

Abstract
Conserved homology-1 (C1) domains are peripheral membrane domains that target their host proteins to diacylglycerol (DAG)-containing membranes. It has been previously shown that a conservative aromatic mutation of a single residue in C1 has a profound effect on DAG affinity. We report that the “DAG-toggling” mutation changes the conformational dynamics of the loop region that forms the binding site for the C1 activators. Moreover, there is a correlation between the residue identity at the mutation site, DAG affinity, and loop dynamics in four C1 variants. We propose that “toggling” of DAG affinity may occur through modulation of both protein-membrane interactions and the geometry of the activator binding cleft, with the loop dynamics being responsible for the latter.
Keywords: Conserved homology-1 domain, C1 domain, protein kinase C, diacylglycerol, diacylglycerol affinity, signal transduction, millisecond-to-microsecond dynamics, NMR spectroscopy, protein dynamics, relaxation dispersion
Graphical abstract
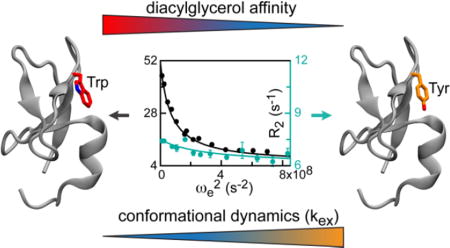
Diacylglycerol (DAG) is a membrane lipid component that serves both as a second messenger and an intermediate in lipid metabolism.1 All signaling proteins that translocate to membranes and directly bind DAG have one or more conserved homology-1 (C1) domains.2,3 C1 domains are “conditional” membrane modules that show weak membrane association in the ligand-free state, but bind membranes with high affinity in the presence of activators, such as DAG and non-endogenous hydrophobic phorbol esters. The folded core of C1 domains consists of 50 amino acids arranged in a treble-clef fold, with two structural Zn2+ sites (Fig. S1). The activators bind in the cleft between the two loop regions, denoted β12 and β34 (Figs. 1A and 3A).
Figure 1.

Slow but not fast dynamics of ligand binding loops in C1Bδ is altered by the W252Y mutation. (A) Sequence alignment of C1B domains from novel and conventional PKC isoforms of R. norvegicus. (B,C) Spectral density J(0.87ωH) (B) and R2,CPMG values (C) plotted against the primary structure of C1Bδ. The position of DAG-toggling mutation is shown with a vertical line.
Figure 3.

Trp252 sidechain is solvent-exposed. (A) Superposition of the apo (1UEJ, backbone:purple, sidechains:orange; only folded core shown) and phorbol 13-acetate-complexed (1PTR, backbone:silver, sidechains/ligand:gray) structures of C1Bδ. (B) Expansion of the loop region showing two ordered water molecules that take place of the phorbol ester oxygens in the apo C1Bδ. (C) CLEANEX-PM buildup curve for the Hε1 of the Trp252 sidechain. The results of two independent experiments are shown with open circles. Solid line is the fit to Eq. S9.
The affinity of C1 domains to DAG has a profound effect on the activation threshold of the host proteins and their sub-cellular localization.4 However, what specific features of C1 domains are responsible for their well-documented differential affinities to DAG5–7 is not well understood. In 2007, Newton’s laboratory discovered a “DAG-toggling” mutation that dramatically altered the affinity of C1 domains to DAG.8 The C1 domains in question belong to Protein Kinase C (PKC), a family of Ser/Thr kinases that regulate cell proliferation, differentiation, apoptosis, and motility. The mutation involves a conservative substitution of an aromatic residue at the hinge of the β34 ligand-binding loop (Fig. 1A). Having a Trp at this position resulted in high DAG affinity, while a Tyr resulted in low affinity. Neither the sidechain nor the backbone atoms of the residue at this position are in direct contact with the ligand.
We found that introducing the DAG-toggling mutation Y123W in the C1B domain from a conventional PKCα isoenzyme (C1Bα) significantly altered the microsecond-timescale dynamics of the loop region of the apo protein, while leaving the structure and sub-nanosecond dynamics essentially unperturbed.9 Here, we set out to determine whether or not the change in conformational plasticity of the ligand-binding site by the DAG-toggling mutation is a signature of a particular C1 domain, or is a general feature of C1 domains in PKC.
