

Abstract
Purpose
Resistance to vascular endothelial growth factor receptor (VEGFR) inhibitors is a major obstacle in the treatment of non-small cell lung cancer (NSCLC). We investigated the cellular mechanisms mediating resistance of NSCLCs to VEGFR tyrosine kinase inhibitors.
Experimental Design
We generated murine models of human NSCLC and performed targeted inhibition studies with the VEGFR TKIs cediranib and vandetanib. We used species-specific hybridization of microarrays to compare cancer (human) and stromal (mouse) cell transcriptomes of TKI-sensitive and -resistant tumors. We measured tumor microvascular density and vessel tortuosity to characterize the effects of therapy on the tumor vascular bed. Circulating cytokine and angiogenic factor levels in patients enrolled in VEGFR TKI trials were correlated with clinical outcomes.
Results
Murine xenograft models of human lung adenocarcinoma were initially sensitive to VEGFR TKIs, but developed resistance to treatment. Species-specific microarray analysis identified increased expression of stromal-derived hepatocyte growth factor (HGF) as a candidate mediator of TKI resistance and its receptor, c-MET, was activated in cancer cells and tumor-associated stroma. A transient increase in hypoxia-regulated molecules in the initial response phase was followed by adaptive changes resulting in a more tortuous vasculature. Forced HGF expression in cancer cells reduced tumor sensitivity to VEGFR TKIs and produced tumors with tortuous blood vessels. Dual VEGFR/c-MET signaling inhibition delayed the onset of the resistant phenotype and prevented the vascular morphology alterations. In cancer patients receiving VEGFR TKIs, high pretreatment HGF plasma levels correlated with poorer survival.
Conclusions
HGF/c-MET pathway mediates VEGFR inhibitor-resistance and vascular remodeling in NSCLC.
Keywords: angiogenesis, NSCLC models, VEGFR TKIs, therapeutic resistance
Introduction
Most efforts to inhibit angiogenesis as a means to control tumor growth have focused on the vascular endothelial growth factor/receptor (VEGF/R) signaling pathway (1-3). Bevacizumab (BV), a recombinant humanized monoclonal antibody that binds to circulating VEGF and inhibits its function, prolongs both progression-free survival (PFS) and overall survival (OS) of patients with advanced/metastatic non-small cell lung cancer (NSCLC) when it is administered in combination with chemotherapy (4). However, the clinical benefits of anti-VEGF therapies in NSCLC are modest and many lung cancers are either intrinsically refractory to therapy or eventually acquire resistance following continued administration (5-7). Therapeutic resistance is also a limitation of VEGFR tyrosine kinase inhibitors (TKIs), which target the catalytic domain of VEGFR. Indeed, VEGFR TKIs have not demonstrated an improvement in OS either as monotherapy (8, 9) or in combination with chemotherapy (10-12) in NSCLC patients. Previous investigations led to the identification of several mechanisms that allow tumors to progress on therapies that target the VEGF protein (13-15). More recent studies have concentrated on an improved understanding of the mechanisms underlying tumor resistance to VEGFR TKIs in order to develop more effective anti-angiogenic therapies.
Accumulating evidence indicates that both cancer cells and stromal cells play a significant role in mediating therapeutic resistance to agents that target the tumor vasculature (16-23). Anti-VEGF therapies are known to reduce oxygen delivery to tumors and the resulting hypoxia stimulates cancer cells and stromal cells to increase their expression of alternative angiogenic proteins (24). Previously, we demonstrated that acquired resistance of experimental murine models of human NSCLC to BV was due, in large part, to upregulation of epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor (FGFR) on tumor-associated stromal cells (13, 25). Antibody blockade of VEGFR1 and VEGFR2 signaling in experimental pancreatic islet tumors also prompted cancer cells and tumor-associated endothelial cells to increase their synthesis and secretion of FGF-related proteins (21). In our studies, we also noted that acquired resistance to BV was associated with a normalized revascularization phenotype in which tumor blood vessels were covered with pericytes that expressed activated EGFR. Moreover, we found that dual EGFR/VEGF pathway inhibition significantly delayed emergence of resistant phenotype. However, less information is known regarding the cellular and molecular mechanisms that mediate the resistance of NSCLC to VEGFR TKIs.
To begin to address this issue, herein we examined the efficacy of two VEGFR TKIs, cediranib (CED; AZD2171, AstraZeneca) (26) and vandetanib (VAN; ZD6474, Caprelsa™, AstraZeneca) (27), in murine models of human NSCLC. We applied species-specific gene expression profiling analysis to NSCLC tumors that were sensitive and resistant to TKIs in order to identify the origin of candidate signaling pathways that may be responsible for the drug-tolerant phenotype. We validated the findings in our models and then assessed their contribution to the human disease in patients with NSCLC and metastatic renal cell carcinoma (mRCC) that were enrolled in clinical trials evaluating VEGFR TKIs.
Materials and Methods
In Vivo Studies
Male athymic nude mice (NCI-nu) were purchased from the National Cancer Institute (NCI). The mice were maintained in facilities approved by the American Association for Accreditation of Laboratory Care (AAALAC) and according to institutional guidelines. H1975 and A549 NSCLC cells (2×106 cells) were injected subcutaneously (sc) into 6-week-old male mice and when tumor size reached ∼270 mm3 animals were randomized to the following treatment groups: vehicle [phosphate buffered saline; PBS] orally (po) daily; VAN 50 mg/kg po daily; or CED 6 mg/kg po daily. Parallel short-term treatment studies (14 days) were conducted in H1975 tumors when the tumor volume average reached ∼350 mm3. The tumor volume average in treated versus control group calculated at the final tumor evaluation was used as index to assess the antitumor activity of CED and VAN. Tumors were considered resistant when their volume tripled when compared to pretreatment volume and PFS was defined as the time from treatment initiation until the tumor volume tripled. Animals were euthanized when tumor burden was reached (progression) or for morbidity, according to the animal's protocol guidelines. We defined the progression vehicle control as the vehicle (PBS)-treated animals that are the control group for the long-term treatment experiment and sensitive vehicle control as the vehicle treated animals for the short-term experiment (14 days). Tumors were excised and processed for immunostaining studies and a portion of each tumor was also snap-frozen in liquid nitrogen. Hematoxylin and Eosin staining was used to confirm the presence of tumor.
Orthotopic lung tumors were produced by implanting H441 lung adenocarcinoma cells (1 × 106) in 50 μl HBSS containing 50 μg growth factor–reduced Matrigel (BD Bioscience) into the left lungs of 6-week-old nude mice, as previously described (13, 28). We euthanized eight mice three weeks after implanting the cancer cells in order to determine the kinetics of tumor growth. Animals were treated with either vehicle (PBS) 100 ul po daily or VAN 50 mg/kg po daily and euthanized when moribund.
For HGF stable transfection in vivo studies, HCC827-vector, -HGF.20, H1975-vector, or -HGF.24 cancer cells (2×106 cells) were implanted sc into 6-week-old male mice. Treatment was initiated when tumor volumes reached ∼300 mm3. Cabozantinib (XL184) 30 mg/kg and BV 10 mg/kg were administered po daily and into peritoneal space (ip) twice a week, respectively. Control mice were treated with PBS administered po daily and ip twice weekly. PFS was defined as time from treatment initiation to tumor volume doubling.
Gene Expression Profiling: Sample Preparation and Analysis
Total RNA was extracted from snap-frozen tissues using mirVana™ miRNA Isolation Kit (Ambion), according to manufacturer's protocol. Biotin-labeled cRNA samples for hybridization were prepared using the Illumina Total Prep RNA amplification kit (Ambion). One microgram of total RNA was used for the synthesis of cDNA, followed by amplification and biotin labeling. Next, 1.5 μg of biotinylated cRNAs were hybridized to human WG-6 v3.0 and mouse WG-6 v2.0 Expression BeadChips (Illumina) for analysis of tumor (human) and stromal (mouse) transcriptomes, respectively. Previous testing of the human and murine microarray platforms determined that they provide sufficient specificity to discriminate expression of mouse and human genes when mRNA from both species is mixed together and that they maintain the necessary sensitivity to detect the expression of species-specific genes even when they only represent 10% of mRNA in the mixture (29). Signals were developed by Amersham fluorolink streptavidin-Cy3 (GE Health Care Bio-Sciences). Gene expression data were collected using the Illumina bead array reader confocal scanner (BeadStation 500GXDW). Quantile normalization was performed on the average signal intensity values of the probes. The normalized expression data were logarithm transformed (base 2) for further analysis. To reveal significant genes within all treatment groups, a linear regression model was fitted and specific contrasts were tested for significance for each probe using the R package limma (30). The comparisons made in our study were: CED-resistant vs. CED-sensitive tumors (“CED prog. vs. CED sens.”) and VAN-resistant vs. VAN-sensitive tumors (“VAN prog. vs. VAN sens.”) for both human and mouse samples. To determine significance, a beta-uniform model was applied to adjust for multiple comparisons (31). We chose a false discovery rate (FDR) of 0.1 to identify any genes that were significantly modulated. Comparisons between specific treatment groups were performed using the same FDR, with an additional fold change cutoff (>1.5-fold). Finally, we applied the method to specific gene lists consisting of genes known to be associated with angiogenesis, hypoxia, and lymphangiogenesis (32). The gene expression data are deposited in GEO-NCBI database under the accession number GSE64472.
