

SUMMARY
The Arp2/3 complex nucleates branched actin, forming networks involved in lamellipodial protrusion, phagocytosis and cell adhesion. We derived primary bone marrow macrophages lacking Arp2/3 complex (Arpc2−/−) and directly tested its role in macrophage functions. Despite protrusion and actin assembly defects, Arpc2−/− macrophages competently phagocytose via FcR and chemotax towards CSF and CX3CL1. However, CR3 phagocytosis and fibronectin haptotaxis, both integrin-dependent processes, are disrupted. Integrin-responsive actin assembly and αM/β2 integrin localization are compromised in Arpc2−/− cells. Using an in vivo system to observe endogenous monocytes migrating toward full-thickness ear wounds we found that Arpc2−/− monocytes maintain cell speeds and directionality similar to control. Our work reveals that the Arp2/3 complex is not a general requirement for phagocytosis or chemotaxis, but is a critical driver of integrin-dependent processes. We demonstrate further that cells lacking Arp2/3 complex function in vivo remain capable of executing important physiological responses that require rapid directional motility.
eTOC/In-Brief blurb
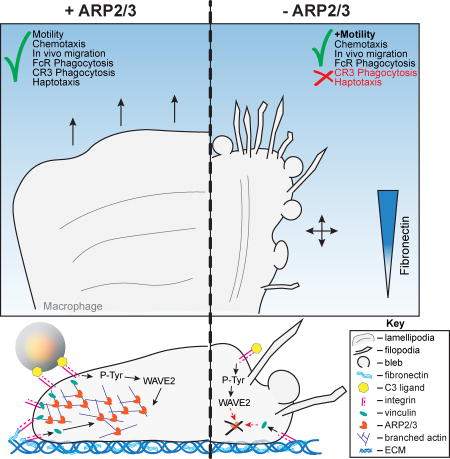
Using a combination of cell culture-based and in vivo mouse experiments, Rotty et al. demonstrate that the actin-nucleating Arp2/3 complex not absolutely required for macrophage FcR phagocytosis, chemotaxis, or in vivo monocyte directional motility. Rather, the complex has a critical role in regulating integrin-dependent macrophage processes.
INTRODUCTION
One fundamental function of the actin cytoskeleton is to exert force against lipid membranes through polymerization (Lemière et al., 2016). The force generated by growing actin filaments (F-actin) helps maintain cell shape, establishes and maintains membrane protrusions (i.e. lamellipodia, filopodia) associated with cell motility, and facilitates vesicular trafficking (Svitkina, 2013). The critical nature of actin’s involvement in these pathways is reflected by its conserved function from yeast to humans. Thus, many dynamic cellular functions require tight spatial and temporal regulation of actin filament production, stabilization and turnover.
The seven subunit Arp2/3 complex is unique in its ability to nucleate actin filament branches from the sides of pre-existing filaments, leading to dense dendritic networks evident in lamellipodia (Svitkina and Borisy, 1999) and phagocytic cups (Machesky et al., 2000). In addition to motility and phagocytosis, the Arp2/3 complex has been implicated in numerous cellular processes from endocytic trafficking to cell-cell and cell-extracellular matrix (ECM) adhesion. Nucleation Promoting Factors (NPFs) (Machesky et al., 1999) bind directly to the Arp2 and Arp3 subunits to induce the conformational change that activates the Arp2/3 complex (Goley et al., 2004), and supply the initial actin monomers that are used by the Arp2/3 complex to nucleate a new actin filament (Boczkowska et al., 2014; Pollard et al., 2001; Ti et al., 2011). Specific NPFs are thought to differentially localize the Arp2/3 complex to the leading edge, podosomes, endocytic vesicles, or phagocytic cups, and to then stimulate its activity in a spatially-defined way.
