

Abstract
Large‐scale sequencing efforts have described the mutational complexity of individual cancers and identified mutations prevalent in different cancers. As a complementary approach, allele‐specific competitive blocker PCR (ACB‐PCR) is being used to quantify levels of hotspot cancer driver mutations (CDMs) with high sensitivity, to elucidate the tissue‐specific properties of CDMs, their occurrence as tumor cell subpopulations, and their occurrence in normal tissues. Here we report measurements of PIK3CA H1047R mutant fraction (MF) in normal colonic mucosa, normal lung, colonic adenomas, colonic adenocarcinomas, and lung adenocarcinomas. We report PIK3CA E545K MF measurements in those tissues, as well as in normal breast, normal thyroid, mammary ductal carcinomas, and papillary thyroid carcinomas. We report KRAS G12D and G12V MF measurements in normal colon. These MF measurements were integrated with previously published ACB‐PCR data on KRAS G12D, KRAS G12V, and PIK3CA H1047R. Analysis of these data revealed a correlation between the degree of interindividual variability in these mutations (as log10 MF standard deviation) in normal tissues and the frequencies with which the mutations are detected in carcinomas of the corresponding organs in the COSMIC database. This novel observation has important implications. It suggests that interindividual variability in mutation levels of normal tissues may be used as a metric to identify mutations with critical early roles in tissue‐specific carcinogenesis. Additionally, it raises the possibility that personalized cancer therapeutics, developed to target specifically activated oncogenic products, might be repurposed as prophylactic therapies to reduce the accumulation of cells carrying CDMs and, thereby, reduce future cancer risk. Environ. Mol. Mutagen. 58:466–476, 2017. © 2017 This article is a U.S. Government work and is in the public domain in the USA. Environmental and Molecular Mutagenesis published by Wiley Periodicals, Inc. on behalf of Environmental Mutagen Society
Keywords: biomarker, cancer susceptibility, tumor heterogeneity, personalized medicine, clonal selection
INTRODUCTION
Large‐scale cancer genome sequencing projects have been valuable in terms of identifying the prevalent cancer driver mutations (CDMs) associated with particular types of cancers [Ciriello et al., 2013; Kandoth et al., 2013; Watson et al., 2013; Stover and Wagle, 2015]. Cancer “drivers” have been defined as genetic events associated with tumor initiation or progression [Alizadeh et al., 2015] and as somatic mutations that increase the fitness of a cell [Fisher et al., 2013]. More broadly, a cancer driver is “a cell autonomous or non‐cell autonomous alteration that contributes to the tumor evolution at any stage—including initiation, progression, metastasis, and resistance to therapy—by promoting a variety of functions including proliferation, survival, invasion, or immune evasion” [Alizadeh et al., 2015]. Some of the most impactful cancer drivers are hotspot point mutations. Hotspot point mutations merit investigation because of their high prevalence in cancer, their established roles as early events in carcinogenesis, and evidence that the mutant proteins can be therapeutic targets and/or determinants of the efficacy of specific anticancer therapies (i.e., biomarkers to inform personalized approaches to cancer treatment) [Lu et al., 2014; Ryan et al., 2015]. There are known hotspot base substitution mutations in the PIK3CA and KRAS genes. The prevalence of these mutations in carcinomas of the breast, colon, lung and thyroid are provided in Supporting Information Table S1 (data from Catalogue of Somatic Mutations in Cancer, COSMIC) [COSMIC, 2016].
Individual tumors are heterogeneous in terms of histology, genetics, epigenetics, phenotypic markers, and gene expression [Bonavia et al., 2011; Marusyk et al., 2012]. It has been established that cancers exhibit spatial and temporal genetic diversity, meaning different mutations have been detected in different sectors of a cancer and that abundance of clonal subpopulations can evolve over time [Martelotto et al., 2014; Renovanz and Kim, 2014]. Investigations into the genetic heterogeneity of several cancers has enabled clinicians to identify patients who will benefit from particular therapies (i.e., personalized cancer treatment) and, conversely, demonstrated that mutant cell subpopulations can derail the process and drive the development of acquired resistance to therapy [Fisher et al., 2013; Burrell and Swanton, 2014; Stover and Wagle, 2015]. Given the extent of tumor heterogeneity, the prevalence of mutant subpopulations in advanced cancers and the difficulties associated with treating such cancers, it has been suggested that the development of cancer preventative modalities may be a more effective approach for reducing cancer deaths [Maresso et al., 2015].
Because tumor heterogeneity is a consequence and a reflection of the mechanisms driving tumor initiation and progression, understanding the origins of tumor heterogeneity is critical for designing therapeutic strategies to block the development of resistance to treatment [Hiley et al., 2014]. In addition, information about the earliest stages of carcinogenesis is needed as a foundation for experimental approaches to abrogate or delay the development of cancer [Kensler et al., 2016]. For these reasons, there is growing interest in describing the mutations present in normal and pre‐neoplastic tissues and tracking genetic changes during tumor progression and treatment [Hiley et al., 2014; Jamal‐Hanjani et al., 2014; Nakamura et al., 2014; Roberts and Gordenin, 2014; Francioli et al., 2015; Martincorena and Campbell, 2015].
It has been asserted that a majority of cancers (65%, with 95% CI of 39–81%) arise due to spontaneous mutations, a claim based on the correlation between the incidence of cancer in different tissue types and the number of stem cell divisions in those tissues [Tomasetti and Vogelstein, 2015]. Others have claimed that intrinsic factors account for <10 to 30% of lifetime cancer risk and “rates of endogenous mutation accumulation due to intrinsic processes are not sufficient to account for the observed cancer rates” [Wu et al., 2016]. Clearly, additional information about the frequencies of spontaneous mutations in cancer driver genes and the selective advantages they may confer is necessary to clarify how spontaneous mutations contribute to cancer risk and whether reducing spontaneous CDM frequencies through medical intervention is a viable approach for reducing cancer deaths [Martincorena and Campbell, 2015].
Our laboratory uses a high sensitivity method called Allele‐specific Competitive Blocker PCR (ACB‐PCR), to quantify specific CDMs in normal and cancerous tissue samples derived from particular organs [Parsons et al., 2010; Myers et al., 2016, 2015, 2014a]. By comparing ACB‐PCR amplification of unknown samples to that of a standard curve, ACB‐PCR can determine the ratio of mutant:wild‐type sequence in a DNA sample, measuring one specific base substitution mutation at a time, as long as the mutant to wild‐type ratio is ≥10−5. The ACB‐PCR limit of detection is considered to be 1 × 10−5 because this is the lowest MF standard that can be discriminated experimentally from a wild‐type only control. An ACB‐PCR MF of 10−5 equates to the detection of three mutant molecules in a total of 300,000. ACB‐PCR is robust in the measurement of MFs between 10−1 and 10−5 (ACB‐PCR may underestimate MFs between 10−1 and 1). ACB‐PCR can identify robustly which samples have MFs below 10−5, but cannot measure MFs below 10−5. This sensitivity is unlikely to be sufficient to detect neutral mutations in normal human tissues. For some CDMs, however, this sensitivity has been shown to be sufficient for the mutant quantification in normal rodent and human tissues, likely due to clonal expansion of cells carrying CDMs that confer a positive selective advantage [Parsons et al., 2005, 2010, 2013; McKinzie et al., 2006; Meng et al., 2010a, 2010b 2011; McKinzie and Parsons, 2011; Banda et al., 2015, 2013]. Information about ultralow frequency mutant cell populations can complement existing characterizations of tumor samples by next‐generation sequencing (NGS, which generally detects variant alleles only at frequencies >1% [Cottrell et al., 2014]), to yield a more complete description of the genetic changes present in cancers. Here we report quantification of levels of the PIK3CA E545K and H1047R hotspot point mutations in normal colonic mucosa, colonic adenocarcinomas, normal lung, and lung adenocarcinomas, as well as levels of the PIK3CA E545K mutation in normal breast, mammary ductal carcinoma, normal thyroid, and papillary thyroid carcinoma (PTC). These mutations were selected because they have the potential to provide a positive selective advantage. The PIK3CA E545K (guanine to adenine mutation at codon 545) results in a glutamine to lysine amino acid change in the p110α kinase binding domain. The PIK3CA H1047R (adenine to guanine mutation at codon 1047) results in a histidine to arginine amino acid change in the p110α kinase binding domain. Both PIK3CA mutations increase activation of PI3K and its binding to the cell membrane, thereby impacting oncogenic transformation, cell proliferation, cell growth, survival, differentiation and altering invasion potential [Myers et al., 2016].
