

Abstract
Glucose transporter type 1 (GLUT1) deficiency syndrome (GLUT1‐DS) leads to a wide range of neurological symptoms. Ketogenic diets are very efficient to control epilepsy and movement disorders. We tested a novel simple and rapid blood test in 30 patients with GLUT1‐DS with predominant movement disorders, 18 patients with movement disorders attributed to other genetic defects, and 346 healthy controls. We detected significantly reduced GLUT1 expression only on red blood cells from patients with GLUT1‐DS (23 patients; 78%), including patients with inconclusive genetic analysis. This test opens perspectives for the screening of GLUT1‐DS in children and adults with cognitive impairment, movement disorder, or epilepsy. Ann Neurol 2017;82:133–138
Glucose transporter type 1 (GLUT1) deficiency syndrome (GLUT1‐DS) is caused by impaired glucose transport across the blood–brain barrier and into astrocytes attributed to heterozygous, likely mostly de novo, mutations in the SLC2A1 gene encoding the glucose transporter, GLUT1.1 GLUT1 is a membrane‐bound glycoprotein expressed in erythrocytes, brain microvessels, and glial cells. SLC2A1 mutations limit brain glucose availability and lead to cerebral energy deficiency. As a consequence, GLUT1‐DS manifests with a wide range of neurological symptoms, including cognitive impairment, epilepsy, and permanent and/or paroxysmal motor disorders that usually start in early childhood.1, 2 Early detection of GLUT1‐DS is critical given that the disease is treatable with ketogenic diets3 and, possibly, novel experimental therapies.4, 5
Any neuropediatrician or neurologist can encounter patients with GLUT1‐DS. Its diagnosis is, however, challenging because of the great heterogeneity of symptoms and ages at onset. Besides the classical severe infantile‐onset epileptic encephalopathy, adult‐onset forms have been described with epilepsy or paroxysmal movement disorders as the sole manifestations.6 The 3‐O‐methyl‐D‐glucose (OMG) uptake assay is an important functional measure of glucose transport across the red blood cell (RBC) membrane,7 but is not available on a clinical basis because it implies the use of radioactivity and precautions in blood sample storage and preanalytical steps.8 In a patient whose phenotype suggests GLUT1‐DS, the diagnosis thus relies on lumbar puncture (LP)—that is, low cerebrospinal fluid (CSF) glucose concentration, supported by low CSF lactate—and SLC2A1 molecular analysis. These approaches have some limitations. LP must be performed after at least 6 hours of fasting, is invasive, and runs the risk of complications.9 Moreover, an unequivocal cutoff has not been set to date for CSF glucose, with an heterogeneity in current practice although the most used value is 2.2mM.8 On the other hand, the analysis of the coding regions of SLC2A1 can be tedious, inconclusive in case of variants of unknown significance, or may fail at identifying variants despite a very suggestive clinical and biochemical phenotype.10
A minimally invasive and rapid diagnostic test for GLUT1‐DS, readily available in clinical practice, would be an invaluable tool for the early detection of GLUT1‐DS. Here, we tested a novel diagnostic test on RBCs, using flow cytometry, on a series of consecutive GLUT1 deficient patients with predominant movement disorders, compared to patients with movement disorders attributed to other genetic defects and healthy controls.
Study Population and Methods
Patients and Controls
We enrolled 30 patients (2–50 years old, 17 males/13 females) with GLUT1‐DS presenting with a highly suggestive clinical phenotype comprising variable degree of developmental delay, movement disorders, and epilepsy (Table), associated with low glucose CSF concentration (2.0mM ± 0.3), and/or a pathogenic SLC2A1 mutation (see Table). CSF lactate concentration was available for 16 patients (see Table) and was consistently low (0.9mM ± 0.2), except for 1 patient (P20). Because the majority of our GLUT1‐DS patients manifested paroxysmal movement disorders, we also enrolled 18 patients (11–73 years old, 12 males/6 females) with paroxysmal movement disorders attributed to other genetic defects—that is, mutations in ATP1A3, ADCY5, or PRRT2—to determine whether the RBC test was able to distinguish them among GLUT1‐DS patients. All participants or their caregivers provided written informed consent for study procedures and data reporting. The study was approved by the local ethics committee (CPPIdF6, La Pitié‐Salpêtrière University Hospital). The French National Blood Service (Etablissement Français du Sang) provided blood samples from healthy donors.
CSF Glucose Concentration
CSF glucose was measured after a fasting period of at least 6 hours using glucose hexokinase methodology.