Phase II and Phase III Study Designs and Plasma Analysis
In this retrospective analysis, we obtained data from three multicenter clinical trials. The first study was a phase II randomized clinical study evaluating VAN alone, carboplatin and paclitaxel, or the combination of VAN plus carboplatin and paclitaxel in patients with advanced/metastatic NSCLC in the first line setting (12). The second study was a randomized study evaluating VAN or erlotinib in patients with refractory NSCLC (8). The third study consisted of an open-label phase 2 trial evaluating pazopanib in patients with metastatic renal cell carcinoma (RCC) (33, 34). Details and results of all three trials have been published previously. Clinical protocols and informed consent documents were approved by participating local institution's review boards and the trials were undertaken in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the amended Declaration of Helsinki. All patients provided written informed consent before study entry. Blood samples were collected prior to treatment, processed, stored and analyzed for HGF concentration as detailed (see Supplementary Materials and Methods for details).
Biostatistics and Standard Methods
Statistical and bioinformatics methods, reagents, cancer cells and cell culture conditions, quantitative real-time PCR, immunostaining, HGF stable transfection and vascular morphology analysis are described in Supplementary Materials and Methods.
Results
NSCLC Xenografts Acquire Resistance to VEGFR TKIs
We evaluated the efficacy of CED and VAN in NSCLC xenograft models. H1975 or A549 NSCLC tumor-bearing animals were treated with vehicle, CED, or VAN until mice were euthanized due to tumor burden (progression). The individual tumor growth curves of H1975 and A549 xenografts that received vehicle and CED are shown in Figure S1A and B, respectively. The individual tumor growth curves of vehicle and VAN treatment are shown in Figure S1A (H1975) and Figure S1B (A549) and in our prior published studies (13). After initial tumor shrinkage, three H1975 xenografts and two A549 xenografts acquired resistance to CED long-term treatment. In H1975 xenografts 2 animals acquired resistance to VAN after 148 days of treatment; in A549 xenografts 1 animal acquired resistance to VAN after 102 days of treatment (13). The grey arrows indicate the resistant xenograft-bearing animals and when they were euthanized due to tumor burden. We obtained tumor tissues from xenograft-bearing animals that were responding to therapy by treating H1975 tumors with CED or VAN for a period of 14 days (short-term treatment). Animals were euthanized after 2 weeks of treatment or earlier, if necessary, due to tumor burden or morbidity. H1975 xenografts treated with CED or VAN were smaller than controls at 14 days (CED induced an 84% tumor growth reduction vs. vehicle; VAN induced a 69% tumor growth reduction vs. vehicle, Figure S1C). The control group has been previously shown (13). These findings suggest that the response of H1975 and A549 NSCLC xenografts model the acquired resistance that takes place in the clinic in which tumors are initially sensitive to VEGFR inhibition, but develop resistance following prolonged treatment.
Angiogenic Genes are Upregulated in Stromal Cell and Cancer Cell Compartments in VEGFR TKI-resistant NSCLC Xenografts
To identify candidate angiogenic factors that may mediate acquired resistance to VEGFR TKIs, we performed species-specific (human: cancer cells; mouse: tumor stroma) gene expression profiling on CED- and VAN-sensitive H1975 tumors (short-term treatment) and to tumors that progressed after prolonged treatment. This analysis revealed that seven stromal (mouse) angiogenic genes were significantly modulated in both VAN- and CED-resistant tumors when compared with VAN- and CED-sensitive tumors (Figure 1A). The heatmaps depicting the expression of these seven commonly modulated stromal angiogenic genes are shown in Figures 1B and C. In tumors that progressed while receiving VAN, stromal Rps7, Eng, Nos3, Hgf, and Cxcl1 genes were upregulated, and Rpl37 and Npy genes were downregulated (Figure 1B, Table S1). In tumors that progressed while receiving CED, stromal Hgf, Cxcl1, Nos3, and Eng genes were upregulated, whereas Rps7, Npy, and Rpl37 genes were downregulated (Figure 1C, Table S2).
Figure 1. VEGFR TKI resistance is associated with increased stromal HGF in H1975 xenografts.
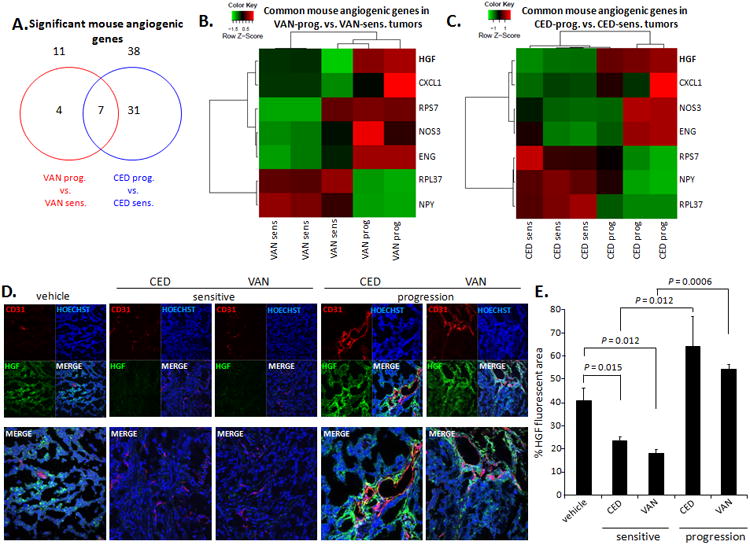
A. Venn diagram of stromal (mouse) angiogenic genes significantly modulated in H1975 tumors with acquired resistance (prog) to VAN (red) or CED (blue) versus VAN- or CED-sensitive tumors (14 days of treatment). B, C. Heatmaps showing 7 common stromal angiogenic genes significantly modulated in both (B) H1975 VAN–prog. vs. –sens. tumors and (C) H1975 CED–prog. vs. –sens. tumors. When there are more than one significant probe for a gene, the mean of the expression values across the probes were used for the heat maps in B, C. D. Representative immunofluorescent staining for CD31 (red), HGF (green), and DAPI (blue) in confocal microscopy (×200) of H1975 tumors treated with vehicle, CED, or VAN for 14 days (sensitive) or until progression; n=2-4 samples/group. E. Quantification of HGF+ staining (% HGF-fluorescent area: green/blue fluorescence) in H1975 tumors (≥5 microscopic fields/sample) treated with vehicle, CED, or VAN. Data graphed as percentage ± SEM. P values are from t test. In panels D and E, vehicle-treated samples were pooled for this analysis from both vehicle short-term and long-term groups.
Fourteen cancer cell (human) angiogenic genes were differentially expressed and shared by tumors that progressed while receiving VAN or CED treatments compared with VAN- and CED-sensitive tumors (Figure S2 A-C, Tables S3 and S4). This group of shared angiogenic genes included IL6 and HIF3A. HIF3A encodes an adaptor protein in transforming growth factor beta signaling modulation of angiogenesis and cell cycle progression, which were upregulated in both sets of resistant tumors. CASP1, an enzyme and member of the caspase family that plays a role in apoptosis, was downregulated in VAN- and CED-resistant tumors.
HGF/c-MET Axis is Overexpressed and Activated in NSCLC Murine Models of VEGFR TKI Resistance
Stromal HGF gene expression was upregulated in both VAN- and CED-resistant tumors and other investigators have reported alterations in HGF signaling in tumors resistant to antiangiogenic agents (35). Therefore, we performed additional experiments to determine the contribution of HGF to VEGFR resistance in our models. First, we validated HGF and c-MET at the protein level in H1975 tumors by immunostaining. The HGF staining area was significantly smaller in CED- or VAN-sensitive tumors compared with control tumors; however, HGF expression increased dramatically in tumors that acquired resistance to CED and VAN treatments (Figures 1D and E). Total c-MET expression levels were significantly increased in resistant tumors when compared to CED- or VAN- sensitive tumors (Figures 2A and B). We observed higher expression of the activated form of c-MET (p-MET) in VAN-resistant vs. VAN-sensitive tumors in both cancer cell and stromal compartments (Figure S3). We also noted that p-MET expression localized to tumor-associated endothelium of H1975 VAN-resistant tumors, but not to endothelial cells of VAN-sensitive and control tumors (Figure 2C). These findings indicate that HGF/c-MET pathway is upregulated in both cancer cells and stromal cells of VEGFR TKI resistant tumors.
