

Abstract
Interplay between tumor cells and host cells in the tumor microenvironment dictates the development of all cancers. In breast cancer, malignant cells educate host macrophages to adopt a pro-tumorigenic phenotype. In this study, we show how the integrin regulatory protein kindlin-2 (FERMT2) promotes metastatic progression of breast cancer through the recruitment and subversion of host macrophages. Kindlin-2 expression was elevated in BC biopsy tissues where its levels correlated with reduced patient survival. Based on these observations, we used CRISPR/Cas9 technology to ablate Kindlin-2 expression in human MDA-MB-231 and murine 4T1 breast cancer cells. Kindlin-2 deficiency inhibited invasive and migratory properties in vitro without affecting proliferation rates. However, in vivo tumor outgrowth was inhibited by >80% in a manner associated with reduced macrophage infiltration and secretion of the macrophage attractant and growth factor CSF-1. The observed loss of CSF-1 appeared to be caused by a more proximal deficiency in TGF-β-dependent signaling in Kindlin-2 deficient cells. Collectively, our results illuminate a Kindlin-2/TGF-β/CSF-1 signaling axis employed by breast cancer cells to capture host macrophage functions that drive tumor progression.
Keywords: Kindlin-2, CSF-1, TGF-β, Breast Cancer, Macrophage, Tumor Microenvironment
Introduction
The complex interactions between cancer and stromal cells creates a microenvironment where tumor cells can thrive and ultimately metastasize (1). Pro-inflammatory macrophages, which are significant cellular components of the tumor microenvironment, contribute to tumor initiation and progression (2). Both autocrine and paracrine signaling loops promote interactions between cancer cells and host cells within the tumor microenvironment (3, 4). In several cancers, including those originating in the breast, a paracrine signaling loop between tumor cells and macrophages involving tumor cell-derived epidermal growth factor (EGF) and macrophage-derived colony-stimulating factor-1 (CSF-1) conspire to promote tumor growth (5, 6). However, the molecular mechanisms that underlie the expression and secretion of both CSF-1 and EGF remain largely unknown.
Kindlins are members of the 4.1- ezrin-ridixin-moesin (FERM) domain containing proteins (7). Kindlins contain F1, F2 and F3 subdomains typical of FERM domain proteins that follow an N-terminal F0 subdomain and a pleckstrin homology (PH) subdomain that transects F2 subdomain. The three mammalian kindlin family members (Kindlin-1:FERMT1; Kindlin-2:FERMT2 and Kindlin-3:FERMT3) are highly homologous, sharing ~60% amino acid sequence identity (7). The adaptor functions of kindlins allow them to regulate numerous cellular responses. Of particular importance is their capacity to regulate integrin activation, and integrin-dependent cell adhesion, spreading and migration (7–11). Kindlin-2, as well as its homologues, Kindlin-1 and Kindlin-3, has been linked to the malignancy of several types of cancers (reviewed in (7, 8)). However, despite a few reports implicating Kindlin-2 in BC (12–15), a molecular mechanism by which Kindlin-2 contributes to the pathogenesis of BC is presumed to be dependent on its activation of tumor cell integrins, but its role in regulating tumor cell-stromal interactions has not been considered.
In the present study, we report that Kindlin-2 is upregulated in human BC cell lines and tumors and that its increased expression correlates with poor prognosis in BC patients and with the metastatic potential of both human and mouse BC progression series. Both human and mouse BC tumors lacking Kindlin-2 grow slowly in mice. Importantly, tumors lacking Kindlin-2 have diminished tumor associated macrophages (TAMs). This is a consequence of Kindlin-2-mediated regulation of expression and secretion of CSF-1 by BC cells, thus supporting macrophage infiltration into tumors. We further show that TGF-β signaling modulates Kindlin-2-mediated regulation of CSF-1 and EGF expression and is required for autocrine and paracrine cross-talk between cancer cells and macrophages that promotes tumor growth.
Materials and Methods
Cell Lines and Reagents
Normal murine mammary gland cells (NMuMG), murine 4T1 cells, and human MCF7, MCF10A, MDA-MB-231, T47D, SKBr3 and BT549 cells were obtained from American Type Culture Collection (ATCC) between 2010 and 2013. We used STR DNA fingerprinting analysis for authentication of these cells in 2014 and again in March 2016. Murine 67NR and 4T07 and human MCF10Ca1h and MCF10Ca1a were obtained from Dr. Fred Miller (Wayne State University). Cells were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% FBS. 4T1 cells were engineered to stably express firefly luciferase by transfection with pNifty-CMV-luciferase and selection (500μg/ml) with Zeocin (Invitrogen). Kindlin-2-deficient cells were produced by pLenti-CRISPRv2 lentiviral transduction using a scrambled sgRNA (i.e., nonsilencing sgRNA) or two independent and verified Kindlin-2-specific sgRNAs for human and mouse Kindlin-2. Stable pools of Kindlin-2-deficient 4T1 or MDA-MB-231 cells were obtained by culture over 14 days in puromycin (5μg/ml). The extent of Kindlin-2 deficiency was determined by immunoblots.
CRISPR/Cas9 gene editing-mediated targeting of Kindlin-2 in cancer cells
LentiCRISPRv2 lentiviral plasmid system (Addgene) was used to specifically knockout Kindlin-2 in human MDA-MB-231 and mouse 4T1 BC cells as described by Cong et al. (16). The human- and mouse-Kindlin-2 specific single guide RNAs (sgRNAs) were identified based on two different predictive algorithms (Chopchop; https://chopchop.rc.fas.harvard.edu, and CRISPR Design, http://crispr.mit.edu). The sgRNAs common to both algorithms were validated against human and mouse GECKOv2 sgRNA libraries (16) and only sgRNAs found in the GECKOv2 libraries were selected. sgRNA oligos were purchased from IDT-DNA and subcloned in lentiCRISPRv2 plasmid (16). Lentivirus production and cancer cell infection were performed as previously described (17–19).
Tumor Growth and Bioluminescence Imaging
Parental (scram) or Kindlin-2-deficient MDA-MB-231 cells (106 cells per mouse, n = 5) were implanted into the mammary fat pads of female NSG mice (Cleveland Clinic). Tumor growth was followed by thrice weekly monitoring of tumor volume with digital vernier calipers.
Luciferase-expressing parental (scram) or Kindlin-2-deficient 4T1 cells (10,000 cells per mouse, n=5) were implanted into the mammary fat pads of female BALB/c mice (Jackson Laboratory). Tumor growth was quantified using bioluminescence imaging as described (17). All animal studies were performed under protocols approved by the Institutional Animal Care and Use Committee.
Isolation of murine peritoneal macrophages
Female BALB/c mice were injected intraperitoneally with 0.5ml of 4% thioglycollate (20). After 72h, cells were isolated from peritoneal lavages, where macrophages constituted ~90% of all cells as determined by flow cytometry using FITC-labeled anti-F4/80 Ab (21).
Three-Dimensional Organotypic Cultures
Three-dimensional (3D) organotypic cultures using the “on-top” method were performed as described (22). Briefly, cells (2000 cells per well) were cultured in 96-well plates onto Cultrex cushions (50μl/well; Trevigen, Gaithersburg, MD) in complete medium supplemented with 5% Cultrex Organoid growth, which was monitored by bright-field microscopy (23).
TGF-β1 and CSF-1 Enzyme Linked Immunosorbent Assay (ELISA)
TGF-β1 and CSF-1 production by 4T1 and MDA-MB-231 cell derivatives was measured using the mouse or human Quantikine TGF-β1 assays (R&D systems MB100B and DB100B for human and mouse TGF-β1, respectively) and the human and mouse CSF ELISA Kits (Thermo Scientific, EHCSF1 and EMCSF1, respectively) according to the manufacturer’s instructions. 4T1 and MDA-MB-231 cell derivatives were cultured for 24h, and the medium was replaced with serum-free DMEM. After an additional 24h, conditioned medium (CM) was collected and centrifuged at 500×g for 3 min. TGF-β1 (latent+active) and CSF-1 were measured in the serum-free CM. Cells remaining on the plate were lysed. Total protein concentrations in lysates were used to normalize total TGF-β1 and CSF-1.
Antibodies and reagents
The following primary antibodies were from Cell Signaling: anti-phospho-Smad3; anti-Smad3; anti-phospho-ERK MAPK; anti-ERK MAPK, and anti-M-CSF Receptor. The FC blocker used was Mouse SeroBlock FcR, which is a rat monoclonal antibody (clone FCR 4G8, BioRad) that specifically recognizes mouse CD16 and CD32, which are cell surface proteins also known as FcRgIII and FcRgII, respectively. Mouse monoclonal anti-K2, clone 3A3 (1:2000, EMD Millipore); anti-F4/80 (1:50, eBioscience); anti-Gr-1 (1:50, AbD Serotec); anti-CD206 (1:50, BioRad); goat horseradish peroxidase-conjugated anti-mouse IgG (1:2,000) and goat horseradish peroxidase-conjugated anti-rabbit IgG (1:2,000) were from Calbiochem. Vecta-shield with 4′,6-diamidino-2-phenylindole was from Vector Laboratories. Gel electrophoresis reagents were from BioRad. CSF-1 was from Thermo Scientific. LPS and CCL2 were from Sigma.
