

Abstract
Cancer immune checkpoint therapy has achieved remarkable clinical successes in various cancers. However, current immune checkpoint inhibitors block the checkpoint of not only the immune cells that are important to cancer therapy but also the immune cells that are irrelevant to the therapy. Such an indiscriminate blockade limits the efficacy and causes the autoimmune toxicity of the therapy. It might be beneficial to use a carrier to target immune checkpoint inhibitors to cancer-reactive immune cells. Here, we explore a method to load the inhibitors into carriers. We used the anti-programmed death-1 antibody (αPD-1) as a model immune checkpoint inhibitor. First, we generated a recombinant single-chain variable fragment (scFv) of αPD-1. Then, we designed and generated a fusion protein consisting of the scFv and an amphiphilic immune-tolerant elastin-like polypeptide (iTEP). Because of the amphiphilic iTEP, the fusion was able to self-assemble into a nanoparticle (NP). The NP was proved to block the PD-1 immune checkpoint in vitro and in vivo. Particularly, the NP exacerbated diabetes development in non-obese diabetic mice as effectively as natural, intact αPD-1. In summary, we successfully expressed αPD-1 as a recombinant protein and linked αPD-1 to a NP, which lays a foundation to develop a delivery system to target αPD-1 to a subpopulation of immune cells.
Keywords: anti-programmed death-1 antibody, nanoparticle, recombinant anti-programmed death-1 antibody fusion, multivalency, diabetes
Graphical abstract
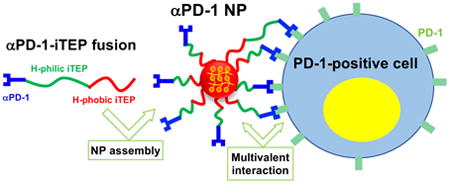
Introduction
Immune checkpoint inhibitors such as the anti-cytotoxic T lymphocyte antigen-4 antibody (αCTLA-4) and the anti-programmed death-1 antibody (αPD-1) have been approved to treat advanced melanoma, lung cancer, head and neck cancer, among others1-5. Some of these inhibitors have been approved by the FDA, such as Pembrolizumab and Nivolumab6. Recently, αCTLA-4 and αPD-1 were combined to further boost their efficacy2, 5. However, the further improvement of the immune checkpoint therapy is hindered by its autoimmune toxicity. For example, in the above combination therapy, 55% of the combination therapy patients suffered from high-grade (grades 3-4) toxicity, and 36% of the patients had to discontinue the therapy due to the toxicity5. In contrast to the pressing need to reduce the toxicity, the current toxicity mitigating method, non-specific immune suppression, is apparently not effective enough because one third of the treated patients had to stop the therapy even after using this method, not to mention that the method has its own side effects (e.g. immune deficiency)7. Previously, intra-tumor injection of the inhibitors was attempted and proven effective8; however, this method is not practical for advanced cancer patients as it is almost impossible to inject inhibitors to metastatic tumors. Therefore, new strategies are needed to reduce the toxicity of immune checkpoint inhibitors.
Intrinsically, immune checkpoints (e.g. PD-1 and CTLA-4) not only protect tumors from immune elimination9-10, but also prevent autoimmune toxicity in healthy tissues11. The root cause of the toxicity is that the checkpoint inhibitors indiscriminately block the checkpoint in all cells that utilize the checkpoints11-16.Thus, to resolve the toxicity of the inhibitors, it would be desirable to target the inhibitors to those cells that are necessary for tumor treatment but suppressed by the checkpoint. The targeting also has potential to boost the efficacy of the inhibitors because it concentrates the inhibitors to those necessary cells for cancer therapy, whereas the current non-specific blockade wastes inhibitors in tumor treatment-unrelated interactions. Recently, a platelet-based carrier was used to target an immune checkpoint inhibitor, anti-PD-L1 antibody, to tumors, which resulted in better prevention of tumor recurrence under a post-surgery setting17. However, it is unclear whether the carrier reduced the toxicity of immune checkpoint inhibitors. Thus, more efforts are needed to establish the strategies and drug carriers that can target immune checkpoint inhibitors and reduce their toxicity.
In this work, we generated a NP carrier for one model immune checkpoint inhibitor, αPD-1. Different from the previously reported carrier of the inhibitors17, αPD-1 and its carrier molecule, iTEP18-20, were generated together as a recombinant fusion protein. The fusion is able to self-assemble into a NP. This NP effectively blocks the PD-1 immune checkpoint in vitro and in vivo. On the basis of these findings, it is feasible to integrate cell targeting moieties to this NP that bind with tumor-reactive immune cells so that the NP is able to target αPD-1 as well as other inhibitors to the cells. In summary, we provided a simple platform to develop cell-targeting carriers for immune checkpoint inhibitors.
