

Abstract
The receptor tyrosine kinase fms-like tyrosine kinase 3 (FLT3), involved in regulating survival, proliferation and differentiation of hematopoietic stem/progenitor cells, is expressed on acute myeloid leukemia (AML) cells in most patients. Mutations of FLT3 resulting in constitutive signaling are common in AML, including internal tandem duplication (ITD) in the juxtamembrane domain in 25% of patients and point mutations in the tyrosine kinase domain in 5%. Patients with AML with FLT3-ITD have a high relapse rate and short relapse-free and overall survival after chemotherapy and after transplant. A number of inhibitors of FLT3 signaling have been identified and are in clinical trials, both alone and with chemotherapy, with the goal of improving clinical outcomes in patients with AML with FLT3 mutations. While inhibitor monotherapy produces clinical responses, they are usually incomplete and transient, and resistance develops rapidly. Diverse combination therapies have been suggested to potentiate the efficacy of FLT3 inhibitors and to prevent development of resistance or overcome resistance. Combinations with epigenetic therapies, proteasome inhibitors, downstream kinase inhibitors, phosphatase activators and other drugs that alter signaling are being explored. This review summarizes the current status of translational and clinical research on FLT3 inhibitors in AML, and discusses novel combination approaches.
Keywords: FLT3 inhibitors, acute myeloid leukemia, FLT3 internal tandem duplication, FLT3 point mutation, resistance
INTRODUCTION
Standard therapy, including intensive chemotherapy with or without allogeneic hematopoietic stem cell transplantation (HSCT), has limited efficacy in acute myeloid leukemia (AML), with a cure rate of only 30 to 40% [1]. Cytogenetic and molecular research has demonstrated the heterogeneity of AML, established prognostic factors for risk stratification and treatment selection, and identified molecular targets for therapy [1]. Among molecular abnormalities, fms-like tyrosine kinase 3 (FLT3) mutations are frequent and well characterized and were the first, and are still one of very few, “actionable” mutations in AML.
TYROSINE KINASE RECEPTOR FLT3
FLT3 structure and function
FLT3, on chromosome 13q12, encodes a receptor tyrosine kinase (RTK) expressed on normal hematopoietic stem/progenitor cells. FLT3 dimerizes and autophosphorylates upon binding of FLT3 ligand (FLT3L), activating the intracellular tyrosine kinase domain (TKD) [2], which causes phosphorylation of downstream molecules, thereby activating signaling cascades that promote transcription of genes regulating survival, proliferation and differentiation [2]. FLT3 is silenced during hematopoietic differentiation [2].
FLT3 mutations
FLT3 is expressed in AML cells of most patients and is mutated in AML cells of approximately 30% [2]. Mutations include internal tandem duplications (ITD), present in AML cells of approximately 25% of patients, and point mutations in the tyrosine kinase domain (TKD), present in approximately 5% [2]. Both ITD and TKD mutations are activating, causing ligand-independent, or constitutive, FLT3 receptor signaling, and thereby promote cytokine-independent AML cell survival and proliferation [2].
In-frame internal tandem duplications within the FLT3 gene (FLT3-ITD) occur most commonly in exon 14, encoding the juxtamembrane (JM) domain. The JM domain inhibits activation of the receptor by steric hindrance, preventing the TKD from assuming an active conformation. Presence of an ITD causes loss of this inhibitory effect, resulting in activation of the TKD [2]. ITDs are of variable size, ranging from 3 to 1,236 nucleotides; loss of FLT3 inhibitory effect is independent of size of the duplication within the receptor [2,3]. Additionally, FLT3 signaling activated by ITDs is aberrant, notably activating signal transducer and activator of transcription (STAT) 5 and its downstream effectors, including Pim-1 kinase [4]. Aberrant signaling occurs in association with partial retention of FLT3-ITD in the endoplasmic reticulum (ER), with trafficking of the receptor out of the ER-Golgi impaired by the presence of the duplicated domain [4].
Point mutations in the TKD are less common; they are present in AML cells of approximately 5% of patients [2]. TKD point mutations cause amino acid substitutions producing changes in the activation loop that favor the active kinase conformation [2].
While both FLT3-ITD and FLT3 TKD mutations result in constitutive activation of FLT3 signaling, signaling pathways differ [5]. FLT3-ITD activates FLT3 signaling through STAT5, in addition to PI3 kinase (PI2K)/Akt and mitogen-activated protein kinase (MEK)/extracellular-signal-regulated kinase (ERK) (Figure 1), while FLT3 TKD mutations activate FLT3 signaling through Akt and ERK, but not STAT5 [5]. Additionally FLT3-ITD suppresses CCAAT/estradiol-binding protein alpha (c/EBPalpha) and Pu.1, transcription factors that promote myeloid differentiation, while FLT3 TKD mutations do not [5].
Figure 1. Signaling pathways in cells with FLT3-ITD and mechanisms of action of drugs studied in combination with FLT3 inhibitors.

The figure shows different mechanisms involved in FLT3 inhibitor resistance, and drugs that can potentially prevent these processes when used in combination with FLT3 inhibitors.
AML with FLT3-ITD usually presents with high blood blast counts and a normal karyotype, and has poor treatment outcomes, with initial treatment response, but high relapse rate and short relapse-free survival (RFS) and overall survival (OS) [2]. ITD locations and allelic ratios vary, as does size, as noted above; higher allelic ratios are associated with lower complete remission (CR) rate and shorter OS [3]. FLT3-ITD is present in CD34+/CD38− AML stem cells [6], the cells that likely generate relapse. New structural cytogenetic abnormalities are frequently present at relapse of AML with FLT3-ITD, consistent with genomic instability [7]. Genomic instability may result from increased DNA double-strand breaks associated with increased reactive oxygen species generation and from error-prone DNA double-strand break repair [8]. HSCT is the preferred treatment for FLT3-ITD AML patients in remission, but outcomes are inferior to those of patients without FLT3-ITD due to a high rate of early relapses, suggesting the potential utility of treatments targeting FLT3 signaling after transplant [9].
In contrast to FLT3-ITD, FLT3 TKD mutations are not associated with leukocytosis and only modestly negatively impact treatment outcomes [2,3]. These clinical differences may be due to the difference in downstream signaling between FLT3-ITD and TKD mutations.
FLT3 INHIBITORS
Preclinical development
Since FLT3 mutations cause ligand-independent cell survival, proliferation and resistance to apoptosis, it was hypothesized that inhibiting FLT3 signaling would produce cytotoxicity and clinical responses. The primary approach has been identification and testing of small molecule inhibitors of FLT3 signaling, but some work has also focused on developing internalizing fully human antagonistic antibodies directed against FLT3 [10].
A number of FLT3 inhibitors have been studied (Table 1). FLT3 inhibitors are classified into first- and second-generation based on their specificity for FLT3, and into type I and type II based on their mechanism of interaction with FLT3.
Table 1. FLT3 inhibitors in clinical trials in AML.