Real-time quantitative RT-PCR
Total RNA was extracted from cancer cell lines or tumor tissue using TRIzol reagent (Invitrogen), following the manufacturer’s instructions. cDNA was generated and used as a template for qRT-PCR as described previously (24–26). Oligonucleotide primers used for qRT-PCR were from SABiosciences.
Cell migration, invasion, and proliferation
Cell migration was assessed by wounding confluent cultures with a micropipette tip and immediately placing them in complete medium. Bright-field images were obtained immediately after wounding and after 18h. Wound closure was quantified by measuring wound areas from ≥6 different fields using ImageJ 1.34s (NIH). For invasion assays, modified Boyden chambers were coated with Matrigel (1:10 dilution; BD Biosciences), the invasion of MDA-MB-231 and 4T1 cells and their Kindlin-2-deficient derivatives in response to 10% serum was measured as described (17, 25). Alterations in cell proliferation of MDA-MB-231 and 4T1 cells and their Kindlin-2-deficient derivatives were determined by counting the number of viable cells. Cells were seeded into six well-plates in complete growth medium, then harvested at one day intervals over 5 days, and counted in a hemocytometer. Cell viability was assessed using trypan blue staining. Assays were performed in triplicates, and the values plotted were the average of two independent experiments.
Immunostaining
Tissues were collected at the designated times and snap-frozen in optimal cutting temperature (OCT) medium (Sakura Finetek), and 8-μm sections were prepared. Macrophages were detected with rat anti-F4/80 (eBioscience). M2 Macrophages were detected with rat anti-F4/80 and rat anti-mouse CD206 (BioRad). Monocytes/neutrophils were detected with anti-Gr1 (AbD Serotec), followed by Alexa 488 or Alexa 568-conjugated goat anti-rabbit IgG. Stained sections were analyzed using fluorescent or bright-field imaging microscopy (Leica) and ImagePro Plus Capture and Analysis software (Media Cybernetics). F4/80-, CD206-, and Gr1-positive areas were quantified in 15 independent fields/section using Image Pro-Plus software (25, 27).
Oncomine cancer microarray database analysis
The expression of Kindlin-2 gene in breast cancer was examined via Oncomine database (www.oncomine.com). The Minn dataset (28) was used to compare expression levels of Kindlin-2 between breast cancer and normal tissues. The expression values of Kindlin-2 were log-transformed, median-centered and normalized to one per array.
Kaplan-Meier overall survival analysis
The effect of Kindlin-2 expression on the prognosis of 3554 breast cancer patients was analyzed using the Kaplan-Meier plotter online software (http://kmplot.com/analysis/). The Kaplan-Meier plotter evaluates the effect of 54,675 genes on survival using 10,188 cancer samples including breast, lung, ovarian and gastric cancer patients. Kindlin-2 expression and survival data was derived from Affymetrix microarray data (ID: 214212_x_at). To analyze the prognostic value of Kindlin-2 gene, the samples were divided into two groups according to the median expression of Kindlin-2. The two patient groups (high and low expression of Kindlin-2) were compared using the Kaplan-Meier survival plot. The hazard ratio (HR) with 95% confidence intervals (CI), and the log rank p value was computed as part of the Kaplan-Meier plotter online software. Similar analysis was performed for CSF-1 expression and survival data using Affymetrix microarray (ID: 210557_x_at).
Statistical analysis
Experiments were done in triplicate and analyzed using the Student’s t-test. In calculating two-tailed significance levels for equality of means, equal variances were assumed for the two populations. Results were considered significant at p<0.05.
Results
Kindlin-2 expression is increased in aggressive BC
Enhanced expression of Kindlin-2 has been reported for several human cancers (7, 8), including BC (12–15). However, a definitive role for Kindlin-2 in tumorigenesis remains to be elucidated. To begin to address its role in BC, we analyzed Kindlin-2 expression across a panel of human BC cell lines. Kindlin-2 levels were 3-fold or higher in aggressive human MDA-MB-231 and BT549 BC cells compared to less aggressive BC lines, T47D, MCF7 or SKBR3 (Fig. 1A). Also, Kindlin-2 was expressed at very low levels in normal human MCF10A and indolent MCF10aT1K cells relative to their high-grade and more aggressive human MCF10aCa1h and MCF10aCa1a counterparts (Fig. 1B). We also found Kindlin-2 expression to be increased in metastatic murine 4T1 and dormant 4T07 cells relative to their indolent 67NR counterparts (Fig. 1C). A similar trend was observed in the murine NMUMG BC series: Kindlin-2 expression was significantly elevated in the LM2 metastatic cells compared to their less aggressive NME cells or non-tumorigenic NMuMG counterparts (Fig. 1C). Next, we addressed whether this trend in Kindlin-2 expression also occurred in human BC tumors by Oncomine microarray expression analyses (http://www.oncomine.org). Kindlin-2 expression was significantly upregulated in BCs compared to normal breast tissue (Fig. 1D; (28)); it was in the top 3% of upregulated genes in BC (Fig. 1D). Likewise, interrogation of the KM-Plotter BC dataset (http://kmplot.com/analysis/) determined that Kindlin-2 expression was correlated with poor outcome and reduced survival in BC patients (Fig. 1E). Collectively, these findings suggest that increased Kindlin-2 expression is characteristic of invasive BC.
Figure 1.
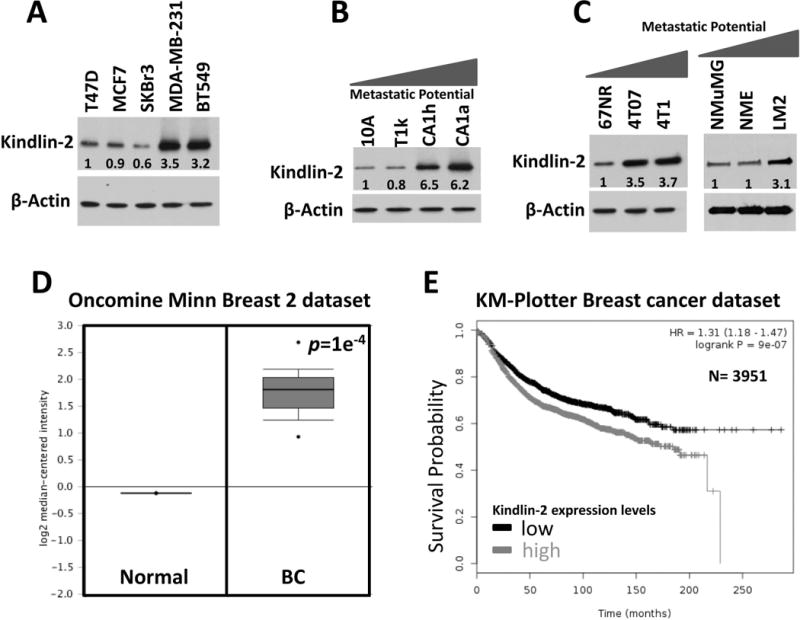
Kindlin-2 expression is increased in aggressive BC.
Western blots with anti-kindlin-2 on lysates of: (A) human breast cancer cell lines, (B) human MCF10A BC progression series, and (C) mouse 4T1 and NMuMG BC progression series. β-Actin is a loading control. The values below the Kindlin-2 panel indicate the fold change of Kindlin-2 levels, determined by densitometry and normalized to the loading control lane (first lane in each panel), and are the average values from three different blots. (D). Oncomine dataset (28) comparing Kindlin-2 mRNA levels of normal and breast cancer tissues. (E) Kaplan-Meier (KM, http://kmplot.com/analysis/) plot correlating survival of 3951 breast cancer patients with kindlin-2 expression levels.
Loss of Kindlin-2 inhibits BC cell migration and invasion
CRISPR/Cas9 technology has emerged as a significant tool for efficient site-specific gene editing and targeting (16, 29). We designed two single-guide RNAs (sgRNAs), sgRNA-1 and -2, to target exon 2 and exon 5 of human Kindlin-2 (FERMT2, Fig. 2A and 2B), and to target exon 2 and exon 3 of mouse Kindlin-2 (Fermt2, Fig. 2C and 2D) and used these in human MDA-MB-231 and murine 4T1 cells as widely used BC models. Both have elevated levels of Kindlin-2 (Fig. 1A and 1C). The sgRNAs designed against human Kindlin-2 were very efficient in human MDA-MB-231 cells compared to cells infected with a scrambled (Scram) sgRNA or the parental cells (Fig. 2B). Likewise, the sgRNAs designed against mouse Kindlin-2 resulted in a very efficient knockdown of Kindlin-2 in mouse 4T1 cells (Fig. 2D). Sequencing of individual PCR clones from the genomic DNA of sgRNA-targeted exon 2 of Kindlin-2 in MDA-MB-231 cells showed several small insertion-deletions (indels) near the target site (Fig. 2E), which resulted in frame shifts in the coding sequence (Fig. 2F).
