

Summary
The mosaic distribution of interbreeding taxa with contrasting ecology and morphology offers an opportunity to study microevolutionary dynamics during ecological divergence. We investigate here the evolutionary history of an alpine and a montane ecotype of Heliosperma pusillum (Caryophyllaceae) in the south‐eastern Alps.
From six pairs of geographically close populations of the two ecotypes (120 individuals) we obtained a high‐coverage restriction site associated DNA sequencing (RADseq) dataset that was used for demographic inference to test the hypothesis of parallel evolution of the two ecotypes.
The data are consistent with repeated ecological divergence in H. pusillum, uncovering up to five polytopic origins of one ecotype from the other. A complex evolutionary history is evidenced, with local isolation‐with‐migration in two population pairs and intra‐ecotype migration in two others. In all cases, the time of divergence or secondary contact was inferred as postglacial. A metagenomic analysis on exogenous contaminant RAD sequences suggests divergent microbial communities between the ecotypes.
The lack of shared genomic regions of high divergence across population pairs illustrates the action of drift and/or local selection in shaping genetic divergence across repeated cases of ecological divergence.
Keywords: coalescent‐based simulations, demography, phyllosphere, RAD sequencing, repeated evolution
Introduction
Ecologically divergent populations, eventually evolving into stable ecotypes, constitute an ideal opportunity to study the pace and mode of neutral and adaptive processes along the speciation continuum (Rundle & Nosil, 2005; Butlin et al., 2008; Schluter & Conte, 2009). The evolution of ecotypes is driven by two fundamentally different processes, that is selection on locally adaptive alleles and random drift, which is especially strong in small populations across fragmented landscapes. Given this complexity, repeated evolution of ecotypes provides natural replicates that add significant power to tackle fundamental evolutionary questions about the trajectory of genomic divergence from locally adapted populations to isolated species, and to infer the adaptive role of different alleles (Seehausen et al., 2014; Lowry, 2012).
Even if recurrent origins of morphological and/or ecological adaptation is a more common phenomenon than originally predicted (Levin, 2001; Wood et al., 2005), only a few cases have been documented up to now. Besides the paradigmatic case of parallel freshwater adaptation in threespine stickleback (Rundle et al., 2000; Colosimo et al., 2005), multiple origins of the same ecotype have been discovered among seaweeds (Pereyra et al., 2013), angiosperms (Brochmann et al., 2000; Berglund et al., 2004; Foster et al., 2007; Roda et al., 2013), invertebrates (Butlin et al., 2014; Soria‐Carrasco et al., 2014) and vertebrates (Østbye et al., 2006). Recurrent de novo evolution can result (1) from independent origins of the underlying molecular modification leading to similar phenotypes via recurrent mutations at the same genomic location, (2) through different alterations in the same gene producing a similar product or (3) through convergent changes in different components of the same molecular pathway (for a review see Stern, 2013). Alternatively, selection can act on already existing variation. Different populations of a species could be exposed to a similar selective pressure in a marginal or patchy habitat in the species range. As a consequence, any trait that is advantageous in that specific habitat and is present in the gene pool of the different populations as shared polymorphism (e.g. standing or flowing variation) will probably increase in frequency (Loh et al., 2013; Stankowski, 2013; Pearse et al., 2014; Lamichhaney et al., 2015; O'Brown et al., 2015).
A mosaic (i.e. interspersed) distribution of ecotypes, with genetic diversity rather structured by geographical proximity (isolation‐by‐distance; IBD) than by ecology (isolation‐by‐ecology; IBE), is often proposed as indirect evidence of repeated ecological divergence (e.g. Nosil et al., 2008; Johannesson et al., 2010). In such cases, a contrasting pattern of higher relatedness by ecology is expected only around ecologically relevant adaptive loci. The lack of such pattern at some genetic traits is not, however, conclusive evidence against parallel ecological divergence. In fact, different mutations at the same locus or at different loci participating towards the same adaptive trait can arise and/or be fixed in different populations independently (Pritchard et al., 2010; Wellenreuther & Hansson, 2016). Signal of local different adaptation could also be diluted over time by gene flow, leaving a signature (i.e. partial or soft sweeps) that is very difficult to detect (Ralph & Coop, 2010; Hermisson & Pennings, 2017). Genome‐wide signature of IBD across interspersed populations of different ecotypes could also be produced by a single origin of the ecological divergence followed by independent range expansion of the two ecotypes and subsequent intensive local introgression at different localities (Bierne et al., 2011, 2013; Welch & Jiggins, 2014). According to this hypothesis, gene flow is expected to homogenize the differentiation accumulated in allopatry across the whole genome but divergence will remain preserved at adaptive loci by selection. Even if secondary contacts followed by admixture and hybridization, driven for example by recurrent climatic shifts, are fundamental processes in evolution (Hewitt, 2001, 2011), the independent colonization (i.e. without any gene flow) of mosaically distributed habitats with contrasting ecology followed by a sudden re‐opening of gene flow between the two habitats at many different localities (‘instantaneous colonization’ model in Bierne et al., 2013) can be considered as arguably less parsimonious.
An excellent model to test the hypothesis of repeated ecological divergence is provided by the perennial caespitose herb Heliosperma pusillum (Caryophyllaceae) in the eastern Alps where two ecotypes, alpine and montane, present a mosaic distribution (Fig. 1). These two ecotypes inhabiting rocky calcareous habitats have also been described as distinct species, namely H. pusillum Vis. and H. veselskyi Janka. The former is widely distributed from the Sierra Cantabrica in the west to the Carpathians in the east (Frajman & Oxelman, 2007), whereas the latter comprises a few disjunct populations restricted to the south‐eastern Alps, usually occurring near to alpine ecotype even if separated by forest‐covered slopes extending over several hundred meters of altitude. The alpine ecotype is characterized by glabrous or sparsely hairy leaves and occasional presence of unicellular glands (Neumayer, 1923; Frajman & Oxelman, 2007; Frajman et al., 2009) and occurs in damp, rocky habitats above the timberline. The montane ecotype has instead a dense indumentum with long multicellular sticky glandular trichomes and is found in canyons and below cliff overhangs with dry soils and poor light conditions far below the timberline. Phenotypic differences between the two ecotypes remain stable after cultivation under uniform conditions for at least three generations but inter‐ecotype crossings produce viable and fertile offspring (Bertel et al., 2016a). Available phylogenies (Frajman & Oxelman, 2007; Frajman et al., 2009) suggest that the relationships among populations of both ecotypes may be governed by geographic proximity rather than by ecological preference, implying that the disjunct populations of the montane ecotype could have originated more than once from the widespread alpine populations.
Figure 1.
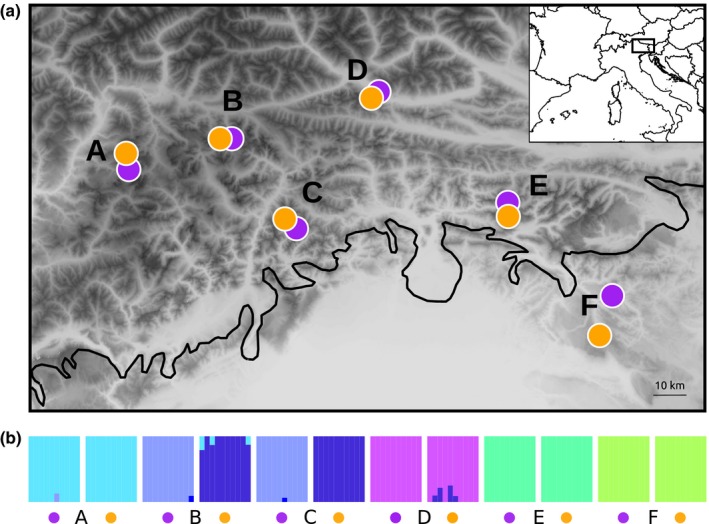
(a) Map of sampled ecotype pairs of Heliosperma pusillum (alpine ecotype, orange; montane ecotype, purple) in south‐eastern Alps. The extension of the ice sheet during the Last Glacial Maximum is shown with a solid black line. The inset shows the position of the sampling area in Europe. (b) Barplots showing proportion of ancestry relative to k = 6 clusters as inferred in faststructure. Colors identifying each cluster are randomly assigned and used only in this figure.