First- and second-generation refers to the phase of FLT3 inhibitor drug discovery, while types I and II refer to the interaction with FLT3, with type I inhibitors active in cells with either FLT3-ITD or FLT3 kinase domain point mutations, while type II inhibitors are active in cells with FLT3-ITD, but not FLT3 kinase domain point mutations.
| FLT3 inhibitors | Type 1 | Type II | ||||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||
| Name | IC50s* (nmol/L) | Other targets | Structure | Name | IC50s* (nmol/L) | Other targets | Structure | |
| First-generation | Sunitinib (SU11248) | ITD 5.4 D835Y >100 |
VEGFR2, PDGFRβ, KIT, RET |
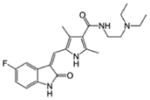
|
Sorafenib (DB00398) | ITD 18.5 D835Y >2000 |
RAF, VEGFR1,2,3, PDGFRβ, KIT, RET |
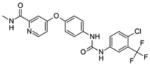
|
| Midostaurin (PKC412) | ITD 9.3 D835Y 10 |
PKC, Syk, Flk-1, Akt, PKA, KIT, Fgr, Src, PDGFRβ, VEGFR1, VEGFR2 |
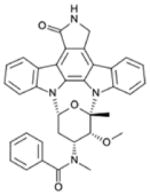
|
Ponatinib (AP24534) | ITD<1 D835Y 92 |
LYN, ABL, PDGFRα, VEGFR2, FGFR1, SRC, KIT, TEK, RET |
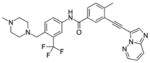
|
|
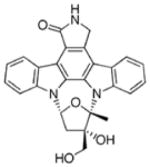
| Lestaurtinib (CEP-701) | ITD 8.6 D835Y 9.8 |
JAK2,3, TrkA,B,C |
|
Tandutinib (MLN518) | ITD 550 D835Y >10,000 |
KIT, PDGFRβ |
|
|
|
| ||||||||
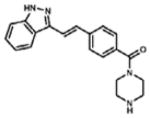
| Second-generation | KW-2449 | ITD 41 D835Y >200 |
ABL, aurora kinase |
|
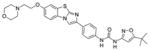
Quizartinib (AC220) | ITD 1.2 D835Y >100 |
KIT, PDGFRβ RET |
|
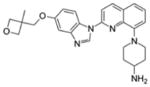
| Crenolanib (CP-868-596) | ITD 57 D835Y 58 |
PDGFRβ |
|
|||||
| Gilteritinib (ASP2215) | ITD 1.6 D835Y 1.4 |
LTK, ALK, AXL |
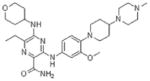
|
|||||
NOTE: First- and second generation refers to the phase of FLT3 inhibitor drug discovery, while types I and II refer to the interaction with FLT3, with type I inhibitors active in cells with either FLT3-ITD or FLT3 kinase domain point mutations, while type II inhibitors are active in cells with FLT3-ITD, but not FLT3 kinase domain point mutations.
References: Nguyen B, Williams AB, Young DJ, Ma H, Li L, Levis M, Brown P, Small D. FLT3 activating mutations display differential sensitivity to multiple tyrosine kinase inhibitors. Oncotarget 2017;8:10931–44; Clark JJ, Cools J, Curley DP, Yu JC, Lokker NA, Giese NA, Gilliland DG. Variable sensitivity of FLT3 activation loop mutations to the small molecule tyrosine kinase inhibitor MLN518. Blood. 2004;104:2867–72. Lee LY, Hernandez D, Rajkhowa T, Smith SC, Raman JR, Nguyen B, Small D, Levis M. Preclinical studies of gilteritinib, a next-generation FLT3 inhibitor. Blood. 2007;129:257–60.
First- and second-generation inhibitors
First-generation inhibitors, including sunitinib, sorafenib, midostaurin, lestaurtinib and tandutinib, lack specificity for FLT3. Inhibition of multiple RTKs may enhance anti-leukemia efficacy by inhibiting targets downstream of FLT3 and/or in parallel signaling pathways, or other targets in AML cells. However, off-target activities also cause toxicities.
In contrast, second-generation FLT3 inhibitors obtained by rational drug development are more specific and potent, and have fewer toxicities associated with off-target effects. However, second-generation FLT3 inhibitors largely only target FLT3, and do not have efficacy against targets downstream of FLT3 or in parallel signaling pathways in AML cells. The second-generation inhibitors quizartinib, crenolanib and gilteritinib are in clinical trials.
Type I and II inhibitors
FLT3 inhibitors are also classified based on their mechanism of interaction with the receptor [11]. Upon activation, FLT3 undergoes a conformational change involving flipping of three residues, Asp-Phe-Gly, or DFG; active and inactive conformations are called DFG-in and DFG-out, respectively. All FLT3 inhibitors interact with the ATP-binding site of the intracellular TKD and competitively inhibit ATP binding, thereby preventing receptor autophosphorylation and activation of downstream signaling. However type I inhibitors bind to the ATP-binding site when the receptor is active, while type II inhibitors interact with a hydrophobic region immediately adjacent to the ATP-binding site that is only accessible when the receptor is in the inactive conformation, and they prevent receptor activation. D835 is the most common site for TKD mutations and D835 mutations favor the active conformation. Consequently type I inhibitors inhibit FLT3 signaling in AML cells with either ITD or TKD mutations, while type II inhibitors inhibit FLT3 with ITD, but not with TKD mutations, though some D835 mutations preserve sensitivity [12]. Importantly, development of D835 mutations in cells with ITD is a mechanism of acquired, or secondary, resistance to type II FLT3 inhibitors [13].
Type I inhibitors include sunitinib, lestaurtinib, midostaurin, crenolanib and gilteritinib, while type II inhibitors include sorafenib, quizartinib and ponatinib.
Clinical trials
FLT3 inhibitors are not yet approved by the United States Food and Drug Administration (FDA). Completed and ongoing clinical trials as monotherapy and with chemotherapy are discussed below.
FLT3 inhibitors may be administered with chemotherapy or after chemotherapy in combination regimens. Administration prior to chemotherapy may decrease chemosensitivity by slowing or arresting cell cycle [14]. This is particularly relevant for cytarabine, with a mechanism of action that requires incorporation into DNA during S-phase. As an additional consideration, concurrent administration of FLT3 inhibitors with chemotherapy may enhance toxicities due to pharmacokinetic interactions. This may be particularly relevant to anthracyclines due to interactions of FLT3 inhibitors and anthracyclines with plasma proteins [14] and, notably, with ATP-binding cassette drug transport proteins [15].
Plasma inhibitory activity (PIA) [16] has been used as a pharmacodynamic assay for FLT3 inhibition in clinical trials. Degree of patient plasma inhibition of FLT3 phosphorylation in FLT3-ITD cell lines is measured by Western blot analysis. In particular, the assay allows measurement of FLT3 inhibition by drugs administered in escalating doses in phase I clinical trials.
First-generation inhibitors
Sorafenib [DB00398; Nexavar® (Bayer)] is a type II FLT3 inhibitor that also inhibits RAF, vascular endothelial growth factor receptors (VEGFR)-1,2,3, platelet-derived growth factor receptor (PDGFR)-β, KIT and RET and is FDA-approved for treatment of renal (2005), hepatocellular (2007) and thyroid (2013) carcinomas.
