

Abstract
New bifunctional hexa- and octadentate analogues of the hydroxamate-containing siderophore desferrichrome (DFC) have been synthesized and evaluated as 89Zr-chelating agents for immunoPET applications. The in vitro and in vivo inertness of these new ligands, Orn3-hx (hexadentate) and Orn-4hx derivatives (octadentate), was compared to the gold standard hexadentate, hydroxamate-containing chelator for 89Zr desferrioxamine (DFO). Density functional theory was employed to model the geometries of the resulting Zr(IV) complexes and to predict their relative stabilities as follows: Zr(Orn4-hx) > Zr(DFC) > Zr(Orn3-hx). Transchelation challenge experiments of the corresponding radiochemical complexes with excess ethylenediaminetetraacetate (EDTA) indicated complex stability in accordance with DFT calculations. Radiolabeling of these ligands with 89Zr was quantitative (0.25 μmol of ligand, pH 7.4, room temperature, 20 min). For antibody conjugation, the isothiocyanate (NCS) functional group was introduced to the N terminus of Orn3-hx and Orn-4hx. An additional trifunctional derivative that bears a silicon-rhodamine fluorophore on the C-terminus and NCS on the N terminus was also furnished. As proof of concept, all NCS derivatives were conjugated to the HER2-targeting antibody, trastuzumab. Radiolabeling of immunoconjugates with 89Zr was accomplished with radiochemical yields of 16 ± 2% to 95 ± 2%. These constructs were administered to naive mice (male, C57BL/6J, n = 4) to assess in vivo inertness, which is inversely correlated with uptake of 89Zr in bone, after 96 h circulation time. We found bone uptake to range from 7.0 ± 2.2 to 10.7 ± 1.3% ID/g, values that compare well to the corresponding DFO conjugate (7.1 ± 0.8% ID/g). In conclusion, we have rationally designed linear, bifunctional and trifunctional desferrichrome analogues suitable for the mild and inert radiolabeling of antibodies with the radionuclide 89Zr.
Keywords: desferrichrome, zirconium-89, multimodality, trastuzumab
Graphical abstract
INTRODUCTION
The discovery of the clinical utility of immunoPET has rekindled the interest in long-lived radioisotopes for imaging and therapy with antibodies.1 89Zr has a half-life (78 h) that is well matched to the in vivo pharmacokinetics of monoclonal antibodies (50–120 h), rendering it a useful alternative to 124I and other long-lived, antibody-compatible SPECT nuclides such as 123I and 111In.2,3 The favorable nuclear emission properties for imaging, its antibody-compatible aqueous coordination chemistry, and the cost-effective, high yielding nuclear reactions for its production4,5 have resulted in an intense interest in this isotope since its first demonstrated application for immunoPET.6,7 The chelation chemistry of 89Zr(IV) is dictated by its strongly oxophilic and highly Lewis acidic properties, in addition to its tendency to form complexes with coordination number six to eight.8,9 To meet these needs, Mejis et al. developed desferrioxamine (DFO) and its bifunctional versions as a chelator for Zr(IV).10 DFO has been utilized for radiolabeling a number of therapeutically used antibodies for imaging applications,11 and has remained the gold standard 89Zr chelator to date. However, extensive preclinical in vivo studies showed that while the 89Zr(DFO) complex is kinetically inert over a short time frame, longer in vivo circulation times of 3–7 days result in substantial increases of uptake of 89Zr in the bone marrow, indicating dissociation of the radionuclide from the complex.12 Release of 89Zr from the imaging probe may represent a considerable dosimetry concern and could reduce the efficacy of the immunoPET probe as an imaging tool especially for bone metastases; however, first clinical studies with 89Zr-labeled antibodies indicate that this may be less of a concern in humans.13,14
Improving the bifunctional chelator by optimizing it to match the preferred coordination environment of the metal ion is a critical step toward developing more stable 89Zr-based immunoPET probes for better preclinical probe validation. Crystallographic, potentiometric, and computational studies on the coordinative preferences of the Zr(IV) metal ion, carried out by Brechbiel and Guérard, show that Zr(IV) coordinated by 4 bidentate hydroxamate donor groups maximizes stability, when compared with Zr species coordinated by 3 or fewer bidentate hydroxamate donor groups.15,16 This trend is also observed with hydroxypyridinone donors, albeit less pronounced. Subsequent investigations of hydroxamate and pyridinone containing octadentate or preorganized hexadentate chelator systems by Brechbiel,15,17 Decristoforo,18 Francesconi,19,20 Donnelly,21 and Mindt22,23 have yielded Zr(IV) complexes with improved stability in comparison with the Zr(DFO) complex. A recent study on metal-templated tetrahydroxamate ligand synthesis by Codd further elaborates on the preference of Zr(IV) for an octadentate coordination environment.24
In the development of an improved Zr(IV) chelator, it must meet three criteria: (1) Quantitative radiolabeling must be achieved at room temperature and pH 7.4 due to the temperature and pH sensitivity of proteins and antibodies that are required for biological targeting. (2) The ligand must form a kinetically inert 89Zr(IV) complex to minimize dissociation of the radionuclide over time. In this regard, octadentate chelators, which satisfy the ideal 8-coordinate coordination geometry of Zr(IV), are preferred. (3) The chelator must be easily functionalizable for conjugation to a peptide or antibody without perturbing its clearance properties or binding affinity to the target.
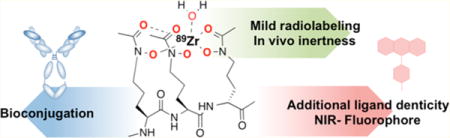
To address these challenges, we looked to nature for inspiration. Desferrichrome (DFC) is a fungal siderophore hexapeptide derived from ornithine. DFC is utilized by both fungi and bacteria as a vehicle for abstraction and absorption of Fe(III) from their host organisms. Albomycin, a naturally occurring sideromycin produced by streptomycetes, is a linear, N-functionalized ornithine tris-hydroxamate also taken up by bacteria.25 The peptidic nature of linear DFC provides high synthetic accessibility, flexibility for modification of the number of donor atoms for coordination, and possibility for the functionalization through the N- or C-termini.26,27 Only few reports describe the investigation of DFC for the coordination of Zr(IV),28,29 but to date, no bifunctional or linear versions have been explored for this purpose. This inspired us to design bifunctional chelators for 89Zr based on linear versions of DFC. Herein, we report the synthesis of the hexadentate, bifunctional chelator Orn3-hx, 3a, the octadentate derivative Orn4-hx, 3b, and their immunoconjugate derivatives, and describe an evaluation of their chelation properties with 89Zr(IV) by transchelation challenge experiments and in vivo biodistribution. Additionally, we evaluated the possibility to generate trifunctional derivatives using the (Orn3-hx) scaffold through C-terminal conjugation to the near-infrared (NIR) fluorophore, silicon rhodamine (SiR),30 while maintaining stable immunoconjugation through the N-terminus (Figure 1).
Figure 1.
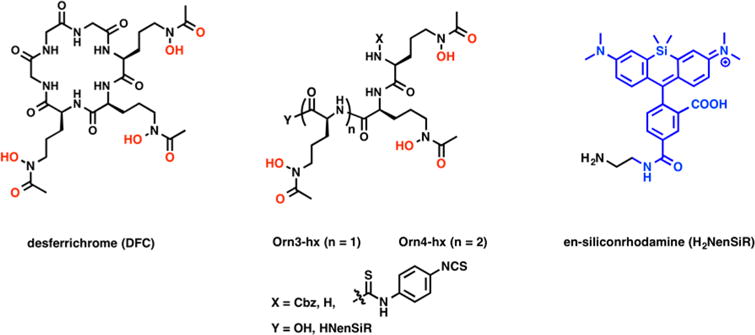
Structures of desferrichrome as well as derivatives of linear polyhydroxamates Orn3-hx/Orn4-hx and their derivatives discussed here.