Methods
Materials
EL4 (ATCC® TIB-39™) cells were purchased from ATCC. The hybridoma RMP1-14 for αPD-1 production was provided by Professor Hideo Yagita at the Juntendo University School of Medicine. DH5α competent E.coli cells were purchased from Thermo Fisher Scientific Inc. (Waltham, MA). SHuffle® T7 Competent E. coli cells were purchased from New England Biolabs (Ipswich, MA). Expression vector pET-25b(+) was purchased from EMD Millipore (Billerica, MA). Restriction endonucleases were purchased from New England Biolabs (Ipswich, MA). LB media were prepared in our lab using the standard formula. Cell culture media and supplements including RPMI-1640, Dulbecco's Modified Eagle Medium (DMEM), and fetal bovine serum (FBS) were purchased from Thermo Fisher Scientific Inc. (Waltham, MA). B6.129S7-Rag1tm1Mom/J mice and NOD/ShiLtJ mice were purchased from the Jackson Laboratory. All the animal experiment protocols were approved by the Institutional Animal Care and Use Committee at the University of Utah.
Design and generation of the expression vectors for αPD-1 scFv and αPD-1-iTEP fusion
The αPD-1 hybridoma clone was sequenced using the variable domain sequencing service from GenScript. The sample submitted for sequencing was prepared following the protocol from GenScript (http://www.genscript.com/mAb-sequencing.html).
To generate the αPD-1 scFv, the variable regions of the αPD-1 heavy (VH) chain and the αPD-1 light chains (VL) were connected by a linker, (GGGSG)4. The resultant scFv is NH2-VH-Linker-VL-COOH. To facilitate the purification, six histidine residues were added to the N-terminus of the scFv. The coding gene of the scFv was synthesized by Thermo Fisher Scientific Inc. The gene was flanked by two BseRI restriction sites at each end to facilitate the ligation of the gene into the pET-25b(+) vector.
The αPD-1(scFv)-iTEP fusion was designed as illustrated in Figure 1. The amphiphilic iTEP, NH2-(GAGVPG)70-(GVLPGVG)56–(GC)4, has a hydrophilic iTEP segment (GAGVPG)70 and a hydrophobic segment (GVLPGVG)5620. The multiple cysteine residual at the hydrophobic end of the iTEP is to crosslink the PD-1-iTEP fusions through disulfide bonds after the fusions self-assemble in a NP. The crosslinking is to stabilize the NP. The gene encoding the iTEP sequence was generated as previously described and inserted into the pET-25b(+) vector20. To generated the expression vector for the αPD-1-iTEP fusion, the gene for the αPD-1 scFv was first digested with BseRI and then ligated into the BseRI-digested iTEP in pET-25b(+).
Figure 1.
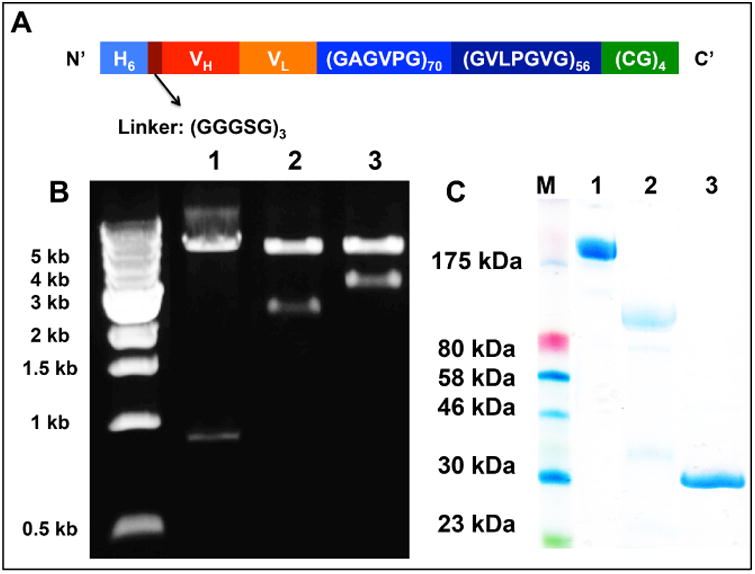
A. The sequence design of αPD-1-iTEP fusion. B. An agarose gel image of DNA digestion products from three plasmids, pET-25b(+) with the scFv coding gene (lane 1), pET-25b(+) with the iTEP coding gene (lane 2), and pET-25b(+) with the fusion coding gene (lane 3). Three plasmids were digested with two restriction enzymes, BamHI and Xbal, which flanked the BseRI sites by which we inserted the genes to the pET-25b(+) vector. The upper bands of each lane represent the pET-25b(+) vectors; the lower bands of each lane represent the coding genes. C. A photo of an SDS-PAGE gel that contains intact αPD-1 (lane 1), αPD-1-iTEP fusion (lane 2), and αPD-1 sc-FV (lane 3). 20 μg of each protein was loaded onto each lane.