SLC2A1 Molecular Analysis
Mutational analysis of all exons and intron‐exon boundaries of the SLC2A1 gene were performed on genomic DNA from patients and their parents by direct Sanger sequencing (primer sequences are available upon request). The GenBank [NM_006516] sequence was used as the reference. Pathogenicity was predicted by in silico analysis with Alamut Visual version 2.7 (Interactive Biosoftware, Rouen, France). When no point mutation was found, a single or multiple exon deletion or duplication was searched by multiple ligation‐dependent probe amplification with SALSA MLPA KIT P138 SLC2A1 (MRC‐Holland, BV, Amsterdam, Netherlands).1
Flow Cytometry Analysis of GLUT1 Surface Expression on Circulating RBCs
At least 0.5ml of nonfasted venous blood was collected on ethylenediaminetetraacetic acid (EDTA) tubes and kept at 4 °C for maximum 7 days before analysis. Whole blood was diluted at 1/3,000 into phosphate‐buffered saline (PBS) containing 0.09% sodium azide, 1mM of EDTA, and 0.33mg/ml of bovine serum albumin (binding buffer). Approximately 80,000 RBCs per sample were transferred into a 96‐well microplate and stained with GLUT1.RBD ligand fused to enhanced green fluorescent protein (eGFP; METAFORA Biosystems, Evry, France) at 37 °C for 20 minutes.11, 12 RBCs were washed twice with PBS and resuspended in binding buffer. Acquisition was performed on a FACSVerse flow cytometer (BD Biosciences, Franklin Lakes, NJ) equipped with 405‐, 488‐, and 640‐nm lasers. Data analysis was performed using Flowjo software (TreeStar, Inc., Ashland, OR). Debris and doublets were excluded from the analysis; eGFP signal was collected for each sample, by taking into account geometric mean of gated, GLUT1.RBD‐stained RBC. Results were available within 24 hours upon sample reception.
Results
Our series reflects the wide range of phenotypes, ages, and ages at onset in GLUT1‐DS—that is, classical early‐onset severe encephalopathy, global developmental delay with dystonia and epilepsy, early‐onset absence epilepsy with cognitive impairment, adult‐onset movement disorders. CSF glucose was measured in 25 of 30 patients because, in two families, only molecular analysis was performed (P11, P13, and P24) once the diagnosis was made in the index case (P12, P25). Twenty of these 25 GLUT1‐DS patients (80%) exhibited a typical low CSF glucose (≤2.2mM). The remaining 5 patients with CSF glucose between 2.3 and 2.5mM harbored either known pathogenic SCL2A1 mutations or novel de novo mutations.
Surface GLUT1 expression level on circulating RBCs was performed blinded from the disease condition in terms of symptoms, biochemistry, and genetics and compared between GLUT1‐DS patients, patients with other genetic defects, and healthy donors. Most patients were analyzed at least twice, with several‐month intervals, and the results were consistent for each patient, showing the stability of GLUT1 deficiency on RBCs. A thorough repeatability and reproducibility study was also performed between two labs, supporting the robustness of the RBC test (Fig 1). A distribution fitting of a normal law to the series of data allowed describing the variability of GLUT1 expression level both in the normal population and among patients (Fig 2). GLUT1 expression on RBCs varied in healthy donors in a limit of 1.5 standard deviations (SDs), corresponding in this population to 15% of change of GLUT1 compared to the mean sample. None of the patients bearing mutations in ATP1A3, ADCY5, or PRRT2 had a reduced GLUT1 expression on RBC (91–118% compared to healthy controls).
Figure 1.
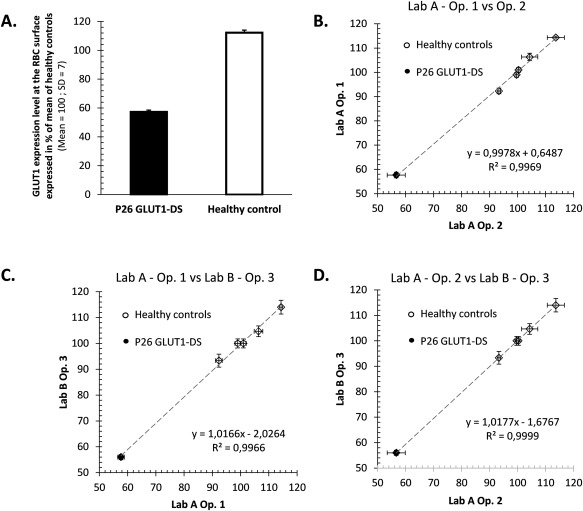
(A) Repeatability: GLUT1‐DS patient P26 was analyzed 12 times along with a healthy control, in the same experiment. Standard deviations are shown, which translate into CVs below 2.5%, pointing out the test's high repeatability. (B,C,D) Reproducibility: GLUT1‐DS patient P26 was analyzed along with 5 healthy control samples. Experiment was repeated three times (3 days) by three operators in two distinct labs. Mean GLUT1 expression and standard deviations are plotted for each sample and each operator. Results were highly correlated between operators (all R 2 > 0.98). No significant difference was found between days, operators, nor labs; results were highly reproducible (Levene's test; all p values, > 0.93). CVs = coefficients of variation; GLUT1 = glucose transporter type 1; GLUT1‐DS = glucose transporter type 1 deficiency syndrome.