Figure 2.
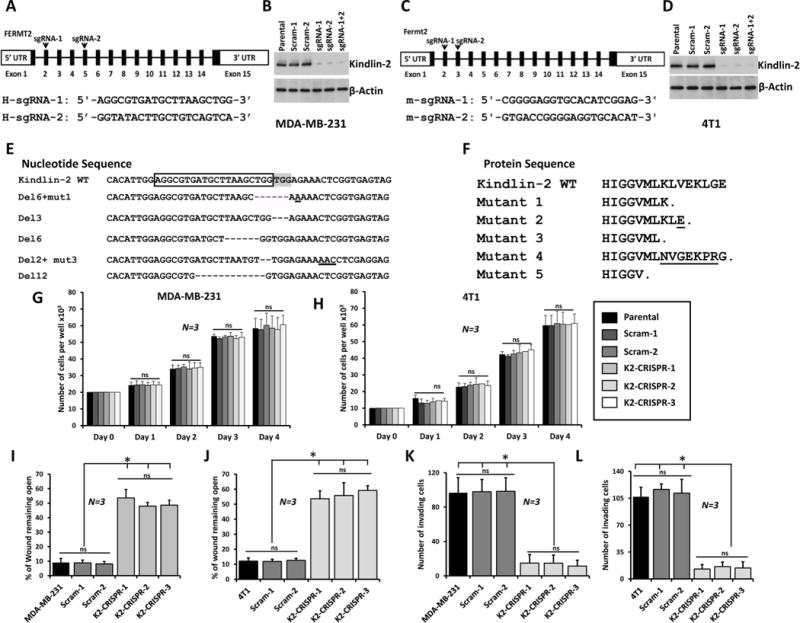
Loss of Kindlin-2 via CRISPR/Cas9 inhibits BC cell migration and invasion in vitro.
Genomic structure of human Kindlin-2 (FERMT2, A) and mouse Kindlin-2 (Fermt2, C) genes showing intron-exon organization and location of sg-RNAs, (arrow-heads) in exon 2, 5; and exon 2, 3 of human and mouse kindlin-2 genes, respectively. Western blots developed with anti-Kindlin-2 antibody of lysates from control MDA-MB-231 (B) and 4T1 (D) transduced with a scrambled sgRNA (Scram), and Kindlin-2-defficient. β-Actin is a loading control. (E) Nucleotide sequence alignment of wild-type (WT) exon 2 of human Kindlin-2 and representative Kindlin-2-deficient clones showing insertions and deletions (Indels, dashes and underlines). sg-RNA-1 is boxed and the Protospacer Adjacent Motif (PAM) sequence is highlighted. (F). Protein sequence alignment of WT exon-2 of the human Kindlin-2 and representative Kindlin-2-deficient clones showing premature stop codons. (G and H) Proliferation over 5 days of parental, 2 pools of Scram and three pools of Kindlin-2-deficient (K2-CRISPR) of MDA-MB-231 (G) and 4T1 (H). (I and J) Cells were induced to migrate into scratch wounds in confluent monolayers over 18h. The unclosed wound (open area) at 18h from 12 different wounds was measured and plotted as the percentage of the wound at time zero for MDA-MB-231 cells (I) and 4T1 cells (J). (K and L) Invasion assays through Matrigel-coated membranes: Invading cells were counted from six different fields and plotted as average number of invading cells per field for MDA-MB-231 cells (K) and 4T1 cells (L). Data are the means ± SD, N=3; ns, not significant; *, p <0.05; Student’s t-test).
Having confirmed the efficiency of Kindlin-2 knockdown using CRISPR/Cas9, we investigated the effect of Kindlin-2 deficiency on the behavior of human and mouse BC cells. First, we found that both the scrambled (Scram) and the K2-sgRNAs (K2-CRISPR), did not have a significant effect on proliferation of either MDA-MB-231(Fig. 2G) or 4T1 (Fig. 2H) cells. Next, in a wound closure assay, we found loss of Kindlin-2 expression (K2-CRISPR) in MDA-MB-231 and 4T1 cells resulted in a significant decrease of migration into wounds as compared to parental and the control (Scram) cells (Fig. 2I and 2J, respectively). In Boyden chamber invasion assays, less MDA-MB-231 and 4T1 Kindlin-2-deficient (K2-CRISPER) cells traversed the Matrigel-coated inserts compared to the parental and Scram cells (Fig. 2K, and 2L, respectively). These results were replicated in two and three different pools of Scram and K2-CRISPR cells, respectively, supporting the reproducibility of the data across multiple pools.
Loss of Kindlin-2 inhibits BC tumor growth in vivo.
To assess the effects of loss of Kindlin-2 on tumor growth in vivo, mammary fat pads of NOD-scid-IL2Rgamma knockout (NSG) mice were inoculated with control (Scram) or Kindlin-2-deficient (K2-CRISPR) MDA-MB-231 cells, and tumor growth was assessed over 8 weeks. Loss of Kindlin-2 inhibited the growth of primary tumors (Fig. 3A to 3C). While every mouse in the control and Kindlin-2 groups developed tumors (100% tumor incidence) after a ∼4-wk latency, tumor burden, as assessed by tumor size (Fig 3A), weight (Fig. 3B), and volume (Fig. 3C), was significantly lower (p<0.05) in the mice implanted with the Kindlin-2-deficient cells. Similarly, loss of the mouse Kindlin-2 in 4T1 cells also delayed tumor initiation and growth in the BALB/c mice (Fig. 3D and 3E). Thus, loss of Kindlin-2 inhibits the rate of primary tumor growth in vivo of both human and mouse models for BC. These differences in tumor burden were not a result of decreased tumor cell proliferation by Kindlin-2 knockdown, as the number of viable cells between the control and Kindlin-2-deficient cells was similar (Fig. 2G and 2H).
Figure 3.
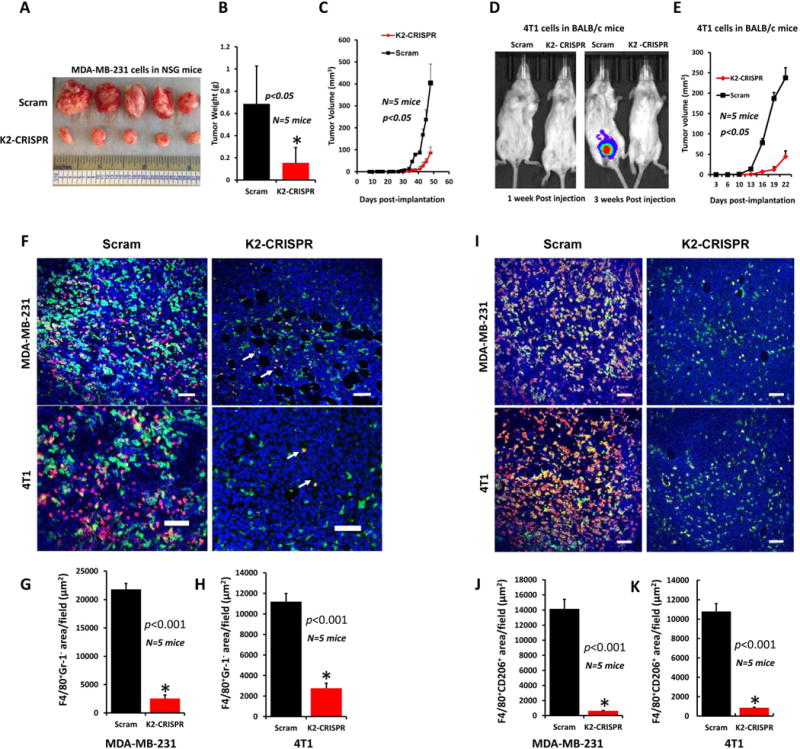
Loss of Kindlin-2 inhibits tumor growth in vivo and macrophage recruitment.
(A) Tumors generated from inoculation of Scram or K2-CRISPR MDA-MB-231 cells into the mammary fat pads of NSG mice. Quantification of weight (B) and volume (C) of tumors shown in A. (D) Luciferase bioluminescence of tumors from inoculation of Scram or Kindlin-2-deficient 4T1 cells into mammary fat pads of BALB/C mice. (E) Quantification of tumor growth. (F) Representative images of MDA-MB-231(Top) and 4T1 (Bottom) tumor sections stained for F4/80 (green) and Gr-1 (red). Most of the F4/80+ cells were macrophages as they were Gr-1−(green cells). White arrowheads point to a few F4/80+Gr-1+ monocytes (yellow). Size bar, 146 μm. (G and H) Quantification of macrophage-specific F4/80+Gr-1− areas in MDA-MB-231(G) and 4T1 (H) tumors. Data are expressed as mean ± SEM. *p<0.001, n=5 mice. (I) Representative images of MDA-MB-231(Top) and 4T1 (Bottom) tumor sections stained for F4/80(green) and CD206(red). Most of the F4/80+ cells were polarized to the M2 phenotype since they were CD206+(yellow+orange cells). Size-bar, 146 μm. (J and K) Quantification of M2 macrophage-specific F4/80+CD206+ areas in MDA-MB-231(J) and 4T1(K) tumors. Data are Data are the means ± SEM. *p<0.001, n=5 mice.