Macrophages play major roles in the innate immune system: sensing and phagocytosing invading microbes, presenting antigen for T cells, and releasing pro-inflammatory factors that can recruit neutrophils, natural killer, B and T cells to sites of infection or damage (Price and Vance, 2014). Dysregulation of actin assembly is a key aspect of the X-linked human disorder Wiskott-Aldrich syndrome (WAS), where a mutation in the Wiskott-Aldrich Syndrome Protein (WASP) (Derry et al., 1994) compromises the function of numerous immune cells including macrophages. WASP, an NPF expressed in cells of the hematological lineage (Machesky and Insall, 1998) localizes to macrophage podosomes and phagocytic cups and has been implicated in chemotaxis, phagocytosis, integrin clustering and immune synapse formation (Thrasher and Burns, 2010). These studies, along with many others (Rougerie et al., 2013), underscore the importance of actin regulation to macrophage function. Current understanding of Arp2/3 complex function in macrophages has often been inferred from its localization pattern and by indirect perturbations focused on NPFs, like those mentioned above. We recently established a conditional mouse model of the Arp2/3 complex where the gene encoding the critical Arpc2 (p34) subunit of the complex can be deleted in a Cre-dependent manner (denoted as Arpc2−/−) (Rotty et al., 2015).
Here, we have used this approach to directly interrogate Arp2/3 complex function in primary bone marrow-derived macrophages. Our findings underscore the importance of Arp2/3 complex function in maintaining cellular structure, but challenge the notion that it is absolutely required for phagocytosis and chemotaxis. We also demonstrate that Arpc2−/− cells are capable of rapid directional motility in vivo. In fact, the major defects in Arpc2−/− macrophages are related to disrupted integrin function. These results refine our understanding of Arp2/3 complex function in macrophages and reveal that the Arp2/3 complex is fundamentally required for integrin-dependent processes.
RESULTS
Arpc2−/− macrophages have reduced F-actin levels, altered cell morphology and protrusion character
To investigate the contribution of the Arp2/3 complex to macrophage biology, we used a mouse containing the recently published Arpc2 conditional allele (Rotty et al., 2015) and CreERT2 driven by the endogenous Rosa26 promoter (Figure S1A). Primary bone marrow-derived macrophages from these mice were treated with 4-OHT to activate CreER. The resulting cells completely lacked Arpc2/p34, as well as multiple other subunits of the Arp2/3 complex (Figure 1A–B). Arpc2−/− macrophages lack the characteristic punctate actin filament staining found in WT cells, instead assembling F-actin in bundled parallel arrays reminiscent of stress fibers (Figure 1B, see inset). Loss of Arp2/3 complex also induced loss of lamellipodia, in favor of filopodial protrusion (Figure 1B). WT macrophages generated small, punctate vinculin structures, as well as large actin-vinculin rosettes (Figure 1C). Arpc2−/− macrophages lacked these large rosettes (Figure S1B) but retained the ability to form vinculin-containing adhesive structures at their periphery (Figure 1C), suggesting that cell adhesion is not totally compromised.
Figure 1. Arpc2−/− macrophage characterization and phenotypes.

A) Expression of Arp2/3 complex subunits in WT and Arpc2−/− macrophages. GAPDH is presented for loading comparison. B) Phalloidin (F-actin) and Arpc2 staining in WT and Arpc2−/− macrophages. Scale (image and inset) = 10 µm. C) Vinculin staining (grey) and merged staining of vinculin (red) and actin (cyan) in WT and Arpc2−/− macrophages. Scale = 3 µm. D) F-actin staining intensity of WT (black bars) and Arpc2−/− (KO, grey bars) macrophages on a range of fibronectin concentrations. Integrated pixel density plotted as mean ± SEM. ***p < 0.0001. A.U., Arbitrary Units. Blot panels display total actin level. N = at least 113 cells/condition, pooled from three separate experiments. E) Spread cell area in µm2, WT (black bars) or Arpc2−/− (KO, grey bars) macrophages on a range of fibronectin concentrations. Plotted as mean ± SEM. ***p < 0.0001. N = at least 113 cells/condition, pooled from three separate experiments. F) WT (black bars) and Arpc2−/− (KO, grey bars) macrophage cell shape on a range of fibronectin concentrations, plotted as mean ± SEM. A.U., Arbitrary Units. N = at least 77 cells/condition, pooled from three separate experiments. G) Scanning electron micrographs of WT and Arpc2−/− macrophages. Scale = 2 µm. See also Figure S1, Movie S1.