We chose to focus on the C1B domain from PKCδ (C1Bδ) because it differs from C1Bα in three important aspects. First, its host protein – the δ PKC isoenzyme – is quite distinct from the α in that it belongs to a class of novel PKC isoenzymes that are Ca2+-independent. Second, the wild-type (wt) C1Bδ is a high-affinity DAG domain with a native Trp at the DAG-toggling position; this is in contrast to wtC1Bα, which has a native Tyr and is a low-affinity DAG domain (Fig. 1A). Third, there are high-resolution crystal structures available for both apo and ligand-bound forms of wtC1Bδ.10,11 The DAG-toggling effect of the W252Y mutation has been demonstrated for C1Bδ in lipid bilayers8 and micelles.12
To determine how the fast (sub-nanosecond) dynamics of the backbone N-H groups is affected by the DAG-toggling W252Y mutation, we used NMR to measure three relaxation parameters: longitudinal (R1) and transverse (R2,CPMG) relaxation rate constants, and {1H}-15N nOe, for all spectrally resolved high signal-to-noise N-H cross-peaks. The relaxation parameters were interpreted using the reduced spectral density mapping formalism (see SI for details). The values of spectral density J(0.87×ωH), where ωH is the Larmor frequency of 1H, report on the nanosecond-timescale motions of the N-H groups. It is evident that the mutation does not significantly affect fast backbone motions of C1Bδ (Fig. 1B). In general, the J(0.87×ωH) values are fairly uniform across the primary structure. Slight elevation is observed in the C-terminal region, likely due to the dynamics of the Cys280-Zn2+ coordination bond.13
In contrast, conformational dynamics is significantly affected by the W252Y mutation. This is evident from the comparison of the R2,CPMG values of C1Bδ variants, which report on the ms-to-µs dynamics (Fig. 1C). There are significant differences in the R2,CPMG values of the β34 ligand-binding loop and its N- and C-terminal hinges. We also noted that the N-H cross-peaks for almost all residues that were broadened beyond detection in the wtC1Bδ reappeared in the spectra of the W252Y variant. This indicates that the DAG-toggling mutation W252Y changes the timescale of the chemical exchange process that affects the loops.
To quantitatively characterize the changes in the µs-timescale dynamics brought about by the W252Y mutation, we conducted a series of near- and off-resonance rotating-frame (R1ρ) 15N relaxation dispersion experiments. The relaxation dispersion curves were constructed by plotting the calculated transverse relaxation rate constants R2 against the squared effective field, ωe2. 15N nuclei whose electronic environment changes as a result of a chemical exchange process have characteristic dispersion curves, in which the R2 values decrease with increasing ωe2 (Figs. 2A–C and S2). The R2 values at ωe=0 represent the sum of the transverse relaxation rate constant in the absence of exchange, R20, and the contribution due to exchange, Rex. For both proteins, the data were fitted with a two-state fast exchange model, in which protein conformations A and B interconvert with rate constants k1 and k−1, . This model is parameterized with the exchange rate kex=k1+k−1, and Φex, which is a composite parameter that depends on the populations of the exchanging species, pA and pB, and on ΔωN, the 15N chemical shift difference between species A and B (see SI for the details).
Figure 2.

DAG-toggling mutation alters the dynamics of ligand binding loops of C1Bδ. (A–C) Representative 15N R1ρ relaxation dispersion curves at 14.1 T shown for three scenarios described in the text. Solid lines are the fits to Eqs. S5 and S7. (D) Rex values of the wt and W252Y mapped onto the 3D structure of C1Bδ (1PTQ) and color-coded according to their magnitude. N-H groups with quantifiable dispersion are represented with spheres. (E) Topology diagram of C1Bδ showing a subset of H-bonds (dotted lines) that connect β-strands and loop hinges and involve amide 1H (orange circles). H-bonds that involve slow-exchanging amide 1H are in black; H-bonds whose presence is not detectable by HX experiments are in red. Residues with quantifiable exchange are underlined.
We found that the W252Y mutation increases the rate of exchange two-fold: the kex values are 8100 ± 300 s−1 and 17600 ± 1400 s−1 for the wtC1Bδ and W252Y, respectively. When comparing the 15N dispersion curves of the wt and W252Y C1Bδ, we encountered three different scenarios (Fig. 2, A–C). In (A), both wt and W252Y show quantifiable dispersion albeit with different amplitudes, as is evident from the comparison of the scale of the left and right axes. In (B), only the W252Y variant data are available, because the corresponding residues in the wt are broadened beyond detection. In (C), the residues in the wt but not the W252Y have quantifiable dispersion. The differences between the dynamics of wt and W252Y are most evident when the Rex values are mapped onto the three-dimensional structure of C1Bδ and color-coded according to their magnitude (Fig. 2D). In addition, many N-H cross-peaks that are either missing or extremely weak in the wtC1Bδ (color-coded pink in Fig. 2D) are brought out of the intermediate exchange regime due to the increase in the W252Y exchange rate.