Production and purification of αPD-1 scFv and αPD-1-iTEP fusion
The expression vectors of the scFv and the αPD-1-iTEP fusion were transformed into the SHuffle® T7 Competent E. coli cells for protein expression. For protein production, the transformed E. coli cells were first cultured in LB medium at 32 °C until the OD600 of the medium reached 0.6 when. Then, IPTG was added into the culture medium at a final concentration of 0.5 mM. After that, the culture was continued at 16 °C overnight before the the cells were harvested from the culture. To purify the scFv and the αPD-1-iTEP fusion from the harvested cells, the cells were lysed in PBS by sonication; the PBS contained 1 mM PMSF (Sigma-Aldrich, St. Louis, MO) for an inhibition of proteolysis. After the cell lysate was centrifuged at 20,000 g for 60 min at 4 °C to remove cell debris, the supernatant of the lysate was collected and loaded onto HisPur Ni-NTA spin columns (Thermo Fisher Scientific Inc). The scFv and the αPD-1-iTEP fusion were purified according to the protocol from Thermo Fisher Scientific Inc. The elute from the columns was dialyzed against PBS at 4°C for 24 hours with three buffer changes. The purity and integrity of the collected proteins were examined by an SDS-PAGE analysis.
Production and purification of intact αPD-1
The αPD-1 was generated from ascetic fluid of the B6.129S7-Rag1tm1Mom/J mice that were inoculated by RMP-1-14 hybridoma cells21. The procedure of the inoculation and the fluid harvest were performed as previously reported22. The αPD-1 was purified from the fluid according to a published protocol23. The yield was 30∼50 mg αPD-1per mouse.
Assembly of αPD-1 NP
The αPD-1-iTEP fusion was incubated at a high concentration (100 μM) at 37 °C for 20 min to promote the self-assembly of the fusion into the NP. Then, H2O2 was added into the sample to reach a final concentration of 0.3%, which was to oxidize cysteines in the fusion and promote the crosslink between the fusion inside the NP for 1 hr. Last, the fusion sample was dialyzed against PBS to remove H2O2. The same approach was applied to the amphiphilic iTEP used in the fusion when an iTEP NP was generated.
Size characterization of protein samples by dynamic light scattering (DLS)
Intact αPD-1, the αPD-1-iTEP fusion and the amphiphilic iTEP used in the fusion were measured using the Malvern Zetasizer Nano (Malvern, Chester County, PA) at 37 °C. The fusion and the iTEP samples were treated to assemble NPs before the measurement. All samples were measured at a concentration of 20 μM. Each sample was measured in triplicate. The instrument settings for the measurement are: material RI=1.59, material absorption=0.010, water dispersant RI=1.330, and viscosity=0.6864 cP. The default value, 4.65 mm, was used as the measurement position. The count rate, duration, and attenuator was automatically optimized by the program of Malvern Zetasizer Nano. Additionally, the αPD-1-iTEP fusion was measured at two concentrations (0.25 μM and 20 μM) at two temperatures (25 °C and 37 °C), and at two different redox status. The oxidization procedure was same as the described above. To reduce the sample, αPD-1-iTEP fusion was incubated with 20 mM TCEP overnight.
The direct binding assay
The assay was used to examine the binding between αPD-1 samples and PD-1-positive EL4 cells. The αPD-1 samples include soluble αPD-1-iTEP fusion, αPD-1 NP, the αPD-1 scFv, and intact αPD-1. First, all αPD-1 samples were labelled with Alexa Fluor 647. Next, each of these samples was incubated with 1 million of EL4 cells on ice for 30 min. The fraction of EL4 cells in each incubation mixture that were Alexa Fluor 647-positive was quantified by flow cytometry on a BD FACSCANTO II (BD Biosciences, San Jose, CA). The percentages were plotted against concentration for each αPD-1 sample. EC50 and the 95% confidence interval (95% CI) of the EC50 was generated for each sample by fitting the curve of the sample to a built-in, Sigmoidal dose-response model of GraphPad V5.0.
The blocking assay of PD-L1 binding
The blocking of the PD-L1 binding to EL4 cells was determined through a competition binding assay. In this assay, soluble αPD-1-iTEP fusion, αPD-1 NP, the αPD-1 scFv, and intact αPD-1 were paired and competed with a PD-L1 sample (PD-L1-human Fc fusion, R&D Systems Inc. Minneapolis, MN, USA), respectively. Specifically, each of the above αPD-1 samples was serially diluted and incubated with 1 million EL4 cells in 5 ml test tubes on ice for 30 min. Next, the PD-L1 fusion was added into the incubation mixtures at the final concentration of 10 μg/mL; the mixtures were kept on ice for additional 30 min. Then, an Alexa Fluor 488-labeled, goat-anti-human Fc antibody (Thermo Fisher Scientific Inc.) was added into the mixtures to stain the PD-L1 fusion; the mixtures were kept on ice for another 30 min. After the incubation, unbound proteins were washed away with a FACS buffer, PBS with 1% FBS; the EL4 cells in the mixtures were collected. The fractions of EL4 cells that were Alexa Fluor 488-positive were quantified using flow cytometry on a BD FACSCANTO II flow cytometer (BD Biosciences, San Jose, CA). In two separate experiments, EL4 cells were treated with an 100% blocking condition (an incubation with the anti-human Fc antibody) and a 0% blocking condition (an incubation with the anti-human Fc antibody plus the PD-L1 fusion); the fractions of Alexa Fluor 488-positive EL4 cells after these two treatments were quantified using flow cytometry. Lastly, all fractions values of Alexa Fluor 488-positive EL4 cells that resulted from the above αPD-1 treatments were transformed into blocking efficiencies (%) through normalization of these values against the fraction values of 100% and 0% blocking. The blocking efficiencies were plotted against the concentrations of the corresponding samples. EC50 and its 95% CI was generated for each sample by fitting the curve of the sample to a Sigmoidal dose-response model using GraphPad V5.0.