EXPERIMENTAL SECTION
General Materials and Methods
1H and 13C NMR spectra were recorded at 25 or 60 °C on a Varian 11.7 T or a 9.4 T NMR system equipped with a 5 mm broadband probe. HPLC purification of intermediates was performed on a Rainin-Dynamax preparative HPLC system (Phenomenex C18 column: 250 mm × 21.2 mm, 10 μm) using method A [0.1% TFA in water with a gradient of 5–95% (0.1% TFA in MeCN) over 27 min, 15 mL/min flow rate]. Semipreparative purification was carried out on a Shimadzu semipreparative HPLC system (Phenomenex Luna C18 column: 250 mm × 10 mm, 100 Å, dual wavelength detection at 220 and 254 nm) using method B [0.1% TFA in water with a gradient of 5–95% (0.1% TFA in MeCN) over 21 min, 3.5 mL flow rate] or method C [0.1% TFA in water with a gradient of 5–95% (0.1% TFA in MeCN) over 29 min, 3.5 mL flow rate]. HPLC purity analyses (both UV and MS detection) were carried out on an Agilent 1260 system (Phenomenex Luna C18(2) column: 100 mm × 2 mm, 0.8 mL/min flow rate) with UV detection at 220, 254, and 280 nm and equipped with an Agilent 6130 series electrospray ionization mass spectrometer (+ESI detection) using the following methods: method D (0.1% TFA in water, solvent B = 0.1% TFA in MeCN; 5–95% B over 10 min; method E (0.1% TFA in water, solvent B = 0.1% TFA in MeCN; 5–95% B over 7 min). Radiochemical complexes were characterized using an Agilent 1100 system (Agilent eclipse plus C18 column: 100 mm × 2 mm, 0.8 mL/min flow rate) with UV detection at 220, 254, and 280 nm and equipped with a radiation detector channel using method F (0.1% TFA in water, solvent B = 0.1% TFA in MeCN; 5–95% B over 13 min, 95–5% B over 1.75 min, 5% B for 5 min). The synthesis of ligand precursors was carried out as shown in Schemes 1 and S1. Chemicals were supplied by Acros Organics, Aldrich Chemical Co., Inc., and Fisher Scientific, and were typically used without further purification. Solvents (HPLC grade) were purchased from various commercial suppliers and used as received. 1a, 2a, and 3a were synthesized using previously published protocols.26 H2NenSiR was synthesized as previously described.31
Scheme 1. Synthetic Scheme of Desferrichrome Derivativesa.
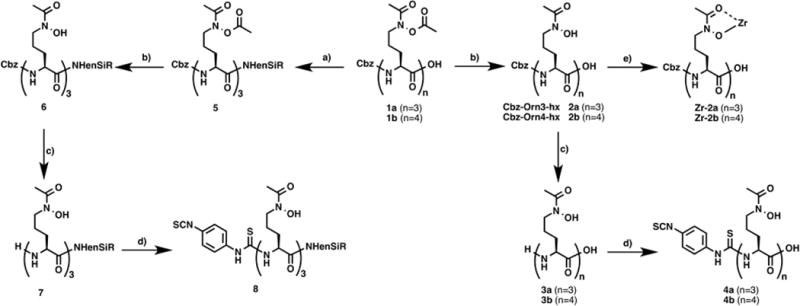
a(a) H2NenSiR, HBTU, NEt3, DMF, 24 h. (b) MeOH, DIPEA, 24 h. (c) Pd/C 10%, MeOH, 1 atm H2, 1 h. (d) p-Phenylene diisothiocyanate, isopropanol/H2O (6:1), chloroform, NEt3, 2 h. (e) Zr(acac)4, MeOH, rt, 1 h.
DFC
1H NMR (DMSO, 500 MHz, ppm): 9.71 (s, 3H, NO–H), 8.45 (s, 1H, NH), 8.29 (s, 1H, NH), 8.20 (s, 1H, NH), 8.03 (s, 2H, NH), 7.78 (s, 1H, NH), 4.25 (s, 1H, αH), 4.09 (s, 1H, αH), 3.95 (s, 1H, αH), 3.77 (d, 1H, αH), 3.67 (d, 1H, αH), 3.55 (d, 1H, αH), 3.47 (s, 6H, δH), 1.98 (s, 9H, CH3), 1.81 (d, 6H, βH), 1.54 (s, 6H, γH). 13C NMR (125 MHz, ppm): 172.4–169.8 (4 signals, amide-C=O), 169.5 (CH3–C=O), 54.5–53.8 (2 signals, αC), 47.1 (δC), 43.1 (αC), 28.8–28.3 (2 signals, βC), 20.8 (γC/CH3).
Zr–DFC
DFC (0.004 g, 0.006 mmol) was dissolved in MeOH (1 mL). To this solution was added Zr(acac)4 (0.002 g, 0.006 mmol). The reaction mixture was stirred at room temperature for 1 h to afford the product quantitatively as a colorless solid after removal of the solvent in vacuo. 1H NMR (DMSO, 500 MHz, ppm): 8.95 (s, 1H, NH), 8.66 (s, 1H, NH), 8.42 (s, 1H, NH), 8.09 (s, 2H, NH), 7.26 (s, 1H, NH), 7.04 (s, 1H, NH), 4.64 (s, 1H, αH), 4.21 (s, 1H, αH), 3.94 (s, 1H, αH), 3.61 (m, 3H, αH), 3.30 (s, 6H, δH), 2.36–1.61 (m, 21H, βH/γH/CH3). 13C NMR (125 MHz, ppm): 173.4 (amide-C=O), 171.7 (amide-C=O), 171.45 (amide-C=O), 170.01 (amide-C=O), 168.26 (amide-C=O), 162.55–162.25 (2 signals, CH3–C=O), 162.25 (CH3–C=O), 53.1–52.2 (2 signals, αC), 50.1–49.3 (3 signals, δC), 44.4–42.3 (3 signals, αC), 27.0–22.5 (6 signals, βC/γC), 17.1–16.8 (2 signals, CH3). LC–ESI-MS (method E), calcd for C27H42N9O12Zr+: 774.2. Found: 774.1 [M]+, tR = 0.77 min.
(5S,8S,11S)-5,8,11-Tris(3-(N-hydroxyacetamido)-propyl)-3,6,9-trioxo-1-phenyl-2-oxa-4,7,10-triazadodecan-12-oic Acid, 2a
2a was synthesized according to the previously published procedure by Miller et al.26 1H NMR (DMSO, 500 MHz, ppm): 11.38 (s, 1H, COOH), 10.00 (s, 3H, NO–H), 8.36–8.17 (m, 2H, NH), 7.66 (s, 6H, Ar–H/NH), 5.32 (s, 2H, Cbz-CH2), 4.60–4.35 (m, 3H, αH), 3.79 (s, 6H, δH), 2.27 (s, 9H, CH3), 1.88 (s, 12H, βH/γH). 13C NMR (DMSO, 125 MHz, ppm): 173.83 (COOH), 172.46–172.04 (3 signals, CH3–C=O), 170.8 (amide-C=O), 156.4 (carbamate-C=O), 137.5 (Ar–C), 128.8–128.1 (3 signals, Ar–C), 65.8 (Cbz-CH2), 54.7 (αC), 52.4–52.2 (2 signals, αC), 47.1–47.0 (2 signals, δC), 29.9–28.7 (3 signals, βC), 23.6–23.4 (2 signals, CH3), 21.5–20.7 (2 signals, γC). LC–ESI-MS (method D), calcd for C29H44N6O12: 668.3. Found: 669.2 [M + H]+, tR = 5.83 min.
Zr–2a
2a (0.020 g, 0.030 mmol) was dissolved in MeOH (1 mL). To this solution was added Zr(acac)4 (0.014 g, 0.030 mmol). The reaction mixture was stirred at room temperature for 1 h to afford the product quantitatively as a colorless solid after removal of the solvent in vacuo. 1H NMR (DMSO, 400 MHz, ppm): 7.32 (s, 4H, Ar–H), 7.02 (s, 1H, Ar–H), 5.65–5.32 (m, 3H, NH), 4.99 (s, 2H, Cbz-CH2), 4.56 (s, 1H, αH), 4.07 (s, 2H, αH), 2.11–1.74 (m, 27H, δH/CH3/βH/γH). 13C NMR (DMSO, 100 MHz, ppm): 206.6 (COOH), 194.39 (CH3–C=O), 192.01 (CH3–C=O), 190.2 (CH3–C=O), 163.4 (amide-C=O), 140.0 (Ar–C), 131.5 (Ar–CH), 130.8 (Ar–CH), 111.35 (Ar–CH), 68.5 (Cbz-CH2), 60.9–60.7 (2 signals, αC), 51.7 (δC), 33.8 (βC), 29.6 (CH3), 27.62 (γC). LC–ESI-MS (method E), calcd for C29H41N6O12Zr+: 755.18. Found: 755.3 [M]+, tR = 2.49 min.
(5 S, 8 S, 11 S, 14 S) - 5, 8, 11, 14 - Tetrakis (3 - (N -hydroxyacetamido)propyl)-3,6,9,12-tetraoxo-1-phenyl-2-oxa-4,7,10,13-tetraazapentadecan-15-oic Acid, 2b
1b (0.050 g, 0.050 mmol) was dissolved in 6% DIPEA in methanol (3 mL). The solution was stirred for 24 h, and the volatiles were removed to yield 2b (0.031 g, 0.0320 mmol, 64%). 1H NMR (DMSO, 500 MHz, ppm): 9.68 (s, 4H, NO–H), 8.02 (s, 1H, NH) 7.87 (s, 1H, NH), 7.35 (s, 5H, Ar–H), 7.31 (s, 1H, NH), 5.02 (s, 2H, Cbz-CH2), 4.26 (s, 2H, αH), 4.13 (s, 1H, αH), 4.01 (s, 1H, αH), 3.48–3.43 (m, 8H, δH), 1.97 (s, 12H, βH), 1.55–1.48 (m, 16H, γH/CH3). 13C NMR (DMSO, 125 MHz, ppm): 176.4 (COOH), 174.9–174.5 (2 signals, amide-C=O), 173.5 (CH3–C=O), 159.1 (carbamate-C=O), 140.1 (Ar–C) 131.5 (Ar–C) 130.8–130.8 (3 signals, Ar–C), 68.5 (Cbz-CH2), 55.1 (broad, δC), 49.6 (αC), 32.4–32.2 (2 signals, βC), 26.1 (γC), 23.4 (CH3). LC–ESI-MS (method D), calcd for C36H56N8O15: 840.9. Found: 841.3 [M + H]+, tR = 5.9 min.