Sorafenib monotherapy at 200 mg to 400 mg twice daily, as tolerated, decreased marrow blasts in 12 of 13 patients with relapsed or refractory AML with FLT3-ITD in a phase II clinical trial, with mean response duration of 72 days; notably, new D835 TKD mutations emerged in 4 of 6 patients studied at progression [17]. Importantly, sorafenib monotherapy has shown efficacy in treating relapse of FLT3-ITD AML, including after allogeneic HSCT [18]. Median total daily sorafenib dose was 486.5 mg following chemotherapy alone and 600 mg following transplant. Main reasons for dose modifications were cytopenias, rash, hand-foot-syndrome and mucositis. Sorafenib can be safely administered as post-HSCT maintenance therapy, with a maximum tolerated dose of 400 mg twice daily [19], and its efficacy in preventing post-HSCT relapse is being studied (ClinicalTrial.gov identifier: NCT02474290).
Data on efficacy of sorafenib with chemotherapy have been inconsistent. In a randomized, double-blind, placebo-controlled phase II trial of sorafenib after induction and consolidation chemotherapy and as 12-month maintenance therapy in AML patients 18 to 60 years with or without FLT3 mutations, median event-free survival (EFS) was 21 versus 9 months with sorafenib versus placebo, and 3-year EFS 40% versus 22% (p=0·013), though differences in FLT3-ITD AML patients were not statistically significant [20]. Sorafenib treatment was associated with increased toxicities, including fever, diarrhea, bleeding, cardiac events, hand-foot syndrome and rash. In a randomized clinical trial in previously untreated AML patients over 60 years, sorafenib after induction and consolidation chemotherapy was associated with increased toxicities and did not improve EFS or OS, including in those with FLT3 mutations [21]. However 1-year OS doubled versus historical controls (62% vs 30%; p <0.0001) in newly diagnosed adults 60 years and older with ITD or TKD mutations receiving sorafenib on days 1–7 of induction and days 1–28 of consolidation chemotherapy and as 1-year maintenance therapy [22]. Sorafenib maintenance therapy was associated with diarrhea, fatigue, transaminitis and hand-foot syndrome. It should be noted again that sorafenib is a type II FLT3 activity and therefore should be active against ITD, but not against most TKD mutations, including D835 mutations.
Sunitinib [SU11248, Sutent® (Pfizer)], a type I FLT3 inhibitor that also targets VEGFR, PDGFR, KIT and RET, is FDA-approved for treatment of renal carcinoma (2006), gastrointestinal stromal tumor (2006) and pancreatic neuroendocrine tumor (2011). In Phase I clinical trials, sunitinib inhibited FLT3 phosphorylation in 5 of 5 ITD or TKD and, at doses of 200 mg and higher, in 10 of 16 of wild-type (WT) FLT3 AML patients [23], and induced short-lived partial responses in patients with refractory AML, especially with FLT3-ITD or TKD mutations [24]. In a phase I/II trial with standard chemotherapy in previously untreated FLT3-mutated AML patients older than 60 years, sunitinib was tolerable only at 25 mg on days 1–7, due to cytopenias and hand-foot syndrome [25]. Eight of 14 patients with ITD and 5 of 8 with TKD mutations achieved CR or CR with incomplete hematologic recovery (CRi); RFS and OS were 1.0 and 1.6 years, respectively. FLT3 mutations were lost in 4 of 5 patients studied at relapse.
Lestaurtinib (CEP-701), a staurosporine analog, is a type I FLT3 inhibitor with broad specificity. It was active in early clinical trials [26], but did not improve CR rate or OS in a randomized trial versus placebo after chemotherapy in patients with AML with FLT3 ITD or TKD mutations in first relapse [27]. Higher plasma lestaurtinib levels were associated with more effective FLT3 inhibition, but also with greater toxicity. Importantly, FLT3L levels increased following chemotherapy [27], and AML cells remain responsive to growth stimulation by FLT3L despite constitutive FLT3 activation [28], so that increased FLT3L levels may have stimulated AML regrowth and thus caused resistance to lestaurtinib. Lestaurtinib administered to newly diagnosed patients with AML with FLT3-ITD or TKD mutations following induction and consolidation chemotherapy courses, without maintenance therapy, also did not improve remission rate, 5-year overall or relapse-free survival compared to placebo, though outcomes were better in patients with sustained >85% inhibition of FLT3 in vivo, as measured by the PIA assay [29]. FLT3L levels increased over successive treatment courses, but increases in FLT3L levels did not correlate with loss of in vivo FLT3 inhibition [29]. Lestaurtinib is no longer in clinical development.
Midostaurin (PKC412; N-benzoylstaurosporin), also a staurosporine analog, is also a type I FLT3 inhibitor with broad specificity. In a randomized phase II trial in patients with relapsed or refractory AML, midostaurin was tolerated at 50 mg or 100 mg twice daily, with mild to moderate nausea and vomiting as the major toxicity, and reduced blood or marrow blasts by ≥ 50% in 71% of 35 patients with FLT3 ITD or TKD and 42% of 60 patients with WT FLT3, with median durations of 60 and 83 days, respectively [30]. In a subsequent phase Ib trial in newly diagnosed AML patients, midostaurin was well tolerated at 50 mg twice daily for 14 days after cytarabine and daunorubicin induction and high-dose cytarabine consolidation therapy [31]. CR rates were 92% and 80% in FLT3-mutated and -WT patients, respectively, and 2-year OS was 62% and 52%.
A randomized, double-blind multinational phase III trial then compared OS in newly diagnosed AML patients 18 to 60 years old with FLT3-ITD or TKD mutations treated with midostaurin 50 mg versus placebo twice daily on days 8 through 22 after cytarabine and daunorubicin induction and high-dose cytarabine consolidation therapy, and as one-year maintenance therapy. Randomization was stratified by high (>0.7) or low ITD allelic ratio or TKD. The CR rate did not differ for midostaurin vs. placebo, but OS was significantly longer with midostaurin, 74.7 vs. 26.0 months (p= 0.007). The survival benefit was consistent for all three stratification groups and persisted when data were censored at HSCT for transplanted patients (p= 0.047). Reported at the 2015 American Society of Hematology annual meeting [32], this trial was the first documentation of improved outcomes in patients with AML with FLT3 mutations treated with a FLT3 inhibitor. Midostaurin received Breakthrough Therapy designation in February 2016.
The AMLSG 16-10 phase II trial evaluated midostaurin 50 mg twice daily combined with induction chemotherapy and as single-agent maintenance therapy after HSCT in patients 18–70 years with newly diagnosed AML with FLT3-ITD, with outcomes better than in historical controls [33].
Tandutinib (MLN518), a first-generation type II FLT3 inhibitor with promising results in vitro and in vivo, had a good safety profile and showed moderate benefit in Phase I clinical testing [34], but is no longer in development.
The overall disappointing clinical results of treatment with first-generation FLT3 inhibitors were attributed in part to inability to dose drugs optimally due to toxicities associated with off-target effects. Prominent toxicities included gastrointestinal intolerance, prolonged cytopenias and hand-foot syndrome, as detailed above. Therefore it was thought that second-generation FLT3 inhibitors selected for narrow targeting of FLT3 might have greater clinical efficacy.
Second-generation inhibitors
KW-2449, an inhibitor of FLT3, ABL and aurora kinase, had unfavorable pharmacokinetic properties and is no longer in clinical development [35].