Using genome‐wide single nucleotide polymorphisms (SNPs) produced by restriction site‐associated DNA sequencing (RADseq) of six pairs of geographically close populations of the two ecotypes, our main aim here was to test whether the montane–alpine ecotypic differentiation is the result of a single divergence event or if it originated independently multiple times. We first characterized the observed patterns of genetic divergence, by evaluating the strength of IBD vs IBE among localities, by estimating the amount of admixture between populations, and by assessing the level of parallel genetic divergence between the two ecotypes across localities. We finally assessed the relative probability of three demographic models representing different historical processes behind the observed pattern of divergence: strict isolation, isolation‐with‐migration and secondary contact following a period of allopatric separation.
Materials and Methods
Sampling and RADseq library preparation
Leaf material from 10 individuals each was collected from six pairs of geographically close populations (120 individuals in total) of the alpine and montane ecotypes of H. pusillum (in the following termed ‘ecotype pairs’, comprising one alpine and one montane population each, and identified with uppercase letters A–F) across the distribution range of the montane ecotype (Fig. 1; Supporting Information Notes S1, Table S1). In all localities, the two ecotypes were separated by several hundred meters of altitude (between 500 and 1200 m) and linear distance (between 1.3 and 15 km), mostly occupied by forest. Sampled material from each individual was collected in the field and immediately dried in silica gel in separate bags to avoid cross‐contamination. As the plants do not propagate vegetatively, the separation of individuals was straightforward.
Single‐digest RAD libraries were prepared using the SbfI restriction enzyme (New England Biolabs, Ipswich, MA, USA) and a protocol adapted from Paun et al. (2016) with modifications. In short, we started with 125 ng DNA per individual and ligated 125 mM P5 barcoded adapters to the restricted samples overnight at 16°C. Shearing by sonication was performed with a Bioruptor Pico (Diagenode, Seraing, belgium) with two cycles of 45 s ‘on’ and 60 s ‘off’ at 6°C, targeting a size range of 200–800 bp. Four libraries of individually barcoded samples were finally sequenced on an Illumina HiSeq at VBCF (http://www.vbcf.ac.at/facilities/next-generation-sequencing/) as 100 bp paired‐end reads.
Identification of RAD loci and SNP calling
The raw reads were quality filtered and demultiplexed using process_radtags.pl included in the Stacks1.19 package (Catchen et al., 2013). As medium to large plant genomes largely consist of transposable elements (TEs), which may interfere with the locus‐by‐locus assembly in the Stacks analysis, we checked for the presence of TEs in the raw reads. For this, we Blasted all paired‐end reads to the RepBase database of plant TEs from the Giri repository (http://www.girinst.org/). Each individual sample was analyzed independently and the abundance of TEs was compared among populations and between species (Notes S2; Figs S1, S2). However, as SbfI has a recognition site with 75% GC content, we expected a generally low representation of TEs, which are rather AT‐rich.
The 95‐bp‐long RAD loci were further assembled and SNPs were called using denovo_map.pl in Stacks. A preliminary dataset was built using a minimum coverage to identify a stack (−m) of 20×, a maximum number of differences between two stacks in a locus in each sample (−M) of seven, and a maximum number of differences among loci to be considered as orthologous across multiple samples (−n) of nine. In this dataset, a bimodal distribution of the GC content across loci was discovered, with one peak below 45% GC and a second one above 60% (Notes S3; Fig. S3a). We suspected the latter to be due to exogenous bacterial DNA contamination, expected to result in a relatively high number of loci present in only a few individuals and having much lower coverage than the loci of the target organism. We further used a metagenomic approach on this dataset to identify microbiome contamination (see Notes S4; Figs S4–S6; Table S2). We then increased our coverage threshold for a locus to be assembled in each individual sample by setting the −m parameter in denovo_map.pl to 100. Results then showed a unimodal distribution of GC content peaking at 40–45% (Fig. S3a).
The function export_sql.pl in Stacks was used to extract loci information from the catalog filtering for a maximum number of missing samples per locus of 67% and a maximum number of SNPs per locus of 10. Custom Python scripts were used to further filter the loci for downstream analyses. A locus was discarded if (1) the proportion of heterozygous individuals was > 0.65, an arbitrary cut‐off used to reduce the risk of including paralogs; (2) the GC content was > 65%, to further minimize the risk of including bacterial sequences; (3) any of the samples was scored as triallelic, again reducing the risk of including paralogs in the dataset; or (4) it was invariant. We observed an increase in the occurrence of SNPs in the last 10 positions of the reads (Fig. S3b). As we could not assess their validity, we discarded all SNPs found in this portion of the reads. To check for the proportion of loci containing coding regions, filtered loci were Blasted against the NCBI nt database and an available Silene vulgaris transcriptome (http://silenegenomics.biology.virginia.edu/; Sloan et al., 2012).
Structure of genetic diversity
Summary statistics of genetic diversity were estimated in each population using the final common catalog of loci (1097 variable loci, containing 3401 SNPs): expected heterozygosity (H e), individual observed heterozygosity normalized by coverage (H o ind; Trucchi et al., 2016), Watterson's θ and π.
To assess the effect of distance among localities on genetic divergence, we tested for IBD across all populations by comparing matrices of genetic and geographic distances. Nei's genetic distances among localities, including a sample size correction factor (Nei, 1978), were estimated using the gstudio package in R (Dyer & Nason, 2004). In addition, we tested the effect of IBE (alpine vs montane ecology) on the genetic structure by comparing matrices of ecological (converted to a binary factor) and genetic distance. To assess statistical correlation among matrices we first applied Mantel tests with 9999 randomizations between geographic and genetic distance and between ecological and genetic distance. We then applied a partial Mantel test (mantel.partial function in vegan R package; Oksanen et al. 2013) with 9999 randomizations between ecological and genetic distance but controlling for geographic distance. In addition, using the dist_amova function in the gstudio R package and the adonis function in the vegan R package, a multilocus MANOVA was used to test for statistical differentiation among populations (i.e. 12 groups), ecotype pairs (i.e. six groups) and ecotypes (i.e. two groups).
To assess the level of parallel genetic divergence between the two ecotypes across localities we applied a hierarchical approach as presented by Ravinet et al. (2016) identifying highly divergent loci in each ecotype pair and then comparing the co‐occurrence of such highly divergent loci in multiple ecotype pairs with a null‐expectation based on random overlap. Using the information provided by all linked SNPs at each 95‐bp locus, we first searched for highly divergent loci in each ecotype pair applying a coalescent‐based method (Beaumont & Nichols, 1996). lositan (Antao et al., 2008) was used to perform 500 000 coalescent simulations and produce joint distributions of F ST and expected heterozygosity in a simple island model without selection for each ecotype pair. The simulations aimed at the observed mean F ST value estimated for each ecotype pair. The observed loci with joint distribution of F ST and expected heterozygosity above the 99th quantile (false discovery rate (FDR) = 0.01) were considered as local highly divergent loci. The six lists of such loci (one for each ecotype pair) were compared to each other looking for shared highly divergent loci among two or more ecotype pairs. None of these highly divergent loci was shared by five or more ecotype pairs. Using the exact number of screened loci and the number of highly divergent loci scored in each ecotype pair, we simulated (by 1000 randomizations) the expected random distribution of shared loci for each comparison. Observed values were then compared with their expected random distribution. In the case of more overlap between ecotype pairs than expected by chance, we investigated the contribution of each ecotype to this signal by comparing the differentiation within one ecotype to the differentiation within the other ecotype. For this analysis we plotted the joint distribution of between‐localities intra‐ecotype F ST (see Fraïsse et al., 2016).