Diabetes exacerbation
10-week-old female NOD/ShiLtJ mice were separated into four groups. Each group of the mice were intraperitoneally injected five times with one of the four samples: soluble αPD-1-iTEP fusion, αPD-1 NP, intact αPD-1, or PBS. The first dose was 0.5 mg αPD-1 equivalent per mouse on day 0 except for the PBS group; the remaining four doses were 0.25 mg αPD-1 equivalent per mouse on day 2, 4, 6, and 8. Blood was drawn from the tails of these mice every other day from day 0. Glucose concentrations in these blood samples were measured by a OneTouch UltraMini meter (LifeScan, Inc., Milpitas, CA). The sampling and monitoring were continued for every mouse until that mouse was confirmed for diabetes. Our criterion of diabetes is that blood glucose concentration reached or surpassed 250 mg/deciliter (dL) for three consecutive measurements24. The first date that confirmed diabetes was observed was recorded and used to calculate diabetes-free survival days. Diabetes-free survival was analyzed by the Kaplan-Meier method, and the median survival of each treatment group was compared using the Log rank test with GraphPad V5.0.
Results and Discussion
The design and generation of αPD-1 scFv and αPD-1-iTEP fusion as recombinant proteins
We sequenced both the heavy chain and light chain cDNA of the αPD-1 (RMP1-14 clone). According to the sequencing results, the three complementarity-determining regions (CDRs) of the heavy chain are: SSYRWN, YINSAGISNYNPSLKR, and SDNMGTTPFTY; the three CDRs of the light chain are: RSSKSLLYSDGKTYLN, WMSTRAS, and QQGLEFPT. Based on the CDR information, we designed an scFv of the αPD-1, NH2-Histag(H6)-(GGGSG)3-VH-(GGGSG)4-VL-COOH, and synthesized a coding gene to express the scFv as a recombinant protein. Next, we designed an αPD-1(scFv)-iTEP fusion as illustrated in Figure 1A. The amphiphilic biblock iTEP, NH2-(GAGVPG)70-(GVLPGVG)56–(GC)4-COOH, was included to drive the fusion to self-assemble into a micelle-like NP. We insert the coding genes of the scFv and the fusion into the pET-25b(+) expression vector (Figure S1). We examined sizes of the coding genes using gel electrophoresis after we cleaved the genes from their host pET-25b(+) vector. On the basis of the gel image (Figure 1B), the size of the scFv gene is between 0.5 kb and 1.0 kb; the size of the fusion gene is between 3.0 kb and 4.0 kb. These estimated sizes are consistent with the theoretical sizes of the scFv gene and the fusion gene, 792 bp and 3252 bp, respectively. These two coding genes were also fully sequenced to confirm their accuracy. Amino acid residual numbers, theoretical sizes of their coding genes, and theoretical molecular weight of the scFv and the fusion were listed in Table 1.
Table 1.
A summary amino acid residual numbers, theoretical sizes of the coding genes, theoretical molecular weight of the αPD-1 scFv, the αPD-1-iTEP fusion.
| Number of residues | Sizes of coding genes | Molecular weight/kDa | |
|---|---|---|---|
| αPD-1 scFv | 264 | 792 | 28.2 |
| αPD-1-iTEP fusion | 1084 | 3252 | 91.9 |
We chose the SHuffle T7 E. coli strain to express the scFv and the fusion because the scFv has the two disulfide bonds critical to its structure and the strain was engineered to express fully functional, disulfide bond-containing proteins25. While we were producing the scFv and the fusion, we also generated intact αPD-1 from RPM1-14 hybridoma inoculated mice. After we purified these proteins, we used SDS-PAGE to analyze the sizes and the purity of these proteins. On the SDS-PAGE gel, the intact αPD-1 showed a band migrating slower than the 175 kDa marker, indicating that the MW of the intact αPD-1 is larger than 175 kDa. The SDS-PAGE results also confirmed the purity of the intact αPD-1 sample. It is noted that the intact αPD-1 sample was not reduced before being loaded on the gel so that the two heavy chains and two light chains of the antibody (IgG) migrated together. On the gel, the fusion sample showed a major band migrating slower than the 80 kDa marker; the scFv sample showed a major band migrating slightly faster than the 30 kDa marker (Figure 1C). These migration results are consistent with their theoretical MWs, 91.9 kDa for the fusion and 28.2 kDa for the scFV, respectively (Table 1).