Quizartinib (AC220) was identified as a highly selective FLT3 inhibitor with low nanomolar potency in compound library screening, and was found to have favorable pharmacokinetics [36]. In a phase I trial in relapsed and refractory AML patients [37], the maximum tolerated dose was 200mg daily, with prolonged QT interval as the dose-limiting toxicity. Quizartinib produced CR or CRi in 53% and 14% of patients with FLT3-ITD and WT FLT3, respectively, with median response duration and survival of 13.3 and 14 weeks. Quizartinib was then evaluated in a phase II trial in patients with relapsed or refractory AML with FLT3-ITD [38]. Blast counts decreased sufficiently to allow HSCT in 35% of patients, and HSCT prolonged OS. Quizartinib is being further studied in combination with chemotherapy in older (NCT01892371) and younger (NCT01390337) newly diagnosed AML patients with FLT3-ITD, as maintenance therapy after HSCT (NCT01468467), and in a randomized, open-label, phase III clinical trial comparing its efficacy as monotherapy vs. salvage chemotherapies in relapsed or refractory AML patients with FLT3-ITD (NCT02039726). Development of FLT3 point mutations, most commonly at D835, is a mechanism of acquired resistance to quizartinib, which is a type II FLT3 inhibitor [13].
Crenolanib (CP-868-596) is a second-generation type I FLT3 inhibitor with potent cytotoxicity toward both FLT3-ITD and FLT3 D835 leukemia cell lines; it is also a PDGFR inhibitor [39]. Clinical trials are evaluating crenolanib in relapsed/refractory FLT3-mutated AML after chemotherapy with or without another FLT3 inhibitor (NCT01657682), and crenolanib with sorafenib (NCT02270788), based on in vitro crenolanib activity in FLT3-ITD AML cells resistant to sorafenib [40]. Crenolanib is also being studied with chemotherapy in newly diagnosed (NCT02283177) and relapsed/refractory (NCT02400281) AML patients, and with and without azacitidine following HSCT (NCT02829840).
Gilteritinib (ASP2215), a small molecule type I FLT3 and AXL inhibitor has in vivo efficacy alone and prior to and combined with chemotherapy [41]. In a phase I/II clinical trial in refractory or relapsed AML, gilteritinib ≥80 mg showed good tolerability and overall response rates of 55%, 17% and 62% in patients with ITD, TKD and both, respectively, regardless of prior TKI treatment [42]. Gilteritinib is being studied in newly diagnosed AML patients in combination with chemotherapy (NCT02236013) and with azacitidine, compared to azacitidine alone (NCT02181660). It is also being studied in refractory or relapsed AML with FLT3 mutations in a phase III randomized trial compared to salvage chemotherapies (NCT02421939).
Ponatinib (AP23534; Iclusig (Ariad)) was designed to target BCR-ABL, but is also a type II FLT3 inhibitor with potent cytotoxicity against leukemia cells with FLT3-ITD and, to a lesser extent, FLT3 TKD mutations [43]. Based on these preclinical results, a phase I/II clinical trial is evaluating safety and efficacy in combination with cytarabine consolidation for patients younger than 70 years with ITD (NCT02428543) and, with or without azacitidine, for untreated FLT3-ITD AML patients unfit for chemotherapy (NCT02829840).
Finally, ibrutinib (PCI-32765; Imbruvica (Pharmacyclics)), a TKI approved for treatment of lymphoid malignancies, targets cells with FLT3-ITD in vitro, and appears to be a type II FLT3 inhibitor [44].
FLT3 INHIBITOR RESISTANCE
FTL3 inhibitors induce responses in patients with AML with FLT3 mutations, but responses are not durable, and AML progresses in virtually all patients.
Mechanisms of resistance
Mechanisms of primary and secondary resistance to FLT3 inhibitors in FLT3-mutated AML cells are summarized in Table 2. Intrinsic resistance may be primary or secondary.
Table 2. Mechanisms of resistance of FLT3-mutated AML cells to FLT3 inhibitors.
Primary resistance occurs before treatment, whereas secondary resistance is induced by FLT3 inhibitor therapy. Intrinsic mechanisms occur within AML cells, while extrinsic processes are external to AML cells.
| INTRINSIC | EXTRINSIC | |
|---|---|---|
|
| ||
| PRIMARY | SECONDARY | |
|
|
|
An important mechanism of primary intrinsic resistance to FLT3 inhibitors is lack of addiction of AML with a FLT3 mutation to FLT3 signaling due to coexistence of multiple leukemic clones and low allelic burden of the FLT3 mutation, especially at diagnosis of AML, whereas a dominant clone with FLT3 mutation tends to emerge at relapse [45]. This may imply greater efficacy of inhibitors with broader specificity at diagnosis of AML. A second mechanism of primary intrinsic resistance is presence of mutations that prevent interaction with specific drugs, notably TKD mutations conferring resistance to type II FLT3 inhibitors [13]. Additionally, a FLT3-ITD627E mutation has been identified that confers primary resistance to FLT3 inhibitors by upregulating the anti-apoptotic protein Mcl-1 [46], and upregulation of the anti-apoptotic protein Bcl-xL has been demonstrated as a mechanism of FLT3 inhibitor and chemotherapy resistance in FLT3-ITD-TKD dual mutant cells [47]. Upregulation of the anti-apoptotic protein Bcl-2 is also reported as a FLT3-independent mechanism of resistance to FLT3 inhibitors in AML cells with FLT3 mutations at diagnosis [48].
Induction of new TKD mutations occurs as a very frequent secondary intrinsic mechanism of resistance to type II FLT3 inhibitors, present, for example, in FLT3-ITD AML relapsed following quizartinib treatment in 8 of 8 patients in one series [13], and also documented in 4 of 6 patients studied at progression following sorafenib therapy in an early series [17]. Genomic instability also appears to be a common phenomenon, with new structural chromosome changes documented at relapse of cytogenetically normal AML with FLT3-ITD in 10 of 12 patients, several of whom were treated with FLT3 inhibitors, in a published series [8]; this is a general mechanism of disease progression, rather than a specific mechanism of FLT3 inhibitor resistance. The oncogenic serine/threonine kinase Pim-1 is transcriptionally upregulated downstream of FLT3-ITD, and potentiates FLT3 signaling in a positive feedback loop [49,50]. In samples from seven FLT3-ITD AML patients with acquired resistance to sorafenib in one series, Pim-1 and the Pim kinase isoform Pim-2 were upregulated in 2 and 4 samples, respectively, compared to pre-treatment, while new TKD mutations were present in 4, without correlation with Pim upregulation [50]. Importantly, pharmacological or genetic inhibition of Pim kinases restored sensitivity of FLT3-ITD cells to FLT3 inhibitors in a mouse model [50]. Increased phosphorylation of the RTK AXL is seen in cells with FLT3-ITD after treatment with FLT3 inhibitors; moreover, FLT3 inhibitor resistance is associated with increased phosphorylated AXL, and AXL inhibition overcomes resistance [51]. Notably, gilteritinib inhibits AXL, in addition to FLT3 [41]. The cytoplasmic kinase spleen tyrosine kinase (SYK) transactivates FLT3 by direct binding and SYK activation is a mechanism of resistance of FLT3-ITD cells to FLT3 inhibitors in a mouse model [52], but frequency of this phenomenon in clinical samples is not known.