To investigate the level of admixture among populations we first used the find.clusters function in the R package adegenet (Jombart & Ahmed, 2011), which estimates the likelihood of structure in the sample according to a range of clusters (here k‐means analysis ranged from 1 to 20). A Bayesian information criterion (BIC) is applied to choose the best k. The Bayesian algorithm implemented in faststructure (Raj et al., 2014) was then used to estimate the cluster's contribution in each individual. As only independent biallelic loci are allowed, we selected one SNP at random from each RAD locus. The results of population structure analysis were plotted using python. The structure of genetic diversity across all individuals was further investigated by principal component analysis (PCA) using the glPca function in the R package adegenet (Jombart & Ahmed, 2011). Individuals from A, B, C and D were also analyzed separately. To reconstruct the relationships among populations of the two ecotypes taking into account admixture, we used treemix (Pickrell & Pritchard, 2012). The result of this analysis includes a maximum‐likelihood (ML) tree of population ancestry, showing also estimated migration events. For the input file, SNP data were converted from diploid genotype calls for each individual into population‐level allele counts using a custom python script (Notes S5). Migration edges were progressively added to the tree and exit log‐likelihood and Akaike's Information Criterion (AIC) were recorded.
Testing alternative demographic scenarios of local divergence
The demographic scenario underlying the observed pattern of local divergence was investigated in the four population pairs for which plain local divergence of the two ecotypes has been inferred in our previous analyses (i.e. A, D–F). Pairs B and C were excluded from this analysis as it became clear from treemix and faststructure results that they have not simply diverged locally from one another. We compared three different demographic models: (1) strict isolation with no gene flow (SI), (2) isolation‐with‐migration (IM) and (3) secondary contact after allopatry (SC). We used a composite‐likelihood maximization method to estimate the best likelihood by simulating joint site frequency spectra under a continuous‐time Markovian coalescent model in fastsimcoal2 (Excoffier et al., 2013). For all models, we let the algorithm estimate the effective size of each population (N a and N m for the alpine and montane ecotype, respectively; ancestral N was rescaled on N a) and the mutation rate (μ). Substitution rate is estimated by using an observed site frequency spectrum based on one selected SNP per 95‐bp locus and the resulting value needs to be scaled by two orders of magnitude to represent an average genome‐wide substitution rate. Migration rates in both directions (m am and m ma) were estimated in the IM and SC models, time since divergence (T d) of the two ecotypes in SI and IM, and time since secondary contact (T c) between the two ecotypes in SC. In the last, the time of the initial divergence was fixed to 50 000 generations ago, before the last Würm glaciation peak (Ivy‐Ochs et al., 2008). Generation time in this species can be considered 1 yr. After excluding the SNP potentially under selection, the folded two‐dimensional site frequency spectrum (2D‐SFS) for each of the four ecotype pairs was used as summary statistic selecting one random SNP per locus (Notes S6; Fig. S7). For each locus, the global frequency of the two alternative alleles across all investigated populations was used to determine the minor allele (i.e. probably the derived one) before estimating its frequency (normalized for missing data) at a local scale. Hence, a minor allele can have a frequency > 0.5 in a specific population (Fig. S7). As variable loci were selected to be present across all populations, invariant loci in both populations of any ecotype pair were the most represented category in the 2D‐SFS. To overcome this issue and further validate our results, analyses were overrepresented in the 2D‐SFS of each pair for each ecotype pair separately. fastsimcoal2 was run without using fixed sites in the spectrum (−0 option). We performed a maximum of 60 expectation/conditional maximization (ECM) cycles with each step requiring the generation of 100 000 simulated joint‐spectra. We ran 10 replicates of each model for each ecotype pair. Convergence of ML and parameter estimates was checked and different models were then compared using AIC.
We further tested the goodness‐of‐fit of the combination ‘scenario + posterior estimates of the parameters’ to the data generating pseudo‐observed datasets and comparing them with the observed datasets through summary statistics (see Bertorelle et al., 2010 for applications of this test in Approximate Bayesian Computation). Average values of N a, N m, m am, m ma, μ, and T d or T c estimated above were used in pseudo‐observed dataset simulations for each ecotype pair. We used π as a summary statistic. For each ecotype pair, we simulated 1000 100‐bp‐long loci on which we estimated the distribution of π calculated on all individuals of both ecotypes (combined π) under the IM and the SC models. The statistical difference between the simulated distribution of π for each model and the observed distribution was assessed by a Kolmogorov–Smirnov two‐samples test.
We then estimated the 95% confidence intervals of the estimates of the demographic parameters in those models passing both the AIC and the goodness‐of‐fit criteria. For each of the successful models, we used the point estimates of the best‐likelihood run to simulate 100 2D‐SFS, which were then used to re‐estimate the demographic parameters through 30 likelihood‐maximization runs each. These 100 estimates were then used to build the 95% confidence intervals for each parameter in each of the selected models (see also https://groups.google.com/forum/#!topic/fastsimcoal/N956Af31iA4).
Data accessibility
Data and additional results are available from the Dryad Digital Repository (doi: 10.5061/dryad.391q5). Raw genomic data are available from the NCBI Short Reads Archive (accession nos. SRP065672, SRP068291).
Results
Genomic datasets
The average number of raw pairs of reads per sample retained after quality filtering was 2.1 (SD = 0.8) million. The proportion of paired‐end reads Blasting to the Viridiplantae Transposable Elements database was very low for all individuals, 0.5–4.1%, with a higher proportion (two‐samples Kolmogorov–Smirnov P = 0.047) in the montane ecotype (Notes S2). The bacterial and fungal contamination in our dataset was significantly different between the two ecotypes (PERMANOVA P = 0.002; Notes S4) with most of the fungal taxa and the bacteria Xanthomonadales occurring across most of, but only in, the montane populations (Table S2; Fig. S5).
After de novo catalog building and SNP calling, we selected 1719 high‐coverage loci present in at least 40 samples. A total of 172 invariant loci were discarded. Average coverage per allele was c. 300× in the alpine individuals and c. 200× in the montane ones (Fig. S3a). The lower coverage in the trichome‐rich montane ecotype was due to higher exogenous DNA contamination. It was possible to map 36% of the retained loci to the S. vulgaris transcriptome and an additional 5% to coding sequences of other plant species in the NCBI database. Four of the loci matched two fungal sequences (poplar leaf fungus), one bacterial sequence (Borrelia) and one nematode sequence (Ascaridae). These loci were removed from the dataset. We further filtered the final dataset according to the criteria detailed in the Methods section to a total of 1097 variable loci, containing 3401 SNPs and occurring on average in 102 individuals (Fig. S3b,c).
Structure of genetic diversity
Expected heterozygosity (H e), Watterson's θ and π for each population of the two ecotypes are given in Table S3 (Notes S7). Boxplots of observed heterozygosity (H o ind) comparing the two ecotypes at each locality are shown in Fig. S8.
The IBD model (Fig. S9) was confirmed (r = 0.69, P = 0.0002) whereas IBE had no effect on the genetic structure (Mantel test ecological vs genetic distance: r = −0.08, P = 0.89; partial Mantel test ecological vs genetic distance controlling for geographical distance: r = 0.01, P = 0.34). The three alternative MANOVAs indicated that the most variance is explained by differences between populations, or between ecotype pairs, but not between the two ecotypes (differentiation among 12 populations: R 2 = 0.91, P = 0.001; six ecotype pairs: R 2 = 0.83, P = 0.001; two ecotypes: R 2 = 0.02, P = 0.14).
Analyzing the six ecotype pairs separately with lositan, we found between 79 and 116 loci with an extreme F ST (FDR = 0.01) taking into account 500 000 simulations of the joint distribution of F ST and H e (Fig. 2a). Ecotype pair mean F ST ranged from 0.17 in A to 0.43 in F. The number of highly divergent loci shared among different ecotype pairs was not different from random expectations (Notes S8; Figs S10–S12), except for the overlap between B and C, and among B, C and D (Fig. 2b). The joint F ST distribution between montane B vs C populations and alpine B vs C populations (Fig. 2c) revealed a much higher differentiation between the montane populations than between the alpine ones (slope of linear regression = 0.06, R 2 = 0.009). The same was found in the comparison B vs D and C vs D (slope of linear regression = 0.13 and 0.2, R 2 = 0.04 and 0.07, respectively). At the global level (Fig. S13), the differentiation among montane populations was slightly stronger than among the alpine ones (slope of linear regression = 0.44, R 2 = 0.2).
Figure 2.