In vitro characterization of the αPD-1-iTEP fusion
According to the DLS data, the fusion had a hydrodynamic diameter of 45.02 ±12.77 nm at 37°C; The amphiphilic iTEP used in the fusion had a hydrodynamic diameter of 35.62 ±10.16 nm (Figure 2A). Thus, both the fusion and the iTEP appeared to form NPs. We termed the NP form of the fusion as αPD-1 NP hereafter. αPD-1 NP has the capacity to multi-display αPD-1 on its surface as αPD-1 was located at the hydrophilic terminus of the fusion. In contrast, intact αPD-1 had hydrodynamic diameters of 11.63 ±3.76 nm respectively (Figure 2). The size of intact αPD-1 is consistent with reported sizes of natural, intact IgGs26. We further measured hydrodynamic diameters of the αPD-1-iTEP fusion at different temperatures, concentrations, and redox conditions. The results of these measurements are summarized in Table 2. The hydrodynamic diameters of the oxidized sample did not change significantly between two tested concentrations, 25 μM and 0.25 μM, suggesting that the NP assembled from the fusion was stabilized by oxidization and cross-linking. The diameters of the reduced sample were very different between the two tested concentrations, suggesting that the NP, without cross-linking through disulfide bonds, dissociated upon dilution. The above two conclusions are valid at the both temperatures used, 37 °C and 25 °C. In addition, diameter values of the fusion did not change between the two temperatures.
Figure 2.
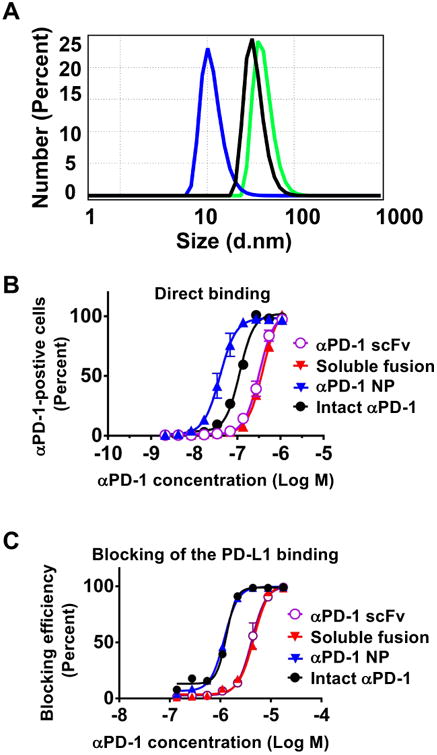
In vitro characterization of the αPD-1-iTEP fusion. (A) The DLS spectra of intact αPD-1 (blue), the αPD-1-iTEP fusion after NP assembly (green), and the amphiphilic iTEP after NP assembly (black). 20 μM of each protein was used for the DLS analysis at 37 °C. The included table lists hydrodynamic diameters of each protein. (B) Direct binding of αPD-1 scFv, the soluble αPD-1-iTEP fusion, αPD-1 NP, and intact αPD-1 to EL4 cells. The cells were incubated with different concentrations of labeled sample before being analyzed by flow cytometry. (C) Blocking the PD-L1 binding to EL4 cells. The blocking efficiencies were plotted against sample concentrations. Various concentrations of each listed αPD-1 sample were used to compete with the PD-L1-human Fc fusion (10 μg/mL) for binding to EL4 cells. The bound PD-L1 fusion was detected by an Alexa Fluor 488-labeled, anti-human Fc antibody. In both B and C, the x-axis labels are αPD-1 equivalent concentrations in each sample.
Table 2.
Hydrodynamic diameters of αPD-1-iTEP under different redox status, concentrations, and temperatures.
| Redox status | Concentration (μM) | Temperature | |
|---|---|---|---|
| 37 °C | 25 °C | ||
| Oxidized | 25 | 44.1±12.8 | 38.3±11.3 |
| 0.25 | 43.9±12.8 | 35.2±10.7 | |
| Reduced | 25 | 42.9±12.3 | 39.2±11.4 |
| 0.25 | 8.9±2.2* | 5.7±1.5* | |
Note: The values of the hydrodynamic diameters are mean±standard deviation.
These small hydrodynamic diameter values suggest that sample does not have a NP structure.
We next examined the binding of the αPD-1-iTEP fusion, in both its soluble form and its NP form, with EL4 cells, a PD-1-positive cell line27. According to the results of a direct binding assay (Figure 2B), the soluble fusion and the scFv have comparable binding avidities to EL4 cells (EC50=0.40 μM, 95% CI 0.38∼0.41 μM vs. EC50=0.32 μM, 95% CI 0.30∼0.35 μM). Thus, adding the amphiphilic iTEP to the scFv did not significantly compromise the binding of the scFv to its antigens. However, the avidities of both the soluble fusion and the scFv are clearly weaker than intact αPD-1 (EC50=0.11 μM, 95% CI 0.10∼0.12 μM), suggesting that scFv loses some of its binding avidity as compared to its intact, parental antibody. Such loss is not uncommon for scFvs28. αPD-1 NP, in contrast, possesses a 4-times stronger avidity than intact αPD-1 (EC50=0.039 μM with 95% CI 0.036∼0.043 μM). The stronger avidity may be attributed to a potentially multivalent display of the scFvs by the NP and a synergistic effect between the binding of first scFv on the NP and the binding of another scFv on the NP. This effect was described as a area of influence previously29.