Figure 2.

GLUT1 expression levels on RBC quantified by flow cytometry, expressed as % of mean of healthy controls. Twenty‐three GLUT1‐DS patients, of 30 (black dots), clustered in a specific group, clearly distinct from 18 patients with other gene‐related movement disorders (white squares) and 346 healthy controls—the black curve represents the distribution fitting of a normal law to the series of data (healthy controls only). GLUT1 = glucose transporter type 1; GLUT1‐DS = glucose transporter type 1 deficiency syndrome; RBC = red blood cells; SD = standard deviation.
The RBC test identified 23 GLUT1‐DS patients of 30 (78%) with at least 20% decrease (23–59% decrease) of GLUT1 expression and no overlap between their group and the controls (Fig 2). The RBC test did not correlate with CSF glucose, nor age or age at onset. Three GLUT1‐DS patients were detected with this new test despite a CSF glucose concentration >2.2mM (Table, P15–21–23). Two patients with a clinical phenotype very suggestive of GLUT1‐DS, and low CSF glucose and lactate, but no SLC2A1 mutation had an abnormal RBC test (Table 1, P16–27), thus further confirming the diagnosis of GLUT1‐DS. An OMG uptake assay was previously performed on P16's RBCs and showed 63% of normal uptake, consistent with the 63% GLUT1 expression on RBCs measured here. Conversely, 1 patient presenting with a severe nonepileptic encephalopathy, low CSF glucose (2.2mM), and a frameshift mutation in exon 10 (p.Gly471Glufs*37), but inconclusive familial segregation, exhibited a normal RBC test (data not shown) supporting that he does not have GLUT1‐DS. Seven GLUT1‐DS patients displayed normal GLUT1 expression on RBCs (Fig 2) and mainly carried SLC2A1 mutations in exons 6 and 7. Two mutations (P3 Gly130Ser and P4 Thr295Met) have been reported in the literature, but were accompanied by a mild decrease in OMG uptake compared to most mutations—75% of normal uptake versus 50% meanwhile.13 Two mutations (P19‐24 p.Ala275Thr and P1‐P20 p.Gly314Ser) were shown to affect glucose uptake, but only in Xenopus oocytes.14
Discussion
This proof‐of‐concept study shows that GLUT1‐DS can be accurately detected through a minimally invasive and rapid (24 hours) diagnostic test. We have been able to define the physiological variation of GLUT1 expression level at the surface of RBCs in the general population, and can conclude that plus or minus 15% GLUT1 expression change fell within the physiological range, whereas more than 20% decrease is a hallmark of GLUT1 deficiency related to SLC2A1 mutations. Currently, the diagnostic rate of this new test is comparable to CSF glucose and promises to be more specific. Indeed, in a large series of 109 patients suspected of GLUT1‐DS with low CSF glucose and lactate, only 68% displayed abnormal OMG uptake and 64% harbored a pathogenic SCL2A1 mutation.7 Furthermore, the RBC test should be more cost‐effective as compared to LP and genetics, and the result comes back in less than 3 days in current clinical practice. It has been designed so as to be automated from sample preparation to data analysis, and is CE (“Conformité Européene” [“European Conformity”]) marked opening easy diffusion in central testing labs.
The RBC test was able to detect GLUT1‐DS patients regardless of their age and disease severity, including 3 patients with CSF glucose concentration >2.2mM, but known pathogenic SCL2A1 mutations and/or de novo mutations. The detection of GLUT1 deficiency on RBCs in 2 patients presenting with a very suggestive clinical and biochemical phenotype, but without SLC2A1 exonic mutation suggests that this assay should identify GLUT1‐DS patients with mutations in introns or regulating regions. Conversely, patients not detected by the RBC test may correspond either to patients with mutations affecting mildly glucose uptake, as shown for four mutations13, 14 harbored by 6 of the 7 patients with a normal RBC test, but not GLUT1 expression, or a brain‐restricted GLUT1 deficiency, possibly attributed to alterations of specific SLC2A1 isoforms.