The bone marrow microenvironment mediates extrinsic mechanisms of FLT3 inhibitor resistance. Increased FLT3L secretion by the bone marrow microenvironment after chemotherapy stimulates AML cells with FLT3 mutations, which remain responsive to FLT3L despite constitutive FLT3 activation, and decreases sensitivity to FLT3 inhibitors [27,28]. Similarly, increased fibroblast growth factor 2 (FGF2) secretion by the bone marrow microenvironment after chemotherapy or FLT3 inhibitor therapy stimulates AML cells with FLT3 mutations via binding to fibroblast growth factor receptor (FGFR) 1 on AML cells [53]. Bone marrow stroma-mediated resistance also results from enhanced CXCL12-CXCR4-mediated homing [54], at least in part due to Pim-1 overexpression, as Pim-1 phosphorylates CXCR4, enabling its cell surface translocation and expression [55]. Finally, both FLT3L and bone marrow stromal cells activate ERK downstream of FLT3-ITD, thereby overcoming the effects of FLT3 inhibitors [56].
As an additional extrinsic mechanism of acquired resistance to a specific FLT3 inhibitor, induction of hepatic enzyme activity reducing drug bioavailability causes acquired resistance to midostaurin [57].
In summary, first-generation inhibitors, such as midostaurin [30–33], have broad activity and may therefore be less susceptible to resistance mediated by activation of kinases in FLT3 downstream or parallel pathways, compared to second-generation inhibitors, with greater specificity. This may be particularly relevant to AML at diagnosis, which is characterized by coexistence of multiple leukemic clones and low allelic burden of the FLT3 mutation [45], whereas a dominant clone with FLT3 mutation tends to emerge at relapse [45] and may be better targeted by the second-generation FLT3 inhibitors, which are more specific and also more potent. Indeed the randomized trial of a FLT3 inhibitor showing a survival benefit was the midostaurin trial in newly diagnosed AML patients [32]. Type I inhibitors, such as midostaurin, gilteritinib and crenolanib, are effective in cells with either TKD or ITD mutations, and should therefore not induce resistance through new TKD mutations, while type II inhibitors such as quizartinib and sorafenib are inactive against TKD mutations and induce TKD mutations as a mechanism of acquired resistance [13].
Combination treatments
Given limited and transient efficacy of FLT3 inhibitors, combination therapies are being explored, including a number of agents with diverse mechanisms of action thought to enhance the efficacy of FLT3 inhibitors or synergize with them. Drugs used in combination treatments with FLT3 inhibitors and their mechanisms of action are shown diagrammatically in Figure 1.
Combinations of FLT3 inhibitors with epigenetic therapies show promise. Synergistic induction of apoptosis in vitro by histone deacetylase inhibitors (HDACis) and FLT3 inhibitors has been demonstrated, with enhanced proteolytic cleavage of both FLT3-ITD and STAT5 protein by caspase-3 [58], as well as Mcl-1 downregulation [59]. Sorafenib and vorinostat (Zolinza®, Merck) were combined in a phase I trial (NCT00875745), and sorafenib, vorinostat and bortezomib are combined in a current phase I/II trial (NCT01534260). Combinations of FLT3 inhibitors and hypomethylating agents also demonstrate synergistic anti-leukemic effects in vitro, including enhanced apoptosis, growth inhibition and differentiation, with simultaneous administration most efficacious [60]. Azacitidine and sorafenib combination therapy has shown clinical activity [61]. Enhanced expression of the tumor suppressor Src homology-2 (SH2)-containing protein-tyrosine phosphatase 1 (SHP-1), a negative regulator of JAK/STAT signaling, is one proposed mechanism of efficacy of azacitidine and FLT3 inhibitor combination therapy [62]. Finally, the bromodomain antagonist JQ1 synergizes with FLT3 inhibitors to enhance apoptosis of cells with FLT3-ITD [63], with effects of the combination including decreased levels of c-MYC, Bcl-2, and CDK4/6, increased levels of p21, BIM, and cleaved PARP, and decreased p-STAT5, p-AKT and p-ERK1/2 in FLT3-ITD AML blast progenitor cells.
Proteasome inhibitors, such as bortezomib (Velcade®, Takeda), have been shown to induce autophagosomal degradation of FLT3-ITD and to be cytotoxic to cells with FLT3-ITD in vitro and in vivo, including FLT3-ITD cells with resistance to quizartinib associated with FLT3 D835 mutations [64]. Bortezomib and sorafenib are being evaluated together (NCT01371981) and also in combination with decitabine (NCT01861314) in Phase I trials. Bortezomib and midostaurin combined with chemotherapy had activity, but also toxicities [65].
Another approach to overcoming FLT3 inhibitor resistance is targeting signaling molecules downstream of FLT3-ITD. The STAT5 inhibitor pimozide reduces viability of cells with FLT3-ITD, and synergistic cytotoxicity was demonstrated with the FLT3 inhibitors midostaurin and sunitinib [66]. Pim-1, a serine/threonine kinase involved in cell survival and proliferation, is upregulated transcriptionally through STAT5 activation downstream of FLT3-ITD [67], and Pim-1 phosphorylates and stabilizes FLT3 and promotes its signaling in a positive feedback loop in cells with FLT3-ITD [49,50]. Pim kinase inhibitors and FLT3 inhibitors show synergistic cytotoxicity in AML cells with FLT3-ITD [49,50], and Pim inhibitors restore sensitivity to FLT3 inhibitors in resistant cells [50]. The data support combining Pim kinase inhibitors, which are currently in Phase I clinical trials, with FLT3 inhibitors. The PI3K/Akt/mTOR pathway is also a promising target in AML with FLT3 mutations. mTOR, downstream of FLT3, is upregulated in FLT3 inhibitor-resistant AML cells and promotes cell survival and proliferation [68]; inhibition of both mTOR and FLT3 leads to synergistic suppression of cell proliferation [69]. A clinical trial is evaluating the safety of the mTOR inhibitor everolimus (Afinitor®, Novartis) in combination with midostaurin (NCT00819546). Combined FLT3 and Akt inhibitors are also synergistic, including in the presence of bone marrow stroma [70]. Finally, in vitro data support combining sorafenib with metformin, a drug widely used as an antidiabetic, to downregulate the mTOR/p70S6K/4EBP1 pathway and promote apoptosis and autophagy [71].
FLT3 activation inhibits activity of the tumor suppressor serine/threonine phosphatase protein phosphatase 2A (PP2A), and PP2A activating drugs, including the immunomodulating agent fingolimod (FTY720), FDA-approved for relapsing multiple sclerosis, are cytotoxic toward cells with FLT3-ITD and produce synergistic cytotoxicity with FLT3 inhibitors in cells with FLT3-ITD in vitro [72, 73], including in the presence of bone marrow stroma [73]. PP2A activating drugs do not decrease phosphorylation of FLT3-ITD and actually increase phosphorylation of STAT5, but significantly decrease phosphorylation of AKT and ERK [72].
Combinations of other small molecules with FLT3 inhibitors also produce synergistic efficacy. Hedgehog (Hh) signaling was found to be upregulated in cells with FLT3-ITD, and combined FLT3 and Hh inhibitors decreased growth of leukemia cells with FLT3-ITD in vitro and in vivo [74]. Moreover, all-trans-retinoic acid synergized with FLT3 inhibitors to not only enhance apoptosis of cells with FLT3-ITD, but also deplete FLT3/ITD+ stem cells, through downregulation of the anti-apoptotic protein BCL6, which is upregulated by FLT3 inhibitor treatment [75].