Probing the actin cytoskeleton further, we found that although total actin levels did not differ significantly in whole cell lysates, Arpc2−/− macrophages generated significantly lower levels of F-actin compared to WT cells (Figure 1D). These differences in actin filament organization and level led to smaller spread cell area (Figure 1E) and altered cell shape (Figure 1F), across a range of substrate fibronectin concentrations. We used scanning electron microscopy (SEM) to gain further insight into the nature of Arpc2−/− cell protrusion. SEM images revealed that Arpc2−/− cells generate large surface blebs that were not obvious from confocal imaging of fixed cells, as well as filopodia that appear capable of adhering to and spreading over ECM-coated surfaces (Figure 1G, Movie S1). WT macrophages show no evidence of either blebs or filopodia and instead form large lamellipodia and dorsal ruffles (Figure 1G). Since the actin cytoskeleton and protrusive structures of Arpc2−/− cells were profoundly altered, we wondered whether processes dependent upon actin-based protrusion would be similarly affected.
The Arp2/3 complex is not required for FcR-mediated phagocytosis
Macrophage phagocytosis is initiated by a variety of surface receptors that detect an array of distinct engulfment cues deposited on targets ranging from the nanoscale (viruses), the microscale (bacteria, yeast) and even tens of microns (apoptotic cellular fragments) (Flannagan et al., 2012). In order to probe some of this broad phagocytic capacity in a systematic way, we employed several sizes of latex beads (2, 6, or 10 microns in diameter) opsonized with IgG to assess the contribution of the Arp2/3 complex to FcR-mediated phagocytosis. We assessed phagocytic index using differential antibody labeling of opsonized beads (Figure S1C). To our surprise, initial SEM experiments revealed that Arpc2−/− macrophages readily form phagocytic structures around IgG-opsonized beads (Figure 2A). In addition, F-actin staining and fully internalized IgG beads of all sizes were detected in Arpc2−/− cells even at early time points (Figure 2B, arrows; Figure S1D). Measurements taken at early time points (20 min.) indicate reduced FcR phagocytosis in Arpc2−/− cells, especially when challenged with large beads (Figure 2C). Surprisingly, Arpc2−/− phagocytic index increased at later time points with internalization being partially (6 micron, 10 micron beads) or completely rescued (2 micron beads; Figure 2C, Figure S1D, Figure 2B, arrows). To exclude the possibility that this result is an aberrant response arising only in Arpc2−/− cells maintained in culture, we also acutely treated WT cells with the small molecule Arp2/3 complex inhibitor CK-666 (Nolen et al., 2009). As with the genetically null cells, CK666-treated macrophages demonstrated FcR phagocytic capability similar to DMSO-treated controls (Figure 2D). Though the overall trend is similar to the Arpc2−/− cells, CK-666 treatment yielded a milder phenotype. We noted that like their Arpc2−/− counterparts, CK-666 treated cells elongate and lack lamellipodia (Figure S1E, ventral), indicating that the drug is affecting Arp2/3 complex function. However, we were also able to detect p34 staining at FcR phagocytic cups (Figure S1E), suggesting a residual pool of active Arp2/3 complex. Incomplete inhibition of Arp2/3 complex activity by CK-666 likely explains the contrast between results generated with the drug and our genetic null system. Next, we treated WT and Arpc2−/− macrophages with the actin-disrupting drug Cytochalasin D (CytoD) and observed significant and dose-dependent decreases in their ability to internalize IgG-opsonized beads (Figure 2E, Figure S1F). WT macrophage internalization of 2 micron beads was surprisingly resilient and required significantly higher doses of CytoD than Arpc2−/− (4 µM, Movie S2). These data demonstrate that the Arp2/3 complex is not strictly required for FcR-mediated phagocytosis, and that a compensatory, actin-dependent pathway operates in Arp2/3 complex-deficient cells.