KRAS is a downstream mediator of growth factor receptor signaling, with critical roles in cell proliferation, survival, and differentiation. The KRAS G12D (GGT to GAT at codon 12 resulting in a glycine to aspartic acid amino acid change) and KRAS G12V (GGT to GTT at codon 12 resulting in a glycine to valine amino acid change) mutations alter the phenotype of KRAS, providing a context‐dependent selective advantage [Parsons and Myers, 2013]. Here, we also report additional measurements of KRAS G12D and G12V in normal colonic mucosa. These new data, integrated with that published previously, creates a dataset of four hotspot point mutations (KRAS G12D, KRAS G12V, PIK3CA E545K, and PIK3CA H1047R) in normal and malignant tissues from four different organs. Analysis of these data demonstrates that interindividual variability in cancer driver gene mutant fraction (MF) within normal tissues is correlated with the tissue‐specific prevalence of carcinomas carrying the corresponding mutations.
MATERIALS AND METHODS
Sample Collection
Procedures for the acquisition and analysis of anonymous human tissues were reviewed by the FDA's IRB (Research Involving Human Subjects Committee, FWA 00006196). The National Disease Research Interchange (NDRI, Philadelphia, PA) and the National Cancer Institute's Cooperative Human Tissue Network (CHTN) were identified as the primary sources of normal and malignant tissues, respectively. Fresh‐frozen, normal breast, normal colonic mucosa, normal lung, and normal thyroid tissue samples, as well as one fresh‐frozen papillary thyroid carcinoma (PTC), were purchased from the NDRI [Parsons et al., 2010; Myers et al., 2016, 2015, 2014a]. Fresh‐frozen, primary breast ductal carcinomas (DCs), colonic adenomas, colonic adenocarcinomas, lung adenocarcinomas, and PTCs, along with normal colonic mucosa samples, were provided by the CHTN [Parsons et al., 2010; Parsons and Myers 2013; Myers et al., 2016, 2015, 2014a]. These samples were collected at autopsy from tissue donors (one sample per donor) who died from causes unrelated to cancer or diseases affecting the relevant target organ. The numbers of samples analyzed for the different mutations are given in Supporting Information Table S2. All tumor specimens were histologically evaluated and classifications were confirmed by board certified pathologists. The extent of information provided regarding the smoking history of the subjects was inconsistent across subjects (and in some instances absent), only allowing for stratification of subjects as ever‐ and never‐smokers.
DNA Isolation and First‐Round PCR
DNA isolation was performed as described previously [Parsons et al., 2010; Myers et al., 2016, 2015, 2014a], using sufficient tissue to ensure that the characteristic cellular content of each tissue type would be represented proportionally in the genomic DNA. The average weights of the tissue samples processed for breast, colonic mucosa (separated from submucosa), lung and thyroid were 4.0, 0.4, 2.3, and 1.9 g, respectively. A high fidelity, first‐round PCR amplification of a 306 bp sequence encompassing human PIK3CA codon 545 was performed for each DNA sample, using 1 µg of EcoRI‐digested genomic DNA in a 200 μl PCR reaction mix of: 10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris‐HCl (pH 8.75), 2 mM MgSO4, 0.1% Triton X‐100, 0.1 mg/ml bovine serum albumin, 0.2 mM dNTPs, and 10 units of cloned PfuUltra Hotstart DNA Polymerase (Agilent Technologies, Santa Clara, CA). The primers were 5′‐GGGAAAATGACAAAGAACAG‐3′ and 5′‐AATGTGCCAACTACCAATGT‐3′. The thermocycler reaction conditions were 2 min at 94°C, followed by 31 cycles of 1 min at 94°C, 2 min at 56°C, and 1 min at 72°C, followed by a final 7 min at 72°C extension and 4°C hold.
A high fidelity, first‐round PCR amplification of a 265 bp sequence encompassing human PIK3CA codon 1047 was performed for each DNA sample, using 1 µg of EcoRI‐digested genomic DNA in a 200 μl PCR reaction mix of: 10 mM KCl, 10 mM (NH4)2SO4, 20 mM Tris‐HCl (pH 8.75), 2 mM MgSO4, 0.1% Triton X‐100, 0.1 mg/ml bovine serum albumin, 0.2 mM dNTPs, and 10 units of cloned PfuUltra Hotstart DNA Polymerase (Agilent Technologies). The primers were 5′‐CCAAACTGTTCTTATTACTTATAG‐3′ and 5′‐GAAGATCCAATCCATTTTTGT‐3′. Thermocycler reaction conditions were 2 min at 94°C, followed by 33 cycles of 1 min at 94°C, 1 min at 52°C, and 1 min at 72°C, followed by a final 6 min at 72°C extension and 4°C hold.
The high fidelity, first‐round PCR of a 384 bp sequence encompassing human KRAS codon 12 was described previously [Myers et al., 2014a]. Each first‐round PCR employed one intron‐specific primer to avoid amplification of pseudogenes.
Synthesis of PIK3CA and KRAS WT and Mutant Standards
Plasmids carrying the mutant or wild‐type sequences (for the two different amplicons of the PIK3CA gene) were used as template to synthesize mutant and wild‐type standards. Plasmids carrying G12D mutant KRAS, G12V mutant KRAS, and wild‐type KRAS sequences were used as template to synthesize standards. Each of the standards was synthesized using conditions identical to those used to amplify the unknown genomic DNAs, except that the amount of input plasmid DNA was calibrated prior to PCR to ensure approximately equivalent amplification in the synthesis of standards and unknowns.
Purification and Quantification of Standards and Unknowns
Purification of PIK3CA E545K WT, mutant, and unknown PCR products was accomplished by ion‐pair reverse phase chromatography using non‐denaturing conditions and a Transgenomic WAVE Nucleic Acid Fragment Analysis and Collection system (Omaha, NE). PIK3CA H1047R PCR products and KRAS PCR products were gel purified as described previously [Myers et al., 2016]. The DNA concentration of each sample was determined using an Epoch Spectrophotometer Model (Biotek, Winooski, VT), calculated as the average of three measurements that varied by ≤10% from the group mean.