BC Tumors lacking Kindlin-2 fail to recruit macrophages
Cross-talk between the tumor and stromal microenvironment is a prominent mechanism in primary tumor growth and subsequent invasion and metastasis (2, 6, 30, 31). We determined whether loss of Kindlin-2 affected the recruitment of tumor-associated macrophages (TAMs) into in MDA-MB-231 and 4T1 tumors. Since F4/80 mAb we used for staining (32) can also react with monocytes from TAM, we double-stained tumor sections for F4/80 (green) and Gr-1 (red), a marker of not only neutrophils but also inflammatory monocytes. The staining revealed abundant TAMs (F4/80+-Gr-1− cells-green) in Scram as compared K2-CRISPER MDA-MB-231 (Fig. 3F, top panels and Sup. Fig. 1) and 4T1 (Fig. 3F, bottom panels and Sup Fig. 1) tumors. In contrast, as indicated by white arrowheads, we detected very few monocytes (F4/80+-Gr-1+-yellow cells) in all tumors. Quantification of F4/80+-Gr-1− areas (green) in tumor sections showed a ~7-fold and ~3.5-fold increase in TAMs in Scram-MDA-MB-231 (Fig. 3G) and Scram-4T1 (Fig. 3H) tumors, respectively, as compared to their K2-CRISPER counterparts (p<0.001, n=5). In addition, F4/80+-Gr-1+ monocytes (yellow) did not comprise more than 5% of the F4/80+ population in the tumors. We also double stained tumor sections for F4/80 (green) and CD206 (red), a marker for polarized M2 mouse macrophage phenotype (33). Tumors derived from Scram-MDA-MB-231 and Scram-4T1 showed massive infiltration with immunosuppressive M2 macrophages (F4/80+-CD206+, yellow and orange), compared their K2-CRISPR counterparts (Fig. 3I, top panels and bottom panels, respectively, and Sup. Fig. 2). Quantification of F4/80+-CD206+ areas (yellow and orange) in tumor sections show that majority of macrophages are of the M2 phenotype and their counts are enhanced by approximately 10-fold in the MDA-MB-231 and 4T1 Scram-tumors compared to the K2-CRISPR tumors (Fig. 3J and 3K, respectively). Similar results were found when comparing Scram-MDA-MB-231 and K2-CRISPR-MDA-MB-231 tumors of roughly similar sizes (Sup. Fig. 3). In addition, since the average volume of tumors derived from K2-CRISPR-4T1 did not exceed 55 mm3, compared to more than 240 mm3 for the Scram-4T1 tumors (Fig. 3E), we considered whether K2-CRISPR-4T1 tumors were too small to initiate macrophage recruitment. Accordingly, we repeated this experiment allowing the tumors from the 4T1-K2-CRISPR to grow for an additional 3 weeks before excision so that K2-CRISPR tumors reached comparable sizes (~300 mm3, Sup. Fig. 4A) to the Scram 4T1 tumors excised three weeks earlier. Tumors derived from Scram-4T1 showed increased infiltration with immunosuppressive M2 macrophages (F4/80+-Cd206+, yellow and orange), compared their K2-CRISPR counterparts (Sup. Fig. 4B), similar to the data shown in Fig. 3I. Quantification of F4/80+-CD206+ areas (yellow and orange) in tumor sections also show that majority of macrophages were of the M2 phenotype in the 4T1 Scram-tumors compared to the K2-CRISPR tumors (Sup. Fig. 4C). Thus, K2 deficiency in cancer cells inhibits tumor infiltration by macrophages independent of the tumor size.
Kindlin-2 enhances macrophage-mediated stimulation of tumor growth in 3-dimentional organotypic cultures
To further explore the interrelationship between Kindlin-2 in BC tumor cells and macrophages, 3D organotypic outgrowth was used system. Scram or K2-CRISPR MDA-MB-231 cells were seeded onto 3D organotypic cultures, either alone or pre-mixed with mouse peritoneal macrophages. To distinguish the cell populations within the 3D organoids, MDA-MB-231 cells were labeled with GFP. When Scram MDA-MB-231 cells were seeded alone, they readily formed 3D organoids, an established characteristic of aggressive BC cells (Fig. 4A-1, top and bottom panels). Pre-mixing the cancer cells with macrophages (Scram+Mɸ) resulted in a ~3-fold increase of the 3D outgrowth (Fig. 4A-2, top and bottom panels & 4B). These 3D organoids mainly originated from MDA-MB-231 BC cells, not from the macrophages, as indicated by fluorescence labeling of these structures (Fig 4A, top panels). Seeding of macrophages alone onto 3D organotypic cultures did not support any 3D outgrowth (not shown), confirming that cancer cells give rise to these structures. Interestingly, Kindlin-2 deficiency significantly repressed the outgrowth of BC organoids in 3D cultures (Fig 4A-3 top and bottom panels & 4B); and inclusion of macrophages with the Kindlin-2-deficient BC cells (K2-CRISPR+Mɸ) did not overcome the repression of 3D outgrowth caused by lack of Kindlin-2 in the cancer cells (Fig. 4A, top and bottom panels & 4B).
Figure 4.
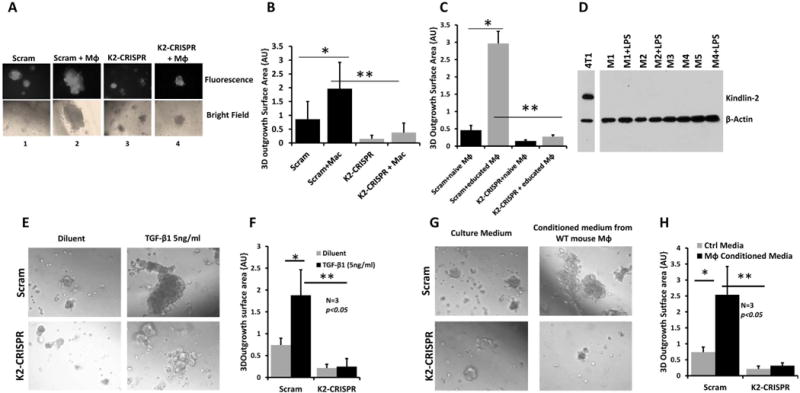
Tumor growth in 3-dimentional organotypic cultures depends on Kindlin-2 and Kindlin-2-dependent macrophage-secreted factors.
(A) Representative micrographs of Scram and Kindlin-2-deficient MDA-MB-231 cells induced to form organoids, in presence of culture medium alone (panel 1 and 3) or when mixed with mouse peritoneal macrophages (panel 2 and 4). Control and the Kindlin-2-deficient MDA-MB-231 cells express EGFP to differentiate them from macrophages. Confocal micrographs are either fluorescence (upper panels) or bright field (lower panels). (B) Quantification of surface area occupied by 3D organoids. (C) Quantification of surface area occupied by 3D organoids of control and Kindlin-2-deficient MDA-MB-231 cells in 3D cultures when mixed with untreated macrophages (naïve Mɸ) or macrophages pre-incubated for 48h with conditioned medium of MDA-MB-231 cells cultures (educated Mɸ). (D) Immunoblotting with anti-Kindlin-2 of macrophage lysates from five mice (M1 to M5), with or without LPS stimulation (100ng/ml) for 48h. 4T1 cells were used as a positive control for Kindlin-2 expression and β-Actin as a loading control. (E) Micrographs of Scram (upper panels) and Kindlin-2-deficient (lower panels) of MDA-MB-231 cells that were induced to form tumor organoids in 3D cultures in the presence of medium alone (left panels) or medium supplemented with TGF-β1 (5ng/ml, right panels). (F) Quantification of surface area occupied by the 3D organoids under the indicated conditions as described in (E). (G) Representative micrographs of control (upper panels) and Kindlin-2-deficient (lower panels) MDA-MB-231 cells that formed tumor organoids in 3D cultures in the presence of plain culture medium (left panels) or culture medium from LPS-stimulated mouse peritoneal macrophages. (H) Quantification of surface area occupied by the 3D organoids under the indicated conditions as described in (G). Data are the means ± SD (n=3,*and**,p<0.05; Student’s t-test).