Figure 2. The Arp2/3 Complex is dispensable for FcR phagocytosis.

A) Scanning electron micrograph of WT and Arpc2−/− macrophages with 10 micron IgG-coated beads. Scale = 1 µm. B) WT and Arpc2−/− macrophages at early (20 min.) and later (60 min.) time points bound to 6 micron beads. Staining (L to R): F-actin, external beads, all beads. Arrows denote internalized beads. Scale = 5 µm. C) Phagocytic index (internalized beads/total beads) of WT (black bars) or Arpc2−/− (KO, grey bars) macrophages at early (20 min.) or later (60 min.) time points with 2, 6, or 10 micron IgG-opsonized beads. Data is plotted as mean ± SEM. **p ≤ 0.0014, *p ≤ 0.03. Means represent compiled data from three separate experiments. D) Phagocytic index (internalized beads/total beads) of WT macrophages acutely treated with DMSO (−, black bars) or 150 µM CK-666 (+, grey bars) at early (20 min.) or later (60 min.) time points with 2, 6, or 10 micron IgG-opsonized beads. Data is plotted as mean ± SEM. None of the p-values under these conditions were statistically significant. Means represent compiled data from three separate experiments. E) Phagocytic index (internalized beads/total beads) of WT (black bars) or Arpc2−/− (KO, grey bars) macrophages in the absence (−) or presence (0.5, 1) of 0.5 or 1 µM Cytochalasin D (Cyto.D) after 60 minutes with 2, 6, or 10 micron IgG-opsonized beads. Data is plotted as mean ± SEM. ***p ≤ 0.001, **p = 0.0019, *p ≤ 0.0282. Means represent compiled data from three separate experiments. See also Figure S1, Movie S2.
CR3-mediated phagocytosis is more severely affected by loss of Arp2/3 complex function
We next turned to CR3 phagocytosis to test whether phagocytic ability in Arpc2−/− cells is maintained with different opsonization schemes. Unlike FcR, CR3 is an integrin pair (αMβ2) with an entirely different set of intracellular effectors thought to generate a phenotypically different phagocytic structure than the type generated by FcR (Freeman and Grinstein, 2014). WT cells generate classical phagocytic cups in response to C3bi-opsonized beads, while Arpc2−/− macrophages showed a marked deficiency in both cup protrusion (Figure 3A) and bead internalization (Figure 3B, Figure S2A). In fact, Arpc2−/− macrophages displayed significant, general and long-lasting defects in CR3 phagocytosis (Figure 3C) that were almost perfectly recapitulated by acute treatment of WT cells with CK-666 (Figure 3D). Indeed, we found that CR3 phagocytosis is sensitive to much lower doses of CK-666 (Figure S2B), indicating that even partial Arp2/3 complex inhibition may compromise this process.
Figure 3. The Arp2/3 complex is critical for CR3 phagocytosis.