ACB‐PCR
Mutant (either PIK3CA E545K, PIK3CA H1047R, KRAS G12D, or KRAS G12V) and WT reference DNA samples were mixed to generate standards with mutant:WT ratios (i.e., MFs) of 10−1, 10−2, 10−3, 10−4, 10−5, and 0 (containing only the WT sequence), at a concentration of 5 × 107 copies/μl for the PIK3CA E545K mutation or 1 × 108 copies/µl for the PIK3CA H1047R mutation. Each ACB‐PCR reaction employed 10 μl of each DNA mixture, for a total of 5 × 108 or 1 × 109 copies per reaction for the E545K and H1047R assays, respectively. Each MF standard was analyzed in duplicate, along with a no‐DNA control. ACB‐PCR reactions were performed in 96‐well PCR plates (Thermo Fisher Scientific, Waltham, MA) using three primers: a fluorescein‐labeled mutant‐specific primer (MSP), a non‐extendable blocker primer (BP, to reduce background amplification of WT PIK3CA), and an upstream/downstream primer [Myers et al., 2014b]. The ACB‐PCR measurement of KRAS G12D and G12V was performed as described previously [Myers et al., 2015].
For PIK3CA E545K mutation, each 50 µl ACB‐PCR reaction contained: 1× Universal Taq Buffer [New England BioLabs, Inc. (NEB), Beverly, MA), 1.5 mM MgCl2, 0.1 mg/ml gelatin, 1.0 mg/ml Triton X‐100, 80 µM dNTPs, 400 nM each primer [MSP, 5′‐fluorescein‐TCCTCTCTCTGAAATCACCA‐3′; BP, 5′‐TCCTCTCTCTGAAATCACCdG′3′ (i.e., 3′‐deoxyG modification); and downstream primer, 5′‐CAGAGAATCTCCATTTTAGC‐3′], 3 ng/µl ET SSB (NEB), 2.8 mU/µl Perfect Match PCR Enhancer (Stratagene, La Jolla, CA), and 0.08 µl Hemo KlenTaq DNA polymerase (NEB). Cycling conditions were 2 min at 94°C, followed by 36 cycles of 94°C for 30 sec, 41°C for 90 sec, and 68°C for 1 min. The PIK3CA E545K ACB‐PCR product is 77 bp in length.
For PIK3CA H1047R mutation, each 50 µl ACB‐PCR reaction contained: 1× Universal Taq Buffer, 0.75 mM MgCl2, 0.1 mg/ml gelatin, 1.0 mg/ml Triton X‐100, 40 µM dNTPs, 270 nM MSP (5′‐fluorescein‐TGTTGTCCAGCCACCATGTC‐3′), 500 nM BP (5′‐TGTTGTCCAGCCACCATGTdT‐3′), 1.5 µM downstream primer (5′‐GATGCTTGGCTCTGGAATGC‐3′), 1.2 mU/µl Perfect Match PCR Enhancer, and 0.3 µl Hemo KlenTaq DNA polymerase. Cycling conditions were 2 min at 94°C, followed by 41 cycles of 94°C for 30 sec, 49°C for 45 sec, and 72°C for 1 min. The PIK3CA H1047R ACB‐PCR product is 148 bp in length. Blocker primers were purchased from Sigma Genosys (The Woodlands, TX), all others were purchased from Integrated DNA Technologies, Inc. (Coralville, IA).
Vertical Polyacrylamide Gel Electrophoresis, Image Analysis, and Data Collection
Following ACB‐PCR, 10 µl of 6× ficoll loading buffer/dye were added to each 50 µl reaction, and 10 µl of each ACB‐PCR product were analyzed on 8% nondenaturing, polyacrylamide gels. A PharosFX Molecular Imager with an external blue laser (Bio‐Rad) was used to visualize the fluorescent ACB‐PCR products. Quantity One software (Bio‐Rad), with a locally averaged background correction, was used to quantify the pixel intensities of the correct‐sized bands. For the PIK3CA E545K, log‐log plots relating MF to fluorescence were constructed and fit with a power function. For the PIK3CA H1047R, KRAS G12D, and G12V mutations, log‐linear plots relating MF to fluorescence were constructed and fit with a logarithmic function. Using the function of the standard curve and the pixel intensities of the ACB‐PCR products, the MF (ratio of mutant to wild‐type sequence) of each unknown sample was calculated.
Statistical Analyses
For each sample, MF was calculated as the arithmetic average of three independent ACB‐PCR measurements. The average MF measurement for each sample was log10‐transformed. For each mutational target, the geometric mean MF was calculated as the average log10‐transformed MF measured for a particular tissue type. Statistical analyses were performed using log10‐transformed data.
Log‐transformed datasets were examined for normality using the D'Agostino and Pearson omnibus normality test. Nonparametric approaches were applied to log‐transformed data that were not normally distributed. The Mann‐Whitney rank sum test was used when a quantitative ranking of MFs was possible. Contingency analyses were used to test for statistical significance when datasets contained multiple samples with MFs below the limit of accurate ACB‐PCR quantification (i.e., <10−5). Specifically, the numbers of samples with MFs >10−5 and <10−5 were examined using χ 2 or Fisher's exact test. Pearson correlation analyses were performed on log‐transformed, normally‐distributed data (Spearman's rank correlation analyses were performed when data were not normally‐distributed). Two‐tailed P values <0.05 were considered significant. All statistical analyses were performed using GraphPad Prism 5 Software (GraphPad Software, Inc., La Jolla, CA).
RESULTS
The goals of this study were to elucidate the role of spontaneous CDMs in tissue‐specific carcinogenesis and to define the prevalence of low frequency CDMs within cancers. Therefore, unlike many studies that examine tumor tissue and normal tumor adjacent sample, this study examined the levels of hotspot CDM in “normal” autopsy samples from men (except breast) and women without cancer or disease in the relevant organ.
Levels of PIK3CA E545K mutation (codon 545 GAG→AAG) were measured in normal breast, colonic mucosa, lung, and thyroid DNA samples, as well as in DNA isolated from ductal carcinomas, colonic adenomas, colonic adenocarcinomas, lung adenocarcinomas, and papillary thyroid carcinomas. Each unknown was quantified in three independent experiments, generating a dataset of 441 PIK3CA E545K MF measurements. The numbers of samples analyzed for each tissue type are presented in Supporting Information Table S2. Following ACB‐PCR, products were run on vertical polyacrylamide gels. Replicate examples of PIK3CA E545K ACB‐PCR output are provided in Supporting Information Figure S1. MF quantification was achieved by interpolation of the fluorescent intensities of unknown samples with that of a standard curve constructed using samples with defined ratios of mutant:WT alleles (i.e., duplicate 10−1, 10−2, 10−3, 10−4, 10−5, and 0 standards). A representative example of a PIK3CA E545K ACB‐PCR standard curve is shown in Supporting Information Figure S2. The average coefficient of determination (r 2) for the PIK3CA E545K standard curves were as follows: breast, 0.9730 (range 0.9662–0.9828); colon, 0.9628 (range 0.9334–0.9831); lung, 0.9595 (range 0.9316–0.9726); and thyroid, 0.9670 (range 0.9595–0.9724). The measured MFs are given in Supporting Information Table S3. The average coefficient of variation for the triplicate PIK3CA E545K MF measurements obtained from individual breast, colon, lung, and thyroid samples were 0.36, 0.55, 0.35, and 0.43, respectively.