FUTURE DIRECTIONS FOR FLT3-TARGETED THERAPY
The major current questions in the field are which FLT3 inhibitor(s) are most effective in different settings, and which combination regimens will enhance the efficacy of FLT3 inhibitors.
The first-generation type I FLT3 inhibitor midostaurin given to newly diagnosed AML patients 18 to 60 years old with FLT3-ITD or TKD mutations after induction and consolidation chemotherapy and as maintenance therapy showed significant efficacy in prolonging survival, compared to placebo [32]. Therefore midostaurin will likely become the first FLT3 inhibitor to be approved by the FDA, and treatment with midostaurin may therefore become the standard of care, in conjunction with chemotherapy, for newly diagnosed patients with AML with FLT3 mutations. It is possible that midostaurin has particular efficacy in the newly diagnosed setting because of broad activity against AML with multiple leukemic clones and low FLT3 mutation allelic burden [45]. Nevertheless, it is also possible that a more potent and better tolerated inhibitor such as gilteritinib might be even more efficacious, and unfortunately answering this question would require another large randomized trial with long follow-up. It will be challenging to test new inhibitors against midotaurin with chemotherapy in the newly diagnosed setting. Additionally, HSCT in first CR has become the standard or care, and it will be important to determine which FLT3 inhibitors are well tolerated following HSCT and have efficacy in preventing relapse in that setting.
In contrast, the first-generation type I FLT3 inhibitor lestaurtinib was ineffective, compared to placebo, in relapsed patients with FLT3-ITD or TKD mutations after reinduction chemotherapy [27]. It is likely that more potent and specific FLT3 inhibitors will be more efficacious following reinduction chemotherapy, given the common presence of a dominant clone with FLT3 mutation at relapse [45]. Diverse FLT3 inhibitors will need to be tested against placebo, and then potentially against each other, following reinduction chemotherapy in the relapsed/refractory setting.
Numerous drug combinations with FLT3 inhibitors are being explored, and will be essential for patients who are not candidates for chemotherapy or whose AML is refractory to chemotherapy. Combinations may also be effective post HSCT.
CONCLUSION
FLT3 is an important target in AML due to the high incidence of mutations resulting in constitutive signaling, and associated poor outcomes. The first-generation type I inhibitor midostaurin has shown benefit, and second-generation type I inhibitors such as gilteritinib show promise, as do novel combinations. The current status of this rapidly evolving field was summarized in this review, but new preclinical and clinical data continue to be rapidly generated, with the ultimate goal of successful targeted therapy for this common subset of AML patients who currently continue to have poor treatment outcomes.
Acknowledgments
Financial support: The authors gratefully acknowledge a Fulbright Program scholarship awarded to M. Larrosa-Garcia and Merit Review grant BX002184 from the Department of Veterans Affairs and Leukemia and Lymphoma Society Translational Research Award 6346-11 to M.R. Baer.
References
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373:1136–52. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 2.Gilliland DG, Griffin JD. The roles of FLT3 in hematopoiesis and leukemia. Blood. 2002;100:1532–42. doi: 10.1182/blood-2002-02-0492. [DOI] [PubMed] [Google Scholar]
- 3.Schnittger S, Bacher U, Haferlach C, Alpermann T, Kern W, Haferlach T. Diversity of the juxtamembrane and TKD1 mutations (exons 13–15) in the FLT3 gene with regards to mutant load, sequence, length, localization, and correlation with biological data. Genes Chromosomes Cancer. 2012;51:910–24. doi: 10.1002/gcc.21975. [DOI] [PubMed] [Google Scholar]
- 4.Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Böhmer FD, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36:326–39. doi: 10.1016/j.molcel.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 5.Choudhary C, Schwäble J, Brandts C, Tickenbrock L, Sargin B, Kindler T, et al. AML-associated Flt3 kinase domain mutations show signal transduction differences compared with Flt3 ITD mutations. Blood. 2005;106:265–73. doi: 10.1182/blood-2004-07-2942. [DOI] [PubMed] [Google Scholar]
- 6.Levis M, Murphy KM, Pham R, Kim KT, Stine A, Li L, et al. Internal tandem duplications of the FLT3 gene are present in leukemia stem cells. Blood. 2005;106:673–80. doi: 10.1182/blood-2004-05-1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gourdin TS, Zou Y, Ning Y, Emadi A, Duong VH, Tidwell ML, et al. High frequency of rare structural chromosome abnormalities at relapse of cytogenetically normal acute myeloid leukemia with FLT3 internal tandem duplication. Cancer Genet. 2014;207:467–73. doi: 10.1016/j.cancergen.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fan J, Li L, Small D, Rassool F. Cells expressing FLT3/ITD mutations exhibit elevated repair errors generated through alternative NHEJ pathways: Implications for genomic instability and therapy. Blood. 2010;116:5298–305. doi: 10.1182/blood-2010-03-272591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Song Y, Magenau J, Li Y, Braun T, Chang L, Bixby D, et al. FLT3 mutational status is an independent risk factor for adverse outcomes after allogeneic transplantation in AML. Bone Marrow Transplant. 2016;51:511–20. doi: 10.1038/bmt.2015.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams B, Atkins A, Zhang H, Lu D, Jimenez X, Li H, et al. Cell-based selection of internalizing fully human antagonistic antibodies directed against FLT3 for suppression of leukemia cell growth. Leukemia. 2005;19:1432–8. doi: 10.1038/sj.leu.2403825. [DOI] [PubMed] [Google Scholar]
- 11.Ke YY, Singh VK, Coumar MS, Hsu YC, Wang WC, Song JS, et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci Rep. 2015;5:11702. doi: 10.1038/srep11702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith CC, Lin K, Stecula A, Sali A, Shah NP. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia. 2015;29:2390–2. doi: 10.1038/leu.2015.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–3. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levis M, Pham R, Smith BD, Small D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: sequence of administration is important to achieve synergistic cytotoxic effects. Blood. 2004;104:1145–50. doi: 10.1182/blood-2004-01-0388. [DOI] [PubMed] [Google Scholar]
- 15.Sen R, Natarajan K, Bhullar J, Shukla S, Fang HB, Cai L, et al. The novel BCR-ABL and FLT3 inhibitor ponatinib is a potent inhibitor of the MDR-associated ATP-binding cassette transporter ABCG2. Mol Cancer Ther. 2012;11:2033–44. doi: 10.1158/1535-7163.MCT-12-0302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levis M, Brown P, Smith BD, Stine A, Pham R, Stone R, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477–83. doi: 10.1182/blood-2006-04-015743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Man CH, Fung TK, Ho C, Han HH, Chow HC, Ma AC, et al. Sorafenib treatment of FLT3-ITD(+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent nonresponsiveness associated with the emergence of a D835 mutation. Blood. 2012;119:5133–43. doi: 10.1182/blood-2011-06-363960. [DOI] [PubMed] [Google Scholar]