Figure 2. c-MET is overexpressed and activated in VEGFR TKI-resistant NSCLC murine models.
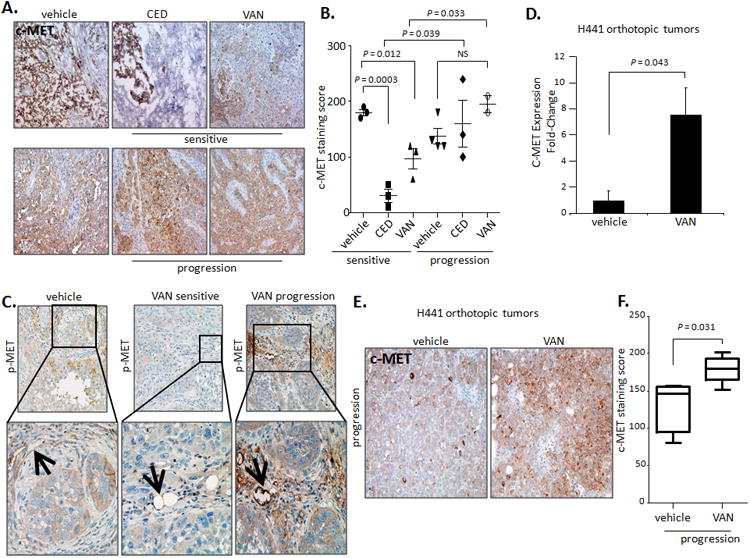
A. Representative IHC staining (×200) and B. Quantification of total c-MET in H1975 tumors treated with vehicle, CED, or VAN. Data graphed as mean score (overall intensity) ± SEM. C. Representative IHC images (×200 top; ×400 bottom) for phosphorylated c-MET (p-MET) in H1975 tumors treated with vehicle (n=3), VAN for 14 days (n=3), or until progression (n=2). At least 3 photomicrographs were collected per sample. Data graphed as mean score ± SEM. P values are from t test. The black arrows indicate the localization of positive p-MET staining on tumor-associated endothelium. D. Quantitative reverse-transcriptase PCR shows human c-MET mRNA expression levels in H441 orthotopic tumors treated with vehicle or VAN until progression (n=4/group). Data are normalized relative to mRNA levels in vehicle-treated samples and graphed as mRNA relative levels ± SEM. P values are from t test. E. Representative IHC staining (×100) for c-MET in H441 orthotopic tumors treated with vehicle or VAN until progression (≥5 photomicrographs/sample, n=4-5/group). F. Quantification of the c-MET immunostaining in H441 tumors treated with vehicle or VAN until progression (≥3 core fields/sample quantified). Data shown as mean score ± SEM. P values are from t test.
Given that the organ microenvironment plays a significant role in determining a cancer cells responsiveness to treatment (5), we examined the contribution of c-MET signaling to the VEGFR TKI resistant phenotype in an orthotopic model of lung adenocarcinoma (13). We analyzed H441 orthotopic tumors that were treated with either vehicle or VAN until progression and found that cancer cell (human) c-MET mRNA expression levels, as well as total c-MET staining area, significantly increased in VAN-resistant tumors compared with controls (Figure 2 D-F).
VEGFR-Inhibitor Resistance is HGF-Dependent
To determine whether HGF/c-MET signaling is sufficient to confer resistance of NSCLC to the VEGFR TKIs, we transfected HCC827 and H1975 human lung adenocarcinoma cells (36) with the human HGF gene (Figure S4A and B). The resulting HCC827-vector, H1975-vector, and HGF-overexpressing (HCC827-HGF.20 and H1975-HGF.24) cells were implanted into nude mice and once tumors were established, the animals were treated with VEGF/R inhibitors. In HCC827-vector and –HGF.20 xenografts, CED decreased tumor growth after 28 days of treatment compared with vehicle (CED vs. vehicle -vector P = 0.057; CED vs. vehicle -HGF.20 P = 0.015); however, reduced sensitivity to therapy was noted in tumors overexpressing HGF and treated with both CED and BV compared with vector-controls receiving the same treatments (CED HGF.20 vs. CED vector-control; P = 0.023; BV HGF.20 vs. BV vector-control P = 0.035, Figure 3A). These results indicate that high HGF expression may render tumors less sensitive to VEGF/R pathway inhibitors.
Figure 3. Ectopic HGF overexpression reduces sensitivity of NSCLC xenografts to VEGF/R inhibitors and dual VEGFR2/c-MET blockade prolongs PFS of NSCLC murine models.
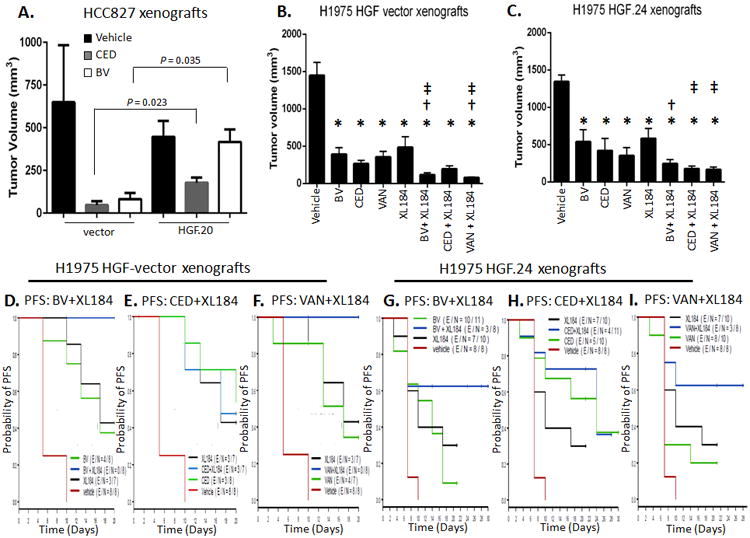
A. Mean tumor volume differences (± SEM) following BV and CED 28-day treatment in HCC827-vector and -HGF.20 xenografts (at 28 day point: vector xenografts n=3-4/group; HGF.20 xenografts: n=5/group). P is from Mann-Whitney test. B, C. Tumor growth inhibition following 17 days of treatment with BV, CED, VAN, or XL184 alone or in combination in (B) H1975-vector (n=4-7/group) and (C) -HGF.24 xenografts (n=7-10/group). Data graphed as mean tumor volume at day 17 of treatment ± SEM. * P < 0.05 in the indicated treatment group vs. vehicle; † P ≤ 0.05 in BV + XL184 vs. BV and in VAN + XL184 vs. VAN in vectors, and in BV + XL184 vs. BV in HGF.24 xenografts; ‡ P < 0.05 in BV + XL184 vs. XL184, CED + XL184 vs. XL184 and VAN + XL184 vs. XL184. P values are from Mann-Whitney test. D-I. Kaplan-Meier curves show probability of PFS (time to tumor doubling) of mice bearing H1975 vector control tumors (D, E and F) or H1975-HGF.24 overexpressor tumors (G-I) treated with vehicle, BV, CED, VAN, and cabozantinib (XL184) or combination treatments. H1975 vector xenografts: overall P < 0.0001; BV + XL184 vs. BV, P = 0.015 (D); BV + XL184 vs. XL184, P 0.024 (D); CED + XL184 vs. CED, P = 0.73 (E); CED + XL184 vs. XL184, P = 0.95 (E); VAN + XL184 vs. VAN, P = 0.017 (F); VAN + XL184 vs. XL184, P = 0.033 (F). H1975 HGF.24 xenografts: overall P = 0.004; BV + XL184 vs. BV, P = 0.062 (G); BV + XL184 vs. XL184, P = 0.23 (G); CED + XL184 vs. CED P = 0.64 (H); CED + XL184 vs. XL184 P = 0.074 (H); VAN + XL184 vs. VAN P = 0.064 (I); VAN + XL184 vs. XL184, P = 0.19 (I). The P values are from log-rank test.