Quantifying GLUT1 at the RBC surface opens new perspectives for GLUT1‐DS, especially to avoid current considerable diagnosis delays. It is a major issue in light of the great phenotypic variability and availability of efficient treatments. Although more studies are required to establish the diagnostic gain of the RBC test on a larger cohort, such a simple diagnostic test, readily available in clinical practice, ought to greatly enlarge the screening of GLUT1‐DS in any patient, child, or adult, presenting with cognitive impairment, epilepsy, ataxia and/or dystonia, or paroxysmal movement disorder.
Author Contributions
D.G., L.D.C., V.P., O.B.T., E.R., and F.M. were responsible for conception and design of the study. D.G., C.C., C.K., C.W.F., S.A., N.E., B.H.C., E.H., M.P.L., V.P., S.V., E.R., and F.M. were responsible for acquisition and analysis of data. D.G., V.P., and F.M. were responsible for drafting a significant portion of the manuscript or figures.
Potential Conflicts of interest
Vincent Petit is the CEO of Metafora Biosystems, which developed the test and performed the flow cytometry experiments during the study. The company is commercializing the RBC test to testing labs.
Table 1.
Main Clinical, Biochemical, and Molecular Characteristics of Patients With GLUT1 Deficiency
| Patient | Sex | Age | Age at Onset | LD | Motor Symptoms | Epilepsy | Paroxysmal Dyskinesia | CSF Glucose | CSF Lactate | RBC Test | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | M | 9y | 2y | + | A | — | ++ | 1.9 | 1.1 | 100 | p.Gly314Ser |
| P2 | F | 21y | 6m | + | D | — | ++ | 2.0 | Nd | 66 | p.Met1Thr |
| P3 | F | 25y | 3m | + | A,D | AA | ++ | 2.0 | 0.9 | 109 | p.Gly130Ser |
| P4 | M | 19y | 2.5y | + | A,D | AA | ++ | 2.4 | 1.3 | 110 | p.Thr295Met |
| P5 | M | 2y | 9m | + | — | AA | — | 1.8 | 1.1 | 41 | p.Gly286Hisfs*94 |
| P6 | M | 5y | 6m | +++ | D | S | + | 1.7 | 0.9 | 60 | del 1p34.2‐p34.1 |
| P7 | F | 4y | 2y | +++ | A, D | S | ++ | 1.5 | 1.0 | 53 | p.Lys183Glnfs*54 |
| P8 | F | 26y | 1.5y | + | A | — | ++ | 2.2 | 0.7 | 51 | p.Ser294Pro |
| P9 | F | 22y | 1y | + | A, D | — | ++ | 2.2 | Nd | 59 | p.Met142Lys |
| P10 | F | 17y | 6m | +++ | A | GE | — | 2.2 | 0.7 | 62 | p.Ser313Thrfs*23 |
| P11 | M | 50y | 14y | — | D | AA | ++ | Nd | Nd | 52 | p.Leu12Phe |
| P12 | M | 18y | 2.5y | + | A | — | ++ | 2.2 | Nd | 61 | p.Leu12Phe |
| P13 | M | 16y | 2.5y | + | A, D | — | + | Nd | Nd | 66 | p.Leu12Phe |
| P14 | M | 3y | 3m | ++ | A | S | ++ | 1.4 | Nd | 52 | p.Met96Cysfs*2 |
| P15 | M | 15y | 9m | ++ | A | — | ++ | 2.3 | 1.1 | 54 | p.Gly18Arg |
| P16 | M | 14y | 1.5y | + | A | S | ++ | 1.9 | 0.7 | 63 | not found |
| P17 | M | 6y | 3m | +++ | D | GE | — | 1.5 | 1.2 | 75 | large del (1.97 Mb) |
| P18 | F | 29y | 3m | +++ | A, D, S | AA | — | 1.9 | 0.6 | 54 | p.Gln282* |
| P19 | F | 4y | 3m | + | A, D | AA | ++ | 1.9 | 0.9 | 86 | p.Ala275Thr |
| P20 | F | 20y | 1.5y | — | — | GE | ++ | 2.5 | 1.8 | 106 | p.Gly314Ser |
| P21 | F | 18y | 8y | + | — | AA | — | 2.3 | Nd | 70 | p.Val328Leu |
| P22 | M | 13y | 3y | + | A, D | ++ | 1.8 | 0.9 | 97 | p.Thr310Asn | |
| P23 | M | 25y | 1.5y | + | D | AA | ++ | 2.4 | Nd | 45 | p.Arg153Cys |
| P24 | F | 25y | 5y | — | — | AA | + | Nd | Nd | 88 | p.Ala275Thr |
| P25 | M | 16y | 4y | + | — | S | ++ | 2.2 | Nd | 77 | p.Ala275Thr |
| P26 | M | 13y | 1m | +++ | — | S | — | 1.7 | 1.0 | 57 | p.Glu41* |
| P27 | F | 10y | 1.5y | ++ | D | AA | + | 1.5 | Nd | 54 | not found |
| P28 | M | 13y | 6m | ++ | A, D | S | + | 1.8 | Nd | 44 | p.Arg126Cys |
| P29 | F | 18y | 19m | ++ | — | AA | ++ | Nd | Nd | 60 | p.Ser324Leu |
| P30 | M | 50y | 6y | — | — | AA | + | Nd | Nd | 76 | p.Ser324Leu |
Age at onset refers to the first noticeable motor or cognitive symptom.