- 18.Metzelder SK, Schroeder T, Finck A, Scholl S, Fey M, Gotze K, et al. High activity of sorafenib in FLT3-ITD-positive acute myeloid leukemia synergizes with allo-immune effects to induce sustained responses. Leukemia. 2012;26:2353–9. doi: 10.1038/leu.2012.105. [DOI] [PubMed] [Google Scholar]
- 19.Chen YB, Li S, Lane AA, Connolly C, Del Rio C, Valles B, et al. Phase I trial of maintenance sorafenib after allogeneic hematopoietic stem cell transplantation for fms-like tyrosine kinase 3 internal tandem duplication acute myeloid leukemia. Biol Blood Marrow Transplant. 2014;20:2042–8. doi: 10.1016/j.bbmt.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Röllig C, Serve H, Hüttmann A, Noppeney R, Müller-Tidow C, Krug U, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691–9. doi: 10.1016/S1470-2045(15)00362-9. [DOI] [PubMed] [Google Scholar]
- 21.Serve H, Krug U, Wagner R, Sauerland MC, Heinecke A, Brunnberg U, et al. Sorafenib in combination with intensive chemotherapy in elderly patients with acute myeloid leukemia: Results from a randomized, placebo-controlled trial. J Clin Oncol. 2013;31:3110–8. doi: 10.1200/JCO.2012.46.4990. [DOI] [PubMed] [Google Scholar]
- 22.Uy GL, Mandrekar SJ, Laumann K, Marcucci G, Zhao W, Levis MJ, et al. A phase 2 study incorporating sorafenib into the chemotherapy for older adults with FLT3-mutated AML: CALGB 11001 (Alliance) Blood Adv. 2017;1:331–340. doi: 10.1182/bloodadvances.2016003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Farrell AM, Foran JM, Fiedler W, Serve H, Paquette RL, Cooper MA, et al. An innovative phase I clinical study demonstrates inhibition of FLT3 phosphorylation by SU11248 in acute myeloid leukemia patients. Clin Cancer Res. 2003;9:5465–76. [PubMed] [Google Scholar]
- 24.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell AM, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986–93. doi: 10.1182/blood-2004-05-1846. [DOI] [PubMed] [Google Scholar]
- 25.Fiedler W, Kayser S, Kebenko M, Janning M, Krauter J, Schittenhelm M, et al. A phase I/II study of sunitinib and intensive chemotherapy in patients over 60 years of age with acute myeloid leukaemia and activating FLT3 mutations. Br J Haematol. 2015;169:694–700. doi: 10.1111/bjh.13353. [DOI] [PubMed] [Google Scholar]
- 26.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 27.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294–301. doi: 10.1182/blood-2010-08-301796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato T, Yang X, Knapper S, White P, Smith BD, Galkin S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117:3286–93. doi: 10.1182/blood-2010-01-266742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Knapper S, Russell N, Gilkes A, Hills RK, Gale RE, Cavenagh JD, et al. A randomised assessment of adding the kinase inhibitor lestaurtinib to 1st-line chemotherapy for FLT3-mutated AML. Blood. 2016 Nov 21; doi: 10.1182/blood-2016-07-730648. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28:4339–45. doi: 10.1200/JCO.2010.28.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stone RM, Fischer T, Paquette R, Schiller G, Schiffer CA, Ehninger G, et al. Phase IB study of the FLT3 kinase inhibitor midostaurin with chemotherapy in younger newly diagnosed adult patients with acute myeloid leukemia. Leukemia. 2012;26:2061–8. doi: 10.1038/leu.2012.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone RM, Mandrekar S, Sanford BL, Geyer S, Bloomfield CD, Döhner K, et al. The multi-kinase inhibitor midostaurin (M) prolongs survival compared with placebo (P) in combination with daunorubicin (D)/cytarabine (C) induction (ind), high-dose C consolidation (consol), and as maintenance (maint) therapy in newly diagnosed acute myeloid leukemia (AML) patients (pts) age 18–60 with FLT3 mutations (muts): An international prospective randomized (rand) P-controlled double-blind trial (CALGB 10603/RATIFY [Alliance]) Blood. 2015;126(suppl):23. abstr 6. [Google Scholar]
- 33.Schlenk R, Döhner K, Salih H, Kündgen A, Fiedler W, Salwender H-J, et al. Midostaurin in combination with intensive induction and as single agent maintenance therapy after consolidation therapy with allogeneic hematopoietic stem cell transplantation or high-dose cytarabine ( NCT01477606) Blood. 2015;126(suppl):23. abstr 322. [Google Scholar]
- 34.DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: Safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108:3674–81. doi: 10.1182/blood-2006-02-005702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiotsu Y, Kiyoi H, Ishikawa Y, Tanizaki R, Shimizu M, Umehara H, et al. KW-2449, a novel multikinase inhibitor, suppresses the growth of leukemia cells with FLT3 mutations or T315I-mutated BCR/ABL translocation. Blood. 2009;114:1607–17. doi: 10.1182/blood-2009-01-199307. [DOI] [PubMed] [Google Scholar]
- 36.Zarrinkar PP, Gunawardane RN, Cramer MD, Gardner MF, Brigham D, Belli B, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML) Blood. 2009;114:2984–92. doi: 10.1182/blood-2009-05-222034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31:3681–7. doi: 10.1200/JCO.2013.48.8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levis MJ, Martinelli G, Perl AE, Dombret H, Steffen B, Rousselot PH, et al. The benefit of treatment with quizartinib and subsequent bridging to HSCT for FLT3-ITD(+) patients with AML. J Clin Oncol. 2014;32(suppl):5s. abstr 7093. [Google Scholar]
- 39.Galanis A, Ma H, Rajkhowa T, Ramachandran A, Small D, Cortes J, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014;123:94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman EI, Turner DC, Buaboonnam J, Hu S, Orwick S, Roberts MS, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013;122:3607–15. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ueno Y, Kaneko N, Saito R, Kondoh Y, Shimada I, Mori M, et al. ASP2215, a novel FLT3/AXL inhibitor: Preclinical evaluation in combination with cytarabine and anthracycline in acute myeloid leukemia (AML) J Clin Oncol. 2014;32(suppl):5s. abstr 7070. [Google Scholar]
- 42.Perl AE, Altman JK, Cortes J, Smith C, Litzow M, Baer MR, et al. Results of the CHRYSALIS TRIAL: A first-in-human phase 1/2 dose-escalation, dose-expansion study of gilteritinib (ASP2215) in patients with relapsed/refractory acute myeloid leukemia (R/R AML) Blood. 2016;128(suppl):22. abstr 1069. [Google Scholar]
- 43.Gozgit JM, Wong MJ, Wardwell S, Tyner JW, Loriaux MM, Mohemmad QK, et al. Potent activity of ponatinib (AP24534) in models of FLT3-driven acute myeloid leukemia and other hematologic malignancies. Mol Cancer Ther. 2011;10:1028–35. doi: 10.1158/1535-7163.MCT-10-1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Hu C, Wang A, Weisberg EL, Wang W, Chen C, et al. Ibrutinib selectively targets FLT3-ITD in mutant FLT3-positive AML. Leukemia. 2016;30:754–7. doi: 10.1038/leu.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pratz KW, Sato T, Murphy KM, et al. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010;115:1425–32. doi: 10.1182/blood-2009-09-242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Breitenbuecher F, Markova B, Kasper S, Carius B, Stauder T, Bohmer FD, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009;113:4063–73. doi: 10.1182/blood-2007-11-126664. [DOI] [PubMed] [Google Scholar]