Combined VEGFR2/c-MET Targeting Inhibits Tumor Growth and Prolongs Survival in Murine Models of NSCLC
To determine the therapeutic efficacy of combined inhibition of VEGFR2 and c-MET signaling, we treated mice bearing established H1975-vector and -HGF.24 tumors with BV, CED, or VAN alone or in combination with cabozantinib (XL184), a c-MET and VEGFR2 TKI (37). H1975-vector xenografts were sensitive to BV, CED, VAN and XL184 treatments and the combinations BV + XL184 or VAN + XL184 had a more profound antitumor effect than each agent monotherapy (Figure 3B). Combination treatment with BV or VAN plus XL184 also significantly prolonged the median PFS compared with each agent alone (Figure 3D and F). H1975-HGF.24 tumors were relatively less sensitive to VEGFR inhibitors than vector controls (Figure 3C). XL184 combined with BV, CED, or VAN inhibited tumor growth of HGF.24 xenografts more than any single agent (Figure 3C). Combined treatments resulted in a trend to significantly longer PFS than mice receiving BV, VAN or XL184 alone (Figure 3G and I). Combination with CED plus XL184 was slightly more effective compared with each single agent in HGF-overexpressor tumors (Figure 3H) and compared with CED plus XL184 in HGF-vector tumors (Figure 3E). These findings suggest that dual VEGFR2/c-MET axis blockade delays tumor growth of NSCLCs that overexpress HGF.
HGF Overexpression Increases Vessel Tortuosity and Combined VEGFR2/c-MET Targeting Normalizes the Tumor-Associated Vasculature in NSCLC Xenografts
To determine the effects of therapy on the tumor vascular bed, we measured the microvascular density (MVD) of VEGFR TKI-treated H1975 tumors and determined that MVD was significantly reduced in CED- and VAN-sensitive tumors (Figure 4A and B, left). However, the MVD of resistant tumors (progression) actually increased compared with vehicle-treated and VEGFR TKI-sensitive tumors (Figure 4B, right) and the blood vessels perfusing the resistant tumors were more tortuous and disorganized (Figure 4C).
Figure 4. Tumor vasculature morphology is altered in NSCLC xenograft models that are sensitive or resistant to VEGFR TKIs.
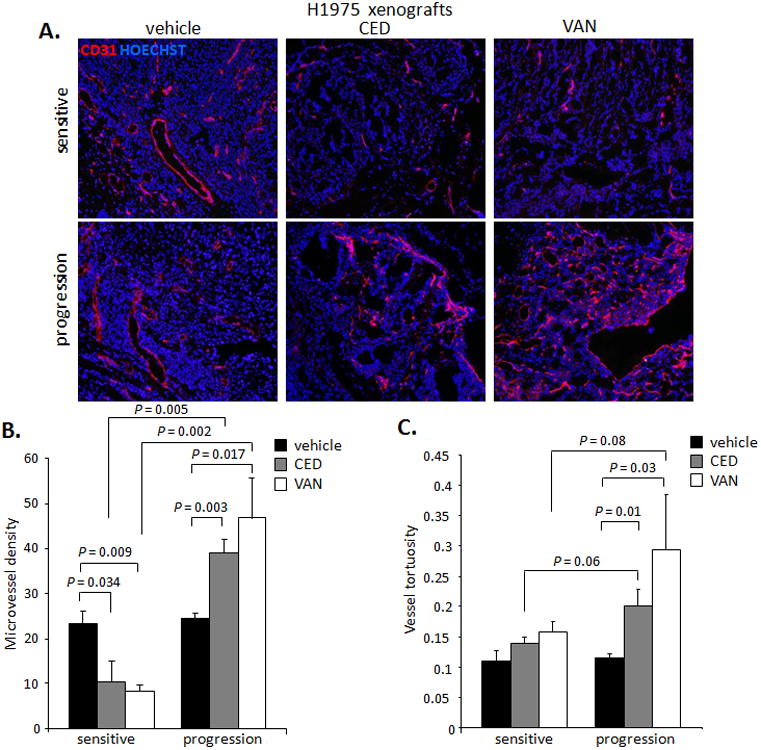
A. Representative photomicrographs (×100) of CD31+ tumor vessels (red) in H1975 xenografts treated with vehicle, CED, or VAN for 14 days or until progression (≥5 microscopic fields/sample). B, C. Quantification of MVD (B) and vessel tortuosity (C) based on CD31-stained tumor sections (≥5 microscopic fields/sample at ×200 for MVD and ≥5 microphotographs/sample at ×100 for vessel tortuosity) in H1975 xenografts treated with vehicle, CED, or VAN for 14 days or until progression (n=2-4/group). Data shown as mean ± SEM. P values are from a t test.
Next, we determined the effect of HGF on tumor vessels by comparing the tumor vasculature patterns of H1975-HGF.24 tumors and vector controls. There were no significant differences in the MVD between vehicle-treated H1975-vector tumors and HGF.24 tumors (Figure 5A and B). However, we noted that the vessel tortuosity was significantly increased in vehicle-treated HGF-overexpressing tumors (P < 0.05; Figure 5C). In H1975-vector xenografts, the administration of CED, VAN, XL184, and VAN + XL184 treatments significantly decreased MVD compared with vehicle (P < 0.05; Figure 5D, left), whereas in vehicle-treated HGF.24 xenografts no MVD changes were observed. Both CED and VAN treatments induced revascularization in HGF-overexpressing tumors (Figure 5D, right). Dual VEGFR2/c-MET inhibition with XL184 or CED + XL184 reduced the MVD in HGF.24 xenografts compared with CED or VAN alone (P < 0.05, Figure 5D). Vessels supplying vehicle-treated HGF.24 xenografts were more tortuous and disorganized than the blood vessels of vehicle-treated vector xenografts (P = 0.01; Figure 5C and E), and c-MET blockade alone or in combination with VEGFR inhibition significantly reduced vessel tortuosity (P < 0.05; Figure 5E) of HGF overexpressing tumors.
Figure 5. Altered patterns of tumor vasculature are HGF-dependent in NSCLC models of VEGFR TKI resistance.
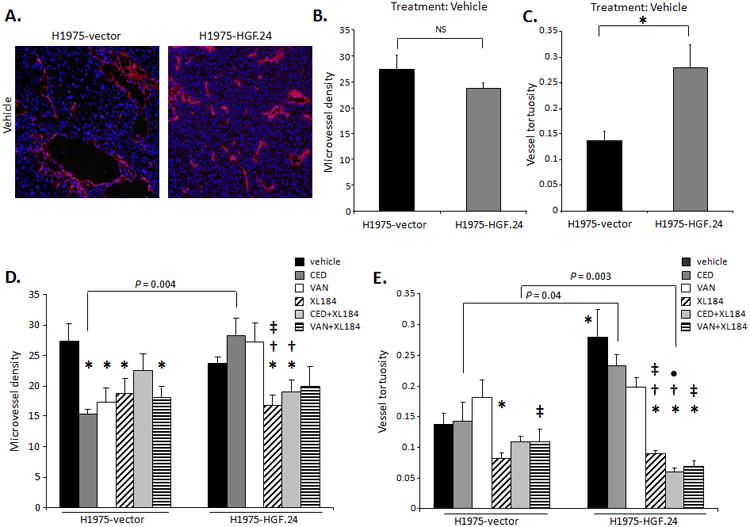
A. Representative IF staining (×100) of CD31+ (red) and nuclei (blue), B. MVD and C. vessel tortuosity quantification in H1975-vector and -HGF.24 xenografts treated with vehicle for 17 days, which was the time point by when all the animals in vehicle group had been euthanized. D. MVD and E. vessel tortuosity quantification in H1975-vector and -HGF.24 xenografts treated with vehicle, CED, VAN, or XL184 alone or in combination. In all panels data shown as mean ± SEM. P values are from t test. In HGF-vectors: *P < 0.05 in indicated group vs. vehicle; †P < 0.05 in indicated group vs. CED; ‡P < 0.05 in indicated group vs. VAN. In HGF.24 xenografts: *P < 0.05 in vehicle vs. HGF-vector vehicle group, and in indicated group vs. vehicle; †P < 0.05 in indicated group vs. CED; ‡P < 0.05 in indicated group vs. VAN. •P < 0.05 in indicated group vs. XL184 (n=6-10/group).
Intratumoral Hypoxia Increases in NSCLC Tumors Following VEGFR Inhibition
The studies above indicate that administration of VEGFR TKIs significantly reduces the vascular surface area of H1975 tumors. To determine any functional consequences of the reduced MVD, we quantified levels of the hypoxic biomarker carbonic anhydrase IX (CAIX) in experimental H1975 tumors. We noted that CAIX levels significantly increased in H1975 tumors that were sensitive to VAN when compared with vehicle-treated tumors (P = 0.02; Figure S5A and B) and remained elevated once tumors acquired resistance to VAN treatment.