A) Scanning electron micrograph of WT and Arpc2−/− macrophages with 10 micron C3bi-coated beads. Scale = 1 µm. B) WT and Arpc2−/− macrophages at early (20 min.) and later (60 min.) time points bound to 6 micron beads. Staining (L to R): F-actin, external beads, all beads. Arrows denote internalized beads. Scale = 5 µm. C) Phagocytic index (internalized beads/total beads) of WT (black bars) or Arpc2−/− (KO, grey bars) macrophages at early (20 min.) or later (60 min.) time points with 2, 6, or 10 micron C3bi-opsonized beads. Data is plotted as mean ± SEM. **p = 0.0063, *p ≤ 0.0258. Means represent compiled data from three separate experiments. D) Phagocytic index (internalized beads/total beads) of WT macrophages acutely treated with DMSO (−, black bars) or 150 µM CK-666 (+, grey bars) at early (20 min.) or later (60 min.) time points with 2, 6, or 10 micron C3bi-opsonized beads. Data is plotted as mean ± SEM. ***p = 0.0005, **p = 0.0028, *p < 0.05. Means represent compiled data from three separate experiments. E) Flow cytometry analysis of αM and β2 integrin surface levels in WT (black bars) and Arpc2−/− (KO, grey bars) macrophages. Average Mean Fluorescence Intensity (MFI) of αM and β2 integrin on cultured macrophages were plotted ± SEM. Data were combined from three independent datasets. Negative control (−) cells were incubated with secondary antibody only. No statistically significant p-values were detected. A.U., Arbitrary Units. F) αM and β2 integrin surface levels on spread WT (black bars) and Arpc2−/− (KO, grey bars) macrophages in culture plated on 1 µg/mL collagen. Mean integrated pixel densities are plotted ± SEM. No statistically significant p-values were detected. A.U., Arbitrary Units. N = at least 132 cells. G) Confocal images (L to R, with insets) of αM integrin, F-actin and p34 in WT (top row) or Arpc2−/− macrophages (bottom row) plated on 1 µg/mL collagen. Scale = 20 µm. H) Scanning electron micrograph of WT or Arpc2−/− macrophages with bound C3bi-opsonized 2 micron beads. Scale = 500 nm. See also Figure S2.
One potential explanation for these data could be that Arpc2−/− macrophages have fewer surface-exposed integrins. However, both αM and β2 integrin surface levels are similar between WT and Arpc2−/− in suspension and when cells are grown on collagen-coated coverslips (Figure 3E, 3F; for flow cytometry gating strategy see Figure S2C). Confocal microscopy revealed that αM and β2 integrins localize to the substrate-bound ventral surface of macrophages (where they co-localize with the Arp2/3 complex, Figure S2D), as well as to F-actin-Arp2/3 complex-containing dorsal ruffles present on the upper surface of WT cells (Figure 3G). CR3 integrins are much more dispersed in Arpc2−/− macrophages and do not co-localize with actin filaments (Figure 3G). Based on SEM images, dorsal ruffles appear to play an active role in phagocytosis and are absent in Arpc2−/− cells (Figure 3H). These data reveal the Arp2/3 complex as a critical effector of CR3 phagocytosis and suggest that Arp2/3-generated actin structures organize the CR3 receptor for optimal phagocytosis.
Arp2/3 complex-generated branched actin drives integrin-directed protrusion at CR3 phagocytic cups
Actin is a key effector of the membrane protrusion critical for phagocytic cup formation, as well as forming a physical barrier that organizes the signaling induced by target binding (Freeman et al., 2016). Our bead uptake and SEM data led us to hypothesize that the Arpc2−/− CR3 phagocytic defect is rooted in an inability to efficiently generate integrin-directed actin structures. Superresolution imaging revealed that WT cells generate actin-rich structures around both IgG and complement-opsonized beads (Figure 4A) reminiscent of the membrane protrusions seen in the SEM images (Figure 2A, 3A). In line with their differential phagocytic ability, Arpc2−/− macrophages generate protrusive F-actin containing phagocytic cups only via the FcR pathway, while C3bi-opsonized beads elicit little response (Figure 4A). Quantification of F-actin levels at phagocytic cups demonstrates that Arpc2−/− cells produce F-actin at IgG opsonized beads comparable to WT levels (Figure 4B) in spite of the reduced whole cell F-actin levels observed in Arpc2−/− macrophages (Figure 1D). In direct contrast to FcR phagocytosis, F-actin levels at Arpc2−/− CR3 phagocytic cups were significantly lower than WT and remained low even at later time points and with all bead sizes (Figure 4B).
Figure 4. The Arp2/3 complex promotes actin assembly and coordinates cellular signaling at CR3 phagocytic cups.