We lastly examined how well the soluble αPD-1-iTEP fusion and αPD-1 NP inhibit the binding of PD-L1 to the PD-1-positive cells, the working mechanism of PD-1 immune checkpoint therapy30-31. To accomplish the examination, we designed and employed a PD-L1 binding inhibition assay. Specifically, we took advantage of the facts that PD-L1-human Fc, a fusion protein of mouse PD-L1 fusion and human IgG Fc, bound with EL4 cells, and that the bound PD-L1-human Fc can be detected by an anti-human Fc antibody. According to results of the binding inhibition assay (Figure 2C), the soluble fusion and the scFv have the same inhibition capacity (EC50=4.59 μM with 95% CI 4.26∼4.94 μM vs. EC50=4.16 μM with 95% CI 3.87∼4.48 μM). However, both the soluble fusion and the scFv have a three times lower inhibition capacity than intact αPD-1 (EC50=1.32 μM with 95% CI 1.25∼1.38 μM), a result consistent with the result of the direct binding assay. On the other hand, αPD-1 NP possesses a three times higher inhibition capacity than the soluble αPD-1-iTEP fusion, (EC50=1.19 μM with 95% CI 1.15∼1.23 μM). Indeed, the inhibition capacity of the NP is slightly but significantly higher than intact αPD-1. Again, these results reinforce the advantage of multi-displaying antibodies by the NP.
In vivo characterization of αPD-1-iTEP fusion
αPD-1 exacerbates diabetes development in non-obese diabetic (NOD)/ShiLtJ mice because it blocks the PD-1 immune checkpoint and worsens the autoimmune disorders of the mice24. We utilize this effect to examine whether the αPD-1-iTEP fusion is functional in vivo (Figure 3A) and use diabetes-free survival as an outcome to evaluate the effect (Figure 3B). According to the survival data, both the soluble αPD-1-iTEP fusion and αPD-1 NP significantly accelerated diabetes development in NOD mice as compared to PBS (median survival time, Log rank test, p=0.049 and p=0.049). The median diabetes-free survival time of the soluble fusion- and the αPD-1 NP-treated mice are 16 days and 21 days, respectively. In contrast, none of the PBS-treated mice developed diabetes before all these mice were censored on day 30 after the treatment initiation. Further, the effect of the soluble fusion and αPD-1 NP on diabetes development are not statistically different from intact αPD-1 (P=0.771 and p=0.900). The median diabetes-free survival time for intact αPD-1-treated mice is 19 days. Last, the effect of the soluble fusion and αPD-1 NP are not different (p=0.775). Together, these results suggest that the αPD-1-iTEP fusion, either in its soluble form or in its NP form, is functional in vivo and is as effective as intact αPD-1 in blocking the PD-1 immune checkpoint and promoting diabetes in NOD mice.
Figure 3.

A. Blood glucose concetrations of all mice treated with intact αPD-1, the soluble αPD-1-iTEP fusion, and αPD-1 NP. Each line represents glucose concentration changes of one mouse. Blood glucose concetrations were monitored up to 30 days after the initial treatment. Line colors reflect treatments. Data of PBS-treated mice were not included for the simplicity of the figure. In addition, none of PBS-treated mice showed glucose levels higher than 250 mg/dL, a threshold level of diabetes (red dash line), during the observation period. B Diabetes-free survival of the mice that received PBS, intact αPD-1, the soluble fusion, and αPD-1 NP. The diabetes-free survival data were analyzed by the Kaplan-Meier method. Red arrows indicate the date of treatments.
Discussion
In the current study, we successfully generated an scFv of αPD-1 and a NP that deliver αPD-1. We further showed that αPD-1 on the NP carrier is able to block the PD-1 immune checkpoint. This study has two major contributions to immune checkpoint therapy.
We generated the first recombinant αPD-1, the αPD-1 scFv. We also generated a fusion protein of αPD-1 and iTEP as well as an αPD-1 NP. These deliverables of the project constitute a set of tools that may facilitate the improvement of αPD-1 immune therapy. For example, these tools could be instrumental when developing delivery systems to realize a cell-specific αPD-1 therapy, which is required to resolve the root cause of the αPD-1 toxicity. Additionally, the success of generating these tools offers important insights on how to produce other immune checkpoint inhibitors as recombinant proteins as all reported inhibitors thus far are antibodies. This success also provides a simple yet effective strategy to create carriers to deliver these inhibitors.