PIK3CA H1047R (codon 1047 CAT→CGT) MF was measured in normal colonic mucosa and lung DNA samples, as well as in DNA isolated from colonic adenomas, colonic adenocarcinomas, and lung adenocarcinomas (see Supporting Information Table S2). Each unknown was quantified in three independent experiments (276 PIK3CA H1047R MF measurements). Replicate examples of PIK3CA H1047R ACB‐PCR output are provided in Supporting Information Figure S1. A representative example of a PIK3CA H1047R ACB‐PCR standard curve is shown in Supporting Information Figure S2. The average coefficient of determination (r 2) for the standard curves used to measure PIK3CA H1047R MF in colon and lung were 0.9849 (range 0.9732–0.9928) and 0.9802 (range 0.9738–0.9840), respectively. The PIK3CA H1047R MFs are given in Supporting Information Table S4. The average coefficient of variation for the triplicate PIK3CA H1047R MF measurements obtained from individual colon and lung samples were 0.40 and 0.16, respectively.
Fifteen normal colonic mucosa samples were analyzed for KRAS G12D (codon 12 GGT→GAT) and G12V (codon 12 GGT→GTT) MFs as described above (90 ACB‐PCR MF measurements), adding to the previously published ACB‐PCR analysis of six colonic mucosa samples [Parsons et al., 2010]. Replicate examples of the KRAS G12D and G12V ACB‐PCR output are provided in Supporting Information Figure S1. Representative examples of KRAS G12D and G12V standard curves are shown in Supporting Information Figure S2. The average coefficient of determination (r 2) for the standard curves used to measure KRAS G12D and G12V MF in colon were 0.9895 (range 0.9735–0.9969) and 0.9889 (range 0.9744–0.9973), respectively. The KRAS G12D and G12V MFs are given in Supporting Information Table S5. The average coefficient of variation for the triplicate KRAS G12D and G12V MF measurements obtained from individual colon samples were 0.52 and 0.17, respectively.
Comparison of PIK3CA E545K, PIK3CA H1047R, KRAS G12D, and KRAS G12V MFs Between Normal and Malignant Tissues
A summary of MF measurements for normal tissues and tumors is given in Table 1. No significant differences were observed between the levels of PIK3CA E545K mutation in normal tissues and tumor samples, for any of the organs examined (see Fig. 1), although the increased MFs in colonic adenomas compared to normal colonic mucosa were of borderline significance (Fisher's exact test, P = 0.0741). No significant differences were observed between the levels of PIK3CA H1047R mutation in normal tissue and tumors of the colon or lung (see Fig. 1). For KRAS G12D and G12V, the 15 ACB‐PCR measurements from normal colonic mucosa were compared to those previously published for colonic adenomas and adenocarcinomas [Parsons et al., 2010]. This analysis confirmed an earlier report conducted using just six normal samples. Significant differences in MF were observed among sample types (Kruskal‐Wallis test, P = 0.0013), with colonic adenomas having significantly greater KRAS G12D mutant levels than normal colonic mucosa. Significant differences in KRAS G12V MFs were also observed (χ 2 test, P = 0.0220).
Table 1.
Summary of ACB‐PCR MF Measurements
| Sample type | Gene mutation | Tissue | Median MF | Geomean MF | No. samples >10−5 | No. samples <10−5 | Mutation positive samples (%) |
|---|---|---|---|---|---|---|---|
| Normal | PIK3CA H1047R | Colon | 1.57 × 10−5 | 1.98 × 10−5 | 20 | 0 | 100 |
| PIK3CA H1047R | Lung | 2.45 × 10−5 | 3.96 × 10−5 | 19 | 0 | 100 | |
| PIK3CA E545K | Breast | 6.06 × 10−6 | 1.78 × 10−5 | 4 | 6 | 40 | |
| PIK3CA E545K | Colon | 2.67 × 10−6 | 2.69 × 10−6 | 0 | 20 | 0 | |
| PIK3CA E545K | Lung | 5.35 × 10−6 | 6.75 × 10−6 | 3 | 16 | 16 | |
| PIK3CA E545K | Thyroid | 6.52 × 10−6 | 7.02 × 10−6 | 5 | 14 | 26 | |
| KRAS G12D | Colon | 2.88 × 10−5 | 4.90 × 10−5 | 15 | 0 | 100 | |
| KRAS G12V | Colon | 7.66 × 10−6 | 1.08 × 10−5 | 6 | 9 | 40 | |
| Adenoma | PIK3CA H1047R | Colon | 1.73 × 10−5 | 4.56 × 10−5 | 8 | 1 | 89 |
| PIK3CA E545K | Colon | 2.15 × 10−6 | 2.38 × 10−6 | 2 | 6 | 25 | |
| Carcinoma | PIK3CA H1047R | Colon | 1.63 × 10−5 | 3.46 × 10−5 | 19 | 1 | 95 |
| PIK3CA H1047R | Lung | 2.54 × 10−5 | 3.48 × 10−5 | 24 | 0 | 100 | |
| PIK3CA E545K | Breast | 3.92 × 10−5 | 8.36 × 10−5 | 7 | 2 | 78 | |
| PIK3CA E545K | Colon | 3.07 × 10−6 | 7.89 × 10−6 | 3 | 17 | 15 | |
| PIK3CA E545K | Lung | 4.17 × 10−6 | 7.13 × 10−6 | 4 | 20 | 8 | |
| PIK3CA E545K | Thyroid | 8.39 × 10−6 | 8.78 × 10−6 | 7 | 11 | 39 |
The median and geomean MFs shown in italics are below the limit of accurate ACB‐PCR quantification (10−5).
Figure 1.
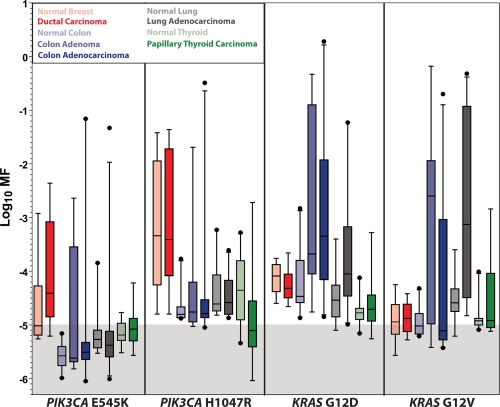
The frequency distributions are shown for PIK3CA E545K, PIK3CA H1047R, KRAS G12D, and KRAS G12V in paired datasets of normal and malignant tissues. The data presented include that reported in the current publication, integrated with that reported previously [Parsons et al., 2010; Parsons and Myers, 2013; Myers et al., 2016, 2015, 2014a]. Measurements within the shaded area of the graph are below the lowest MF standard used and are considered below the limit of accurate ACB‐PCR quantification.
The data developed in the current study, combined with that previously published [Parsons et al., 2010; Myers et al., 2016, 2015, 2014a], constitutes a data set of more than 1,800 ACB‐PCR MF measurements on 606 different DNA samples. The mutant frequency distributions observed in normal/tumor tissue pairs are presented in Figure 1, for each mutation analyzed. Figure 1 illustrates for the first time: (1) the tissue‐specific differences in ultralow frequency KRAS and PIK3CA mutation levels, (2) the remarkable prevalence of low‐frequency mutant populations, and (3) in some cases, a suprising overlap in the frequency distributions between normal and tumor.