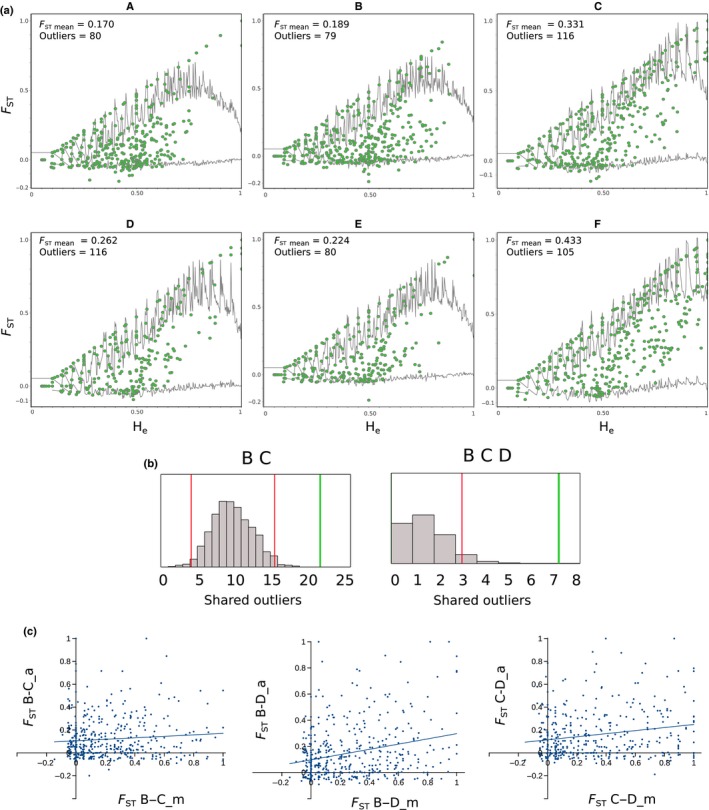
Shared highly divergent loci across six pairs of the alpine and the montane ecotype of Heliosperma pusillum. (a) Joint distribution of FST and expected heterozygosity (H e) across all loci (i.e. haplotypes) between the two ecotypes in each pair. Average FST and number of highly divergent loci in each ecotype pair are indicated. Green dots: observed loci; gray lines: upper and lower bounds of the joint distribution of FST and H e estimated by 500 000 coalescence‐based simulations. (b) Significant overlap of highly divergent loci is found between B and C and among B, C and D. Gray bars, null distribution of shared highly divergent loci (1000 randomizations) under neutrality; red vertical lines, 95% quantiles of the null distribution; green vertical lines, observed number of shared highly divergent loci. See Figs S9–S11 for all possible comparisons with two, three and four ecotype pairs. (c) Joint distribution of FST between the alpine and the montane ecotypes between ecotype pairs with significantly over‐shared highly divergent loci.
BIC in find.clusters analysis started plateauing after k = 6 (Fig. S14). Few individuals with putatively admixed ancestry were found by faststructure in populations A, B, C and D (Fig. 1). PCAs clearly clustered individuals by locality and not by ecology with the exception of populations B and C (Fig. S15). The topology of the ML population tree reconstructed with treemix (Fig. 3) corresponded to the structure inferred by faststructure and PCA. After adding more than three migration edges the likelihood reached a plateau.
Figure 3.
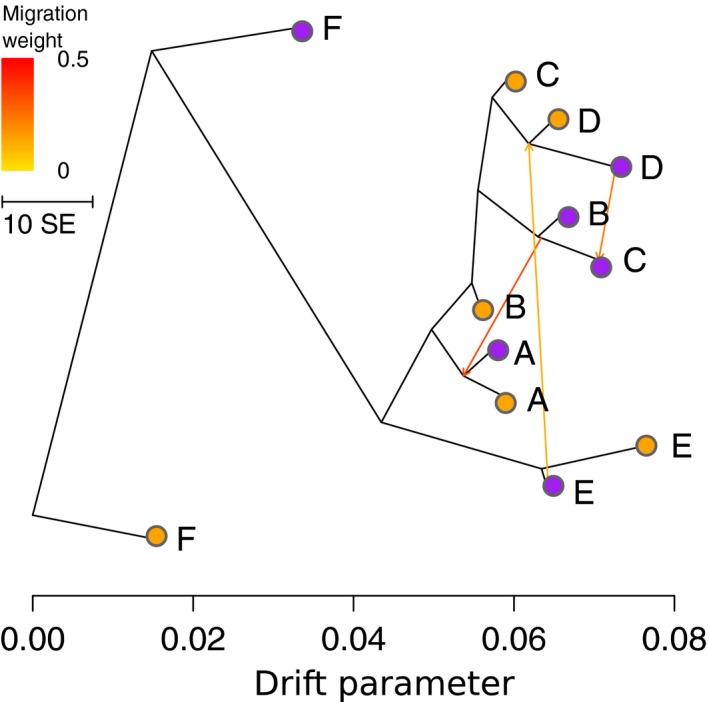
Maximum‐likelihood tree and migration events among six pairs of the alpine and the montane ecotype of Heliosperma pusillum. Population tree and migration edges have been inferred by treemix. Orange, alpine ecotype; purple, montane ecotype; population labels as in Fig. 1.
Testing alternative demographic scenarios
In all tested ecotype pairs, the scenario of local divergence with no migration (SI) was rejected, whereas the isolation‐with‐migration (IM) and the secondary contact (SC) scenarios were not distinguishable due to too similar AIC scores (Table 1). In all cases, the time of the split between the two ecotypes was estimated after the Last Glacial Maximum (the oldest split is estimated in F between 4651 and 19 189 yr ago (95% confidence intervals) according to the IM model) and the mutation rate, estimated on one selected SNP per 95‐bp locus, was c. 5 × 10−7 substitutions per site per generation. Higher migration rate from the alpine to the montane ecotype was estimated in E whereas higher rates in the other direction were inferred in D and F (Table 1). Further comparing the combined distribution of π in each ecotype pair as simulated under the two best‐fit models (SC and IM) fixing the parameters to the average ML estimates (Fig. 4; pseudo‐observed dataset simulation), the IM scenario obtained a higher goodness‐of‐fit in the case of A (Kolmogorov–Smirnov, Obs − SCpods: P = 4.3 × 10−5, Obs − IMpods: P = 0.55) and E (Kolmogorov–Smirnov, Obs − SCpods: P = 1.6 × 10−6, Obs − IMpods: P = 0.33) ecotype pairs. As the SI model was within four AIC points from the best‐fit model (SC) in E, we generated a pseudo‐observed SI dataset and, after assessing its goodness‐of‐fit as above, we definitely discarded this model (Kolmogorov–Smirnov, Obs − SIpods: P = 0). In the remaining ecotype pairs (D and F), the distribution of combined π produced under the two competing models was indistinguishable from the observation (Fig. 4). Replicated analyses building the 2D‐SFS in each ecotype pair separately, thus using a higher number of variable loci and a lower proportion of missing data, fully confirmed these results.
Table 1.