Zr–2b
2b (0.002 g, 0.002 mmol) was dissolved in MeOH (1 mL). To this solution was added Zr(acac)4 (0.014 g, 0.030 mmol). The reaction mixture was stirred at room temperature for 1 h to afford the product quantitatively as a colorless solid after removal of the solvent in vacuo. 1H NMR (DMSO, 500 MHz, ppm): 7.81–7.05 (m, broad, 4H, NH), 7.35 (s, 5H, Ar–H), 5.02 (s, 2H, Cbz-CH2), 4.59–4.08 (m, broad, 4H, αH), 3.68–3.17 (m, broad, 8H, δH), 2.08–1.23 (m, 28H, βH/γH/CH3). 13C NMR (DMSO, 60 °C, 125 MHz, ppm): 203.2 (COOH), 191.4 (amide-C=O), 171.4 (CH3–C=O), 160.9 (carbamate-C=O), 128.7–127.9 (3 signals, Ar–C), 65.9 (Cbz-CH2), 58.3 (αC), 49.0 (δC), 30.9 (CH3), 27.8 (βC), 23.0 (γC). LC–ESI-MS (method D), calcd for C36H52N8O15Zr: 926.3. Found: 927.4 [M + H]+, tR = 3.22 min.
(S)-2-((S)-2-((S)-2-Amino-5-(N-hydroxyacetamido)-pentanamido)-5-(N-hydroxyacetamido)pentanamido)-5-(N-hydroxyacetamido)pentanoic Acid, 3a
3a was synthesized as previously reported.26 In brief, 2a (0.02 g, 0.03 mmol) was dissolved in a mixture of methanol and water (5:1, 5 mL). To this solution was added 10% Pd/C (0.01 g) suspended in methanol. The flask was evacuated, charged with H2 (1 atm), and then stirred for 1 h, until no more starting material was detected by LC–MS. The reaction mixture was filtered, and the volatiles were removed from the filtrate to yield 3a (0.01 g, 0.02 mmol, 63%).
(S)-5-(N-Hydroxyacetamido)-2-((S)-5-(N-hydroxyace-tamido)-2-((S)-5-(N-hydroxyacetamido)-2-(3-(4-isothiocyanatophenyl)thioureido)pentanamido)-pentanamido)pentanoic Acid, 4a
3a (0.013 g, 0.024 mmol) was dissolved in 3.5 mL of isopropanol/H2O (6:1). To this solution was added p-phenylendiisothiocyanate (0.019 g, 0.099 mmol) in chloroform (2 mL), followed by 5 μL of triethylamine. The solution was stirred for 2 h. The volatiles were removed, and the resulting residue was purified using semipreparative HPLC and method C, with the product eluting at 13.8 min. The fractions containing product were pooled, and the solvent was removed in vacuo to afford 4a (0.009 g, 0.012 mmol, 17%) as a white solid. 1H NMR (DMSO, 500 MHz, ppm): 9.97 (s, 1H, Bn–NH), 9.69 (s, 3H, NO–H), 8.18–7.90 (m, 3H, NH), 7.66–7.65 (d, 2H, Ar–H), 7.38–7.36 (d, 2H, Ar–H), 4.93 (s, 1H, αH), 4.30–4.13 (m, 2H, αH), 2.62 (s, 6H, δH), 1.97 (s, 9H, CH3) 1.78–1.56 (m, 12H, βH/γH). 13C NMR (DMSO, 125 MHz, ppm): 226.09 (C=S), 186.9 (COOH), 170.8 (amide-C=O), 158.2 (CH3–C=O), 139.7 (Ar–C), 133.1 (Ar–C), 126.6 Ar–C), 123.2 (Ar–C), 118.6 (Ar–C), 109.9 (Ar–C), 46.5 (δC), 23.5 (βC), 20.7 (CH3), αC n.o. LC–ESI-MS (method E), calcd for C29H42N8O10S2: 726.3. Found: 727.2 [M + H]+, tR = 3.17 min.
(2S,5S,8S,11S)-11-Amino-15-hydroxy-2,5,8-tris(3-(N-hydroxyacetamido)propyl)-4,7,10,16-tetraoxo-3,6,9,15-tetraazaheptadecanoic Acid, 3b
2b (0.010 g, 0.012 mmol) was dissolved in methanol (1 mL). To this solution was added 10% Pd/C (0.002 g) suspended in methanol. The flask was evacuated, charged with H2 (1 atm), and then stirred for 1 h, until no more starting material was detected by LC–MS. The reaction mixture was filtered, and the volatiles were removed from the filtrate to yield 3b (0.007 g, 0.010 mmol, 83%). 1H NMR (DMSO, 400 MHz, ppm): 9.73–9.70 (d, 4H, NO–H), 8.45–8.44 (d, 2H, NH2), 8.06 (s, 3H, NH), 4.32–4.25 (m, 2H, αH) 4.11 (s, 1H, αH) 3.76 (s, 1H, αH), 3.45 (s, 8H, δH), 1.94 (s, 12H, CH3), 1.57–1.53 (m, 16H, βH/γH). 13C NMR (DMSO, 100 MHz, ppm): 176.4 (amide-C=O), 174.6–173.4 (3 signals, amide-C=O), 171.5 (CH3–C=O), 55.0 (broad, αC), 49.8 (δC), 32.5–31.7 (2 signals, βC), 26.1 (γC), 23.4 (CH3). LC–ESI-MS (method E), calcd for C28H50N8O13: 706.7. Found: 707.3 [M + H]+, tR = 2.06 min.
(2S, 5S, 8S, 11S)-15-Hydroxy-2,5,8-tris(3-(N-hydroxyacetamido) propyl) - 11 - (3 - (4 -isothiocyanatophenyl)thioureido)-4,7,10,16-tetraoxo-3,6,9,15-tetraazaheptadecanoic Acid, 4b
3b (0.001 g, 0.001 mmol) was dissolved in 900 μL of isopropanol/H2O(8:1). To this solution was added p-phenylene diisothiocyanate (0.0015 g, 0.008 mmol) dissolved in 100 μL of chloroform, followed by triethylamine (0.5 μL). The solution was stirred for 2 h. The volatiles were removed, and the resulting residue was purified using semipreparative HPLC and method C, with the product eluting at 12.4 min. The fractions containing product were pooled, and the solvent was removed in vacuo to afford 4b (0.0005 g, 0.0006 mmol, 39%) as a white solid. 1H NMR (DMSO, 400 MHz, ppm): 9.93 (s, 1H, Bn–NH), 9.67 (s, 3H, NO–H), 8.15–7.92 (m, 4H, NH), 7.63–7.61 (d, 2H, Ar–H), 7.35–7.32 (d, 2H, Ar–H), 4.89 (s, 1H, αH), 4.25 (s, 2H, αH), 4.09 (s, 1H, αH), 1.93 (s, 12H, CH3), 1.52 (m, 16H, βH/γH), δH peak is hidden by H2O peak. LC–ESI-MS (method E), calcd for C36H54N10O13S2: 898.33. Found: 899.3 [M + H]+, tR = 3.12 min.
N - (10 - (4 - (((8 S, 11 S, 14 S) - 11, 14 - Bis (3 - (N -acetoxyacetamido)propyl)-4-acetyl-8-(((benzyloxy)-carbonyl)amino)-2,9,12,15-tetraoxo-3-oxa-4,10,13,16-tetraazaoctadecan-18-yl)carbamoyl)-2-carboxyphenyl)-7-(dimethylamino)-5,5-dimethyldibenzo[b,e]silin-3(5H)-ylidene)-N-methylmethanaminium, 5
1a (0.013 g, 0.016 mmol) was dissolved in DMF (1 mL) with HBTU (0.0075 g, 0.020 mmol). In a separate vial, H2NenSiR (0.010 g, 0.031 mmol) was dissolved in DMF (1 mL) with triethylamine (10 μL). The two solutions were combined, and the reaction mixture was stirred for 18 h at room temperature. The reaction mixture was filtered and purified using semipreparative HPLC and method B, with the product eluting at 12.4 min. The fractions containing product were pooled, and the solvent was removed in vacuo to afford 5 (0.010 g, 0.008 mmol, 47%) as a blue solid. LC–ESI-MS (E), calcd for C64H83N10O17Si+: 1291.57. Found: 646.4 [M + H]2+, tR = 3.047 min.