MF Measurements in Normal Tissues
PIK3CA H1047R MF in normal colonic mucosa and lung, along with PIK3CA E545K MF in normal colonic mucosa, lung, breast, and thyroid, were examined for correlations with age, smoking status, and gender. No statistically significant associations with age or smoking status were observed (comparing ever smoker vs. never‐smokers). For colonic mucosa, however, the PIK3CA H1047R mutation was significantly more abundant in DNA from men than from women (Fig. 2). This novel finding is consistent with the higher age‐adjusted incidence rates for colorectal cancer among men than among women [Abotchie et al., 2012]. In addition, KRAS G12D MF in normal colonic mucosa was positively correlated with age, although this correlation was statistically significant at only the 90% confidence level (Spearman r = 0.4553, P = 0.0576).
Figure 2.
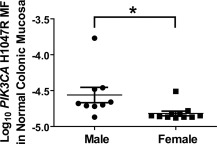
Significantly greater PIK3CA H1047R MFs were observed in DNA samples isolated from normal male colonic mucosa as compared to normal female colonic mucosa. The asterisk denotes statistical signficance, Mann Whitney test, P = 0.0068.
Table 1 and Figure 3 show there are differences in mutation prevalence and variability across normal tissue types. For example, all colonic mucosa samples had PIK3CA H1047R and KRAS G12D MFs >10−5, whereas none of the normal colonic mucosa DNA samples had PIK3CA E545K MFs >10−5. MF measurements in normal tissues were analyzed statistically for tissue‐specific differences (see Table 1 and Fig. 3). Because many PIK3CA E545K mutations were below the limit of accurate ACB‐PCR quantification (10−5), statistical differences between normal tissue types were examined using a χ 2 test (comparing the numbers of samples with MFs greater than and less than 10−5 across tissues). This analysis detected a significant difference in levels of PIK3CA E545K mutation across the normal tissue types (P = 0.0328). Current PIK3CA H1047R MF measurements in normal tissues were combined with those published previously. Because all PIK3CA H1047R MFs were >10−5, analysis of variance was performed using a Kruskal‐Wallis test, with a Dunn's Multiple Comparison post‐test. This analysis identified significant differences in PIK3CA H1047R MF across tissue types (P = 0.0001), and established that PIK3CA H1047R MFs in breast DNA samples were significantly greater than those of DNA from colonic mucosa.
Figure 3.
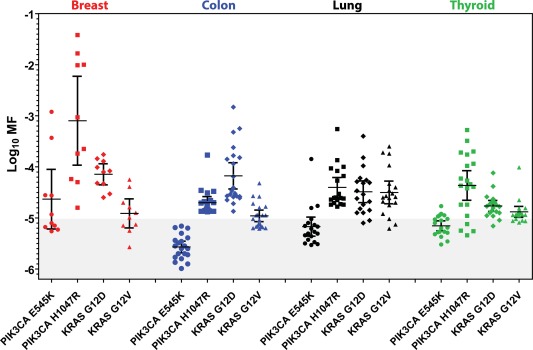
Variability in CDM frequency is tissue‐ and mutant target‐specific. Individual ACB‐PCR measurements were plotted for four different mutations (PIK3CA E545K, PIK3CA H1047R, KRAS G12D, and KRAS G12D) in normal DNA samples isolated from four different tissues (breast, colonic mucosa, lung, and thyroid). Measurements within the shaded area of the graph are below the lowest MF standard used and are considered below the limit of accurate ACB‐PCR quantification.
This dataset was used to investigate relationships between tissue‐specific MF measurements and the importance of particular mutations in tissue‐specific carcinogenesis. Frequencies of the PIK3CA E545K, PIK3CA H1047R, KRAS G12D, and KRAS G12V mutations in carcinomas of the breast, large intestine, lung, and thyroid were collected from the expertly curated data in the COSMIC database (v76) [COSMIC, 2016] (see Supporting Information Table S1). We determined that the log10 geomean MFs from the 16 sets of ACB‐PCR MF measurements collected from normal tissues (PIK3CA E545K, PIK3CA H1047R, KRAS G12D, and KRAS G12V in breast, colonic mucosa, lung, and thyroid) were not correlated with the frequencies with which these mutations are reported in carcinomas from each organ (Spearman r = 0.3676, P = 0.1612).
Visual inspection of the data, however, suggested that mutations reported to occur frequently in particular cancers showed large inter‐individual variability in the ACB‐PCR MFs in normal tissues (e.g., PIK3CA H1047R for breast, KRAS G12D for colon, see Figs. 1 and 3) [COSMIC, 2016]. Therefore, the relationship between interindividual variability (as log10 MF standard deviation) and the frequency of mutation within the various target organs was examined. Calculated MFs were used, except when the calculated MFs were below the theoretical limit of detection 3.33 × 10−6 (a log10 MF of −5.477), which is equivalent to one mutant molecule in a background of 3 × 105 WT molecules. In order to standardize the analysis of standard deviation across the data set and avoid overestimation of MF standard deviation due to non‐detects, samples with a MF below 3.33 × 10−6 were ascribed a MF of 3.00 × 10−6 (log10 MF −5.481, less than 1 mutant molecule per 300,000). The non‐detects included 2/10 breast PIK3CA E545K measurements, 13/20 colon PIK3CA E545K measurements, 3/19 lung PIK3CA E545K measurements, 1 thyroid PIK3CA E454K measurement, and 1 breast KRAS G12D measurement. A strong correlation was observed between the log10 MF standard deviation and the frequencies of the mutations in carcinomas from the corresponding organs (Spearman r = 0.7265, P = 0.0014). The correlation was significant with and without the correction for non‐detects, but the correlation was stronger with the correction [Spearman r (without correction) = 0.6382, P = 0.0078]. Furthermore, the correlation was not driven by a single data point. Even when the breast PIK3CA H1047R data point (log10 MF standard deviation = 1.2418; COSMIC tumor prevalence = 13.5%) was removed from the analysis, a strong correlation was observed (r = 0.6679, P = 0.0065). The log10 MF variance within normal tissue samples also correlated significantly with the frequency of mutations in tumors of the cognate target organ (Spearman r = 0.5088, P = 0.0441). In Figure 4, linear regression analysis was used to depict the relationship between log10 MF standard deviation in normal tissue and the corresponding prevalence by tumor type for the mutations examined.
Figure 4.
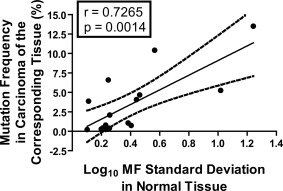
Correlation between the log10 MF standard deviation for PIK3CA and KRAS hotspot point mutations in normal tissues and the frequencies with which the mutations occur in corresponding carcinomas (breast ductal carcinomas, colonic adenocarcinomas, lung adenocarcinomas, and papillary thyroid carcinomas) according to the COSMIC database (v76) [COSMIC, 2016]. Calculated MFs were used, except when calculated MFs were below the theoretical limit of detection 3.33 × 10−6 (log10 MF = −5.477, or one mutant molecule in a background of 3 × 105 WT molecules).
DISCUSSION
The PIK3CA and KRAS mutations analyzed in this study are recognized hotspot point mutations, meaning they are overrepresented in tumor sequence databases compared to most other mutations. For example, the PIK3CA H1047R mutation is the most prevalent somatic mutation detected in DCs. KRAS G12D is the most prevalent somatic mutation detected in colonic adenocarcinomas. Beyond that, the signaling dysfunctions caused by the mutant proteins are well‐described. Thus, these mutations are functional drivers of carcinogenesis.