Demographic inference for each model tested with fastsimcoal2 in four population pairs of the alpine and the montane ecotypes of Heliosperma pusillum
| Model | L | AIC | Fit | N a (ind) | N m (ind) | Time (yr) | m ma (% of N a) | m am (% of N m) | μ* (substitutions per site per generation) | |
|---|---|---|---|---|---|---|---|---|---|---|
| A | IM | −1011 | 2032 | Yes | 110–9746 | 47–4321 | 3841–14 044 | 2.0e‐5–4.5e‐3 | 2.1e‐4–1.8e‐2 | 1.2e‐7–8.1e‐7 |
| SI | −1021 | 2050 | – | |||||||
| SC | −1010 | 2032 | No | |||||||
| D | IM | −732 | 1476 | Yes | 102–887 | 646–5633 | 3276–14 104 | 3.9e‐4–4.1e‐3 | 1.2e‐5–4.5e‐4 | 2.1e‐7–8.6e‐7 |
| SI | −747 | 1501 | – | |||||||
| SC | −732 | 1476 | Yes | 144–743 | 852–4444 | 3467–14 263 | 6.6e‐4–2.8e‐3 | 1.5e‐5–1.7e‐4 | 7.0e‐8–8.7e‐7 | |
| E | IM | −1052 | 2116 | Yes | 103–22 761 | 13–2135 | 3996–15 503 | 1.2e‐5–1.0e‐3 | 2.7e‐4–5.2e‐2 | 1.4e‐7–9.3e‐7 |
| SI | −1055 | 2117 | – | |||||||
| SC | −1050 | 2113 | No | |||||||
| F | IM | −1388 | 2789 | Yes | 102–1718 | 223–3990 | 4651–19 189 | 8.3e‐5–1.7e‐3 | 3.4e‐5–7.4e‐4 | 1.3e‐8–8.9e‐7 |
| SI | −1416 | 2840 | – | |||||||
| SC | −1388 | 2787 | Yes | 105–610 | 374–2182 | 3378–12 560 | 3.2e‐4–1.6e‐3 | 2.4e‐5–4.6e‐4 | 1.8e‐7–8.6e‐7 |
Mean values of likelihood (L) and Akaike Information Criterion (AIC) across 10 maximum‐likelihood runs for each model are given in the respective columns. Summary results (i.e. yes/no passing the criterion) of goodness‐of‐fit of the combination ‘scenario + posterior estimates of the parameters’ is reported in the column ‘fit’ (see Fig. 4 and the Results section). The 95% confidence intervals for the demographic parameters are reported for the models passing both the AIC and the goodness‐of‐fit criteria. N a, effective population size of the alpine ecotype; N m, effective population size of the montane ecotype; m am, migration rate from the alpine to the montane ecotype within ecotype pair; m ma, migration rate from the montane to the alpine ecotype within ecotype pair; Time, time since split or secondary contact; μ*, mutation rate (estimated selecting one variable SNP in each 95‐bp‐long locus); IM, isolation‐with‐migration; SI, strict divergence; SC, secondary contact.
Figure 4.
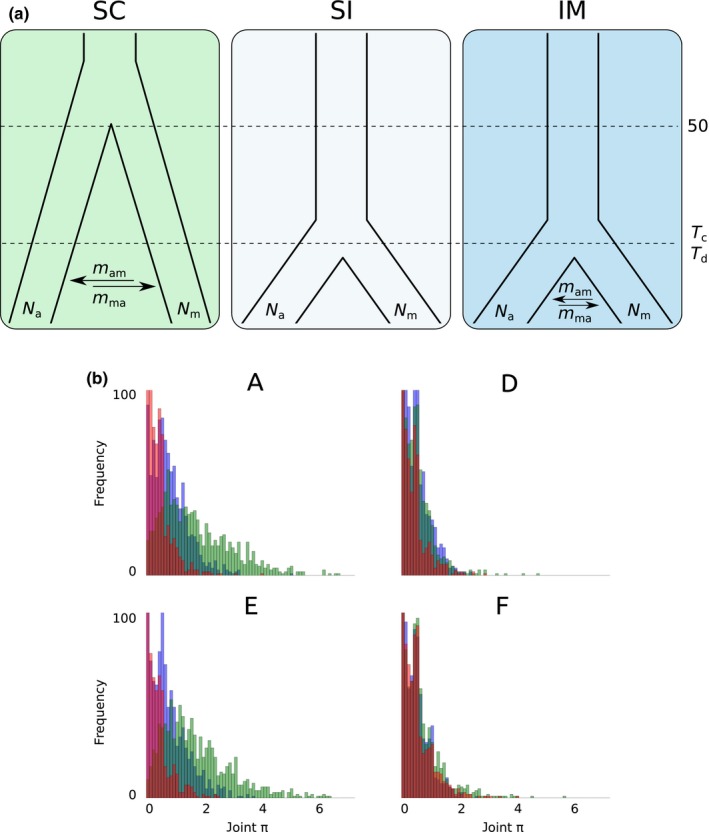
Demographic scenario potentially leading to local divergence between the alpine and the montane ecotype in Heliosperma pusillum. (a) Three alternative demographic models are compared: allopatric divergence followed by secondary contact (SC), strict isolation with no gene flow (SI) and isolation‐with‐migration (IM). Effective population size of the montane and the alpine ecotype (N a and N m), migration rates in both directions (m am and m ma) and time since onset of gene flow (T c) or since the split (T d) have been estimated in fastsimcoal2 using the two‐dimensional site frequency spectrum (2D‐SFS) in each ecotype pair. The SD model was rejected in all cases. (b) Goodness‐of‐fit of the combination ‘scenario + posterior estimate of the parameters’ for SC and IM have been further assessed by pseudo‐observed dataset simulation using the distribution of combined π as the summary statistic. Maximum frequency has been cut to 100 for graphical clarity. In ecotype pairs A and E, the SC model was rejected. See the text for Kolmogorov–Smirnov test results. Green bars, SC model; purple bars, IM model; red bars, observation.
Discussion
Demographic scenarios underlying the mosaic distribution of H. pusillum ecotypes
Using genome‐wide molecular data of 120 individuals from 12 populations we show here that the alpine and montane ecotypes of H. pusillum, usually classified as distinct species (H. pusillum Vis. and H. veselskyi Janka; Frajman & Oxelman, 2007; Fischer et al., 2008), are not independent evolutionary units and their populations cluster more often by locality than by ecology (Figs 1, 3, S15). Despite clear parallel differences in anatomical, morphological and ecological traits (Bertel et al., 2016b,c) and in the phyllosphere biotic community (Table S2; Fig. S5), genome‐wide RADseq loci presented a divergence pattern rather shaped by neutral processes (i.e. IBD, hierarchical population structure and local drift) than by similar selective environments (i.e. IBE). The retrieved mosaic distribution suggests up to five repeated instances of ecological divergence between the six alpine and montane population pairs analyzed here. Recent genomic investigations suggested parallel ecological divergence as common to several organisms (e.g. Hohenlohe et al., 2010; Roda et al., 2013; Butlin et al., 2014; Soria‐Carrasco et al., 2014; Feulner et al., 2015; Rougemont et al., 2017). In the case of H. pusillum, coalescent simulations suggested a scenario of isolation‐with‐migration (IM) in two out of four instances of local divergence of ecotype pairs (Fig. 4). In the other two locally diverging pairs, the two best‐fit scenarios (IM and secondary contact after allopatry, SC) had very similar likelihoods (Table 1). In these two cases, observed distributions of combined π (Fig. 4) are compatible with either a recent IM scenario or late SC even in the case of the putatively oldest split in F (the only pair currently found outside the maximum ice sheet extension at the LGM; Ivy‐Ochs et al., 2008). A scenario of strict isolation without gene flow has been discarded in the four ecotype pairs tested here. Migration, even if at a low level (Table 1), appears as an important component to be taken into account in the evolution of the alpine and montane ecotypes of H. pusillum. Indeed, in all populations, π was higher than Watterson's θ (Table S3), suggesting migration between diverging populations as an important process in this biological system.
The time of divergence estimated in each ecotype pair (Table 1) consistently postdates the last glaciation (Ivy‐Ochs et al., 2008). In the isolation‐with‐migration scenario, ecological divergence could have been triggered by the rapid spread of forests induced by Holocene warming (e.g. Magri et al., 2006), separating montane stands below overhanging cliffs from alpine populations. Globally, the structure among the populations of the two ecotypes reflects a clear IBD pattern (Fig. S9). This may be related to the limited time elapsed since the colonization of recently de‐glaciated mountains or to gene flow maintaining genetic contiguity. Indeed, some traces of admixture or – alternatively, but difficult to disentangle – shared ancestry (i.e. incomplete lineage sorting) between the alpine populations are visible across the western localities, as supported by the low differentiation (expressed as drift parameter in treemix analysis) between B, C and D, in particular (Fig. 3). In addition, the higher than expected proportion of shared outliers among these localities (Fig. 2b) is probably due to a lower differentiation between the alpine populations, as also supported by the joint distribution of F ST analysis (Fig. 2c). On average, differentiation is lower among all alpine populations when compared with the montane ones (Fig. S13). This can be a consequence of the insular, disjunct habitat preferred by the montane ecotype, whereas the alpine populations are more continuously distributed (Poldini, 2002; Wilhalm et al., 2014).