Tumor-educated macrophages switch from an anti-inflammatory to pro-inflammatory phenotype that can encourage tumor growth (34). We investigated whether manipulating Kindlin-2 in cancer cells would regulate macrophage function and thereby affect the 3D outgrowth of tumor cells. Untreated macrophages (naïve Mɸ) or macrophages pre-incubated with the conditioned medium of MDA-MB-231 cells for 48 h (tumor-educated Mɸ) were pre-mixed with either Scram or K2-CRISPR MDA-MB-231 cells. Control MDA-MB-231 cells that were mixed with tumor-educated macrophages (Scram+tumor-educated Mɸ) formed 3D organoids that were ~6-fold larger (p<0.05) than those formed by the same tumor cells mixed with untreated (Scram+naïve Mɸ) macrophages (Fig. 4 C). The 3D organoids that developed from the K2-CRISPR tumor cells remained significantly (p<0.05) smaller than those generated from the Scram tumor cells, regardless whether they were mixed with naïve or educated macrophages (Fig. 4C). These data clearly indicate that Kindlin-2 in cancer cells is required for both 3D outgrowth, for regulation of macrophage function, and for their ability to stimulate 3D outgrowth of cancer cells. Kindlin-2 from the tumor cells most likely mediate these biological effects since neither resting nor LPS-activated macrophages expressed Kindlin-2 as assessed by Western blots (Fig. 4D). These findings also indicate that macrophages, as a part of the tumor microenvironment, play an important role in stimulating tumor growth, as suggested from Fig. 3 and 4 and consistent with the literature (6).
We considered whether the macrophage-mediated stimulation of tumor growth depends on direct contact with the cancer cells or through secreted chemotactic and stimulatory factors. MDA-MB-231 cells were treated with transforming growth factor-beta1 (TGF-β1), which stimulates outgrowth of tumor cells in 3D organotypic cultures (17, 18). While TGF-β1 stimulated 3D outgrowth of the Scram cells (Fig. 4E, top panel), it failed to exert the same effect on the K2-CRISPR cells (Fig 4E bottom panel). The size of the 3D organoids was ~4-fold (p<0.05) smaller in the Kindlin-2 deficient cells compared to Scram cells (Fig. 4F). Next, we investigated the effect of conditioned medium of macrophages on 3D outgrowth. The size of the 3D organoids from the Scram MDA-MB-231 cells was > 4-fold larger (p<0.05) when incubated with the conditioned medium of LPS-stimulated macrophages (Fig. 4G, top panels and 4H), compared to those incubated with plain culture medium. The 3D outgrowth of the K2-CRISPR cells was significantly smaller (p<0.05) than the control cells under both culture conditions (Fig. 4G, lower panels and 4H). These findings suggest that factors secreted from activated macrophages stimulate cancer cell growth in the 3D-organotypic cultures; i.e., a paracrine signaling loop. Since Kindlin-2-deficient MDA-MB-231 cells incubated with the conditioned medium of LPS-activated macrophages failed to stimulate 3D outgrowth, cancer cells possess a kindlin-2 dependent autocrine signaling loop, which is also involved in stimulation of 3D outgrowth.
Kindlin-2 in cancer cells is required for expression of CSF-1 and its chemotactic attraction of macrophages
Both colony-stimulating factor-1 (CSF-1) and epidermal growth factor (EGF) have been implicated the interaction between tumor cells and macrophages (4); tumor cells secrete CSF-1 and respond to EGF, while macrophages secrete EGF and respond to CSF-1 (4). In a transwell migration assay, naïve macrophages were allowed to migrate towards control culture medium, medium supplemented with chemotactic agents, or conditioned medium from control or Kindlin-2-deficient tumor cells. Macrophages attracted by CSF-1 was >7-fold higher (p<0.05) than macrophages attracted to control medium (Fig. 5A and 5B), consistent with the known property of CSF-1 as a macrophage chemoattractant (3, 4). Similar numbers were obtained with CCL2, another known chemotactic agent (35), and with the macrophage-activator LPS (Fig. 5A and 5B). The number of macrophages attracted by the conditioned medium from the K2-CRISPR-231 cells was 5-times lower (p<0.05) than attracted to the conditioned medium from control cells (231-Scram; Fig. 5C and 5D), confirming that Kindlin-2 is required for the production and secretion of chemoattractants by cancer cells. These results were duplicated with murine 4T1 BC cells (Fig. 5E and 5F). Kindlin-2 itself was not detected in the conditioned medium of the cancer cells by Western blotting (not shown).
Figure 5.
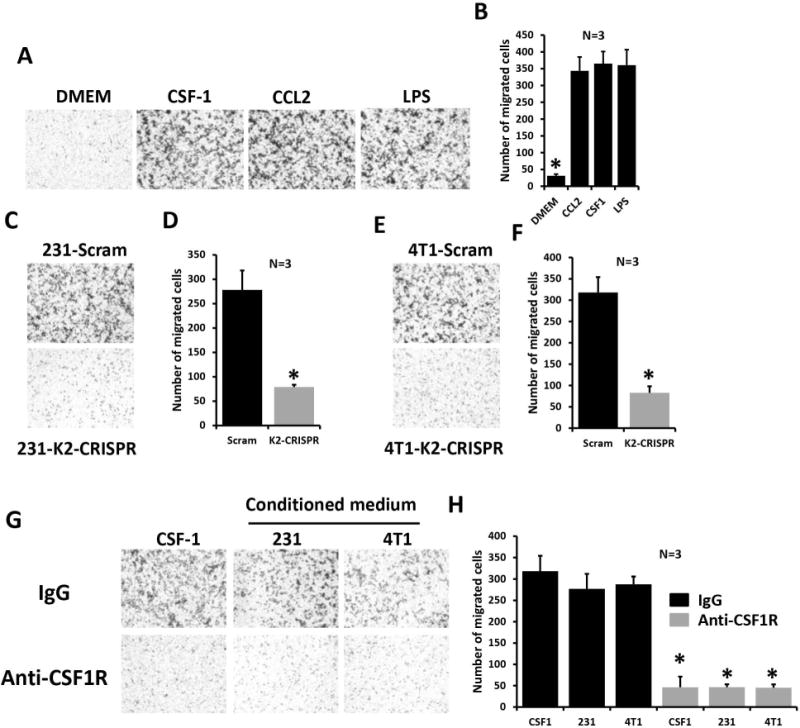
Kindlin-2 in cancer cells is required for CSF-1 expression and chemoattraction of macrophages.
(A) Representative micrographs of mouse peritoneal macrophages in transwell migration assays overnight in response to CSF-1 (100ng/ml), CCL-2 (500ng/ml) and LPS (100ng/ml). (B) Quantification of macrophage migration. (C and E) Representative micrographs of mouse macrophages migration in transwell assays in response to conditioned medium of control (upper panels) and Kindlin-2-deficient (lower panels) from MDA-MB-231 cells (C) or 4T1 cells (E). (D and F) Quantification of migrating macrophages from experiments shown in C and E, respectively. (G) Representative micrographs of mouse macrophages in transwell assays in the presence of control IgG (upper panels) or anti-CSF1R antibody (lower panels). Macrophages were pre-incubated with an FC blocker prior to adding anti-CSF1R. A representative of three experiments is shown. (H) Quantification of migrating macrophages under conditions shown in G. Data are the means ± SD (n=3,*,p<0.05; Student’s t-test).
To directly implicate tumor cell-secreted CSF-1 in macrophage chemotaxis and the requirement of Kindlin-2 for release of CSF-1 from cancer cells, we pre-incubated macrophages with a blocking anti-CSF1 receptor (CSF1R). Macrophages pre-incubated with control IgG successfully migrated towards medium supplemented with CSF-1 or the conditioned medium from MDA-MB-231 or 4T1 cells (Fig. 5G, top panels and 5H), but anti-CSF1R decreased macrophage recruitment by more than 6-fold (p<0.05) under all experimental conditions (Fig. 5G, bottom panels and 5H). Thus, Kindlin-2 in tumor cells is required for the production and secretion of CSF-1, which in turn, mediates chemoattraction of macrophages.