Mutations within these genes are even more prevalent in tumors than previously appreciated, when the mutation detection method employed is capable of detecting mutations within low‐frequency mutant cell subpopulations (see Supporting Information, Table S6). The current study demonstrates PIK3CA mutant cells are prevalent as subpopulations within colon adenomas, colon adenocarcinomas, and lung adenocarcinomas. Further, some breast DCs carry subpopulations of PIK3CA E545K mutant cells, as well as previously reported subpopulations of PIK3CA H1047R [Myers et al., 2016].
We conclude that these particular mutations are able to impact tumor initiation and progression as relatively small subpopulations of cells. An early driver mutation, in a tumor‐initiating clone, is expected to result in a predominant mutation (variant allele frequency of ∼10–50%). Such events would be detected by DNA sequencing and account for the percentages of tumors reported in the COSMIC database (Supporting Information Table S6). The low variant allele frequencies (10−5 to 10−2), detected in significant fractions of the tumors analyzed, could arise in a number of different ways. First, the mutation could occur late in tumor development. Second, the mutation could occur in an initiating clone, whose growth is outpaced by a cooperating initiating clone or clones. Or, third, the mutation might confer a selective advantage early, but not late in tumor progression. Because these mutations are not sufficient for carcinogenesis, each of these potential paths is expected to be impacted by the accumulation of additional mutations in the tumor mass as a whole.
There is growing evidence that tumors (including breast and colon tumors) can have polyclonal or multiancestral architecture [Parsons, 2008; Zahm et al., 2016]. We propose that some mutations are able to function as “transdriver” mutations, meaning small subpopulations of cells can contribute to (or drive) the initiation and progression of a different clone (or clones) of cells, which carry complementing genetic or epigenetic lesions. Such clonal interactions have been documented [Inda et al., 2011, 2010; Marusyk et al., 2014; Altrock et al., 2015] and are believed to operate through paracrine or non‐cell‐autonomous mechanisms [Hobor et al., 2012]. In a portion of such cases of multiclonal initiation, a PIK3CA or KRAS mutant cell may acquire an additional genetic hit and progress to become the predominant tumor clone. The alternative pathway is that the co‐initiating clone(s) acquire additional genetic or epigenetic damage and progress to become the predominant tumor clone(s), generating carcinomas with PIK3CA or KRAS mutant subpopulations.
ACB‐PCR quantification of PIK3CA and KRAS mutations demonstrated there is a remarkable amount of interindividual variability in the levels of these mutations in some normal tissue types (most notably PIK3CA mutations in breast and KRAS mutations in colon). Importantly, we report for the first time a significant correlation exists between the degree of interindividual variability in PIK3CA and KRAS MFs in normal tissues and the tissue‐specific prevalence of the mutations in carcinomas. Conversely, the log10 geomean MF did not correlate with the corresponding tumor mutation frequency. Because standard deviation is influenced by the magnitude of the measured variable, the variability among individuals samples combined with the magnitude of the tissue‐specific normal MF measurements may be contributing to the observed correlation (Fig. 4). These observations suggest that some level of mutagenesis (an ACB‐PCR measureable background MF) combined with tissue‐specific clonal expansion (inter‐sample variability in MF measurements) are characteristics of cancer driver mutations within a particular tissue type. These observations have important implications in terms of establishing the relative importance of different mutations as biomarkers of susceptibility for particular human tissues. Further, this suggests that interindividual variation in MF in normal tissues can be used as a metric to assess the context‐dependent, selective advantage of cells carrying oncogenic point mutations.
According to Rozhok and DiGrigori, “oncogenic mutations should have vastly different fitness effects on somatic cells dependent on the tissue microenvironment in an age‐dependent manner” [Rozhok and DeGregori, 2015]. ACB‐PCR MF measurements provide evidence that PIK3CA and KRAS mutations impact mutant cell fitness in a manner that is context dependent. In the current study, we observed significant differences in PIK3CA E545K and H1047R mutation abundance across the different normal tissue types. We also observed a gender difference in PIK3CA H1047R MF in colonic mucosa. The significantly greater levels of PIK3CA H1047R mutation in male versus female colonic mucosa are consistent with men having a greater risk of developing colon cancer than women. PIK3CA H1047R mutation increases in abundance with age in normal breast, suggesting it confers a positive selective advantage in breast [Myers et al., 2016]. KRAS G12D and G12V mutations are more abundant in colonic adenomas, as compared to normal colonic mucosa, suggesting these mutations confer a selective advantage early in carcinogenesis. However, there is a significant decrease in KRAS G12V during adenoma to adenocarcinoma progression (see Fig. 1) [Parsons et al., 2010], and there is a significant inverse correlation between KRAS G12V MF and maximum tumor dimension for colon tumors and papillary thyroid carcinomas [Parsons and Myers, 2013], but not for lung adenocarcinomas [Myers et al., 2015]. These data suggest that KRAS G12V is neutral or selected against in some large/advanced cancers. This negative selection in large/advanced cancers could explain the prevalence of pre‐existing KRAS mutant tumor cell subpopulations in the colorectal cancers that develop resistance to anti‐EGFR monoclonal antibody therapy [Diaz et al., 2012; Misale et al., 2012].
There are a few reports of KRAS mutations being detected in normal colon and lung tissues [Ronai et al., 1994; Yakubovskaya et al., 1995; Sudo et al., 2006; Parsons et al., 2010]. Recently, Gao et al. [2017] reported detecting KRAS mutations in 52/156 histologically normal bronchial biopsies from former lung cancer patients. To our knowledge, there is only one report of PIK3CA mutation in normal tissue (breast) [Myers et al., 2016], although PIK3CA mutations have been detected in breast hyperplasia [COSMIC, 2016] and ductal hyperplasia with columnar cell change [Ang et al., 2012]. Data from the current and previous ACB‐PCR analyses [Parsons et al., 2010; Myers et al., 2016, 2015, 2014a], allow us to conclude that cells carrying PIK3CA and KRAS hotspot point mutations are prevalent in normal tissues. The prevalence of PIK3CA and KRAS mutations in normal tissues is consistent with the mathematically‐derived conclusion that “half or more of the somatic mutations in cancers of self‐renewing tissues originate prior to tumor initiation” [Tomasetti et al., 2017, 2013].
There is an ongoing debate about the degree to which spontaneous mutation determines human cancer risk. Recent publications by Tomasetti and Vogelstein highlight the importance of spontaneous mutation in tissue‐specific carcinogenesis [Tomasetti et al., 2013]; they suggest that two‐thirds of cancers are attributable to random mutations in normal stem cells due to replication errors [Tomasetti and Vogelstein, 2015; Tomasetti et al., 2017]. Others contend that “cancer risk is heavily influenced by extrinsic factors,” with only 10 to 30% of cancers due to intrinsic factors [Wu et al., 2016]. Our results refute arguments that spontaneous mutation is insufficiently frequent to have an impact on carcinogenesis, as the combination of spontaneous mutation plus subsequent cell selection must give rise to the relatively high levels of mutations that we observed in normal tissues.
The concept of hotspot transdriver mutations has the potential to unify the opposing schools of thought regarding the importance of intrinsic versus extrinsic factors in carcinogenesis. Spontaneous transdriver mutations can be viewed as substrates that contribute to carcinogenesis, potentially though interaction with exogenously‐induced genetic or epigenetic changes. Our analysis does not discriminate spontaneous from exogenously‐induced mutation. Given the deficits in the smoking histories for the normal tissue samples, the lack of significant associations between MF and smoking history should be interpreted cautiously and revisited in the future. Nevertheless, given the relative insensitivity of epidemiological studies and the relatively small sample sizes analyzed by ACB‐PCR, it seems unlikely that the significant correlation we observed between inter‐individual variability in cancer driver MF and tissue‐specific prevalence of the corresponding mutations in carcinomas is due solely to exogenous exposures. That said, the correlation depicted in Figure 4 may provide some insights into which cancers are impacted by exogenous exposures. The three data points where the cancer incidence is greater than the linear regression expectation correspond to PIK3CA E545K, KRAS G12D, and KRAS G12V in colorectal cancer, a cancer whose incidence is considered to have an extrinsic exposure component.