To probe the change in solvent exposure upon dynamic process, we conducted hydrogen exchange (HX) experiments. The deprotection of hydrogens due to structural fluctuations results in an increase of HX rates with the solvent, which provides a probe of structural dynamics.14,15 Because the loop dynamics is fast, the HX rates measured in our system are averaged over the populations of existing conformers. We found that the pattern and identity of residues with quantifiable exchange behavior of amide hydrogens is essentially identical for the wt and W252Y (Fig. S3). This holds for both 1H-2H and 1H-1H amide-water exchange rates that were measured using 2H2O solvent exchange and CLEANEX-PM16 experiments, respectively. These data suggest that the DAG-toggling mutation does not significantly alter the nature of the exchange process but rather its thermodynamic and kinetic parameters.
We used the topology diagram of C1Bδ to relate the 1H-2H exchange rates to a subset of H-bonds that stabilize the loop region (Fig. 2E). The inter-strand H-bonds (black dotted lines) are protected from the solvent, indicating that the protein core remains stable. In contrast, amide hydrogens located in the hinge regions of loops β12 and β34 undergo very fast exchange with 2H2O, despite being involved in the H-bonds in the crystal structure (red dotted lines). The hinge residues are either broadened beyond detection or show quantifiable dispersion in the wt and W252Y C1Bδ. Taken together, these data are consistent with partial “unzipping” of the hydrogen bonds at the β14 and β23 loop hinges that would give rise to the conformational fluctuation of the loops. Molecular dynamics simulations conducted by us9 and others17 on the C1 domains from PKCα support this idea: in the absence of activators such as DAG or phorbol esters, the loop tip distances varied as much as ~5 Å in water and ~4–6 Å in a lipid bilayer.
Armed with the C1Bδ dynamics data, we then compared the DAG affinities (in micelles) and exchange rates with those obtained for the C1Bα (Table 1). There is a clear correlation between the identity of the aromatic residue at the N-terminal β34 hinge, DAG affinity, and dynamics of the loop regions in the apo proteins. When the hinge residue is Trp, the DAG affinity is high and the dynamics is fast. When the residue is Tyr, DAG affinity is low and the loop dynamics is slower, as reflected in the kex values.
Table 1.
Correlation between exchange parameters and DAG affinity in C1B domains of PKC.
| C1B domain | Residue @ β34 hinge |
kex,s−1 | Kd, µMa |
|---|---|---|---|
|
| |||
| wtC1Bδ | Trp | 8100 ± 300 | < 0.2 |
| W252Y C1Bδ | Tyr | 17600 ± 1400 | 5.2 ± 0.5 |
| Y123W C1Bα | Trp | 9600 ± 200 | < 0.2 |
| wtC1Bα | Tyr | 15400 ± 400 | 24 ± 2 |
To gain insight into the role of Trp/Tyr at the β34 hinge position, we inspected the crystal structures of C1Bδ (Fig. 3A). In the complex of C1Bδ with phorbol 13-acetate, the ligand stabilizes the loop region by forming hydrogen bonds with backbone atoms of Thr242, Leu251, and Gly253.10 In the apo C1Bδ, this role is fulfilled by a network of ordered water molecules, two of which take place of the oxygen atoms of the phorbol ester (Fig. 3B).11 Given the small difference in the loop tip distances, we speculate that both structures correspond to the more “closed” loop conformation (see section S2.1 for discussion). Despite facing away from the ligand binding cleft, the Trp252 sidechain may influence its geometry by “propping up” the β34 loop through a π-π stacking interaction with His269, and a cation-π interaction with the amine group of Lys271, which is ~4 Å away (Fig. 3A and S3). Replacing Trp with a smaller aromatic system of the Tyr sidechain may destabilize the “closed” loop conformation because these interactions could not be effectively formed. If by analogy with C1Bα9 we assign conformations A and B to the “open” (major) and “closed” (minor) states in the exchange scheme , then Trp→Tyr replacement would increase k−1 (see section S2.2 for discussion). As a result, Tyr-containing variants of C1 domains would have larger kex values, which is consistent with our experimental data (Table 1). The Trp252 sidechain is solvent-exposed, based on the Hε1-1H(water) exchange rate of 1.6 ± 0.1 s−1 (Fig. 3C); for comparison, the random-coil exchange values for Hε1 are 0.7–1.5 s−1.18 The relaxation dispersion profile of Hε1 is flat (Fig. S4), indicating that the chemical shift of Hε1 does not change. We conclude that in the apo C1Bδ Trp252 maintains the solvent exposure during the exchange process and is unlikely to populate the inter-loop space.19
Based on the accumulated functional, structural, and dynamical information about C1 domains, we offer the following interpretation of the correlation between residue identity (Trp vs. Tyr), DAG affinity, and dynamics (Table 1). First, the presence of a suitable hydrophobic environment, such as membranes/membrane mimics, is a prerequisite for high-affinity interactions of C1 with activators. For the wtC1Bδ/W252Y pair, the Trp at the hinge position enhances protein-micelle interactions ~15-fold in the absence of DAG.12 The wtC1Bα/Y123W pair qualitatively shows a similar trend.9 Therefore, stronger interactions of Trp-containing variants with membranes are one factor that contributes to the correlation between the residue identity and DAG affinity.