N-(10-(2-Carboxy-4-(((5S,8S,11S)-5,8,11-tris(3-(N-hydroxyacetamido)propyl)-3,6,9,12-tetraoxo-1-phenyl-2-oxa-4,7,10,13-tetraazapentadecan-15-yl)carbamoyl)-phenyl)-7-(dimethylamino)-5,5-dimethyldibenzo[b,e]-silin-3(5H)-ylidene)-N-methylmethanaminium, 6
5 (0.010 g, 0.008 mmol) was dissolved in methanol (3 mL) with DIPEA (20 μL). The solution was stirred for 18 h, and the volatiles were removed to afford 6 (0.009 g, 0.008 mmol, quantitative). LC–ESI-MS (E), calcd for C58H77N10O14Si+: 1165.54. Found: 583.3 [M + H]2+, tR = 3.30 min.
N-(10-(4-(((5S,8S,11S)-11-Amino-15-hydroxy-5,8-bis-(3-(N-hydroxyacetamido)propyl)-4,7,10,16-tetraoxo-3,6,9,15-tetraazaheptadecyl)carbamoyl)-2-carboxy-phenyl)-7-(dimethylamino)-5,5-dimethyldibenzo[b,e]-silin-3(5H)-ylidene)-N-methylmethanaminium, 7
6 (0.009 g, 0.008 mmol) was dissolved in methanol (2 mL). To this solution was added Pd/C (10% w/w, 0.004 g) suspended in methanol (2 mL). The flask was evacuated, charged with H2 (1 atm), and then stirred for 1 h, until no more starting material was detected by LC–MS. The reaction mixture was filtered, and the volatiles were removed from the filtrate to afford 7 (0.008 g, 0.008 mmol, quantitative). LC–ESI-MS (E), calcd for C50H71N10O12Si+: 1031.50. Found: 516.3 [M + H]2+, tR = 2.83 min.
N-(10-(2-Carboxy-4-(((5S,8S,11S)-15-hydroxy-5,8-bis-(3 - (N - hydroxyacetamido) propyl) - 1 1 - (3 - (4 -isothiocyanatophenyl)thioureido)-4,7,10,16-tetraoxo-3,6,9,15-tetraazaheptadecyl)carbamoyl)phenyl)-7-(di-methylamino)-5,5-dimethyldibenzo[b,e]silin-3(5H)-yli-dene)-N-methylmethanaminium, 8
7 (0.008 g, 0.008 mmol) was dissolved in 3 mL of isopropanol/H2O (6:1). To this solution was added p-phenylene diisothiocyanate (0.012 g, 0.063 mmol) dissolved in 0.5 mL of chloroform, followed by 6.5 μL of triethylamine. The solution was stirred for 2 h. The volatiles were removed, and to the resulting residue was added 2 mL of H2O/acetonitrile (1:1) + 0.1% TFA. The precipitate was filtered off, and the filtrate was purified using semipreparative HPLC and method B, with the product eluting at 11.5 min. The fractions containing product were pooled, and the solvent was removed in vacuo to afford 8 (0.005 g, 0.004 mmol, 49%). LC–ESI-MS (E), calcd for C58H75N12O12S2Si+: 1224.51. Found: 613.3 [M + H]2+, tR = 3.10 min.
Antibody Conjugation
Trastuzumab (100 μL of 5 mg/mL solution) was diluted with pH = 9 NaHCO3 solution (500 μL, 0.1 M) followed by addition of 20 equiv of NCS-functionalized chelator in DMSO. The reaction was gently mixed for 18 h. Subsequently, the solution was purified by passage over ZebaSpin size exclusion column in order to separate nonconjugated small molecules from the antibody. Conjugates were characterized using MALDI-MS, gel electrophoresis, and isotope dilution assay.
Computational Studies
DFT calculations were carried out using the ORCA quantum chemistry program, version 3.0.3.32 Geometries of Zr(IV) complexes were optimized in the gas phase employing the hybrid B3LYP functional. Carbon, hydrogen, nitrogen, and oxygen atoms were assigned the Pople-style 6-31+G(d) basis set;33,34 Zr(IV) was treated using the LANL2DZ effective core potential and the associated double-ζ basis set.35 This choice of functional and basis sets was previously demonstrated to provide reliable geometries and energies for Zr(IV) complexes.9 Coordinates for optimized geometries are given in Tables S1–S5.
Radiolabeling
89Zr(oxalate)4 was received from Washington University at an average specific activity of (15 mCi/mL). For a typical radiolabeling procedure, an aliquot of stock solution was removed and the pH of the solution was adjusted with 1 M Na2CO3 (105 μL) to 7.4–7.6 and used as the neutralized working stock solution of 89Zr(oxalate)4. For the labeling of nonfunctionalized molecules DFO, DFC, 2a, and 2b, an aliquot of 89Zr(oxalate)4 (45 μCi, 15 μL, pH ∼ 7.2) was thoroughly mixed with a solution of ligand (250 μL, 1 mM) in Chelex-resin treated water. Complexation was monitored by radio-HPLC, method F. 89Zr(oxalate)4 elutes with the mobile phase front (tR = 3 min). 89Zr–DFO: tR = 7 min. 89Zr–DFC: tR = 6.6 min. 89Zr–2a: tR = 8.7 min. 89Zr–2b: tR = 8.8 min. For assessment of radiochemical yield as a function of ligand concentration, a 100 μL aliquot of ligand solution in Chelex-resin treated water was incubated with 89Zr(oxalate)4 (5 μCi, 5 μL, pH ∼ 7.2) for 12 h. Subsequently, the radiochemical yield was determined by radio-TLC. Ligand concentrations of 0.015–1500 μM were investigated. For the radiolabeling of immunoconjugates, an aliquot of 89Zr(oxalate)4 was mixed with 200 μg of trastuzumab conjugate in 300 μL of saline and reacted under slow mixing for 90 min. The reaction was monitored using radio-TLC (solid phase: Sigma-Aldrich, silica gel on TLC aluminum foils, 10 × 70 mm; mobile phase; 50 mM EDTA, pH 7). Under these conditions, 89Zr(oxalate)4 migrates with the solvent front, while chelated 89Zr remains at the origin. Complex solutions for in vivo administration were purified by size exclusion chromatography using the ZebaSpin size exclusion column protocol. The labeled product was characterized via radio-TLC and autoradiography of the corresponding electrophoresis gel. Isotope dilution assay was carried out as previously reported.19
In Vivo Study and Biodistribution
All animal experiments were conducted according to the guidelines of the Institutional Animal Care and Use Committee (IACUC). For the evaluation of in vivo behavior through imaging and biodistribution, healthy, male C57BL6 mice (6 weeks old, Charles River Laboratories, Cambridge, MA) were intravenously injected with 89Zr-labeled antibody (10–40 μCi, n = 4, 0.1–0.3 nmol of mAb per mouse) through a tail vein catheter. Mice were sacrificed at 96 h postinjection. Select organs were harvested and collected; radioactivity was counted by using a gamma counter. For calibration of counts in organs, known dose quantities were measured to generate a standard curve. Radioactivity associated with each organ was expressed as % ID/g. Biodistribution data were assessed by unpaired t tests by using GraphPad Prism (version 7 for Mac OS X Graph-Pad Software, San Diego, CA, USA) to determine if differences between groups were statistically significant (p < 0.05).
RESULTS AND DISCUSSION
Synthesis
Synthetic sideromycins (siderophore–antibiotic conjugates) have been widely explored as Trojan horse systems for the development of new antibiotics with less susceptibility to bacterial resistance. Among the siderophores explored, a number of polyhydroxamates, such as linear desferrichrome analogues, have been synthesized and evaluated for their ability to act as delivery vehicles for antibiotics. Desferrichrome analogues are typically accessed through synthesis of the N-protected tris-ornithine, followed by deprotection of the primary amine and stepwise amine group transformation to the corresponding hydroxamate. We followed and adapted the protocol by Miller and co-workers26 to synthesize 1a and 1b starting from the single amino acid building blocks Cbz-Orn(Boc)–OH and H-Orn(Boc)–OH. We found that preformation and coupling of ornithine dipeptides increases yields for the Orn4-hx precursor significantly (Supporting Information). After assembly of the N-Boc-protected poly-ornithine, a 5-step functional group transformation starting with deprotection, formation of a benzyl-imine, oxidation with mCPBA to the oxaziridine, and hydrolysis to the hydroxylamine followed by acetylation produces the key intermediates 1a and 1b (Scheme 1). Partial deacetylation affords the model ligands Cbz-Orn3-hx–OH (2a) and Cbz-Orn4-hx–OH (2b). Debenzylation and thiourea bond formation with 1,4-diisothiocyanatobenzene provides the bifunctional versions 4a and 4b. The peptidic nature of 3a/3b is well suited for bilateral functionalization to produce trifunctional derivatives. To further explore this, we carried out C-terminal functionalization using the ethylenediamine-functionalized near-IR dye silicon-rhodamine30 (H2NenSiR) on fully acetylated precursor 1a to avoid decomposition of the hydroxamate.36 Subsequent deacetylation of 5 followed by debenzylation afforded 7 with quantitative yields. This species was reacted with 1,4-diisothiocyanatobenzene, providing 8 as a light blue solid indicating the presence of SiR within the molecule.