Collectively, these data suggest that short-term administration of VEGFR TKIs inhibits tumor angiogenesis in VEGFR TKI-sensitive tumors. The reduction in tumor microvascular surface area then leads to declining oxygen tensions in the tumor microenvironment resulting in upregulation and activation of proangiogenic pathways, such as the HGF/c-MET axis, that restores tumor progression.
High Baseline Circulating HGF Levels Are Associated with Poor Outcomes in Patients with NSCLC and metastatic Renal Cell Carcinoma (mRCC)
Next, we validated our preclinical findings in samples from patients with NSCLC and RCC enrolled in clinical trials evaluating VEGFR TKIs. We analyzed levels of circulating HGF in patients from three clinical studies and compared PFS among groups (8, 12, 33). The first study was a phase II randomized study of patients with advanced NSCLC treated with VAN alone, VAN plus carboplatin and paclitaxel (VCP), or CP alone (control group) (12). After controlling for sex and smoking status in all groups, we determined that elevated plasma HGF was predictive of reduced PFS benefit for VAN versus CP (P for interaction = 0.036, Figure 6A and B). Table S5 shows median PFS by treatment arm for patients with low and high baseline plasma HGF levels. Among patients who received VAN, low baseline plasma HGF levels were associated with superior PFS versus high baseline levels (hazard ratio [HR] = 1.437 per 2-fold increase, 95% confidence interval [CI] = 1.038-1.988, P = 0.029). However, among patients treated with CP alone, those with low baseline plasma HGF had equivalent or inferior PFS compared with patients with high plasma HGF levels (Figure 6A and B).
Figure 6. Plasma HGF levels predict poor outcome in NSCLC patients treated with VEGFR TKIs.
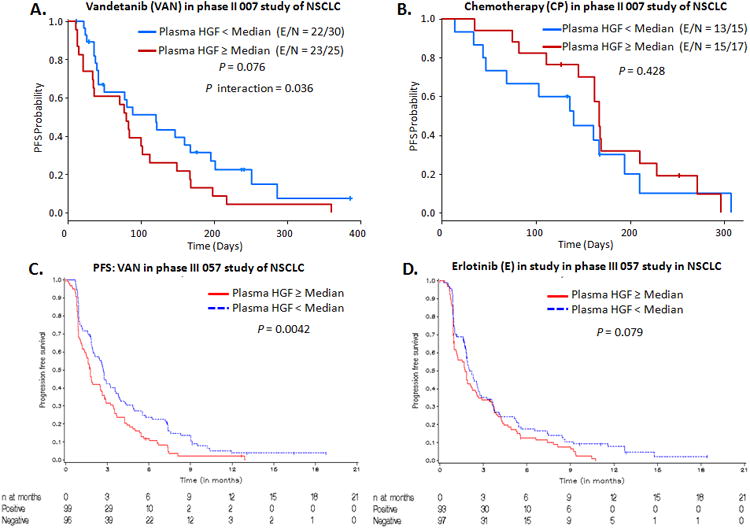
A, B. Kaplan-Meier (KM) plots showing PFS probability according to baseline plasma HGF levels in NSCLC patients treated with CP or VAN. The P interaction compares the risk of progression (HR) in patients with high plasma HGF levels between CP and VAN arms. C, D. KM plots showing PFS probability according to baseline plasma HGF levels in NSCLC patients treated with VAN or E in the phase III of NSCLC. CP: carboplatin and paclitaxel, E: erlotinib. P is from log-rank test.
The second study was a phase III trial evaluating the efficacy of VAN versus erlotinib (E) in unselected patients with advanced NSCLC who had progressed after receiving one to two prior chemotherapy regimens (8). We performed an exploratory analysis for 623 patients treated with VAN (300 mg po daily) and 617 patients treated with E (150 mg po daily) by HGF status; patients were defined as positive if their pretreatment (baseline) HGF levels were greater than or equal to the median value at baseline, negative if their pretreatment HGF levels did not meet the criteria for positive, and unknown if they did not have a baseline HGF value. Patients with positive HGF levels experienced slightly poorer outcomes in response to VAN vs. E treatment (median PFS: 7.7 vs. 7.6 weeks, respectively; events/number of patients: 93/99 vs. 88/93, HR 1.1 (95% CI 0.82, 1.49; Table S6). However, positive patients receiving VAN had a significantly poorer PFS compared with negative patients treated with VAN (median PFS: 7.7 vs. 11.7 weeks, respectively; events/number of patients: 93/99 vs. 88/96, P = 0.0042), as shown in Figure 6C and Table S6. No significant differences in PFS were noted in patients receiving E by HGF status (P = 0.079; Figure 6D). These findings confirm the predictive role of HGF as biomarker of resistance to treatment with VAN in NSCLC.
We then questioned whether our findings could be applied to patients treated with VEGFR inhibitors in other malignancies. In a separate open-label, single-arm phase II study (VEG102616, NTC00244764), pazopanib (Votrient, GlaxoSmithKline), a multi VEGFR TKI, had significant clinical activity in mRCC patients, demonstrating an overall RR of 35%, a disease control rate (ORs + SD) of 80%, and median PFS of 52 weeks (33). Analysis of pretreatment plasma samples revealed that low (relative to median) plasma HGF levels correlated with increased tumor shrinkage (34). Cox regression analysis demonstrated a significant association between low baseline plasma HGF and increased PFS (low plasma HGF levels [n = 108], median PFS = 53 weeks; high plasma HGF levels [n = 108], median PFS = 28 weeks; P = 0.016; Figure S6). These results indicate that HGF has a role of predictive biomarker of resistance to treatment with VEGFR TKIs in solid tumors other than NSCLC.
Discussion
Biologic agents targeting the VEGF pathway have produced significant clinical benefit in some cancers, including those that originate in the lung. However, therapeutic responses are usually short-lived and resistance inevitably emerges. Several investigations concluded that both cancer cells and tumor-associated stromal cells can play critical roles in determining the sensitivity of a tumor to therapy (5, 13, 22, 25, 38). Nevertheless, elucidating the molecular mechanisms underlying NSCLC progression and its resistance to anti-angiogenesis therapies has proved challenging. NSCLC is an extremely heterogeneous disease (39) and the fact that there are several classes of anti-angiogenic therapies, each possessing a different mechanism of action, adds to the complexity. Recently, we identified the mechanisms that mediate resistance of experimental NSCLC tumors to BV. However, less information is known regarding the mechanisms involved in the evolution of tumor resistance to therapies that bind to catalytic site of VEGFR. Here, we show that continued administration of the VEGFR TKIs CED and VAN to mice harboring established human lung cancers results in upregulation and activation of HGF/c-MET signaling networks in both the cancer cell and stromal cell compartments, which allows tumors to become refractory to therapy after an initial phase of response. Target cells for the stromal-derived HGF include the tumor-associated endothelial cells, which respond to the HGF signal by forming tortuous vascular networks. Combined inhibition of VEGFR and HGF receptor pathways prevents the vascular alterations and, moreover, delays the onset of therapeutic resistance in our models. In addition, our clinical analyses suggest that HGF is a predictive biomarker of VEGFR TKI resistance in that cancer patients with low baseline plasma concentrations of HGF are more likely to benefit from VEGFR TKIs compared with patients with high HGF concentrations, who have a poorer PFS in multiple clinical trials.
We were able to identify stromal-derived HGF as a mediator of VEGFR TKI resistance by performing cross species-specific hybridization of microarrays on VEGFR TKI-treated tumors and comparing the patterns of gene expression of tumors that were sensitive to short-term VEGFR TKI treatment with tumors that were resistant to prolonged therapy. We elected to use 14 days as our time point for short-term treatment because H1975 tumors were responding to therapy at this time, but were still sufficiently large enough for analyses. Time-matched comparisons between resistant and responding tumors were prohibited because the sensitive tumors were too small to characterize and, in some cases, not detectable. In addition, the emergence of the resistant phenotype was sporadic and this prevented us from selecting one set time point for comparisons of resistant and sensitive tumors.
