beta-tropomyosin is a new cause of escobar syndrome associated with nemaline myopathy. Neuromuscul Disord. 2009;19:118–123. doi: 10.1016/j.nmd.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Morgan NV, Brueton LA, Cox P, Greally MT, Tolmie J, Pasha S, Aligianis IA, van Bokhoven H, Marton T, Al-Gazali L, Morton JE, Oley C, Johnson CA, Trembath RC, Brunner HG, Maher ER. Mutations in the embryonal subunit of the acetylcholine receptor (chrng) cause lethal and escobar variants of multiple pterygium syndrome. Am J Hum Genet. 2006;79:390–395. doi: 10.1086/506256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Kubo T, Mizoguchi A, Carlson CG, Endo K, Ohnishi K. Spontaneous muscle action potentials fail to develop without fetal-type acetylcholine receptors. EMBO Rep. 2002;3:674–681. doi: 10.1093/embo-reports/kvf128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theroux MC, Akins RE, Barone C, Boyce B, Miller F, Dabney KW. Neuromuscular junctions in cerebral palsy: Presence of extrajunctional acetylcholine receptors. Anesthesiology. 2002;96:330–335. doi: 10.1097/00000542-200202000-00017. [DOI] [PubMed] [Google Scholar]
- Theroux MC, Oberman KG, Lahaye J, Boyce BA, Duhadaway D, Miller F, Akins RE. Dysmorphic neuromuscular junctions associated with motor ability in cerebral palsy. Muscle Nerve. 2005;32:626–632. doi: 10.1002/mus.20401. [DOI] [PubMed] [Google Scholar]
- Theroux MC, Olivant A, Akins RE. C histomorphology of neuromuscular junction in duchenne muscular dystrophy. Paediatr Anaesth. 2008;18:256–259. doi: 10.1111/j.1460-9592.2008.02411.x. [DOI] [PubMed] [Google Scholar]
- Vogt J, Harrison BJ, Spearman H, Cossins J, Vermeer S, ten Cate LN, Morgan NV, Beeson D, Maher ER. Mutation analysis of chrna1, chrnb1, chrnd, and rapsn genes in multiple pterygium syndrome/fetal akinesia patients. Am J Hum Genet. 2008;82:222–227. doi: 10.1016/j.ajhg.2007.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Morgan NV, Marton T, Maxwell S, Harrison BJ, Beeson D, Maher ER. Germline mutation in dok7 associated with fetal akinesia deformation sequence. J Med Genet. 2009;46:338–340. doi: 10.1136/jmg.2008.065425. [DOI] [PubMed] [Google Scholar]
- Vogt J, Morgan NV, Rehal P, Faivre L, Brueton LA, Becker K, Fryns JP, Holder S, Islam L, Kivuva E, Lynch SA, Touraine R, Wilson LC, MacDonald F, Maher ER. Chrng genotype-phenotype correlations in the multiple pterygium syndromes. J Med Genet. 2012;49:21–26. doi: 10.1136/jmedgenet-2011-100378. [DOI] [PubMed] [Google Scholar]