The αPD-1 NP effectively blocks the PD-1 checkpoint, a success that underscores the importance of multivalency in interactions between antibodies and the cells expressing the corresponding antigens. The αPD-1 scFv has a reduced avidity to PD-1-positive cell and a weaker inhibition on the PD-L1 binding to the cells as compared to intact αPD-1. A similar deficiency has been reported for scFv previously29, 32. The possible reasons of the deficiency include that (1) scFv is monovalent while intact αPD-1 is divalent; (2) scFv may has a lower thermodynamic stability than αPD-1, which comprises its binding with its antigens32. The deficiency was, nevertheless, resolved by the αPD-1 NP. The NP indeed showed stronger binding to PD-1-positive cells and greater PD-L1 binding inhibition than intact αPD-1, which clearly demonstrated the impact of multivalency as the NP can display multiple scFvs on its surface. It is notable that the EC50 of the NP is about three times smaller than that of intact αPD-1 according to the direct binding results; however, the EC50 of the NP is only slightly smaller than that of intact αPD-1 according to the PD-L1 binding inhibition results. This apparent discrepancy may be due to the different methodologies of the two experiments. Another plausible reason for the discrepancy is that not all of the αPD-1 scFVs on the NP that bound with PD-1-positive cells actually engaged with PD-1 on the cell surface. These unengaged scFv, therefore, were able to inhibit the PD-L1 binding. The existence of these “unengaged” scFvs may be caused by a steric effect between scFvs on the NP and accessibility of adjacent PD-1 on the cells. To ambiguously elucidate avidity difference between the scFv, intact αPD-1, and αPD-1 NP, and the mechanism underlying the difference, more studies are needed in the future.
In summary, we generated an αPD-1 NP that is functional and possess the advantage of multivalency. This NP could serve as a foundation to develop carriers for αPD-1 and other immune checkpoint inhibitors that target the inhibitors to a specific subpopulation of PD-1-positive cells.
Supplementary Material
Acknowledgments
We thank Dr. Shawn C. Owen and Dr. Andrew Dixon for the help in designing and expressing the αPD-1 scFv. We than Dr. James N. Herron for his advice in the production and purification of intact αPD-1.
The work has been supported by research funding from the University of Utah, including Pilot Grant for the University of Utah, Center for Clinical and Translational Science and the Program in Personalized Health Collaborative and The Huntsman Cancer Institute, the Experimental Therapeutics Program (ET) Grant Award Number: 170301.
References
- 1.Michielin O, Hoeller C. Gaining momentum: New options and opportunities for the treatment of advanced melanoma. Cancer Treat Rev. 2015;41(8):660–70. doi: 10.1016/j.ctrv.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 2.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, Burke MM, Caldwell A, Kronenberg SA, Agunwamba BU, Zhang X, Lowy I, Inzunza HD, Feely W, Horak CE, Hong Q, Korman AJ, Wigginton JM, Gupta A, Sznol M. Nivolumab plus ipilimumab in advanced melanoma. The New England journal of medicine. 2013;369(2):122–33. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Administration, U. S. F. a. D. pembrolizumab (KEYTRUDA) http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/125514s009lbl.pdf.
- 4.Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161(2):205–14. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, Ferrucci PF, Hill A, Wagstaff J, Carlino MS, Haanen JB, Maio M, Marquez-Rodas I, McArthur GA, Ascierto PA, Long GV, Callahan MK, Postow MA, Grossmann K, Sznol M, Dreno B, Bastholt L, Yang A, Rollin LM, Horak C, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. The New England journal of medicine. 2015 doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swaika A, Hammond WA, Joseph RW. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Molecular Immunology. 2015;67(2, Part A):4–17. doi: 10.1016/j.molimm.2015.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Tarhini A. Immune-mediated adverse events associated with ipilimumab ctla-4 blockade therapy: the underlying mechanisms and clinical management. Scientifica (Cairo) 2013;2013:857519. doi: 10.1155/2013/857519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fransen MF, van der Sluis TC, Ossendorp F, Arens R, Melief CJ. Controlled local delivery of CTLA-4 blocking antibody induces CD8+ T-cell-dependent tumor eradication and decreases risk of toxic side effects. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(19):5381–9. doi: 10.1158/1078-0432.CCR-12-0781. [DOI] [PubMed] [Google Scholar]
- 9.Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Current Opinion in Immunology. 2012;24(2):207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol. 2015;42(3):363–77. doi: 10.1053/j.seminoncol.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Pentcheva-Hoang T, Corse E, Allison JP. Negative regulators of T-cell activation: potential targets for therapeutic intervention in cancer, autoimmune disease, and persistent infections. Immunological reviews. 2009;229(1):67–87. doi: 10.1111/j.1600-065X.2009.00763.x. [DOI] [PubMed] [Google Scholar]