Zr(IV) Complexation
To study the coordination of linear desferrichrome analogues with Zr(IV), we selected 2a and 2b as N-functionalized model systems. We also hypothesized that the Cbz protective group would increase the lipophilicity of the Zr(IV) complex for easier chromatographic distinction from hydrophilic noncomplexed Zr species in our radiochemical studies. The treatment of the ligands with ZrCl4 to form the complex resulted in insoluble products in all instances. As an alternative to ZrCl4 we explored Zr(acac)4 as a starting material. These compounds were characterized by NMR spectroscopy, which revealed the presence of residual acetylacetonate (Figure 2). Desferrichrome, 2a, and 2b were mixed with Zr(acac)4 in methanol at room temperature for 2 h. The complexation reactions proceeded rapidly and quantitatively as assessed by the disappearance of the diagnostic UV absorption corresponding to the free ligand and appearance of the resulting product peak. The Zr(IV) complexes Zr–2a and Zr–2b were further characterized using mass spectrometry and NMR spectroscopy. The 1H NMR spectra show the disappearance of characteristic NO–H shifts upon complexation, as well as a wider chemical shift dispersion of amide protons of the peptide backbone. The alpha-carbon protons of ornithine experience a change in chemical shift when the ligand is coordinated to Zr(IV), which is consistent with previous NMR studies on the Al(III) and Ga(III) desferrichrome complexes by Llinas and co-workers (Figures 2A, 2B).37 We note peak broadening for diastereotopic α carbon and amide protons for the linear chelates Zr–2a (Figure S2) and Zr–2b (Figures 2C, 2D) with no significant changes to the NMR spectrum within the studied temperature range (25–60 °C), indicating a greater degree of conformational motion that is otherwise restricted by the preorganized peptide macrocycle in the corresponding DFC complexes.
Figure 2.

1H NMR spectra of (a) desferrichrome, (b) Zr(DFC), (c) 2b, and (d) Zr(2b) at 500 MHz in d6-DMSO at 60 °C. Chemical shift changes are indicated for amide and α carbon protons. Impurities are denoted with aserisks (*), as well as residual acetylacetonate (acac) and solvent.
Radiolabeling
An aliquot of pH-neutralized 89Zr(oxalate)4 (45 μCi, 15 μL, pH ∼ 7.2) was mixed with a solution of DFC, 2a, or 2b (250 μL, 1 mM) in Chelex-treated water. Complexation reactions were found to be quantitative within 20 min at room temperature, producing a single labeled species in each instance (Figure 3A) as characterized by radio-HPLC. Residual 89Zr(oxalate)4 elutes with the mobile phase front. 89Zr–2a and 89Zr–2b elute with near identical retention time as polarity of these complexes is mostly dominated by the Cbz-protective group and less by the overall charge. A study of radiolabeling yield as a function of varying ligand concentration was undertaken. For DFC and 2b, radiochemical yields of 99% were achieved at chelator concentrations of 15 μM. For 2a, radiochemical yields of 99% were achieved at chelator concentrations of 1.5 μM (Table S7, Figure 3B). At these concentrations, radiochemical yields follow the order 89Zr–DFC < 89Zr–2b < 89Zr–2a.
Figure 3.
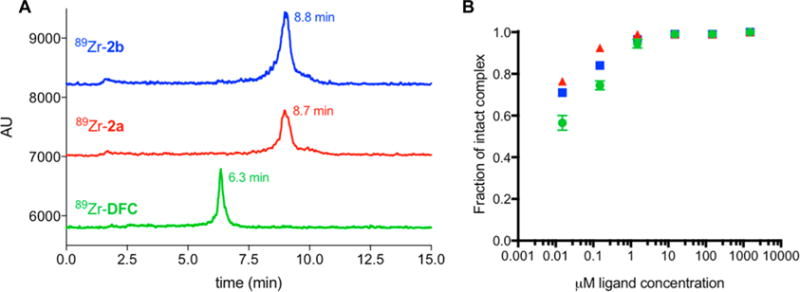
(A) Radio-HPLC traces of 89Zr complexes 89Zr–DFC, 89Zr–2a, 89Zr–2b. (B) Concentration dependence of radiochemical yield with DFC, 2a, and 2b as a function of ligand concentration.
Complex Challenge Experiments
In order to assess inertness of the complexes toward transchelation and to predict in vivo stability, we carried out ligand exchange challenge experiments with a ∼1000-fold excess ethylenediaminetetraacetate (EDTA) and compared complex performance with 89Zr–DFO.17 Desferrichrome derivatives behaved as follows: 89Zr–2b ∼ 89Zr–DFO > 89Zr–DFC > 89Zr–2a (Figure 4, Table S6). We hypothesize that the peptide macrocycle of DFC imparts considerable complex inertness when compared with the acyclic analogue 2a. The octadentate 2b offers a better saturation of the coordination environment around the Zr(IV) center than both hexadentate ligands and thus exhibits greater stability. 89Zr–2b and 89Zr–DFO appear to have both comparable kinetic and thermodynamic behavior when challenged with large excess of EDTA, which suggests that the bifunctional versions of both 2a and 2b, with emphasis on 2b, may demonstrate in vivo inertness competitive with that of 89Zr–DFO complexes.
Figure 4.

1000-fold excess EDTA challenge experiment: Complex integrity is expressed as a function of fraction of intact complex and time upon incubation with EDTA.
DFT Calculations
To investigate the structures of the resulting Zr(IV)–hydroxamate complexes, DFT calculations were employed. Because Zr(IV) typically forms complexes of coordination number eight, we attempted to optimize geometries of the six-coordinate DFC and 3a ligand complexes with two inner-sphere water molecules. A similar approach was carried out in a computational investigation of [Zr(DFO)-(OH2)2]+ complex.38 During the course of the optimizations, however, only a single water remained within the coordination sphere of the Zr(IV) center. We take these results to indicate that the backbone organization of the hydroxamates on DFC and 3a allow for only a single water molecule to bind to Zr(IV), giving rise to seven-coordinate complexes. Such coordination numbers are established for Zr(IV) bearing three bidentate ligands and one monodentate ligand.39 The Zr(IV) complex of 3b converged to a stable 8-coordinate geometry with all four hydroxamate arms interacting with the Zr center. These optimized structures are shown in Figure 5, and relevant structural parameters are collected in Table 1. For comparison, the geometries of [Zr(AHA)4] and [Zr(AHA)3 (OH2)]+ (AHA = N-methyl acetohydroxamic acid) were optimized, and the interatomic distances are given in Table 1. These structures represent the relaxed coordination of the hydroxamate ligand with no constraints imposed by the peptide backbone. The [Zr(AHA)4] complex was previously characterized by X-ray crystallography.15 The crystallographic results showed disorder of the N–O and C–O groups of the hydroxamate ligands, indicating the presence of several stereoisomers in the solid state. For these calculations, we optimized only the stereoisomer of [Zr(AHA)4] where all C–O groups reside at the square vertices of the distorted twisted square antiprismatic coordination polyhedron. This stereoisomer is optimal for comparison to the 3b and 3a ligands, which constrain the Zr coordination sphere in this manner. The Zr–ligand distances are similar for the complexes with multidentate ligands and the relaxed AHA ligands. The 8-coordinate Zr(IV) complexes generally exhibit longer Zr–Ohydroxamate distances, which range from 2.151 to 2.299 Å, compared to those for the 7-coordinate complexes, which range from 2.083 to 2.269 Å. For comparison, in the crystal structure of [Zr(AHA)4], Zr–O distances vary from 2.163 to 2.233 Å,15 reflecting the accuracy of the DFT calculations. Previous quantum chemical calculations on [Zr(AHA)4] revealed longer Zr–OC distances than Zr–ON distances. Although a different basis set was used, the longer Zr–OC (2.268–2.274 Å) and shorter Zr–ON distances (2.182–2.219 Å) are reproduced in the calculations reported here.
Figure 5.
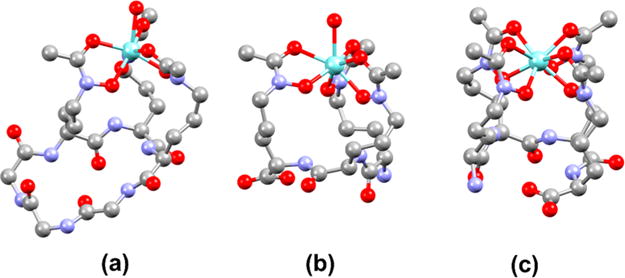
DFT-optimized structures of (a) [Zr(DFC)(OH2)]+, (b) [Zr(3a)(OH2)]+, and (c) [Zr(3b)].
Table 1.