- 47.Bagrintseva K, Geisenhof S, Kern R, Eichenlaub S, Reindl C, Ellwart JW, et al. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L) Blood. 2005;105:3679–85. doi: 10.1182/blood-2004-06-2459. [DOI] [PubMed] [Google Scholar]
- 48.Kohl TM, Hellinger C, Ahmed F, Buske C, Hiddemann W, Bohlander SK, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007;21:1763–72. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 49.Natarajan K, Xie Y, Burcu M, Linn DE, Qiu Y, Baer MR. Pim-1 kinase phosphorylates and stabilizes 130 kDa FLT3 and promotes aberrant STAT5 signaling in acute myeloid leukemia with FLT3 internal tandem duplication. PLoS One. 2013;8:e74653. doi: 10.1371/journal.pone.0074653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Green AS, Maciel TT, Hospital MA, Yin C, Mazed F, Townsend EC, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Sci Adv. 2015;1:e1500221. doi: 10.1126/sciadv.1500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park IK, Mundy-Bosse B, Whitman SP, Zhang X, Warner SL, Bearss DJ, et al. Receptor tyrosine kinase axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015;29:2382–9. doi: 10.1038/leu.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Puissant A, Fenouille N, Alexe G, Pikman Y, Bassil CF, Mehta S, et al. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell. 2014;25:226–42. doi: 10.1016/j.ccr.2014.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Traer E, Martinez J, Javidi-Sharifi N, Agarwal A, Dunlap J, English I, et al. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016;76:6471–82. doi: 10.1158/0008-5472.CAN-15-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Onishi C, Mori-Kimachi S, Hirade T, Abe M, Taketani T, Suzumiya J, et al. Internal tandem duplication mutations in FLT3 gene augment chemotaxis to Cxcl12 protein by blocking the down-regulation of Rho-associated kinase via the Cxcl12/Cxcr4 signaling axis. J Biol Chem. 2015;290:28356. doi: 10.1074/jbc.A114.568287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grundler R, Brault L, Gasser C, Bullock AN, Dechow T, Woetzel S, et al. Dissection of PIM serine/threonine kinases in FLT3-ITD-induced leukemogenesis reveals PIM1 as regulator of CXCL12-CXCR4-mediated homing and migration. J Exp Med. 2009;206:1957–70. doi: 10.1084/jem.20082074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J Haematol. 2014;164:61–72. doi: 10.1111/bjh.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin OQ, Wang Y, Schran H. A mechanism-based population pharmacokinetic model for characterizing time-dependent pharmacokinetics of midostaurin and its metabolites in human subjects. Clin Pharmacokinet. 2008;47:807–16. doi: 10.2165/0003088-200847120-00005. [DOI] [PubMed] [Google Scholar]
- 58.Pietschmann K, Bolck HA, Buchwald M, Spielberg S, Polzer H, Spiekermann K, et al. Breakdown of the FLT3-ITD/STAT5 axis and synergistic apoptosis induction by the histone deacetylase inhibitor panobinostat and FLT3-specific inhibitors. Mol Cancer Ther. 2012;11:2373–83. doi: 10.1158/1535-7163.MCT-12-0129. [DOI] [PubMed] [Google Scholar]
- 59.Lin WH, Yeh TK, Jiaang WT, Yen KJ, Chen CH, Huang CT, et al. Evaluation of the antitumor effects of BPR1J-340, a potent and selective FLT3 inhibitor, alone or in combination with an HDAC inhibitor, vorinostat, in AML cancer. PLoS One. 2014;9:e83160. doi: 10.1371/journal.pone.0083160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chang E, Ganguly S, Rajkhowa T, Gocke CD, Levis M, Konig H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia. 2016;30:1025–32. doi: 10.1038/leu.2015.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ravandi F, Alattar ML, Grunwald MR, Rudek MA, Rajkhowa T, Richie MA, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013;121:4655–62. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Al-Jamal HA, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC Cancer. 2015;15:869. doi: 10.1186/s12885-015-1695-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fiskus W, Sharma S, Qi J, Shah B, Devaraj SG, Leveque C, et al. BET protein antagonist JQ1 is synergistically lethal with FLT3 tyrosine kinase inhibitor (TKI) and overcomes resistance to FLT3-TKI in AML cells expressing FLT-ITD. Mol Cancer Ther. 2014;13:2315–27. doi: 10.1158/1535-7163.MCT-14-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Larrue C, Saland E, Boutzen H, Vergez F, David M, Joffre C, et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016;127:882–92. doi: 10.1182/blood-2015-05-646497. [DOI] [PubMed] [Google Scholar]
- 65.Walker AR, Wang H, Walsh K, Bhatnagar B, Vasu S, Garzon R, et al. Midostaurin, bortezomib and MEC in relapsed/refractory acute myeloid leukemia. Leuk Lymphoma. 2016:1–9. doi: 10.3109/10428194.2015.1135435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nelson EA, Walker SR, Xiang M, Weisberg E, Bar-Natan M, Barrett R, et al. The STAT5 inhibitor pimozide displays efficacy in models of acute myelogenous leukemia driven by FLT3 mutations. Genes Cancer. 2012;3:503–11. doi: 10.1177/1947601912466555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim KT, Baird K, Ahn JY, Meltzer P, Lilly M, Levis M, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005;105:1759–67. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- 68.Lindblad O, Cordero E, Puissant A, Macaulay L, Ramos A, Kabir NN, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016;35:5119–31. doi: 10.1038/onc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mohi MG, Boulton C, Gu TL, Sternberg DW, Neuberg D, Griffin JD, et al. Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc Natl Acad Sci U S A. 2004;101:3130–5. doi: 10.1073/pnas.0400063101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Weisberg E, Liu Q, Zhang X, Nelson E, Sattler M, Liu F, et al. Selective akt inhibitors synergize with tyrosine kinase inhibitors and effectively override stroma-associated cytoprotection of mutant FLT3-positive AML cells. PLoS One. 2013;8:e56473. doi: 10.1371/journal.pone.0056473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang F, Liu Z, Zeng J, Zhu H, Li J, Cheng X, et al. Metformin synergistically sensitizes FLT3-ITD-positive acute myeloid leukemia to sorafenib by promoting mTOR-mediated apoptosis and autophagy. Leuk Res. 2015;39:1421–7. doi: 10.1016/j.leukres.2015.09.016. [DOI] [PubMed] [Google Scholar]
- 72.Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, et al. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clin Cancer Res. 2014;20:2092–103. doi: 10.1158/1078-0432.CCR-13-2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smith AM, Dun MD, Lee EM, Harrison C, Kahl R, Flanagan H, et al. Activation of protein phosphatase 2A in FLT3+ acute myeloid leukemia cells enhances the cytotoxicity of FLT3 tyrosine kinase inhibitors. Oncotarget. 2016;7:47465–78. doi: 10.18632/oncotarget.10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim Y, Gondek L, Li L, Wang Q, Ma H, Chang E, et al. Integration of hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci Transl Med. 2015;7:291ra96. doi: 10.1126/scitranslmed.aaa5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ma HS, Greenblatt SM, Shirley CM, Duffield AS, Bruner JK, Li L, et al. All-trans retinoic acid synergizes with FLT3 inhibition to eliminate FLT3/ITD+ leukemia stem cells in vitro and in vivo. Blood. 2016;127:2867–78. doi: 10.1182/blood-2015-05-646786. [DOI] [PMC free article] [PubMed] [Google Scholar]