HGF is a plasminogen-like protein that mediates its effects by binding to the receptor tyrosine kinase encoded by the MET proto-oncogene (40, 41), referred to as c-MET, or hepatocyte growth factor receptor. c-MET is preferentially expressed in epithelial tissues and its phosphorylation activates transduction of a number of intracellular signaling pathways, including RAS/RAF, PI3K/AKT, and STAT3 (42, 43). Thus, phosphorylation of c-MET can activate programs that signal for cell survival, cell mobility, invasion, angiogenesis, epithelial-to-mesenchymal transition (EMT) (44), and metastasis (45). HGF and c-MET are also localized at sites of pathological angiogenesis and are a potent endothelial mitogens (46-48). Indeed, we found that tumors treated with short-term VEGFR TKIs had reduced levels of c-MET compared with sensitive tumors, while c-MET was upregulated in tumors that progressed on long-term treatment. Ligand-induced activation of receptor tyrosine kinases (RTKs), including VEGFR1-3, PDGFR-β, RET and EGFR, has been shown to increase HIF-1α expression in neuroblastoma models, and pharmacological inhibition of RTK activity can abrogate HIF-1α expression in cancer cells (49). We have shown that EGFR-mutant NSCLC cells, such as H1975 cells, express higher levels of c-MET and p-MET when compared with EGFR wild-type cells, and that EGFR inhibition in EGFR-mutant NSCLC models reduces c-MET expression and activation through a HIF-1α-dependent mechanism (50). We recently extended this finding to show that VEGFR TKIs, including VAN and CED, can modulate HIF-1α and c-MET in other subsets of NSCLC (51). Hence, there may be two opposing influences on c-MET levels via HIF-1α pathway: a short-term decrease in RTK-driven HIF-1α and c-MET levels in tumor cells, and a longer-term upregulation of c-MET, potentially in response to tumor hypoxia induced by the antiangiogenic effect of the drugs. This is consistent with our prior publications using the same NSCLC models and studies from others showing eventual induction of hypoxia after antiangiogenic therapy administered over time (52). c-MET is also enriched on dividing endothelial cells and is a marker of the angiogenic phenotype (48). The tumor vascular bed of H1975 tumors that were treated with short-term TKI therapy was reduced in comparison to vehicle-treated tumors and this contributed, at least partially, to the reduction in total c-MET expression. As tumors are exposed to long-term RTK inhibition and acquire resistance, we observed an increase in tumor MVD and upregulation and activation of the HGF/c-MET axis, as compared with VEGFR TKI-sensitive tumors.
VEGF blockade has been shown to normalize the tumor vasculature by reducing interstitial pressure, thereby increasing perfusion, oxygenation, and drug delivery (53). However, there is some evidence that suggests these events are short-lived due to the excessive vessel pruning, which triggers hypoxia and prompts cancer cells to utilize alternative pathways for rapid tumor regrowth and rebound vascularization (54). Indeed, in other tumor models, antiangiogenic therapy was found to produce sustained hypoxia and impaired tumor vascular function, despite moderate vessel maturation (52). These findings support our observation that tumor hypoxia levels, as determined by CAIX staining on tumor tissues, increased in tumors that were sensitive to VEGFR TKIs compared with vehicle-treated tumors and remained elevated in the phase of acquired resistance to long-term anti-angiogenic therapy. The tumor blood vessels supporting the CED and VAN resistant tumors were significantly more tortuous than vessels supplying tumors that were sensitive to TKI therapy. This finding is in contrast to our earlier report on NSCLC tumors that become resistant to BV following a prolonged period of antibody administration had a less tortuous vasculature (13). Indeed, we found that tumors that had acquired resistance to BV had a normalized tumor vasculature in that the tumor-associated blood vessels were covered in pericytes.
In addition to its effects on tumor and microvascular endothelial cells, HGF/c-MET signaling has also been implicated as central regulator of the drug-tolerant phenotype. In melanoma, HGF was found to play a role in mediating resistance to BRAF inhibition by activating MAPK and PI3K/AKT signaling through the c-MET receptor (55). In that cell-based study, stromal cells were the source of HGF secretion and resistance could be overcome by dual targeting of RAF and either HGF or c-MET. In a similar type of study, MET amplification was found to activate ERBB3/PI3K/AKT signaling in EGFR mutant lung cancers and to enable the cancer cells to become resistance to EGFR TKIs (36). In another cell-based system, fibroblast-derived HGF was found to mediate EGFR TKI resistance in triple-negative breast cancer cells (56). Our observation that stromal HGF was upregulated in TKI-resistant tumors is in agreement with recent reports identifying the HGF/c-MET axis as an alternative mediator of resistance to sunitinib in preclinical tumor models (35). In our report, the strength of our technical approach using species-specific hybridization of microarrays on resistant and sensitive tumors lies in the ability to delineate whether the cancer cell or stromal cell compartment (or both) is being modified by the therapeutic intervention. Dual VEGFR/c-MET pathway blockade delayed resistance compared with single signaling inhibition and reduced vessel sprouting. In animal models of spontaneous pancreatic neuroendocrine tumors VEGF blockade promoted tumor invasion and metastasis, which could be inhibited by concurrent c-MET blockade (57). We acknowledge that as it is the case with other multityrosine kinase inhibitors, off-target effect of VAN and CED must be taken in consideration when interpreting the improved efficacy observed in our in vivo models with the combinations of VEGF/R inhibitors BV, CED or VAN plus XL184. However, our contention that the HGF/c-MET pathway contributes to VEGF/R inhibitor-resistance is supported by the use of the HGF overexpressor models and our clinical findings. Our findings suggest that hypoxia-induced upregulation of the stromal HGF/c-MET axis may promote tumor growth and vascular changes observed in VEGFR TKI-resistant tumors through signaling activation in stromal cells. These observations reinforce the notion that the tumor-associated stroma plays an important—and in some cases dominant—role in NSCLC models of resistance to angiogenesis inhibitors.
Finally, we assessed the clinical relevance of our preclinical findings in patients with advanced/metastatic and refractory NSCLC treated with VAN, and patients with metastatic RCC treated with pazopanib. Prior studies suggest that broad plasma CAF profiling may be used to identify prognostic and predictive markers in cancer patients (58-64). We measured serum levels in patients with advanced/metastatic and refractory NSCLC treated with VAN, and patients with metastatic RCC treated with the multi TKI pazopanib and identified a predictive role for circulating HGF as a marker of poor clinical benefit in mRCC and NSCLC patients treated with VEGFR TKIs in three independent clinical trials (8, 12, 33, 34). Furthermore, our group has previously reported that high pretreatment levels of circulating HGF are associated with shorter PFS compared with placebo (32·1 weeks vs. 13·0 weeks; HR 0·46 [0·32–0·67] P = 0.010) in a phase 3 randomized study evaluating pazopanib in metastatic mRCC (65).
Our data suggest that NSCLC patients with high circulating HGF levels may derive little benefit from the administration of VEGFR TKIs targeting the tumor microenvironment. These findings strengthen our preclinical results and lend credibility to our experimental approach for identifying mechanisms of resistance in NSCLC. Consistent with our preclinical observation, circulating HGF levels increase prior to disease progression in colorectal cancer patients treated with the combination of chemotherapy and anti-angiogenic therapy (66).
Taken together, our results indicate that HGF/c-MET signaling activation may represent a common mechanism of acquired resistance to VEGFR TKIs and may predict poor clinical outcome in patients treated with VEGFR inhibitors. As such, patients with high circulating HGF levels may derive poor relative benefit from VEGFR TKIs targeting the tumor microenvironment, in which both circulating host- and tumor-derived factors may affect therapeutic response. Dual VEGFR and HGF/c-MET targeting may represent a reasonable approach to improve patient outcomes.
Supplementary Material
Statement of Translational Relevance.
VEGFR inhibitors such as the TKIs vandetanib and cediranib have been shown to improve progression-free survival and response rates, respectively, in patients with NSCLC. However, most lung tumors are either indifferent to these agents or eventually acquire resistance following their continued administration. Here, we used species-specific transcriptomic profiling to identify cancer cell- and stromal-specific alterations associated with resistance to VEGFR TKIs. We identified the HGF/c-MET pathway as a mediator of resistance and vascular remodeling in xenograft models of human NSCLC. Dual VEGFR/c-MET pathway inhibition delayed the onset of the resistant phenotype and prevented resistance-associated sprouting revascularization. This study has direct clinical implications and suggests that combined VEGFR and c-MET blockade may provide superior therapeutic benefit for the treatment of NSCLC. Elevated circulating levels of HGF were associated with a poorer outcome in cancer patients treated with VEGFR TKIs from three separate clinical trials.
Acknowledgments
We gratefully acknowledge Donna Reynolds and Denise Wood for technical expertise, Emily B. Roarty, PhD, for expert assistance in the preparation of the manuscript, and Ericka Goodoff (University of Texas MD Anderson Cancer Center) for the scientific editing of the manuscript.