The remarkable prevalence of PIK3CA and KRAS mutations in DNA from normal tissues suggests that there may be significant opportunities for cancer prevention. Therapies that target pre‐existing oncomutations in normal tissues have the potential to decrease future cancer risk. Progress is being made in developing therapeutics that target mutant KRAS [Patricelli et al., 2016]. Therapies that target mutant KRAS or PIK3CA proteins could also be investigated as prophylactic therapies in individuals with Noonan and Cowden syndromes, who have high cancer risk because they carry germline mutations in these genes.
AUTHOR CONTRIBUTIONS
B.L.P. and M.B.M. designed the study. K.L.M., M.B.M., and B.L.P. performed the experiments. B.L.P. prepared figures, tables and manuscript. All authors approved the final manuscript.
Abbreviations
- ACB‐PCR
allele‐specific competitive blocker‐polymerase chain reaction
- CDMs
cancer driver mutations
- DC
ductal carcinoma
- MF
mutant fraction
- NGS
next‐generation sequencing
- PTC
papillary thyroid carcinoma
- WT
wild‐type.
Supporting information
Supporting Information
ACKNOWLEDGMENTS
The authors thank Drs. Page McKinzie, Todd Townsend, and Robert Heflich for their helpful review of the manuscript. Tissue samples were provided by the Cooperative Human Tissue Network which is funded by the National Cancer Institute. Other investigators may have received specimens from the same subjects. The information in these materials is not a formal dissemination of information by FDA and does not represent agency position or policy.
REFERENCES
- Abotchie PN, Vernon SW, Du XL. 2012. Gender differences in colorectal cancer incidence in the United States, 1975–2006. J Womens Health 21:393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh AA, Aranda V, Bardelli A, Blanpain C, Bock C, Borowski C, Caldas C, Califano A, Doherty M, Elsner M, et al. 2015. Toward understanding and exploiting tumor heterogeneity. Nat Med 21:846–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altrock PM, Liu LL, Michor F. 2015. The mathematics of cancer: Integrating quantitative models. Nat Rev Cancer 15:730–745. [DOI] [PubMed] [Google Scholar]
- Ang DC, Corless C, Troxell M. 2012. PIK3CA mutation analysis in breast lesions using LNA‐PCR sequencing. J Mol Diagn 14:729 [DOI] [PubMed] [Google Scholar]
- Banda M, McKim KL, Haber LT, MacGregor JA, Gollapudi BB, Parsons BL. 2015. Quantification of Kras mutant fraction in the lung DNA of mice exposed to aerosolized particulate vanadium pentoxide by inhalation. Mutat Res 789:53–60. [DOI] [PubMed] [Google Scholar]
- Banda M, Recio L, Parsons BL. 2013. ACB‐PCR measurement of spontaneous and furan‐induced H‐ras Codon 61 CAA to CTA and CAA to AAA mutation in B6C3F1 mouse liver. Environ Mol Mutagen 54:659–667. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Inda MDM, Cavenee WK, Furnari FB. 2011. Heterogeneity maintenance in glioblastoma: A social network. Cancer Res 71:4055–4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, Swanton C. 2014. Tumour heterogeneity and the evolution of polyclonal drug resistance. Mol Oncol 8:1095–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. 2013. Emerging landscape of oncogenic signatures across human cancers. Nat Genet 45:1127–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSMIC . 2016. Catalogue of Somatic Mutations in Cancer. Available at: http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/. Accessed March 2016.
- Cottrell CE, Al‐Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, Lockwood CM, Hagemann IS, O'Guin SM, Burcea LC, et al. 2014. Validation of a next‐generation sequencing assay for clinical molecular oncology. J Mol Diagn 16:89–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz LA Jr, Williams RT, Wu J, Kinde I, Hecht JR, Berlin J, Allen B, Bozic I, Reiter JG, Nowak MA, et al. 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 486:537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher R, Pusztai L, Swanton C. 2013. Cancer heterogeneity: Implications for targeted therapeutics. Br J Cancer 108:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francioli LC, Polak PP, Koren A, Menelaou A, Chun S, Renkens I, Van Duijn CM, Swertz M, Wijmenga C, Van Ommen G, et al. 2015. Genome‐wide patterns and properties of de novo mutations in humans. Nat Genet 47:822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Jin J, Yin J, Land S, Gaither‐Davis A, Christie N, Luketich JD, Siegfried JM, Keohavong P. 2017. KRAS and TP53 mutations in bronchoscopy samples from former lung cancer patients. Mol Carcinogen 56:381–388. [DOI] [PubMed] [Google Scholar]
- Hiley C, de Bruin EC, McGranahan N, Swanton C. 2014. Deciphering intratumor heterogeneity and temporal acquisition of driver events to refine precision medicine. Genome Biol 15:453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobor S, Misale S, Crowley E, Scala E, Zanon C, Di Nicolantonio F, Bardelli A. 2012. 166 acquired resistance to anti EGFR therapy in colorectal cancer and paracrine protection by KRAS mutated cells. Eur J Cancer 48:51. [Google Scholar]
- Inda MDM, Bonavia R, Narita A MY, Sah DWY, Vandenberg S, Brennan C, Johns TG, Bachoo R, Hadwiger P, Tan P, et al. 2011. Tumor heterogeneity is an active process maintained by a mutant EGFR‐induced cytokine circuit in glioblastoma. Genes Dev 24:1731–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MDM, Bonavia R, Sah DWY, Vandenberg S, Brennan C, Johns TG, Hadwiger P, Tan P, Cavenee WK, Furnari FB. 2010. Tumor heterogeneity in glioblastoma is an active process driven by a mutant EGFR‐induced paracrine circuit. Cancer Res 70:Abstract 3126. [Google Scholar]
- Jamal‐Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, Middleton G, de Bruin E, Le Quesne J, Shafi S, et al. 2014. Tracking genomic cancer evolution for precision medicine: The Lung TRACERx Study. PLoS Biol 12:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kensler TW, Spira A, Garber JE, Szabo E, Lee JJ, Dong Z, Dannenberg AJ, Hait WN, Blackburn E, Davidson NE, et al. 2016. Transforming cancer prevention through precision medicine and immune‐oncology. Cancer Prev Res 9:2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YC, Yao X, Crystal JS, Li YF, El‐Gamil M, Gross C, Davis L, Dudley ME, Yang JC, Samuels Y, et al. 2014. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res 20:3401–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresso KC, Tsai KY, Brown PH, Szabo E, Lippman S, Hawk ET. 2015. Molecular cancer prevention: Current status and future directions. CA Cancer J Clin 65:345–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, Ng CKY, Piscuoglio S, Weigelt B, Reis‐Filho JS. 2014. Breast cancer intra‐tumor heterogeneity. Breast Cancer Res 16:R48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Campbell PJ. 2015. Somatic mutation in cancer and normal cells. Science 349:1483–1489. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Almendro V, Polyak K. 2012. Intra‐tumour heterogeneity: A looking glass for cancer? Nat Rev Cancer 12:323–334. [DOI] [PubMed] [Google Scholar]
- Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. 2014. Non‐cell‐autonomous driving of tumour growth supports sub‐clonal heterogeneity. Nature 514:54–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinzie PB, Delongchamp RR, Chen T, Parsons BL. 2006. ACB‐PCR measurement of K‐ras codon 12 mutant fractions in livers of Big Blue(registered trademark) rats treated with N‐hydroxy‐2‐acetylaminofluorene. Mutagenesis 21:391–397. [DOI] [PubMed] [Google Scholar]
- McKinzie PB, Parsons BL. 2011. Accumulation of K‐Ras codon 12 mutations in the F344 rat distal colon following azoxymethane exposure. Environ Mol Mutagen 52:409–418. [DOI] [PubMed] [Google Scholar]
- Meng F, Bermudez E, McKinzie PB, Andersen ME, Clewell HJ, Parsons BL. 2010a. Measurement of tumor‐associated mutations in the nasal mucosa of rats exposed to varying doses of formaldehyde. Regul Toxicol Pharmacol 57:274–283. [DOI] [PubMed] [Google Scholar]
- Meng F, Knapp GW, Green T, Ross JA, Parsons BL. 2010b. K‐Ras mutant fraction in A/J mouse lung increases as a function of benzo[a]pyrene dose. Environ Mol Mutagen 51:146–155. [DOI] [PubMed] [Google Scholar]
- Meng F, Wang Y, Myers MB, Wong BA, Gross EA, Clewell HJ, Dodd DE, Parsons BL. 2011. P53 codon 271 CGT to CAT mutant fraction does not increase in nasal respiratory and olfactory epithelia of rats exposed to inhaled naphthalene. Mutat Res 721:199–205. [DOI] [PubMed] [Google Scholar]
- Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, et al. 2012. Emergence of KRAS mutations and acquired resistance to anti‐EGFR therapy in colorectal cancer. Nature 486:532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MB, Banda M, McKim KL, Wang Y, Powell MB, Parsons BL. 2016. Breast cancer heterogeneity examined by high‐sensitivity quantification of PIK3CA, KRAS, HRAS, and BRAF mutations in normal breast and ductal carcinomas. Neoplasia 18:253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MB, McKim KL, Meng F, Parsons BL. 2015. Low‐frequency KRAS mutations are prevalent in lung adenocarcinomas. Personalized Med 12:83–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MB, McKim KL, Parsons BL. 2014a. A subset of papillary thyroid carcinomas contain KRAS mutant subpopulations at levels above normal thyroid. Mol Carcinogen 53:159–167. [DOI] [PubMed] [Google Scholar]
- Myers MB, McKinzie PB, Wang Y, Meng F, Parsons BL. 2014b. ACB‐PCR quantification of somatic oncomutation. Methods Mol Biol 1105:345–363. [DOI] [PubMed] [Google Scholar]
- Nakamura J, Mutlu E, Sharma V, Collins L, Bodnar W, Yu R, Lai Y, Moeller B, Lu K, Swenberg J. 2014. The endogenous exposome. DNA Repair 19:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons BL. 2008. Many different tumor types have polyclonal tumor origin: Evidence and implications. Mutat Res 659:232–247. [DOI] [PubMed] [Google Scholar]
- Parsons BL, Beland FA, Von Tungeln LS, Delongchamp RR, Fu PP, Heflich RH. 2005. Levels of 4‐aminobiphenyl‐induced somatic H‐ras mutation in mouse liver DNA correlate with potential for liver tumor development. Mol Carcinogen 42:193–201. [DOI] [PubMed] [Google Scholar]
- Parsons BL, Manjanatha MG, Myers MB, McKim KL, Shelton SD, Wang Y, Gollapudi BB, Moore NP, Haber LT, Moore MM. 2013. Temporal changes in K‐ras mutant fraction in lung tissue of Big Blue B6C3F1 mice exposed to ethylene oxide. Toxicol Sci 136:26–38. [DOI] [PubMed] [Google Scholar]
- Parsons BL, Marchant‐Miros KE, Delongchamp RR, Verkler TL, Patterson TA, McKinzie PB, Kim LT. 2010. ACB‐PCR quantification of K‐RAS codon 12 GAT and GTT mutant fraction in colon tumor and non‐tumor tissue. Cancer Invest 28:364–375. [DOI] [PubMed] [Google Scholar]
- Parsons BL, Myers MB. 2013. KRAS mutant tumor subpopulations can subvert durable responses to personalized cancer treatments. Personalized Med 10:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Janes MR, Li L‐S, Hansen R, Peters U, Kessler LV, Chen Y, Kucharski JM, Feng J, Ely T, et al. 2016. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discov 6:316–329. [DOI] [PubMed] [Google Scholar]
- Renovanz M, Kim EL. 2014. Intratumoral heterogeneity, its contribution to therapy resistance and methodological caveats to assessment. Front Oncol 4:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts SA, Gordenin DA. 2014. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat Rev Cancer 14:786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronai Z, Luo FC, Gradia S, Hart WJ, Butler R. 1994. Detection of K‐ras mutation in normal and malignant colonic tissues by an enriched PCR method. Int J Oncol 4:391–396. [DOI] [PubMed] [Google Scholar]
- Rozhok AI, DeGregori J. 2015. Toward an evolutionary model of cancer: Considering the mechanisms that govern the fate of somatic mutations. Proc Natl Acad Sci USA 112:8914–8921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MB, Der CJ, Wang‐Gillam A, Cox AD. 2015. Targeting RAS‐mutant Cancers: Is ERK the key? Trends Cancer 1:183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover DG, Wagle N. 2015. Precision medicine in breast cancer: Genes, genomes, and the future of genomically driven treatments. Curr Oncol Rep 17:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudo H, Li‐Sucholeiki XC, Marcelino LA, Gruhl AN, Zarbl H, Willey JC, Thilly WG. 2006. Distributions of five common point mutants in the human tracheal‐bronchial epithelium. Mutat Res 596:113–127. [DOI] [PubMed] [Google Scholar]
- Tomasetti C, Li L, Vogelstein B. 2017. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 355:1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B. 2015. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 347:78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasetti C, Vogelstein B, Parmigiani G. 2013. Half or more of the somatic mutations in cancers of self‐renewing tissues originate prior to tumor initiation. Proc Natl Acad Sci USA 110:1999–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson IR, Takahashi K, Futreal PA, Chin L. 2013. Emerging patterns of somatic mutations in cancer. Nat Rev Genet 14:703–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S, Powers S, Zhu W, Hannun YA. 2016. Substantial contribution of extrinsic risk factors to cancer development. Nature 529:43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakubovskaya MS, Spiegelman V, Luo FC, Malaev S, Salnev A, Zborovskaya I, Gasparyan A, Polotsky B, Machaladze Z, Trachtenberg AC. 1995. High frequency of K‐ras mutations in normal appearing lung tissues and sputum of patients with lung cancer. Int J Cancer 63:810–814. [DOI] [PubMed] [Google Scholar]
- Zahm CD, Szulczewski JM, Leystra AA, Olson TJP, Clipson L, Albrecht DM, Middlebrooks M, Thliveris AT, Matkowskyj KA, Washington MK, et al. 2016. Advanced intestinal cancers often maintain a multi‐ancestral architecture. PLoS One 11:e0150170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information