Local drift and lack of shared genetic divergence
The consistent phenotypic divergence between alpine and montane ecotypes of H. pusillum across the six population pairs is not paralleled by shared genetic divergence within the investigated portion of the genome (i.e. no IBE). In the majority of cases, we did not identify more loci with high F ST shared among pairs than expected by chance (Figs 2, S10–S12). Our result comparing six ecotype pairs suggests caution when interpreting patterns of divergent selection at some loci shared by one or very few contrasting ecotype pairs. In fact, the stochastic outcome of neutral processes, such as drift, could be misinterpreted as the results of a similar selective regime. An investigation of the null expectation as proposed in Fig. S10 together with the reconstruction of the general relationships among the different populations (Fig. 3) should help to assess the false discovery rate in allelic frequency covariation analyses (Figs 3b, S10). In very few cases, highly divergent loci in the F ST−H e scans were more frequent in two to three ecotype pairs than expected by chance, as among B, C and D (Fig. 2b). However, these populations were also characterized by a clear global signature of higher relatedness within ecotype (Figs 2c, 3), suggesting demographic processes such as within‐ecotype gene flow to be a more likely explanation than convergent adaptation due to a similar selective regime. The signature of the latter is, in fact, expected to be found only at adaptive alleles and not spread across the genome. Note, however, that heterogeneity in gene flow along the genome could produce correlated signatures of divergence at certain loci across ecotype pairs. Such signatures could be misinterpreted as the result of a similar selective process independently occurring in different populations (Le Moan et al., 2016; Rougemont et al., 2017).
A lack of a signal of shared genomic divergence across ecotype pairs has also been reported in other biological systems (e.g. Perrier et al., 2013; Roda et al., 2013; Renaut et al., 2014). Interestingly, investigating three pairs of wave and crab ecotypes of the rough periwinkle snail on the Swedish coast, a proportion of shared high F ST loci similar to that found in our study has been scored by a similar analytical approach (Ravinet et al., 2016). Independent and different mutations occurring in the same gene, or polygenic traits underlying ecological adaptations, could explain the lack of shared genomic differentiation. The short divergence time estimated in each Heliosperma ecotype pair renders a significant contribution of novel mutations unlikely. Hence, repetitive divergence from standing genetic variation appears more probable (i.e. collateral evolution by shared ancestry; Barrett & Schluter, 2008; Schluter, 2009; Stankowski, 2013; Stern, 2013). However, as adaptive traits are probably polygenic (Pritchard et al., 2010), the signature of selection may be difficult to detect unless more advanced analytical approaches, not applicable on our data type, are used (Daub et al., 2013).
More importantly, the reduced representation of our RADseq dataset screened only a limited portion of the genome (0.1%) and of the genes (c. 3.5% – assuming c. 20 000 genes in the 2.2 Gb Heliosperma genome and c. 700 RAD loci mapping to the S. vulgaris transcriptome and gene sequences in GenBank). It is thus very likely that adaptive loci, or neutral loci linked to it, were not covered in our genome scan. In addition, if selection is acting on standing genetic variation present in the ancestral population (Pritchard et al., 2010), the signature of divergence around loci under selection is expected to be minimal (Hermisson & Pennings, 2005). These aspects could then exacerbate the difficulties in finding adaptive loci that, in some cases, have been shown to be restricted to a single SNP (e.g. O'Brown et al., 2015).
Different demographic processes (i.e. multiple divergence events and/or independent secondary contacts), probably characterize the evolutionary history of contrasting ecotypes at medium to large geographic scales (e.g. Roda et al., 2013; Rougemont et al., 2017). At the local scale and if effective population size is small, drift can largely overwhelm the mark(s) left by selection. In addition, different instances of contrasting ecotypes could be affected by a diverse range of biotic (e.g. phyllosphere communities, Fig. S5; Table S2; pathogen load, Feulner et al., 2015) and abiotic (e.g. microclimatic conditions; Bertel et al., 2016b) factors of local selection. Further studies involving whole transcriptome and/or genome sequencing are needed to specifically pinpoint the molecular components underlying this iterative ecological divergence and to fully understand the evolutionary forces driving it.
Author contributions
B.F., P.S. and O.P. planned and designed the research. B.F. and P.S. conducted fieldwork. O.P. performed experiments. E.T. analysed data. E.T. and T.H.A.H. performed metagenomic analyses. E.T., P.S. and O.P. wrote the manuscript, and all authors commented on the manuscript.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Proportion of transposable elements in the RADseq dataset.
Fig. S2 Ecotype‐specific proportion of transposable elements.
Fig. S3 RADseq dataset quality control.
Fig. S4 Proportion of loci assigned to putative phyllosphere and contaminant taxa.
Fig. S5 Analysis of putative phyllosphere and contaminant taxa by ecotype.
Fig. S6 Analysis of putative phyllosphere and contaminant taxa by locality.
Fig. S7 Minor 2D‐SFS estimated in each ecotype pair.
Fig. S8 Individual observed heterozygosity normalized by coverage.
Fig. S9 Analysis of isolation‐by‐distance.
Fig. S10 Highly divergent loci shared among two ecotype pairs.
Fig. S11 Highly divergent loci shared among three ecotype pairs.
Fig. S12 Highly divergent loci shared among four ecotype pairs.
Fig. S13 Joint distribution of F ST between alpine and montane populations.
Fig. S14 Bayesian Information Criterion k‐means analysis.
Fig. S15 Principal component analysis.
Table S1 Sample design
Table S2 Microbiome structure and diversity
Table S3 Genetic diversity
Notes S1 Sampling.
Notes S2 Transposable elements.
Notes S3 Dataset quality control.
Notes S4 Analysis of contamination by leaf microbiome.
Notes S5 Custom Python code.
Notes S6 Testing alternative demographic scenarios.
Notes S7 Summary statistics of genomic diversity.
Notes S8 Structure of genetic diversity.
Acknowledgements
We would like to remember here Ruth Flatscher who was initially involved in this study, but sadly passed away. Maria Lorenzo Romero is acknowledged for constructing some of the RADseq libraries. We thank the Editor (Leonie Moyle), two anonymous reviewers and Nicolas Bierne for commenting and discussing earlier versions of the manuscript. This work was supported by the Austrian Climate Research Programme (ACRP5‐EpiChange‐KR12AC5K01286 to P.S.), and the Austrian Science Fund (FWF, Y661‐B16 to O.P.).