Financial Support: This research was funded by The University of Texas Lung SPORE P50 CA070907 (J.V.H.); NIH Cancer Center Support Grant P30 CA016672 (J.V.H.); The University of Texas MD Anderson Cancer Center Head and Neck SPORE 5 P50 CA097007 (J.V.H.); 1 R01 CA168484 (J.V.H.); the LUNGevity's Career Development Award for Translational Research (J.V.H.); the Bruton Endowed Chair in Tumor Biology (J.V.H.); research support from AstraZeneca (J.V.H.).
Footnotes
Disclosure of Potential Conflicts of Interest: J.V. Heymach has served on advisory boards for Genentech, GlaxoSmithKline, AstraZeneca, and received research support from AstraZeneca and GlaxoSmithKline. Y.Liu is a former employee of GlaxoSmithKline. A. J. Ryan and J. M. Jürgensmeier are former employees of AstraZeneca.
Authors' Contributions: T.C, M.B.N. and J.V.H conceived the project, designed experiments and wrote the manuscript; T.C., L.X, M.A.C, U.G., B.S. and Y.Y.P. performed experiments, data acquisition and data analysis; H.Y.L, W.L., H.T.T., Y.L., K.H., V.H., E.H., H.K., J.W. I.W., performed data analysis and manuscript editing; W.P., J.S.L., A.J.R., J.M.J., R.S.H., R.R.L., and J.J.L. provided scientific discussion and manuscript editing.
References
- 1.Kerbel RS. Tumor angiogenesis. The New England journal of medicine. 2008;358(19):2039–49. doi: 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 17(11):1359–70. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473(7347):298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. The New England journal of medicine. 2006;355(24):2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 5.Crawford Y, Ferrara N. Tumor and stromal pathways mediating refractoriness/resistance to anti-angiogenic therapies. Trends Pharmacol Sci. 2009;30(12):624–30. doi: 10.1016/j.tips.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. The New England journal of medicine. 2004;350(23):2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 7.Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. The New England journal of medicine. 2007;357(26):2666–76. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 8.Natale RB, Thongprasert S, Greco FA, Thomas M, Tsai CM, Sunpaweravong P, et al. Phase III trial of vandetanib compared with erlotinib in patients with previously treated advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(8):1059–66. doi: 10.1200/JCO.2010.28.5981. [DOI] [PubMed] [Google Scholar]
- 9.Lee JS, Hirsh V, Park K, Qin S, Blajman CR, Perng RP, et al. Vandetanib Versus placebo in patients with advanced non-small-cell lung cancer after prior therapy with an epidermal growth factor receptor tyrosine kinase inhibitor: a randomized, double-blind phase III trial (ZEPHYR) Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30(10):1114–21. doi: 10.1200/JCO.2011.36.1709. [DOI] [PubMed] [Google Scholar]
- 10.Herbst RS, Sun Y, Eberhardt WE, Germonpre P, Saijo N, Zhou C, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11(7):619–26. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Boer RH, Arrieta O, Yang CH, Gottfried M, Chan V, Raats J, et al. Vandetanib plus pemetrexed for the second-line treatment of advanced non-small-cell lung cancer: a randomized, double-blind phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29(8):1067–74. doi: 10.1200/JCO.2010.29.5717. [DOI] [PubMed] [Google Scholar]
- 12.Heymach JV, Paz-Ares L, De Braud F, Sebastian M, Stewart DJ, Eberhardt WE, et al. Randomized phase II study of vandetanib alone or with paclitaxel and carboplatin as first-line treatment for advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26(33):5407–15. doi: 10.1200/JCO.2008.17.3138. [DOI] [PubMed] [Google Scholar]
- 13.Cascone T, Herynk MH, Xu L, Du Z, Kadara H, Nilsson MB, et al. Upregulated stromal EGFR and vascular remodeling in mouse xenograft models of angiogenesis inhibitor-resistant human lung adenocarcinoma. The Journal of clinical investigation. 2011;121(4):1313–28. doi: 10.1172/JCI42405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McIntyre A, Patiar S, Wigfield S, Li JL, Ledaki I, Turley H, et al. Carbonic anhydrase IX promotes tumor growth and necrosis in vivo and inhibition enhances anti-VEGF therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(11):3100–11. doi: 10.1158/1078-0432.CCR-11-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17(11):1359–70. doi: 10.1038/nm.2537. [DOI] [PubMed] [Google Scholar]
- 16.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 8(4):210–21. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shojaei F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Cancer Lett. 320(2):130–7. doi: 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, et al. G-CSF-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-VEGF therapy in mouse models. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(16):6742–7. doi: 10.1073/pnas.0902280106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol. 2007;25(8):911–20. doi: 10.1038/nbt1323. [DOI] [PubMed] [Google Scholar]
- 20.Ebos JM, Lee CR, Kerbel RS. Tumor and host-mediated pathways of resistance and disease progression in response to antiangiogenic therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(16):5020–5. doi: 10.1158/1078-0432.CCR-09-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Casanovas O, Hicklin DJ, Bergers G, Hanahan D. Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell. 2005;8(4):299–309. doi: 10.1016/j.ccr.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 22.Chung AS, Wu X, Zhuang G, Ngu H, Kasman I, Zhang J, et al. An interleukin-17-mediated paracrine network promotes tumor resistance to anti-angiogenic therapy. Nat Med. 2013;19(9):1114–23. doi: 10.1038/nm.3291. [DOI] [PubMed] [Google Scholar]
- 23.Shojaei F, Zhong C, Wu X, Yu L, Ferrara N. Role of myeloid cells in tumor angiogenesis and growth. Trends Cell Biol. 2008;18(8):372–8. doi: 10.1016/j.tcb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 24.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15(3):232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casanovas O. The adaptive stroma joining the antiangiogenic resistance front. The Journal of clinical investigation. 2011;121(4):1244–7. doi: 10.1172/JCI46430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wedge SR, Kendrew J, Hennequin LF, Valentine PJ, Barry ST, Brave SR, et al. AZD2171: a highly potent, orally bioavailable, vascular endothelial growth factor receptor-2 tyrosine kinase inhibitor for the treatment of cancer. Cancer research. 2005;65(10):4389–400. doi: 10.1158/0008-5472.CAN-04-4409. [DOI] [PubMed] [Google Scholar]
- 27.Hennequin LF, Stokes ES, Thomas AP, Johnstone C, Ple PA, Ogilvie DJ, et al. Novel 4-anilinoquinazolines with C-7 basic side chains: design and structure activity relationship of a series of potent, orally active, VEGF receptor tyrosine kinase inhibitors. J Med Chem. 2002;45(6):1300–12. doi: 10.1021/jm011022e. [DOI] [PubMed] [Google Scholar]
- 28.Wu W, Onn A, Isobe T, Itasaka S, Langley RR, Shitani T, et al. Targeted therapy of orthotopic human lung cancer by combined vascular endothelial growth factor and epidermal growth factor receptor signaling blockade. Mol Cancer Ther. 2007;6(2):471–83. doi: 10.1158/1535-7163.MCT-06-0416. [DOI] [PubMed] [Google Scholar]
- 29.Park ES, Kim SJ, Kim SW, Yoon SL, Leem SH, Kim SB, et al. Cross-species hybridization of microarrays for studying tumor transcriptome of brain metastasis. Proceedings of the National Academy of Sciences of the United States of America. 108(42):17456–61. doi: 10.1073/pnas.1114210108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smyth GK. In: Limma: linear models for microarray data. Gentleman RCV, Dudoit S, Irizarry R, Huber W, editors. New York: Springer; 2005. [Google Scholar]
- 31.Pounds S, Morris SW. Estimating the occurrence of false positives and false negatives in microarray studies by approximating and partitioning the empirical distribution of p-values. Bioinformatics. 2003;19(10):1236–42. doi: 10.1093/bioinformatics/btg148. [DOI] [PubMed] [Google Scholar]
- 32.Van den Eynden GG, Van Laere SJ, Van der Auwera I, Gilles L, Burn JL, Colpaert C, et al. Differential expression of hypoxia and (lymph)angiogenesis-related genes at different metastatic sites in breast cancer. Clin Exp Metastasis. 2007;24(1):13–23. doi: 10.1007/s10585-006-9049-3. [DOI] [PubMed] [Google Scholar]