- 12.Gelao L, Criscitiello C, Esposito A, Goldhirsch A, Curigliano G. Immune checkpoint blockade in cancer treatment: a double-edged sword cross-targeting the host as an “innocent bystander”. Toxins. 2014;6(3):914–33. doi: 10.3390/toxins6030914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nishino M, Sholl LM, Hatabu H, Ramaiya NH, Hodi FS. Anti–PD-1–Related Pneumonitis during Cancer Immunotherapy. New England Journal of Medicine. 2015;373(3):288–290. doi: 10.1056/NEJMc1505197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kochupurakkal NM, Kruger AJ, Tripathi S, Zhu B, Adams LT, Rainbow DB, Rossini A, Greiner DL, Sayegh MH, Wicker LS, Guleria I. Blockade of the programmed death-1 (PD1) pathway undermines potent genetic protection from type 1 diabetes. PloS one. 2014;9(2):e89561. doi: 10.1371/journal.pone.0089561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frebel H, Oxenius A. The risks of targeting co-inhibitory pathways to modulate pathogen-directed T cell responses. Trends in immunology. 2013;34(5):193–9. doi: 10.1016/j.it.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, Sharpe AH, Powrie F. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. Journal of immunology. 2006;177(7):4376–83. doi: 10.4049/jimmunol.177.7.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Sun W, Ye Y, Hu Q, Bomba HN, Gu Z. In situ activation of platelets with checkpoint inhibitors for post-surgical cancer immunotherapy. Nature Biomedical Engineering. 2017;1:0011. [Google Scholar]
- 18.Zhao P, Dong S, Bhattacharyya J, Chen M. iTEP Nanoparticle-Delivered Salinomycin Displays an Enhanced Toxicity to Cancer Stem Cells in Orthotopic Breast Tumors. Molecular Pharmaceutics. 2014;11(8):2703–2712. doi: 10.1021/mp5002312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao P, Xia G, Dong S, Jiang ZX, Chen M. An iTEP-salinomycin nanoparticle that specifically and effectively inhibits metastases of 4T1 orthotopic breast tumors. Biomaterials. 2016;93:1–9. doi: 10.1016/j.biomaterials.2016.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho S, Dong S, Parent KN, Chen M. Immune-tolerant elastin-like polypeptides (iTEPs) and their application as CTL vaccine carriers. Journal of drug targeting. 2016;24(4):328–39. doi: 10.3109/1061186X.2015.1077847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yamazaki T, Akiba H, Koyanagi A, Azuma M, Yagita H, Okumura K. Blockade of B7-H1 on Macrophages Suppresses CD4+ T Cell Proliferation by Augmenting IFN-γ-Induced Nitric Oxide Production. The Journal of Immunology. 2005;175(3):1586–1592. doi: 10.4049/jimmunol.175.3.1586. [DOI] [PubMed] [Google Scholar]
- 22.Noeman SA, Misra DN, Yankes RJ, Kunz HW, Gill TJ. Growth of rat-mouse hybridomas in nude mice and nude rats. Journal of immunological methods. 1982;55(3):319–326. doi: 10.1016/0022-1759(82)90091-6. [DOI] [PubMed] [Google Scholar]
- 23.Reik LM, Maines SL, Ryan DE, Levin W, Bandiera S, Thomas PE. A simple, non-chromatographic purification procedure for monoclonal antibodies. Isolation of monoclonal antibodies against cytochrome P450 isozymes. J Immunol Methods. 1987;100(1-2):123–30. doi: 10.1016/0022-1759(87)90180-3. [DOI] [PubMed] [Google Scholar]
- 24.Ansari MJI, Salama AD, Chitnis T, Smith RN, Yagita H, Akiba H, Yamazaki T, Azuma M, Iwai H, Khoury SJ, Auchincloss H, Sayegh MH. The Programmed Death-1 (PD-1) Pathway Regulates Autoimmune Diabetes in Nonobese Diabetic (NOD) Mice. The Journal of Experimental Medicine. 2003;198(1):63–69. doi: 10.1084/jem.20022125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Marco A. Strategies for successful recombinant expression of disulfide bond-dependent proteins in Escherichia coli. Microbial Cell Factories. 2009;8:26–26. doi: 10.1186/1475-2859-8-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lavoisier A, Schlaeppi JM. Early developability screen of therapeutic antibody candidates using Taylor dispersion analysis and UV area imaging detection. MAbs. 2015;7(1):77–83. doi: 10.4161/19420862.2014.985544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. Journal of immunology (Baltimore, Md: 1950) 2008;181(7):4832–9. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahmad ZA, Yeap SK, Ali AM, Ho WY, Alitheen NBM, Hamid M. scFv Antibody: Principles and Clinical Application. Clinical and Developmental Immunology. 2012;2012:15. doi: 10.1155/2012/980250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cuesta AM, Sainz-Pastor N, Bonet J, Oliva B, Alvarez-Vallina L. Multivalent antibodies: when design surpasses evolution. Trends Biotechnol. 2010;28(7):355–62. doi: 10.1016/j.tibtech.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 30.Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, Tamada K, Chen L. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65(3):1089–96. [PubMed] [Google Scholar]
- 31.Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12293–7. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deyev SM, Lebedenko EN. Multivalency: the hallmark of antibodies used for optimization of tumor targeting by design. Bioessays. 2008;30(9):904–18. doi: 10.1002/bies.20805. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.