References
- Antao T, Lopes A, Lopes RJ, Beja‐Pereira A, Luikart G. 2008. LOSITAN: a workbench to detect molecular adaptation based on a F ST‐outlier method. BMC Bioinformatics 9: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett RDH, Schluter D. 2008. Adaptation from standing genetic variation. Trends in Ecology & Evolution 23: 38–44. [DOI] [PubMed] [Google Scholar]
- Beaumont MA, Nichols RA. 1996. Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London B: Biological Sciences 263: 1619–1626. [Google Scholar]
- Berglund A‐BN, Dahlgren S, Westerbergh A. 2004. Evidence for parallel evolution and site‐specific selection of serpentine tolerance in Cerastium alpinum during the colonization of Scandinavia. New Phytologist 161: 199–209. [Google Scholar]
- Bertel C, Buchner O, Schönswetter P, Frajman B, Neuner G. 2016b. Environmentally induced and (epi‐)genetically based physiological trait differentiation between Heliosperma pusillum and its polytopically evolved ecologically divergent descendent, H. veselskyi (Caryophyllaceae: Sileneae). Botanical Journal of the Linnean Society 182: 658–669. [Google Scholar]
- Bertel C, Hülber K, Frajman B, Schönswetter P. 2016a. No evidence of intrinsic reproductive isolation between two reciprocally non‐monophyletic, ecologically differentiated mountain plants at an early stage of speciation. Evolutionary Ecology 30: 1031–1042. [Google Scholar]
- Bertel C, Schönswetter P, Frajman B, Holinger A, Neuner G. 2016c. Leaf anatomy of two reciprocally non‐monophyletic mountain plants (Heliosperma spp.): does heritable adaptation to divergent growing sites accompany the onset of speciation? Protoplasma 254: 1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertorelle G, Benazzo A, Mona S. 2010. ABC as a flexible framework to estimate demography over space and time: some cons, many pros. Molecular Ecology 19: 2609–2625. [DOI] [PubMed] [Google Scholar]
- Bierne N, Gagnaire P‐A, David P. 2013. The geography of introgression in a patchy environment and the thorn in the side of ecological speciation. Current Zoology 59: 72–86. [Google Scholar]
- Bierne N, Welch J, Loire E, Bonhomme F, David P. 2011. The coupling hypothesis: why genome scans may fail to map local adaptation genes. Molecular Ecology 20: 2044–2072. [DOI] [PubMed] [Google Scholar]
- Brochmann C, Borgen L, Stabbetorp OE. 2000. Multiple diploid hybrid speciation of the Canary Island endemic Argyranthemum sundingii (Asteraceae). Plant Systematics and Evolution 220: 77–92. [Google Scholar]
- Butlin RK, Galindo J, Grahame JW. 2008. Sympatric, parapatric or allopatric: the most important way to classify speciation? Philosophical Transactions of the Royal Society of London B: Biological Sciences 363: 2997–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butlin RK, Saura M, Charrier G, Jackson B, André C, Caballero A, Coyne JA, Galindo J, Grahame JW, Hollander J et al 2014. Parallel evolution of local adaptation and reproductive isolation in the face of gene flow. Evolution 68: 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. 2013. Stacks: an analysis tool set for population genomics. Molecular Ecology 22: 3124–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of Ectodysplasin alleles. Science 307: 1928–1933. [DOI] [PubMed] [Google Scholar]
- Daub JT, Hofer T, Cutivet E, Dupanloup I, Quintana‐Murci L, Robinson‐Rechavi M, Excoffier L. 2013. Evidence for polygenic adaptation to pathogens in the human genome. Molecular Biology and Evolution 30: 1544–1558. [DOI] [PubMed] [Google Scholar]
- Dyer RJ, Nason JD. 2004. Population Graphs: the graph theoretic shape of genetic structure. Molecular Ecology 13: 1713–1727. [DOI] [PubMed] [Google Scholar]
- Excoffier L, Dupanloup I, Huerta‐Sánchez E, Sousa VC, Foll M. 2013. Robust demographic inference from genomic and SNP data. PLoS Genetics 9: e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feulner PGD, Chain FJJ, Panchal M, Huang Y, Eizaguirre C, Kalbe M, Lenz TL, Samonte IE, Stoll M, Bornberg‐Bauer E et al 2015. Genomics of divergence along a continuum of parapatric population differentiation. PLoS Genetics 11: e1004966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer MA, Oswald K, Adler W. 2008. Exkursionsflora für Österreich, Liechtenstein und Südtirol, 3rd edn Linz, Austria: Biologiezentrum der Oberösterreichischen Landesmuseen. [Google Scholar]
- Foster SA, McKinnon GE, Steane DA, Potts BM, Vaillancourt RE. 2007. Parallel evolution of dwarf ecotypes in the forest tree Eucalyptus globulus . New Phytologist 175: 370–380. [DOI] [PubMed] [Google Scholar]
- Fraïsse C, Belkhir K, Welch JJ, Bierne N. 2016. Local interspecies introgression is the main cause of extreme levels of intraspecific differentiation in mussels. Molecular Ecology 25: 269–286. [DOI] [PubMed] [Google Scholar]
- Frajman B, Eggens F, Oxelman B. 2009. Hybrid origins and homoploid reticulate evolution within Heliosperma (Sileneae, Caryophyllaceae) – a multigene phylogenetic approach with relative dating. Systematic Biology 58: 328–345. [DOI] [PubMed] [Google Scholar]
- Frajman B, Oxelman B. 2007. Reticulate phylogenetics and phytogeographical structure of Heliosperma (Sileneae, Caryophyllaceae) inferred from chloroplast and nuclear DNA sequences. Molecular Phylogenetics and Evolution 43: 140–155. [DOI] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. 2005. Soft sweeps molecular population genetics of adaptation from standing genetic variation. Genetics 169: 2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermisson J, Pennings PS. 2017. Soft sweeps and beyond: understanding the patterns and probabilities of selection footprints under rapid adaptation. Methods in Ecology and Evolution 8: 700–716 [Google Scholar]
- Hewitt GM. 2001. Speciation, hybrid zones and phylogeography – or seeing genes in space and time. Molecular Ecology 10: 537–549. [DOI] [PubMed] [Google Scholar]
- Hewitt GM. 2011. Quaternary phylogeography: the roots of hybrid zones. Genetica 139: 617–638. [DOI] [PubMed] [Google Scholar]
- Hohenlohe PA, Bassham S, Etter PD, Stiffler N, Johnson EA, Cresko WA. 2010. Population genomics of parallel adaptation in Threespine Stickleback using sequenced RAD Tags. PLoS Genetics 6: e1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivy‐Ochs S, Kerschner H, Reuther A, Preusser F, Heine K, Maisch M, Kubik PW, Schlüchter C. 2008. Chronology of the last glacial cycle in the European Alps. Journal of Quaternary Science 23: 559–573. [Google Scholar]
- Johannesson K, Panova M, Kemppainen P, André C, Rolán‐Alvarez E, Butlin RK. 2010. Repeated evolution of reproductive isolation in a marine snail: unveiling mechanisms of speciation. Philosophical Transactions of the Royal Society of London B: Biological Sciences 365: 1735–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart T, Ahmed I. 2011. adegenet 1.3‐1: new tools for the analysis of genome‐wide SNP data. Bioinformatics 27: 3070–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez‐Barrio A, Promerová M, Rubin C‐J, Wang C, Zamani N et al 2015. Evolution of Darwin's finches and their beaks revealed by genome sequencing. Nature 518: 371–375. [DOI] [PubMed] [Google Scholar]
- Le Moan A, Gagnaire PA, Bonhomme F. 2016. Parallel genetic divergence among coastal–marine ecotype pairs of European anchovy explained by differential introgression after secondary contact. Molecular Ecology 25: 3187–3202. [DOI] [PubMed] [Google Scholar]
- Levin DA. 2001. The recurrent origin of plant races and species. Systematic Botany 26: 197–204. [Google Scholar]
- Loh Y‐HE, Bezault E, Muenzel FM, Roberts RB, Swofford R, Barluenga M, Kidd CE, Howe AE, Palma FD, Lindblad‐Toh K et al 2013. Origins of shared genetic variation in African cichlids. Molecular Biology and Evolution 30: 906–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry DB. 2012. Ecotypes and the controversy over stages in the formation of new species. Biological Journal of the Linnean Society 106: 241–257. [Google Scholar]
- Magri D, Vendramin GG, Comps B, Dupanloup I, Geburek T, Gömöry D, Latałowa M, Litt T, Paule L, Roure JM et al 2006. A new scenario for the Quaternary history of European beech populations: palaeobotanical evidence and genetic consequences. New Phytologist 171: 199–221. [DOI] [PubMed] [Google Scholar]
- Nei M. 1978. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89: 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumayer H. 1923. Einige Fragen der speziellen Systematik, erläutert an einer Gruppe der Gattung Silene . Österreichische Botanische Zeitschrift 72: 276–287. [Google Scholar]
- Nosil P, Egan SP, Funk DJ. 2008. Heterogeneous genomic differentiation between walking‐stick ecotypes: ‘Isolation By Adaptation’ and multiple roles for divergent selection. Evolution 62: 316–336. [DOI] [PubMed] [Google Scholar]
- O'Brown NM, Summers BR, Jones FC, Brady SD, Kingsley DM. 2015. A recurrent regulatory change underlying altered expression and Wnt response of the stickleback armor plates gene EDA. eLife 4: e05290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O'Hara RB, Simpson GL, Solymos P, Stevens MHH, Wagner H. 2013. vegan: community ecology package. R package version 2.0‐10. [WWW document] URL http://CRAN.R-project.org/package=vegan [accessed on 15 May 2016].