LD = learning disability; CSF = cerebrospinal fluid; RBC test = red blood cell test (expressed as % of mean of healthy controls; normal values are indicated in italics); M = male; F = female; y = year; m = month; A = ataxia; D = dystonia; AA = atypical absence; GE = generalized epilepsy; S = seizures; n = normal; + = mild; + + = moderate; + ++ = severe; — = none; Nd = not done.
Acknowledgment
Part of this work has been realized with the funding support of the Joshua Hellmann Foundation for Orphan Disease, Hong Kong. It has also received funding from the European Union's Horizon 2020 research and innovation program under Grant Agreement No. 761702 to Metafora Biosystems.
We thank warmly Drs Benedicte Heron, Agathe Roubertie, Anne de Saint‐Martin, Cecile Laroche, Elsa Kaphan, and Claude Cances for patient referral and fruitful collaboration, as well as Drs Naomi Taylor and Marc Sitbon for stimulating discussions. We also thank Karine Bonnet, Nadine Litou, and Eléonore Botton from Laboratoire CERBA for participating in the repeatability and reproducibility study to assess the test's robustness.
References
- 1. Leen WG, Klepper J, Verbeek MM, et al. Glucose transporter‐1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain 2010;133:655–670. [DOI] [PubMed] [Google Scholar]
- 2. De Vivo DC, Trifiletti RR, Jacobson RI, et al. Defective glucose transport across the blood‐brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med 1991;325:703–709. [DOI] [PubMed] [Google Scholar]
- 3. Klepper J, Leiendecker B. Glut1 deficiency syndrome and novel ketogenic diets. J Child Neurol 2013;28:1045–1048. [DOI] [PubMed] [Google Scholar]
- 4. Mochel F, Hainque E, Gras D, et al. Triheptanoin dramatically reduces paroxysmal motor disorder in patients with GLUT1 deficiency. J Neurol Neurosurg Psychiatry 2016;87:550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tang M, Gao G, Rueda CB, et al. Brain microvasculature defects and Glut1 deficiency syndrome averted by early repletion of the glucose transporter‐1 protein. Nat Commun 2017;8:14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gras D, Roze E, Caillet S, et al. GLUT1 deficiency syndrome: an update. Rev Neurol (Paris) 2014;170:91–99. [DOI] [PubMed] [Google Scholar]
- 7. Yang H, Wang D, Engelstad K, et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol 2011;70:996–1005. [DOI] [PubMed] [Google Scholar]
- 8. Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res 2012;100:272–277. [DOI] [PubMed] [Google Scholar]
- 9. Chordas C. Post‐dural puncture headache and other complications after lumbar puncture. J Pediatr Oncol Nurs 2001;18:244–259. [DOI] [PubMed] [Google Scholar]
- 10. Verrotti A, D’Egidio C, Agostinelli S, Gobbi G. Glut1 deficiency: when to suspect and how to diagnose? Eur J Paediatr Neurol 2012;16:3–9. [DOI] [PubMed] [Google Scholar]
- 11. Montel‐Hagen A, Blanc L, Boyer‐Clavel M, et al. The Glut1 and Glut4 glucose transporters are differentially expressed during perinatal and postnatal erythropoiesis. Blood 2008;112:4729–4738. [DOI] [PubMed] [Google Scholar]
- 12. Cretenet G, Clerc I, Matias M, et al. Cell surface Glut1 levels distinguish human CD4 and CD8 T lymphocyte subsets with distinct effector functions. Sci Rep 2016;6:24129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang D, Pascual JM, Yang H, et al. Glut‐1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol 2005;57:111–118. [DOI] [PubMed] [Google Scholar]
- 14. Weber YG, Storch A, Wuttke TV, et al. GLUT1 mutations are a cause of paroxysmal exertion‐induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest 2008;118:2157–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]