- 33.Hutson TE, Davis ID, Machiels JP, De Souza PL, Rottey S, Hong BF, et al. Efficacy and safety of pazopanib in patients with metastatic renal cell carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(3):475–80. doi: 10.1200/JCO.2008.21.6994. [DOI] [PubMed] [Google Scholar]
- 34.Tran HT, Liu Y, Zurita AJ, Lin Y, Baker-Neblett KL, Martin AM, et al. Prognostic or predictive plasma cytokines and angiogenic factors for patients treated with pazopanib for metastatic renal-cell cancer: a retrospective analysis of phase 2 and phase 3 trials. Lancet Oncol. 2012;13(8):827–37. doi: 10.1016/S1470-2045(12)70241-3. [DOI] [PubMed] [Google Scholar]
- 35.Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer research. 2010;70(24):10090–100. doi: 10.1158/0008-5472.CAN-10-0489. [DOI] [PubMed] [Google Scholar]
- 36.Turke AB, Zejnullahu K, Wu YL, Song Y, Dias-Santagata D, Lifshits E, et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer cell. 2010;17(1):77–88. doi: 10.1016/j.ccr.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Timar J, Dome B. Antiangiogenic drugs and tyrosine kinases. Anticancer Agents Med Chem. 2008;8(5):462–9. doi: 10.2174/187152008784533035. [DOI] [PubMed] [Google Scholar]
- 38.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nature reviews Clinical oncology. 2011;8(4):210–21. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levitan N, Dowlati A, Shina D, Craffey M, Mackay W, DeVore R, et al. Multi-institutional phase I/II trial of paclitaxel, cisplatin, and etoposide with concurrent radiation for limited-stage small-cell lung carcinoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2000;18(5):1102–9. doi: 10.1200/JCO.2000.18.5.1102. [DOI] [PubMed] [Google Scholar]
- 40.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science. 1991;251(4995):802–4. doi: 10.1126/science.1846706. [DOI] [PubMed] [Google Scholar]
- 41.Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene. 1991;6(4):501–4. [PubMed] [Google Scholar]
- 42.Galimi F, Brizzi MF, Comoglio PM. The hepatocyte growth factor and its receptor. Stem Cells. 1993;11(2):22–30. doi: 10.1002/stem.5530110805. [DOI] [PubMed] [Google Scholar]
- 43.Hecht M, Papoutsi M, Tran HD, Wilting J, Schweigerer L. Hepatocyte growth factor/c-Met signaling promotes the progression of experimental human neuroblastomas. Cancer research. 2004;64(17):6109–18. doi: 10.1158/0008-5472.CAN-04-1014. [DOI] [PubMed] [Google Scholar]
- 44.Birchmeier C, Birchmeier W, Gherardi E, Vande Woude GF. Met, metastasis, motility and more. Nat Rev Mol Cell Biol. 2003;4(12):915–25. doi: 10.1038/nrm1261. [DOI] [PubMed] [Google Scholar]
- 45.Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer. 12(2):89–103. doi: 10.1038/nrc3205. [DOI] [PubMed] [Google Scholar]
- 46.Rosen EM, Grant DS, Kleinman HK, Goldberg ID, Bhargava MM, Nickoloff BJ, et al. Scatter factor (hepatocyte growth factor) is a potent angiogenesis factor in vivo. Symp Soc Exp Biol. 1993;47:227–34. [PubMed] [Google Scholar]
- 47.Bussolino F, Di Renzo MF, Ziche M, Bocchietto E, Olivero M, Naldini L, et al. Hepatocyte growth factor is a potent angiogenic factor which stimulates endothelial cell motility and growth. J Cell Biol. 1992;119(3):629–41. doi: 10.1083/jcb.119.3.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ding S, Merkulova-Rainon T, Han ZC, Tobelem G. HGF receptor up-regulation contributes to the angiogenic phenotype of human endothelial cells and promotes angiogenesis in vitro. Blood. 2003;101(12):4816–22. doi: 10.1182/blood-2002-06-1731. [DOI] [PubMed] [Google Scholar]
- 49.Nilsson MB, Zage PE, Zeng L, Xu L, Cascone T, Wu HK, et al. Multiple receptor tyrosine kinases regulate HIF-1alpha and HIF-2alpha in normoxia and hypoxia in neuroblastoma: implications for antiangiogenic mechanisms of multikinase inhibitors. Oncogene. 2010;29(20):2938–49. doi: 10.1038/onc.2010.60. [DOI] [PubMed] [Google Scholar]
- 50.Xu L, Nilsson MB, Saintigny P, Cascone T, Herynk MH, Du Z, et al. Epidermal growth factor receptor regulates MET levels and invasiveness through hypoxia-inducible factor-1alpha in non-small cell lung cancer cells. Oncogene. 2010 doi: 10.1038/onc.2010.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nilsson MB, Giri U, Gudikote J, Tang X, Lu W, Tran H, et al. KDR Amplification Is Associated with VEGF-Induced Activation of the mTOR and Invasion Pathways but does not Predict Clinical Benefit to the VEGFR TKI Vandetanib. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016;22(8):1940–50. doi: 10.1158/1078-0432.CCR-15-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM, et al. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer research. 2006;66(7):3639–48. doi: 10.1158/0008-5472.CAN-05-3295. [DOI] [PubMed] [Google Scholar]
- 53.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nature medicine. 2001;7(9):987–9. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 54.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10(6):417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 55.Straussman R, Morikawa T, Shee K, Barzily-Rokni M, Qian ZR, Du J, et al. Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Nature. 2012;487(7408):500–4. doi: 10.1038/nature11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mueller KL, Madden JM, Zoratti GL, Kuperwasser C, List K, Boerner JL. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012;14(4):R104. doi: 10.1186/bcr3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer discovery. 2012;2(3):270–87. doi: 10.1158/2159-8290.CD-11-0240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nikolinakos PG, Altorki N, Yankelevitz D, Tran HT, Yan S, Rajagopalan D, et al. Plasma cytokine and angiogenic factor profiling identifies markers associated with tumor shrinkage in early-stage non-small cell lung cancer patients treated with pazopanib. Cancer research. 2010;70(6):2171–9. doi: 10.1158/0008-5472.CAN-09-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hanrahan EO, Lin HY, Kim ES, Yan S, Du DZ, McKee KS, et al. Distinct patterns of cytokine and angiogenic factor modulation and markers of benefit for vandetanib and/or chemotherapy in patients with non-small-cell lung cancer. J Clin Oncol. 2010;28(2):193–201. doi: 10.1200/JCO.2009.22.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanrahan EO, Ryan AJ, Mann H, Kennedy SJ, Langmuir P, Natale RB, et al. Baseline vascular endothelial growth factor concentration as a potential predictive marker of benefit from vandetanib in non-small cell lung cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(10):3600–9. doi: 10.1158/1078-0432.CCR-08-2568. [DOI] [PubMed] [Google Scholar]
- 61.Montero AJ, Diaz-Montero CM, Millikan RE, Liu J, Do KA, Hodges S, et al. Cytokines and angiogenic factors in patients with metastatic renal cell carcinoma treated with interferon-alpha: association of pretreatment serum levels with survival. Ann Oncol. 2009;20(10):1682–7. doi: 10.1093/annonc/mdp054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pena C, Lathia C, Shan M, Escudier B, Bukowski RM. Biomarkers predicting outcome in patients with advanced renal cell carcinoma: Results from sorafenib phase III Treatment Approaches in Renal Cancer Global Evaluation Trial. Clin Cancer Res. 2010;16(19):4853–63. doi: 10.1158/1078-0432.CCR-09-3343. [DOI] [PubMed] [Google Scholar]
- 63.Zurita AJ, Jonasch E, Wang X, Khajavi M, Yan S, Du DZ, et al. A cytokine and angiogenic factor (CAF) analysis in plasma for selection of sorafenib therapy in patients with metastatic renal cell carcinoma. Ann Oncol. 2012;23(1):46–52. doi: 10.1093/annonc/mdr047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heymach JV, Jonasch E, Wang X, Du DZ, Yan S, Xu L, et al. A cytokine and angiogenic factor (CAF) plasma signature for selection of sorafenib (SR) therapy in patients (pts) with metastatic renal cell carcinoma (mRCC) J Clin Oncol. 2009;27(suppl):15S. doi: 10.1093/annonc/mdr047. abstr 5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, et al. Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(6):1061–8. doi: 10.1200/JCO.2009.23.9764. [DOI] [PubMed] [Google Scholar]
- 66.Kopetz S, Hoff PM, Morris JS, Wolff RA, Eng C, Glover KY, et al. Phase II Trial of Infusional Fluorouracil, Irinotecan, and Bevacizumab for Metastatic Colorectal Cancer: Efficacy and Circulating Angiogenic Biomarkers Associated With Therapeutic Resistance. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(3):453–9. doi: 10.1200/JCO.2009.24.8252. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.