- Østbye K, Amundsen P‐A, Bernatchez L, Klemetsen A, Knudsen R, Kristoffersen R, Naesje TF, Hindar K. 2006. Parallel evolution of ecomorphological traits in the European whitefish Coregonus lavaretus (L.) species complex during postglacial times. Molecular Ecology 15: 3983–4001. [DOI] [PubMed] [Google Scholar]
- Paun O, Turner B, Trucchi E, Munzinger J, Chase MW, Samuel R. 2016. Processes driving the adaptive radiation of a tropical tree (Diospyros, Ebenaceae) in New Caledonia, a biodiversity hotspot. Systematic Biology 65: 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse DE, Miller MR, Abadía‐Cardoso A, Garza JC. 2014. Rapid parallel evolution of standing variation in a single, complex, genomic region is associated with life history in steelhead/rainbow trout. Proceedings of the Royal Society of London B: Biological Sciences 281: 20140012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereyra RT, Huenchuñir C, Johansson D, Forslund H, Kautsky L, Jonsson PR, Johannesson K. 2013. Parallel speciation or long‐distance dispersal? Lessons from seaweeds (Fucus) in the Baltic Sea. Journal of Evolutionary Biology 26: 1727–1737. [DOI] [PubMed] [Google Scholar]
- Perrier C, Bourret V, Kent MP, Bernatchez L. 2013. Parallel and nonparallel genome‐wide divergence among replicate population pairs of freshwater and anadromous Atlantic salmon. Molecular Ecology 22: 5577–5593. [DOI] [PubMed] [Google Scholar]
- Pickrell JK, Pritchard JK. 2012. Inference of population splits and mixtures from genome‐wide allele frequency data. PLoS Genetics 8: e1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poldini L. 2002. Nuovo atlante corologico delle piante vascolari nel Friuli‐Venezia Giulia. Regione Autonoma Friuli‐Venezia Giulia, Azienda Parchi e Foreste Regionali, Udine. Tavagnacco, Italy: Arti Grafiche Friulane. [Google Scholar]
- Pritchard JK, Pickrell JK, Coop G. 2010. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Current Biology 20: R208–R215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Stephens M, Pritchard JK. 2014. fastSTRUCTURE: variational inference of population structure in large SNP datasets. Genetics: 197: 573–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralph P, Coop G. 2010. Parallel adaptation: one or many waves of advance of an advantageous allele? Genetics 186: 647–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet M, Westram A, Johannesson K, Butlin R, André C, Panova M. 2016. Shared and nonshared genomic divergence in parallel ecotypes of Littorina saxatilis at a local scale. Molecular Ecology 25: 287–305. [DOI] [PubMed] [Google Scholar]
- Renaut S, Owens GL, Rieseberg LH. 2014. Shared selective pressure and local genomic landscape lead to repeatable patterns of genomic divergence in sunflowers. Molecular Ecology 23: 311–324. [DOI] [PubMed] [Google Scholar]
- Roda F, Ambrose L, Walter GM, Liu HL, Schaul A, Lowe A, Pelser PB, Prentis P, Rieseberg LH, Ortiz‐Barrientos D. 2013. Genomic evidence for the parallel evolution of coastal forms in the Senecio lautus complex. Molecular Ecology 22: 2941–2952. [DOI] [PubMed] [Google Scholar]
- Rougemont Q, Gagnaire P‐A, Perrier C, Genthon C, Besnard A‐L, Launey S, Evanno G. 2017. Inferring the demographic history underlying parallel genomic divergence among pairs of parasitic and non‐parasitic lamprey ecotypes. Molecular Ecology 26: 142–162. [DOI] [PubMed] [Google Scholar]
- Rundle HD, Nagel L, Boughman JW, Schluter D. 2000. Natural selection and parallel speciation in sympatric sticklebacks. Science 287: 306–308. [DOI] [PubMed] [Google Scholar]
- Rundle HD, Nosil P. 2005. Ecological speciation. Ecology Letters 8: 336–352. [Google Scholar]
- Schluter D. 2009. Evidence for ecological speciation and its alternative. Science 323: 737–741. [DOI] [PubMed] [Google Scholar]
- Schluter D, Conte GL. 2009. Genetics and ecological speciation. Proceedings of the National Academy of Sciences, USA 106: 9955–9962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seehausen O, Butlin RK, Keller I, Wagner CE, Boughman JW, Hohenlohe PA, Peichel CL, Saetre GP, Bank C, Brännström A et al 2014. Genomics and the origin of species. Nature Reviews Genetics 15: 176‐192. [DOI] [PubMed] [Google Scholar]
- Sloan DB, Keller SR, Berardi AE, Sanderson BJ, Karpovich JF, Taylor DR. 2012. De novo transcriptome assembly and polymorphism detection in the flowering plant Silene vulgaris (Caryophyllaceae). Molecular Ecology Resources 12: 333–343. [DOI] [PubMed] [Google Scholar]
- Soria‐Carrasco V, Gompert Z, Comeault AA, Farkas TE, Parchman TL, Johnston JS, Buerkle CA, Feder JL, Bast J, Schwander T et al 2014. Stick insect genomes reveal natural selection's role in parallel speciation. Science 344: 738–742. [DOI] [PubMed] [Google Scholar]
- Stankowski S. 2013. Ecological speciation in an island snail: evidence for the parallel evolution of a novel ecotype and maintenance by ecologically dependent postzygotic isolation. Molecular Ecology 22: 2726–2741. [DOI] [PubMed] [Google Scholar]
- Stern DL. 2013. The genetic causes of convergent evolution. Nature Reviews Genetics 14: 751–764. [DOI] [PubMed] [Google Scholar]
- Trucchi E, Facon B, Gratton P, Mori E, Stenseth NC, Jentoft S. 2016. Long live the alien: is high genetic diversity a pivotal aspect of crested porcupine (Hystrix cristata) long‐lasting and successful invasion? Molecular Ecology 25: 3527–3539. [DOI] [PubMed] [Google Scholar]
- Welch JJ, Jiggins CD. 2014. Standing and flowing: the complex origins of adaptive variation. Molecular Ecology 23: 3935–3937. [DOI] [PubMed] [Google Scholar]
- Wellenreuther M, Hansson B. 2016. Detecting polygenic evolution: problems, pitfalls, and promises. Trends in Genetics 32: 155–164. [DOI] [PubMed] [Google Scholar]
- Wilhalm T, Kranebitter P, Hilpold A. 2014. FloraFaunaSüdtirol (www.florafauna.it). Das Portal zur Verbreitung von Pflanzen‐ und Tierarten in Südtirol. Gredleriana 14: 99–110. [Google Scholar]
- Wood TE, Burke JM, Rieseberg LH. 2005. Parallel genotypic adaptation: when evolution repeats itself. Genetica 123: 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Proportion of transposable elements in the RADseq dataset.
Fig. S2 Ecotype‐specific proportion of transposable elements.
Fig. S3 RADseq dataset quality control.
Fig. S4 Proportion of loci assigned to putative phyllosphere and contaminant taxa.
Fig. S5 Analysis of putative phyllosphere and contaminant taxa by ecotype.
Fig. S6 Analysis of putative phyllosphere and contaminant taxa by locality.
Fig. S7 Minor 2D‐SFS estimated in each ecotype pair.
Fig. S8 Individual observed heterozygosity normalized by coverage.
Fig. S9 Analysis of isolation‐by‐distance.
Fig. S10 Highly divergent loci shared among two ecotype pairs.
Fig. S11 Highly divergent loci shared among three ecotype pairs.
Fig. S12 Highly divergent loci shared among four ecotype pairs.
Fig. S13 Joint distribution of F ST between alpine and montane populations.
Fig. S14 Bayesian Information Criterion k‐means analysis.
Fig. S15 Principal component analysis.
Table S1 Sample design
Table S2 Microbiome structure and diversity
Table S3 Genetic diversity
Notes S1 Sampling.
Notes S2 Transposable elements.
Notes S3 Dataset quality control.
Notes S4 Analysis of contamination by leaf microbiome.
Notes S5 Custom Python code.
Notes S6 Testing alternative demographic scenarios.
Notes S7 Summary statistics of genomic diversity.
Notes S8 Structure of genetic diversity.
Data Availability Statement
Data and additional results are available from the Dryad Digital Repository (doi: 10.5061/dryad.391q5). Raw genomic data are available from the NCBI Short Reads Archive (accession nos. SRP065672, SRP068291).