Kindlin-2 influences both paracrine CSF-1 and autocrine EGF signaling
We used quantitative-real-time PCR (qt-RT-PCR) to measure CSF-1 mRNA levels in parental MDA-MB-231 and 4T1, 2 different pools of Scam and 3 different pools of K2-CRISPR cells and found CSF-1 mRNA levels to be ~4-fold lower (p<0.05) in Kindlin-2-deficient cells (Fig. 6A and 6B). CSF-1R levels were also significantly lower (p<0.05) in Kindlin-2-deficient MDA-MB-231 and 4T1 cells compared to controls (Fig. 6C and 6D, respectively). We used ELISA to quantify the amounts of secreted CSF-1. Kindlin-2-deficient MDA-MB-231 and 4T1 cells released significantly less (~5-fold, p<0.05) CSF-1 than their control counterparts (Fig. 6E and 6F, respectively). Since TGF-β signaling activates CSF-1 and CSF1R expression in tumor cells (3), we considered its role in Kindlin-2-mediated production of CSF-1. TGF-β1 ELISA assays showed that Kindlin-2-deficient MDA-MB-231 cells (Fig. 6G) and 4T1 cells (Fig. 6H) released significantly (p<0.05) less TGF-β1 compared to their Kindlin-2 expressing counterparts. We also found that mRNA expression levels of EGF (Fig. 6I and 6J) and its receptor EGFR (Fig. 6K and 6L) to be ~2-fold and ~40% lower (p<0.05), in the Kindlin-2-deficient MDA-MB-231 and 4T1 cells, respectively. These findings are consistent with the existence of an autocrine loop wherein tumor cells secrete their own EGF (3). Thus, our data suggest that Kindlin-2 is also involved in EGF-EGFR autocrine loop in MDA-MB-231 cells. We further confirmed that EGF-TGF-β signaling pathway is required for the Kindlin-2-mediated regulation of the synthesis and secretion of CSF-1 in cancer cells. We specifically targeted both EGF and TGF-β nodes of this signaling axis in MDA-MB-231 and 4T1 cells and assessed for CSF-1 secretion. TGF-β1 and EGF treatment of MDA-MB-231 cells (Fig. 7A) and 4T1 cells (Fig. 7B) activated the TGF-β and EGF downstream effectors, SMAD3 (~6-fold increase for MDA-MB-231 and ~20-fold increase for 4T1 cells, compared to the diluent; Fig. 7C) and ERK MAP kinase (~10-fold increase for 231 and ~6-fold increase for 4T1 cells compared to diluent; Fig. 7D). These treatments also stimulated secretion of CSF-1 in both cells by ~2-fold (Fig. 7A for MDA-MB-231 231 cells and 7B for 4T1 cells). Importantly, while TGF-β and EGF treatment of K2-CRISPR cells still promoted secretion of CSF-1, the basal levels of secreted CSF-1 were significantly (p<0.05) lower in the Kindlin-2-deficient cells (~2-fold decrease in MDA-MB-231-K2-CRISPR and ~2-fold decrease in 4T1-K2-CRISPR cells, compared to the control cells; Fig. 7A and 7B). Loss of Kindlin-2 appears to inhibit maximal activation of TGF-β1 and EGF signaling and, thereby, also blunts the stimulatory effects of TGF-β1 and EGF on CSF-1 secretion. We confirmed this finding at the signaling levels: the phospho-SMAD3 levels were 4-fold (MDA-MB-231 cells) and ~3-fold (4T1 cells) lower in the K2-deficient (K2-CRISPR) after treatment with TGF-β1, compared to their Scram counterparts (Fig. 7C). Similarly, phospho-ERK levels were 5-fold (MDA-MB-231 cells) and 4-fold (4T1 cells) lower in the K2-deficient (K2-CRISPR) after treatment with EGF, compared to their Scram counterparts (Fig. 7C). Treatment with TGF-β or EGF signaling inhibitors (SB4131542 and ZD1839, respectively) blocked the activation of their respective downstream effectors (Fig. 7C and 7D), and significantly (p<0.05) dampened CSF-1 secretion by MDA-MB-231 cells (Fig. 7A) and 4T1 cells (Fig. 7B), supporting the regulatory role of Kindlin-2 in the stimulatory effects of TGF-β1 and EGF on CSF-1 secretion. We also found both EGF and TGF-β signaling to be significantly lower in the K2-CRISPR-derived tumors compared to their Scram counterparts (Fig. 7E for MDA-MB-231- and 7F for 4T1-derived tumors). Expression levels of CSF-1, CSF1R, EGF and EGFR were also significantly lower in the K2-CRISPR-derived tumors compared to their Scram counterparts (Fig. 7G for MDA-MB-231 – and 7H for 4T1-derived tumors; and Sup Fig. 4D for tumors of similar sizes), therefore, supporting our in vitro findings with in vivo data. Together, our findings confirm the involvement of the EGF-TGF-β signaling in the secretion of CSF-1 by cancer cells (3, 4) and expand this pathway to a Kindlin-2-EGF-TGF-β signaling axis, which regulates CSF-1 release from cancer cells and induces chemotaxis and activation of macrophages within the tumor microenvironment (Fig. 7I).
Figure 6.
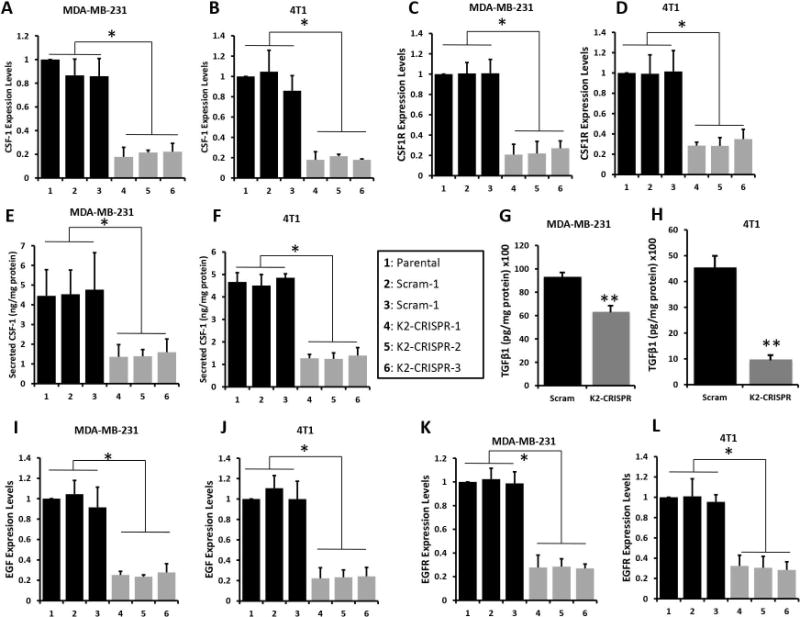
Kindlin-2 is required for the paracrine CSF-1 and the autocrine EGF signaling.
Quantitative-real time RT-PCR of CSF-1 transcripts in control (parental, 1), two different pools of Scram (2 and 3) and or 3 different pools K2-CRISPR (4, 5 and 6) of MDA-MB-231 (A) and 4T1 (B) cells. (C and D) Quantification of CSF1R transcript levels form the same cells as in A and B (C, MDA-MB-231; D, 4T1 cells). GAPDH was used for normalization. (E and F) Quantification of secreted CSF-1 in conditioned medium of control, Scram or K2-CRISPR MDA-MB-231 (E) and 4T1 (F) cells by ELISA. (G and H) ELISA-based quantification of secreted TGFβ1 in conditioned medium of Scram or K2-CRISPR MDA-MB-231 (G) and 4T1 (H) cells. (I and J) Quantification of EGF transcript levels in control, Scram and K2-CRISPR MDA-MB-231 (I) and 4T1 (J) cells. (K and L) Quantification of EGFR transcript levels in control, Scram and K2-CRISPR MDA-MB-231 (K) and 4T1 (L) cells. GAPDH was used for normalization. Data are the means ± SD (n=3,*,p<0.05; Student’s t-test).
Figure 7.