Second, the modulation of loop dynamics by the Trp/Tyr substitution may have a causative relationship to DAG affinity. Loop dynamics alters the geometry of the binding cleft and can therefore directly influence the interactions with DAG. Because the C1-interacting moiety of DAG is the smallest of all known C1 activators, one could then envision that conformational selection – the preferential binding of DAG to the more “closed” loop conformation favored by Trp – contributes to the increased DAG affinity of Trp-containing C1 variants. Atomic-level structural information about C1 complexed to DAG and other activators that support membrane translocation is needed to fully understand how the geometry of the ligand binding cleft adapts to the size and hydrophobicity of the activators.
Supplementary Material
Acknowledgments
This work was supported in part by the NIH R01 GM108998 and NSF CAREER CHE-1151435 awards to T.I.I.
ABBREVIATIONS
- C1B
conserved homology-1 B domain
- DAG
diacylglycerol
- PKC
protein kinase C
- HX
hydrogen exchange.
Footnotes
ASSOCIATED CONTENT
Supporting Information. One pdf file that contains description of experimental procedures and data analysis; nine figures; and one table.
Author Contributions
All authors have given approval to the final version of the manuscript.
References
- 1.Carrasco S, Mérida I. Trends Biochem. Sci. 2007;32:27–36. doi: 10.1016/j.tibs.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 2.Hurley JH, et al. Protein Sci. 1997;6:477–480. doi: 10.1002/pro.5560060228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brose N, et al. Curr. Opin. Neurobiol. 2004;14:328–340. doi: 10.1016/j.conb.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Gallegos LL, Newton AC. IUBMB life. 2008;60:782–789. doi: 10.1002/iub.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Irie K, et al. J. Am. Chem. Soc. 1998;120:9159–9167. [Google Scholar]
- 6.Ananthanarayanan B, et al. J. Biol. Chem. 2003;278:46886–46894. doi: 10.1074/jbc.M307853200. [DOI] [PubMed] [Google Scholar]
- 7.Pu YM, et al. J. Biol. Chem. 2009;284:1302–1312. doi: 10.1074/jbc.M804796200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dries DR, et al. J. Biol. Chem. 2007;282:826–830. doi: 10.1074/jbc.C600268200. [DOI] [PubMed] [Google Scholar]
- 9.Stewart MD, et al. J. Mol. Biol. 2011;408:949–970. doi: 10.1016/j.jmb.2011.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang G, et al. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 11.Shanmugasundararaj S, et al. Biophys. J. 2012;103:2331–2340. doi: 10.1016/j.bpj.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stewart MD, et al. J. Biol. Chem. 2014;289:27653–27664. doi: 10.1074/jbc.M114.585570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart MD, Igumenova TI. Biochemistry. 2012;51:7263–7277. doi: 10.1021/bi300750w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skinner JJ, et al. Protein Sci. 2012;21:987–995. doi: 10.1002/pro.2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skinner JJ, et al. Protein Sci. 2012;21:996–1005. doi: 10.1002/pro.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hwang TL, et al. J. Biomol. NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 17.Li J, et al. J. Am. Chem. Soc. 2014;136:11757–11766. doi: 10.1021/ja505369r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chevelkov V, et al. J. Biomol. NMR. 2010;46:227–244. doi: 10.1007/s10858-010-9398-8. [DOI] [PubMed] [Google Scholar]
- 19.Shen N, et al. Biochemistry. 2005;44:1089–1096. doi: 10.1021/bi0476127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.