Interatomic Distances (Å) of the DFT-Optimized Structures
| [Zr(AHA)3(OH2)]+ | [Zr(DFC)(OH2)]+ | [Zr(3a)(OH2)]+ | [Zr(AHA)4] | [Zr(3b)] | |
|---|---|---|---|---|---|
| Zr–O(C) | 2.178 | 2.153 | 2.157 | 2.268 | 2.240 |
| 2.215 | 2.206 | 2.206 | 2.274 | 2.264 | |
| 2.269 | 2.240 | 2.247 | 2.274 | 2.299 | |
| 2.276 | 2.299 | ||||
| Zr–O(N) | 2.083 | 2.089 | 2.087 | 2.182 | 2.151 |
| 2.115 | 2.134 | 2.131 | 2.183 | 2.192 | |
| 2.127 | 2.157 | 2.156 | 2.218 | 2.212 | |
| 2.219 | 2.216 | ||||
| Zr–OH2 | 2.337 | 2.339 | 2.334 |
To compare structural differences more precisely, the root-mean-square differences (rmsd) of the relaxed AHA structures with the inner coordination sphere of the multidentate Zr(IV) complexes were determined (Table 2). The rmsd values of the inner coordination spheres of [Zr(3a)(OH2)]+ and [Zr(DFC)-(OH2)]+ compared to [Zr(AHA)3(OH2)]+ are 0.1802 and 0.1623 Å, respectively. The smaller rmsd for the DFC complex indicates that this cyclic ligand provides a coordination framework for Zr(IV) that is closer to the relaxed structure and therefore more optimal than that provided by the linear 3a. In comparing the structures of [Zr(AHA)4] and [Zr(3b)], a substantially larger rmsd of 0.8569 Å is revealed, suggesting that the peptide backbone of the 3b ligand arranges the four hydroxamates about the Zr(IV) center in a highly distorted manner.
Table 2.
Rmsd (Å) from Relaxed Structures and ΔFMO (eV) of DFT-Optimized Structures
| compound | rmsd (Å) | ΔFMOa (eV) |
|---|---|---|
| [Zr(DFC)(OH2)]+ | 0.1623b | 4.390 |
| [Zr(3a)(OH2)]+ | 0.1802b | 4.287 |
| [Zr(3b)] | 0.8569c | 4.625 |
Energy gap between the HOMO and LUMO.
Root-mean-square difference between of the inner coordination sphere of the complex with the DFT-optimized geometry of [Zr(AHA)3(OH2)]+
Rmsd between the inner coordination sphere of the complex with the DFT optimized geometry of [Zr(AHA)4].
The EDTA challenge experiments of the Zr(IV)–ligand complexes described above reveal the general trend DFO ≈ 3b > DFC > 3b at equilibrium. The larger structural deviation of the 3a complex from the relaxed [Zr(AHA)3(OH2)]+ complex is consistent with its poorer stability than the DFC complex. The 3b complex, however, is the most distorted among the three structures, indicated by the large rmsd from the relaxed [Zr(AHA)4] complex. Despite this suboptimal coordination geometry, [Zr(3b)] is more stable than the less distorted [Zr(DFC)(OH2)]+ and [Zr(3a)(OH2)]+ complexes. This result suggests that the extra hydroxamate arm of 3b is critical for its stability. The increased electrostatic interaction of the four anionic hydroxamates with the Zr(IV) center and the entropic favorability of this octadentate ligand drive its stable complexation. The energy gap between the HOMO and LUMO, ΔFMO, has recently been proposed to be a simple indicator of Zr(IV) complex stability.9 Consistent with this finding, among the three complexes [Zr(3b)] has the largest ΔFMO value of 4.625 eV. Following with the trend in stability with DFC > 3a, the ΔFMO values of [Zr(DFC)(OH2)]+ and [Zr(3a)(OH2)]+ are 4.390 and 4.287 eV, respectively. We note that the ΔFMO is a thermodynamic measure and therefore may only be compared to results of the EDTA challenge experiments at equilibrium. The DFT calculations reflect the importance of having an octadentate chelation for the formation of stable Zr(IV) complexes, as it appears to more greatly stabilize the complex. These calculations also suggest that more stable complexes may be obtained by tailoring such an octadentate ligand for an optimal coordination geometry for Zr(IV).
Antibody Conjugation and Radiolabeling
To assess radiolabeling properties and kinetic inertness of the bi- and trifunctional 89Zr complexes over an extended period of time in vivo, derivatives 4a, 4b, and 8 were conjugated to the monoclonal antibody trastuzumab, which targets the membrane-associated antigen HER2, a marker highly expressed in breast cancer cell lines. We selected trastuzumab as the proof-of-concept mAb because it has been thoroughly assessed preclinically as an immunoPET probe with radionuclides such as 64Cu,40124I,41 89Zr,42 and 111In.43 Notably, the first phase 1 and 2 clinical immunoPET studies of 89Zr-labeled trastuzumab have been undertaken.11 We used nonspecific thiourea conjugation to the terminal amines (lysines) present on the mAb to introduce the desferrichrome derivatives and form the DFO–NCS conjugate as a reference compound (Scheme 2). We found good conjugation efficiency under conditions involving a 20:1 ratio of chelator to mAb and gentle mixing of the pH 9 NaHCO3-buffered reaction mixtures for 18 h. We assessed conjugation efficiency by radiometric isotope dilution assay; in brief, varying quantities of natZr(oxalate)4 doped with radioactive 89Zr(oxalate)4 are reacted with a constant amount of chelator–antibody conjugate, followed by determination of radiochemical yield. Table 3 summarizes the calculated chelator-to-antibody ratio as well as the radiolabeling yields obtained for the immunoconjugates; we note that the intensity of fluorescence emission at 664–669 nm (λmaxEm for SiR and SiR derivatives, Figure S3) can also be used for direct quantification of the number of fluorophores and chelators incorporated after coupling with 8. We have used a similar approach to quantify conjugation efficiency of fluorogenic BODIPY-DFO derivatives to antibodies.44 The trifunctional trastuzumab conjugate 8–trastuzumab was also utilized for the study of probe uptake in cells using fluorescence microscopy. We incubated BT474 cells (5 × 104) with 15 μg of conjugate for 24 h and subsequently assessed cell uptake by fluorescence microscopy. We observed emission of fluorescence at 694 nm (Cy5.5 channel) in the form of puncta, indicative of uptake and internalization of the conjugate in cells (Figure S4).
Scheme 2.

Sample Synthetic Scheme for a Radiolabeled Linear DFC-Analogue Antibody Conjugate (4a Is Shown)
Table 3.
Summary of Chelator Number and Radiolabeling Yield Obtained with 89Zr for Each Immunoconjugatea
| DFO-NCS–trastuzumab | 4a– trastuzumab | 4b– trastuzumab | 8– trastuzumab | |
|---|---|---|---|---|
| av no. of chelators | 1.03 | 0.54 | 0.81 | 0.45 |
| radiolabeling yield (90 min, 25 °C) | 95 ± 2% | 92 ± 3% | 63 ± 5% | 16 ± 2% |
Determined using radiometric isotope dilution assay.
For labeling with 89Zr, an aliquot of pH-neutralized 89Zr-(oxalate)4 (300 μCi, 100 μL, pH 7.2–7.4) was mixed with a solution of the antibody conjugate (0.300 μL, 0.2 mg, 1.3 nmol) in saline and incubated for 90 min at 25 °C with occasional gentle agitation of the reaction mixture. Radiolabeling was monitored using radio-TLC.45 The radiochemical yields ranged from 16 to 95% after purification (Table 3). The radiolabeling yield for 4b–trastuzumab versus 4a–trastuzumab is somewhat decreased, corresponding well with the observed trend of radiolabeling yields for 89Zr–2a > 89Zr–2b at concentrations below 1 μM. We also note a moderate radiolabeling yield (16%) for 8–trastuzumab when compared to the bifunctional derivatives 4a–trastuzumab and 4b–trastuzumab. We hypothesize that decreased flexibility of the desferrichrome peptide backbone as well as fluorophore-induced interactions with the antibody surface may contribute to strongly decreased radiolabeling kinetics. We purified the corresponding reaction mixture using size exclusion separation and identified the product as the trifunctional immunoconju-gate using gel electrophoresis (Figure S5).
In Vivo Assessment
Whereas the computational studies and transchelation challenge experiments can be used to guide our design of new chelators, they cannot necessarily provide an accurate assessment of in vivo complex inertness. In order to evaluate the radiolabeled immunoconjugates, we followed the approach of Blower and co-workers.46 We intravenously administered purified antibody conjugates to naive C57BL6 mice and assessed biodistribution of the radiolabel after 96 h (10–40 μCi, n = 4, 0.1–0.3 nmol of mAb per mouse).