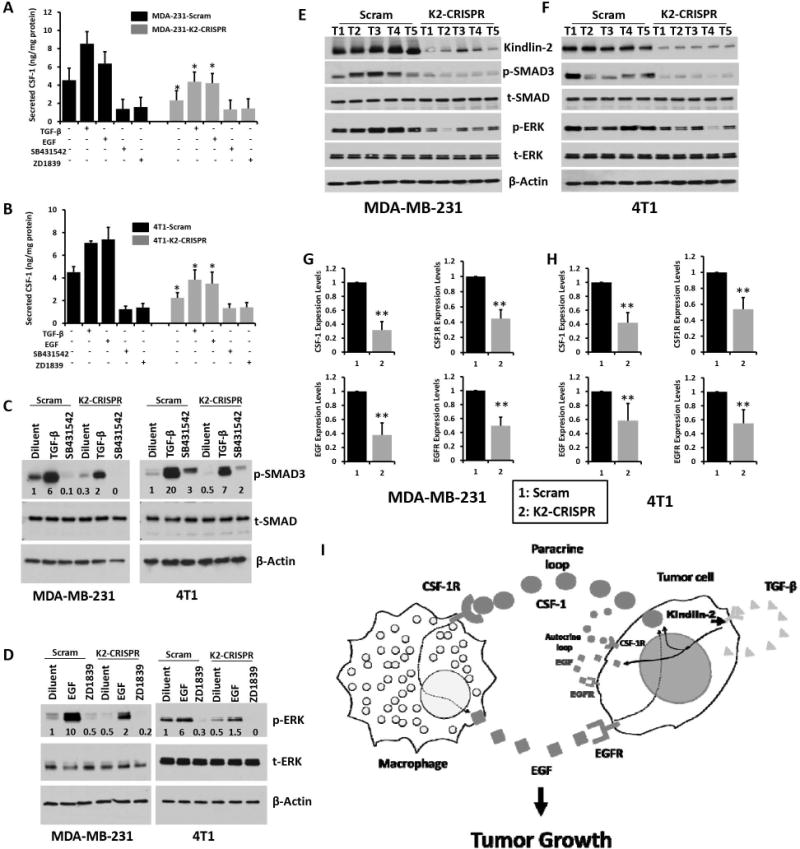
Inhibition of EGF and TGFβ signaling pathways suppresses Kindlin-2-dependent activation of CSF-1 secretion by cancer cells. ELISA-based quantification of secreted CSF-1 in the conditioned medium of Scram or K2-CRISPR MDA-MB-231 (A) and 4T1 (B) cells (*, p<0.05, Scram vs K2-CRISPR). Scram (black) and K2-CRISPR (gray) MDA-MB-231 (A) and 4T1 (B) BC cells were treated with TGFβ-1 (5ng/ml), EGF (100ng/ml), the TGFβR1 inhibitor SB431542 (10μM) or the EGFR inhibitor ZD1839 (5μM) for 48 h and secreted CSF-1 in the conditioned medium was measured by ELISA assay. (C and D) Cell lysates from Scram MDA-MB-231 and 4T1 cells or their K2-CRISPR derivatives with the indicated treatments, were analyzed by immunoblotting with the indicated antibodies. The values below the p-SMAD3 (C) and p-ERK (D) panels indicate the fold change of p-SMAD3 or p-ERK levels measured by densitometry of western blots and normalized to the diluent-treated Scram cells and are average values from three different blots. (E and F) Protein lysates from tumors (T1 to T5) derived from either Scram of Kindlin-2 deficient (K2-CRISPR) MDA-MB-231 (E) or 4T1 (D) cells, were analyzed by immunoblotting with the indicated antibodies. (G and H) Quantitative-real time RT-PCR of CSF-1, CSF1R, EGF and EGFR transcripts in tumors derived Scram (1) or K2-CRISPR (2) of MDA-MB-231 (G) and 4T1 (H) cells. GAPDH was used for normalization. Data are the means ± SD (n=5 mice,**,p<0.05; Student’s t-test). (I) Model describing the role of Kindlin-2 in the regulation of both autocrine and paracrine signaling in CSF-1-mediated recruitment of macrophages to tumors.
Discussion
This study demonstrates that Kindlin-2 supports BC tumor growth and does so at least in part by controlling recruitment of macrophages to the tumor microenvironment. To our knowledge, this is the first study to show that Kindlin-2 influences the tumor microenvironment, thereby providing mechanism by which increased levels of Kindlin-2 in tumors correlates with poor prognosis in BC patients. Additionally, we show that Kindlin-2 enhances the invasive properties of BC cells further promoting the neoplastic properties of tumors. We applied a combination of genetic and pharmacologic manipulations, as well as different biochemical and cell imaging analyses in vitro and in mouse models, to investigate the role Kindlin-2 in the modulation of the growth and progression of BC tumors. This study also appears to be the first to use CRISPR/Cas9 gene editing to delete Kindlin-2 in human and murine BC cell lines. We showed that (i) Kindlin-2 is overexpressed in aggressive BC cell lines, both human and mouse, compared to non-tumorigenic or less aggressive cell lines; and that the levels of Kindlin-2 correlates with the metastatic potential of these cells, as well as correlates with poor prognosis in patients with BC; (ii) in vitro, Kindlin-2 enhances tumor cell migration and invasion; (iii) in in vivo mouse models, Kindlin-2 is required for rapid BC tumor growth; (iv) the extent of tumor growth depends on the amounts of tumor infiltrating macrophages (36–38), which was severely dampened in Kindlin-2-deficient-derived tumors, suggesting that Kindlin-2 regulates recruitment of macrophages to the tumor microenvironment; (v) in tumor cells, Kindlin-2 is required for the production and secretion of CSF-1, that is necessary for macrophage chemotaxis to the tumor microenvironment; (vi) Kindlin-2 plays a key role in both macrophage-tumor cell paracrine and tumor cell autocrine signaling involving EGF and CSF-1; and (vii) this activity depends upon the capacity of Kindlin-2 to regulate TGF-β signaling. Thus, we have established a Kindlin-2-TGF-β signaling axis is a key regulator of CSF-1 secretion by tumor cells, which in turn directs macrophage recruitment.
TGF-β signaling has been found to activate the production and secretion of CSF-1 by tumor cells (34). Our results show that deficiency of Kindlin-2 in cancer cells inhibits the expression and secretion of CSF-1, which is concomitant with a significant decrease in TGF-β1 production and secretion (Fig. 6). These findings clearly suggest that Kindlin-2 regulates TGF-β1 production and its downstream signaling effects on target genes such as CSF-1. Wu and colleagues (39) recently showed that Kindlin-2 binds to the cytoplasmic kinase domain of TGF-β receptor 1 resulting in TGF-β signaling activation and that Kindlin-2 ablation inhibited TGF-β1-induced Smad2 phosphorylation (39). Indeed, our results (Fig. 7) demonstrated that Kindlin-2 deficiency recapitulated pharmacological inhibition of TGF-β receptor and significantly decreased CSF-1 secretion. Therefore, our data identify Kindlin-2 as a new element in a Kindlin-2-TGF-β-CSF-1 signaling axis that is required for macrophage infiltration and tumor growth. Kindlin-2 is a co-activator of integrins (40–42). We and others have shown that loss of Kindlin-2 in many cell types, including BC, inhibits integrin activation (7, 8). Thus, Kindlin-2-mediated regulation of macrophage recruitment to the tumor site may be integrin-dependent, and, hence integrins may intercalate into this signaling axis.
Several studies showed the involvement of a tumor autocrine CSF-1 loop that contributes to tumor growth and metastasis (3, 4). Our data corroborate the importance of this autocrine pathway and show that Kindlin-2 not only regulates CSF-1 expression and secretion (Fig. 6), but also promotes CSF1R, EGF and EGFR, which are all involved in the autocrine loop. KMplotter analysis for the relationship between CSF-1 levels and survival of BC patients showed a similar pattern to that of Kindlin-2: increased levels of CSF-1 correlate with poor outcome in BC patients (p=0.00092, Sup. Fig. 5). Thus, our data support the model shown in Fig. 7I in which Kindlin-2 is involved in both an autocrine and a paracrine CSF-1 signaling loop that activate BC tumor growth.
Cancer tissues with high levels of infiltrating macrophages are associated with poor prognosis and resistance to therapy (43, 44). Our findings demonstrate that Kindlin-2 regulates tumor infiltration by this leukocyte population. Till here, Kindlin-2 modulates the sensitivity to chemotherapeutics in several cancer types (26, 45, 46), thereby providing a link between Kindlin-2-mediated regulation of macrophage and chemoresistance. The hallmarks of cancer as delineated by Hanahan and Weinberg (47) include chemoresistance as well as cancer cell invasion, tumor angiogenesis and interactions between the tumor and its surrounding microenvironment (47). Kindin-2 has now been associated with many of these cancer hallmarks, including tumor survival, growth, progression and metastasis, angiogenesis and chemoresistance (7). Our study now identifies another cancer hallmark regulated by Kindlin-2, i.e., regulation of tumor-stromal interactions through the modulation of macrophage recruitment raises several important directions for future studies. For example, as noted above, Kindlin-2 is likely to insert integrins into the Kindlin-2-TGF-β-CSF-1 signaling pathway; but functions have been ascribed to Kindlin-2 that are independent of integrins (48). Which of the cancer hallmarks involving Kindlin-2 are integrin-dependent and/or integrin-independent, remains to be addressed. Nevertheless, at this juncture, our findings establish an additional interface between Kindlin-2 and tumor biology and suggest that inhibition of Kindlin-2 activity may be a BC therapeutic target. Furthermore, the capacity of Kindin-2 to influence the behavior of cells via paracrine and/or autocrine stimulations broadens its reach even to cells lacking this kindlin and to other biological responses.
Supplementary Material
Acknowledgments
The authors thank Dmitriy Verbovetskiy for his assistance with mouse peritoneal macrophage isolation.
Financial Support: This work was supported in part by NIH grants P01 HL 073311, R01 HL096062 to E.F. Plow.
Footnotes
The authors have no conflicts of interest to disclose.