Specifically, we determined the blood, heart, lung, liver, spleen, kidney, muscle, and bone uptake of the desferrichrome conjugate derivatives in comparison with the gold standard DFO conjugate (Figure 6, Table 4). Furthermore, we were also interested in how functionalization of both the C- and the N-peptide terminus would affect chelate tendency to release the radiometal. Uptake of activity in the bone is characteristic of release of 89Zr(IV) from the chelator. Biodistribution revealed bone uptake to range from 7.0 ± 2.2% ID/g (4b conjugate) to 9.9 ± 1.5% ID/g (4a conjugate) and 10.7 ± 1.3% ID/g (8 conjugate), comparing well to the corresponding DFO conjugate (7.1 ± 0.8% ID/g). Based on bone uptake, which correlates inversely to in vivo complex inertness, the trend of DFO ≈ 3b > 3a is consistent with the DFT calculations and EDTA challenge studies. 89Zr release from 4a–trastuzumab and 8–trastuzumab is comparable as indicated by the very similar uptake of activity in bone in both cohorts and no statistically significant difference for the obtained values. Functionalization of the linear desferrichrome scaffold through the N- or C-terminus thus does not result in significant decrease of the resulting Zr(IV) complex inertness in vivo. We also note a significant difference in blood and liver clearance properties (p < 0.001): All desferrichrome conjugates assessed exhibited significantly accelerated blood clearance in comparison with the DFO conjugate, which may be advantageous for obtaining improved tumor-background ratios in targeted in vivo imaging. However, both 4a–trastuzumab and 4b–trastuzumab exhibit significantly higher liver uptake than DFO–trastuzumab. High liver uptake may be problematic for imaging and discerning of hepatic lesions.
Figure 6.

Summary of biodistribution data 96 h postinjection obtained for immunoconjugates.
Table 4.
% Injected Dose per Gram of 89Zr Obtained 96 h Postinjection Using Immunoconjugates
| DFO-NCS–trastuzumab | 4a–trastuzumab | 4b–trastuzumab | 8–trastuzumab | |
|---|---|---|---|---|
| heart | 8.55 ± 1.68 | 4.75 ± 0.22 | 2.86 ± 0.14 | 2.23 ± 0.32 |
| blood | 26.42 ± 4.51 | 11.82 ± 1.73 | 7.28 ± 0.33 | 5.83 ± 0.48 |
| lung | 12.22 ± 3.00 | 7.99 ± 5.60 | 4.89 ± 0.74 | 3.88 ± 0.32 |
| liver | 9.49 ± 1.52 | 24.91 ± 4.37 | 20.67 ± 1.26 | 5.16 ± 0.44 |
| spleen | 13.22 ± 3.43 | 17.97 ± 3.49 | 6.68 ± 4.57 | 7.34 ± 0.50 |
| kidney | 9.14 ± 1.50 | 5.26 ± 0.65 | 2.34 ± 1.12 | 4.34 ± 0.35 |
| muscle | 2.18 ± 0.41 | 1.29 ± 0.21 | 0.50 ± 0.16 | 0.72 ± 0.04 |
| bone | 7.14 ± 0.79 | 9.97 ± 1.48 | 7.02 ± 2.20 | 10.76 ± 1.25 |
CONCLUSIONS
We have rationally designed bi- and trifunctional linear hexa-and octadentate derivatives of the naturally occurring iron-siderophore desferrichrome and assessed their potential as 89Zr chelators for the radiolabeling of antibodies. DFT calculations suggested enhanced inertness of the octadentate Zr(IV) complex, which was confirmed by in vitro stability testing of radiochemical model complexes. Attachment to the antibody trastuzumab using conventional NCS coupling chemistry produced corresponding immunoconjugates suitable for radiolabeling with 89Zr. The peptidic nature of linear desferrichrome derivatives allows for facile functionalization using both termini without significantly altering complex inertness. All desferrichrome-derived immunoPET probes perform comparably to the gold standard DFO derivative in vivo; specifically, the octadentate derivatives 2b/4b show no significant difference from DFO in terms of EDTA challenge inertness and bone uptake. The in vivo results obtained highlight the great potential of these derivatives for multimodal and multivalent immunoPET probe development with 89Zr. We also note vastly improved blood-clearance properties of all novel derivatives when compared with DFO, a prerequisite to obtain elevated tumor-to-background ratios and enhanced tumor conspicuity in immunoPET. We are confident that linear desferrichrome derivatives will enhance the armamentarium of radiochemists working toward improved immunoPET probes. Bimodal 89Zr-PET/fluorescence imaging studies with the trifunctional immunoconjugate using mouse xenograft tumor models are underway.
Supplementary Material
Acknowledgments
We thank Prof. Peter Caravan for helpful discussions, extensive support, and mentorship. E.B. acknowledges the NHLBI for support (K99HL125728). J.J.W. thanks Cornell University for start-up funding support. Omar Pinkhasov and Clemens K. Probst are acknowledged for their assistance with biodistribution studies. Jonathan C. Carlson and Labros G. Meimetis are thanked for assistance with SiR synthesis and fluorescence spectroscopy. Jing (Jenny) Yang and Chongzhao Ran are acknowledged for assistance with live cell imaging and ex vivo organ imaging. Instrumentation was funded by the National Center for Research Resources and the Ofce of the Director (P41RR14075, S10RR023385, S10OD010650).
Footnotes
Supporting Information
This material is available free of charge at The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.7b00343.
Precursor synthesis, LCMS, NMR, and fluorescence spectra, DFT coordinates, EDTA challenge, radiolabeling, cell uptake images, gel electrophoresis, and ex vivo organ imaging (PDF)
ORCID
Justin J. Wilson: 0000-0002-4086-7982
Eszter Boros: 0000-0002-4186-6586
Notes
The authors declare no competing financial interest.
References
- 1.Boerman OC, Oyen WJ. Immuno-PET of cancer: a revival of antibody imaging. J Nucl Med. 2011;52:1171–1172. doi: 10.2967/jnumed.111.089771. [DOI] [PubMed] [Google Scholar]
- 2.Wright BD, Lapi SE. Designing the magic bullet? The advancement of immuno-PET into clinical use. J Nucl Med. 2013;54:1171–1174. doi: 10.2967/jnumed.113.126086. [DOI] [PubMed] [Google Scholar]
- 3.Wu AM. Antibodies and antimatter: the resurgence of immuno-PET. J Nucl Med. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- 4.Lee FT, Scott AM. Immuno-PET for tumor targeting. J Nucl Med. 2003;44:1282–1283. [PubMed] [Google Scholar]
- 5.Holland JP, Sheh Y, Lewis JS. Standardized methods for the production of high specific-activity zirconium-89. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Link J, Krohn KA, Eary JF, Kishore R, Lewellen T, Johnson M, Badger C, Richter K, Nelp W. 89 Zr for antibody labeling and positron emission tomography. J Labelled Compd Radiopharm. 1986;23:1297–1298. [Google Scholar]
- 7.Dejesus O, Nickles R. Production and purification of 89Zr, a potential PET antibody label. J Labelled Comp Radiopharm. 1990;41:789–790. [Google Scholar]
- 8.Holland JP, Williamson MJ, Lewis JS. Unconventional nuclides for radiopharmaceuticals. Mol Imaging. 2010;9:1–20. [PMC free article] [PubMed] [Google Scholar]
- 9.Holland JP, Vasdev N. Charting the mechanism and reactivity of zirconium oxalate with hydroxamate ligands using density functional theory: implications in new chelate design. Dalton Trans. 2014;43:9872–9884. doi: 10.1039/c4dt00733f. [DOI] [PubMed] [Google Scholar]
- 10.Meijs WE, Herscheid JD, Haisma HJ, Pinedo HM. Evaluation of desferal as a bifunctional chelating agent for labeling antibodies with Zr-89. J Labelled Comp Radiopharm. 1992;43:1443–1447. doi: 10.1016/0883-2889(92)90170-j. [DOI] [PubMed] [Google Scholar]
- 11.Deri MA, Zeglis BM, Francesconi LC, Lewis JS. PET imaging with 89 Zr: from radiochemistry to the clinic. Nucl Med Biol. 2013;40:3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abou DS, Ku T, Smith-Jones PM. In vivo biodistribution and accumulation of 89 Zr in mice. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebhart G, Lamberts L, Wimana Z, Garcia C, Emonts P, Ameye L, Stroobants S, Huizing M, Aftimos P, Tol J. Molecular imaging as a tool to investigate heterogeneity of advanced HER2-positive breast cancer and to predict patient outcome under trastuzumab emtansine (T-DM1): the ZEPHIR trial. Ann Oncol. 2016;27:619–624. doi: 10.1093/annonc/mdv577. [DOI] [PubMed] [Google Scholar]
- 14.Dijkers EC, Oude Munnink TH, Kosterink JG, Brouwers AH, Jager PL, de Jong JR, van Dongen GA, Schröder CP, Lub-de Hooge MN, de Vries EG. Biodistribution of 89Zr-trastuzumab and PET Imaging of HER2-Positive Lesions in Patients With Metastatic Breast Cancer. Clin Pharmacol Ther. 2010;87:586–592. doi: 10.1038/clpt.2010.12. [DOI] [PubMed] [Google Scholar]