Reference List
- 1.Mantovani A, Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J Exp Med. 2015;212:435–45. doi: 10.1084/jem.20150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. doi: 10.1038/nrc1256. [DOI] [PubMed] [Google Scholar]
- 3.Patsialou A, Wang Y, Pignatelli J, Chen X, Entenberg D, Oktay M, et al. Autocrine CSF1R signaling mediates switching between invasion and proliferation downstream of TGFbeta in claudin-low breast tumor cells. Oncogene. 2015;34:2721–31. doi: 10.1038/onc.2014.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patsialou A, Wyckoff J, Wang Y, Goswami S, Stanley ER, Condeelis JS. Invasion of human breast cancer cells in vivo requires both paracrine and autocrine loops involving the colony-stimulating factor-1 receptor. Cancer Res. 2009;69:9498–506. doi: 10.1158/0008-5472.CAN-09-1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193:727–40. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Plow EF, Das M, Bialkowska K, Sossey-Alaoui K. Of Kindlins and Cancer. Discoveries (Craiova) 2016:4. doi: 10.15190/d.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rognoni E, Ruppert R, Fassler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci. 2016;129:17–27. doi: 10.1242/jcs.161190. [DOI] [PubMed] [Google Scholar]
- 9.Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol. 2013;14:503–17. doi: 10.1038/nrm3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. 2009;324:895–9. doi: 10.1126/science.1163865. [DOI] [PubMed] [Google Scholar]
- 11.Ye F, Petrich BG. Kindlin: helper, co-activator, or booster of talin in integrin activation? Curr Opin Hematol. 2011;18:356–60. doi: 10.1097/MOH.0b013e3283497f09. [DOI] [PubMed] [Google Scholar]
- 12.Guo B, Gao J, Zhan J, Zhang H. Kindlin-2 interacts with and stabilizes EGFR and is required for EGF-induced breast cancer cell migration. Cancer Lett. 2015;361:271–81. doi: 10.1016/j.canlet.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 13.Mahawithitwong P, Ohuchida K, Ikenaga N, Fujita H, Zhao M, Kozono S, et al. Kindlin-2 expression in peritumoral stroma is associated with poor prognosis in pancreatic ductal adenocarcinoma. Pancreas. 2013;42:663–9. doi: 10.1097/MPA.0b013e318279bd66. [DOI] [PubMed] [Google Scholar]
- 14.Yu Y, Wu J, Guan L, Qi L, Tang Y, Ma B, et al. Kindlin 2 promotes breast cancer invasion via epigenetic silencing of the microRNA200 gene family. Int J Cancer. 2013;133:1368–79. doi: 10.1002/ijc.28151. [DOI] [PubMed] [Google Scholar]
- 15.Zhao T, Guan L, Yu Y, Pei X, Zhan J, Han L, et al. Kindlin-2 promotes genome instability in breast cancer cells. Cancer Lett. 2013;330:208–16. doi: 10.1016/j.canlet.2012.11.043. [DOI] [PubMed] [Google Scholar]
- 16.Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–23. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor MA, Davuluri G, Parvani JG, Schiemann BJ, Wendt MK, Plow EF, et al. Upregulated WAVE3 expression is essential for TGF-beta-mediated EMT and metastasis of triple-negative breast cancer cells. Breast Cancer Res Treat. 2013;142:341–53. doi: 10.1007/s10549-013-2753-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor MA, Sossey-Alaoui K, Thompson CL, Danielpour D, Schiemann WP. TGF-beta upregulates miR-181a expression to promote breast cancer metastasis. J Clin Invest. 2013;123:150–63. doi: 10.1172/JCI64946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wendt MK, Taylor MA, Schiemann BJ, Sossey-Alaoui K, Schiemann WP. Fibroblast growth factor receptor splice variants are stable markers of oncogenic transforming growth factor beta1 signaling in metastatic breast cancers. Breast Cancer Res. 2014;16:R24. doi: 10.1186/bcr3623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soloviev DA, Hazen SL, Szpak D, Bledzka KM, Ballantyne CM, Plow EF, et al. Dual role of the leukocyte integrin alphaMbeta2 in angiogenesis. J Immunol. 2014;193:4712–21. doi: 10.4049/jimmunol.1400202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ploplis VA, French EL, Carmeliet P, Collen D, Plow EF. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. 1998;91:2005–9. [PubMed] [Google Scholar]
- 22.Taylor MA, Amin JD, Kirschmann DA, Schiemann WP. Lysyl oxidase contributes to mechanotransduction-mediated regulation of transforming growth factor-beta signaling in breast cancer cells. Neoplasia. 2011;13:406–18. doi: 10.1593/neo.101086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8:241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 24.Augoff K, Das M, Bialkowska K, McCue B, Plow EF, Sossey-Alaoui K. miR-31 is a broad regulator of beta1-integrin expression and function in cancer cells. Mol Cancer Res. 2011;9:1500–8. doi: 10.1158/1541-7786.MCR-11-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sossey-Alaoui K, Pluskota E, Davuluri G, Bialkowska K, Das M, Szpak D, et al. Kindlin-3 enhances breast cancer progression and metastasis by activating Twist-mediated angiogenesis. FASEB J. 2014;28:2260–71. doi: 10.1096/fj.13-244004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sossey-Alaoui K, Plow EF. miR-138-Mediated Regulation of KINDLIN-2 Expression Modulates Sensitivity to Chemotherapeutics. Mol Cancer Res. 2016;14:228–38. doi: 10.1158/1541-7786.MCR-15-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pluskota E, Dowling JJ, Gordon N, Golden JA, Szpak D, West XZ, et al. The integrin coactivator kindlin-2 plays a critical role in angiogenesis in mice and zebrafish. Blood. 2011;117:4978–87. doi: 10.1182/blood-2010-11-321182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518–24. doi: 10.1038/nature03799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lokody I. Microenvironment: Tumour-promoting tissue mechanics. Nat Rev Cancer. 2014;14:296. doi: 10.1038/nrc3727. [DOI] [PubMed] [Google Scholar]
- 31.Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15:669–82. doi: 10.1038/nri3902. [DOI] [PubMed] [Google Scholar]
- 32.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11:805–15. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 33.Luo Y, Zhou H, Krueger J, Kaplan C, Lee SH, Dolman C, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest. 2006;116:2132–41. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–67. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Craig MJ, Loberg RD. CCL2 (Monocyte Chemoattractant Protein-1) in cancer bone metastases. Cancer Metastasis Rev. 2006;25:611–9. doi: 10.1007/s10555-006-9027-x. [DOI] [PubMed] [Google Scholar]
- 36.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73:1128–41. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santoni M, Massari F, Amantini C, Nabissi M, Maines F, Burattini L, et al. Emerging role of tumor-associated macrophages as therapeutic targets in patients with metastatic renal cell carcinoma. Cancer Immunol Immunother. 2013;62:1757–68. doi: 10.1007/s00262-013-1487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Y, Zhang J, Li JH, Liu X, Wang JZ, Qu HY, et al. High tumor-associated macrophages infiltration is associated with poor prognosis and may contribute to the phenomenon of epithelial-mesenchymal transition in gastric cancer. Onco Targets Ther. 2016;9:3975–83. doi: 10.2147/OTT.S103112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu C, Jiao H, Lai Y, Zheng W, Chen K, Qu H, et al. Kindlin-2 controls TGF-beta signalling and Sox9 expression to regulate chondrogenesis. Nat Commun. 2015;6:7531. doi: 10.1038/ncomms8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malinin NL, Plow EF, Byzova TV. Kindlins in FERM adhesion. Blood. 2010;115:4011–7. doi: 10.1182/blood-2009-10-239269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meves A, Stremmel C, Gottschalk K, Fassler R. The Kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol. 2009;19:504–13. doi: 10.1016/j.tcb.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 42.Plow EF, Qin J, Byzova T. Kindling the flame of integrin activation and function with kindlins. Curr Opin Hematol. 2009;16:323–8. doi: 10.1097/MOH.0b013e32832ea389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allavena P, Mantovani A. Immunology in the clinic review series; focus on cancer: tumour-associated macrophages: undisputed stars of the inflammatory tumour microenvironment. Clin Exp Immunol. 2012;167:195–205. doi: 10.1111/j.1365-2249.2011.04515.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Quigley DA, Kristensen V. Predicting prognosis and therapeutic response from interactions between lymphocytes and tumor cells. Mol Oncol. 2015;9:2054–62. doi: 10.1016/j.molonc.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong X, An Z, Wang Y, Guan L, Fang W, Stromblad S, et al. Kindlin-2 controls sensitivity of prostate cancer cells to cisplatin-induced cell death. Cancer Lett. 2010;299:54–62. doi: 10.1016/j.canlet.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 46.Ou YW, Zhao ZT, Wu CY, Xu BN, Song YM, Zhan QM. Mig-2 attenuates cisplatin-induced apoptosis of human glioma cells in vitro through AKT/JNK and AKT/p38 signaling pathways. Acta Pharmacol Sin. 2014;35:1199–206. doi: 10.1038/aps.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Pluskota E, Ma Y, Bledzka KM, Bialkowska K, Soloviev DA, Szpak D, et al. Kindlin-2 regulates hemostasis by controlling endothelial cell-surface expression of ADP/AMP catabolic enzymes via a clathrin-dependent mechanism. Blood. 2013;122:2491–9. doi: 10.1182/blood-2013-04-497669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.