- 15.Guérard F, Lee Y-S, Tripier R, Szajek LP, Deschamps JR, Brechbiel MW. Investigation of Zr (IV) and 89 Zr (IV) complexation with hydroxamates: progress towards designing a better chelator than desferrioxamine B for immuno-PET imaging. Chem Commun. 2013;49:1002–1004. doi: 10.1039/c2cc37549d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guerard F, Beyler M, Lee YS, Tripier R, Gestin JF, Brechbiel MW. Investigation of the complexation of natZr(iv) and 89Zr(iv) by hydroxypyridinones for the development of chelators for PET imaging applications. Dalton Trans. 2017;46:4749–4758. doi: 10.1039/c6dt04625h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guérard F, Lee YS, Brechbiel MW. Rational design, synthesis, and evaluation of tetrahydroxamic acid chelators for stable complexation of zirconium (IV) Chem - Eur J. 2014;20:5584–5591. doi: 10.1002/chem.201304115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhai C, Summer D, Rangger C, Franssen GM, Laverman P, Haas H, Petrik M, Haubner R, Decristoforo C. A novel bifunctional cyclic chelator for 89Zr labeling−radiolabeling and targeting properties of RGD conjugates. Mol Pharmaceutics. 2015;12:2142–2150. doi: 10.1021/acs.molpharmaceut.5b00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deri M, Ponnala S, Kozlowski P, Burton-Pye BP, Cicek H, Hu C, Lewis J S, Francesconi L C. p-SCN-Bn-HOPO: A Superior Bifunctional Chelator for 89Zr ImmunoPET. Bioconjugate Chem. 2015;26:2579–259. doi: 10.1021/acs.bioconjchem.5b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deri MA, Ponnala S, Zeglis B M, Pohl G, Dannenberg J, Lewis JS, Francesconi L C. Alternative chelator for 89Zr radiopharmaceuticals: radiolabeling and evaluation of 3, 4, 3-(LI-1, 2-HOPO) J Med Chem. 2014;57:4849–4860. doi: 10.1021/jm500389b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rudd SE, Roselt P, Cullinane C, Hicks RJ, Donnelly PS. A desferrioxamine B squaramide ester for the incorporation of zirconium-89 into antibodies. Chem Commun. 2016;52:11889–11892. doi: 10.1039/c6cc05961a. [DOI] [PubMed] [Google Scholar]
- 22.Patra M, Bauman A, Mari C, Fischer CA, Blacque O, Häussinger D, Gasser G, Mindt TL. An octadentate bifunctional chelating agent for the development of stable zirconium-89 based molecular imaging probes. Chem Commun. 2014;50:11523–11525. doi: 10.1039/c4cc05558f. [DOI] [PubMed] [Google Scholar]
- 23.Vugts DJ, Klaver C, Sewing C, Poot AJ, Adamzek K, Huegli S, Mari C, Visser GWM, Valverde IE, Gasser G, Mindt TL, van Dongen GAMS. Comparison of the octadentate bifunctional chelator DFO*-pPhe-NCS and the clinically used hexadentate bifunctional chelator DFO-pPhe-NCS for 89Zr-immuno-PET. Eur J Nucl Med Mol Imaging. 2017;44:286–295. doi: 10.1007/s00259-016-3499-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tieu W, Lifa T, Katsifis A, Codd R. Octadentate Zirconium(IV)-Loaded Macrocycles with Varied Stoichiometry Assembled From Hydroxamic Acid Monomers using Metal-Templated Synthesis. Inorg Chem. 2017;56:3719–3728. doi: 10.1021/acs.inorgchem.7b00362. [DOI] [PubMed] [Google Scholar]
- 25.Stapley EO, Ormond RE. Similarity of albomycin and grisein. Science. 1957;125:587–589. doi: 10.1126/science.125.3248.587. [DOI] [PubMed] [Google Scholar]
- 26.Lin Y-M, Miller MJ. Practical Synthesis of Hydroxamate-Derived Siderophore Components by an Indirect Oxidation Method and Syntheses of a DIG− Siderophore Conjugate and a Biotin− Siderophore Conjugate. J Org Chem. 1999;64:7451–7458. [Google Scholar]
- 27.Dolence EK, Minnick AA, Lin C-E, Miller MJ, Payne SM. Synthesis and siderophore and antibacterial activity of N5-acetyl-N5-hydroxy-L-ornithine-derived siderophore-beta-lactam conjugates: iron-transport-mediated drug delivery. J Med Chem. 1991;34:968–978. doi: 10.1021/jm00107a014. [DOI] [PubMed] [Google Scholar]
- 28.Petrik M, Zhai C, Novy Z, Urbanek L, Haas H, Decristoforo C. In Vitro and In Vivo Comparison of Selected Ga-68 and Zr-89 Labelled Siderophores. Mol Imaging Biol. 2016;18:344–352. doi: 10.1007/s11307-015-0897-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi K, Oishi S, Kobayashi Y, Ohno H, Tsutsumi H, Hata Y, Fujii N. Synthesis and application of an N δ-acetyl-N δ-hydroxyornithine analog: Identification of novel metal complexes of deferriferrichrysin. Bioorg Med Chem. 2012;20:2651–2655. doi: 10.1016/j.bmc.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 30.Fu M, Xiao Y, Qian X, Zhao D, Xu Y. A design concept of long-wavelength fluorescent analogs of rhodamine dyes: replacement of oxygen with silicon atom. Chem Commun. 2008:1780–1782. doi: 10.1039/b718544h. [DOI] [PubMed] [Google Scholar]
- 31.Koide Y, Urano Y, Hanaoka K, Piao W, Kusakabe M, Saito N, Terai T, Okabe T, Nagano T. Development of NIR Fluorescent Dyes Based on Si−rhodamine for in Vivo Imaging. J Am Chem Soc. 2012;134:5029–5031. doi: 10.1021/ja210375e. [DOI] [PubMed] [Google Scholar]
- 32.Neese F. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci. 2012;2:73–78. [Google Scholar]
- 33.Hehre WJ, Ditchfield R, Pople JA. Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J Chem Phys. 1972;56:2257–2261. [Google Scholar]
- 34.Krishnan R, Binkley JS, Seeger R, Pople JA. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J Chem Phys. 1980;72:650–654. [Google Scholar]
- 35.Hay PJ, Wadt WR. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys. 1985;82:270–283. [Google Scholar]
- 36.Vondenhoff GH, Gadakh B, Severinov K, Van Aerschot A. Microcin C and Albomycin Analogues with Aryl-tetrazole Substituents as Nucleobase Isosters Are Selective Inhibitors of Bacterial Aminoacyl tRNA Synthetases but Lack Efficient Uptake. ChemBioChem. 2012;13:1959–1969. doi: 10.1002/cbic.201200174. [DOI] [PubMed] [Google Scholar]
- 37.Constantine K, De Marco A, Madrid M, Brooks C, Llinas M. The solution conformations of ferrichrome and deferriferrichrome determined by 1H-nmr spectroscopy and computational modeling. Biopolymers. 1990;30:239–256. doi: 10.1002/bip.360300303. [DOI] [PubMed] [Google Scholar]
- 38.Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med. 2010;51:1293–1300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fay RC, VonDreele RB, Stezowski JJ. Crystal and molecular structure of chlorotris (acetylacetonato) zirconium (IV) J Am Chem Soc. 1971;93:2887–2892. [Google Scholar]
- 40.Mortimer JE, Bading JR, Colcher DM, Conti PS, Frankel PH, Carroll MI, Tong S, Poku E, Miles JK, Shively JE. Functional imaging of human epidermal growth factor receptor 2− positive metastatic breast cancer using 64Cu-DOTA-trastuzumab PET. J Nucl Med. 2014;55:23–29. doi: 10.2967/jnumed.113.122630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson MK, Doss M, Shaller C, Narayanan D, Marks JD, Adler LP, Trotter DEG, Adams GP. Quantitative immuno-positron emission tomography imaging of HER2-positive tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005;65:1471–1478. doi: 10.1158/0008-5472.CAN-04-2008. [DOI] [PubMed] [Google Scholar]
- 42.Zhang Y, Hong H, Cai W. PET tracers based on Zirconium-89. Curr Radiopharm. 2011;4:131–139. doi: 10.2174/1874471011104020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costantini DL, Chan C, Cai Z, Vallis KA, Reilly RM. 111In-labeled trastuzumab (Herceptin) modified with nuclear localization sequences (NLS): an Auger electron-emitting radiotherapeutic agent for HER2/neu-amplified breast cancer. J Nucl Med. 2007;48:1357–1368. doi: 10.2967/jnumed.106.037937. [DOI] [PubMed] [Google Scholar]
- 44.Meimetis LG, Boros E, Carlson JC, Ran C, Caravan P, Weissleder R. Bioorthogonal Fluorophore Linked DFO—Technology Enabling Facile Chelator Quantification and Multimodal Imaging of Antibodies. Bioconjugate Chem. 2016;27:257–263. doi: 10.1021/acs.bioconjchem.5b00630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boros E, Holland JP, Kenton N, Rotile N, Caravan P. Macrocycle-based Hydroxamate Ligands for Complexation and Immunoconjugation of 89Zr for PET Imaging. ChemPlusChem. 2016;81:274–281. doi: 10.1002/cplu.201600003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma MT, Meszaros LK, Paterson BM, Berry DJ, Cooper MS, Ma Y, Hider RC, Blower PJ. Tripodal tris (hydroxypyridinone) ligands for immunoconjugate PET imaging with 89 Zr 4+: comparison with desferrioxamine-B. Dalton Trans. 2015;44:4884–4900. doi: 10.1039/c4dt02978j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.