A) Airyscan images of F-actin stains during FcR (left column, 20 min. time point) or CR3 phagocytosis (right column, 60 min. time point) of 10 micron beads. WT cells are in top row, Arpc2−/− in bottom row. Circle drawn on Arpc2−/− CR3 image depicts the diameter of the bead bound to this cell. Scale = 3 µm. B) F-actin staining intensity at WT (black bars) and Arpc2−/− (KO, grey bars) FcR phagocytic cups (left column) or CR3 phagocytic cups (right column). Data with 2 micron (top row), 6 micron (middle row) or 10 micron (bottom row) beads were gathered at 20 or 60 minute time points. Data is plotted as mean integrated pixel density ± SEM. ***p ≤ 0.0004, **p ≤ 0.0068, *p ≤ 0.024. A.U., Arbitrary Units. N = at least 277 (2 micron), 107 (6 micron) or 52 (10 micron) beads analyzed for F-actin, pooled from at least 4 unique experiments. C) WT (top) or Arpc2−/− (bottom) macrophages stably expressing Lifeact-GFP bound to 2 micron pHrodo red-IgG conjugated latex beads. First time point (t = 0) corresponds to initial binding of first bead in panel to the cell surface. Color-coded arrowheads indicate the same bead from frame to frame. Scale = 5 µm. D) Summary table of pHrodo-IgG and pHrodo-C3 experiments. pHrodo-IgG experiments encompassed seven (WT) or eight (Arpc2−/−) experimental repetitions. pHrodo-C3 experiments encompassed four (WT) or three (Arpc2−/−) experimental repetitions. Data are presented as percent of total analyzed beads. E) WT (top) or Arpc2−/− (bottom) macrophages stably expressing Lifeact-GFP were bound to 2 micron pHrodo red-C3 conjugated latex beads. First time point (t = 0) corresponds to initial binding of first bead in panel to the cell surface. Scale = 5 µm. F) Length of time after pHrodo bead binding required for WT or Arpc2−/− (KO) macrophages to produce a shell of F-actin in FcR or CR3 phagocytosis assays. Data is plotted as mean (in minutes)± SEM. **p = 0.0033, ***p < 0.0001. G) Length of time between F-actin peak (tactin) and bead acidification (i.e. phagosome maturation) in WT or Arpc2−/− (KO) macrophages in FcR or CR3 phagocytosis assays. See also Figure S3, Figure S4, Movie S2.
These data prompted us to directly observe both F-actin and bead internalization using live-cell imaging, which allowed us to precisely determine the severity of the Arpc2−/− uptake lag suggested by our bulk internalization data. WT or Arpc2−/− cells stably expressing Lifeact-GFP (LA-GFP) were challenged with 2 micron beads opsonized with IgG or C3bi conjugated to the pH-sensitive dye pHrodo red. WT cells rapidly generated F-actin at bound IgG beads and completed internalization of most beads within five minutes (as judged by increased pHrodo fluorescence indicative of entry into the acidifying phago-lysosome, Figure 4C, 4D, Movie S2). Arpc2−/− macrophages captured IgG beads via filopodial-like protrusions, progressively generated F-actin structures around them, and completed internalization into the phagolysosome (Figure 4C, 4D, Movie S2). Quantification of LA-GFP and pHrodo signal at individual IgG beads revealed a sharp F-actin peak (time to peak actin accumulation = tactin) in WT cells that declined rapidly as acidification occurred (time from peak actin to peak acidification = tacid; Figure S3A). F-actin accumulation at IgG beads in Arpc2−/− macrophages occurred more gradually, taking about 5.6-fold longer to peak compared to WT (11.77 minutes for Arpc2−/− v. 2.084 minutes for WT; Figure 4F, Figure S3A). Arpc2−/− macrophage phagolysosome maturation also took longer (8.59 minutes for Arp2−/− v. 2.69 minutes for WT; tacid, Figure 4G). Overall, Arpc2−/− macrophages complete phagocytosis 4.26 times slower than their WT counterparts (20.35 minutes v. 4.776 minutes), in line with our bulk data showing an uptake lag.
We also used the LA-GFP/pHrodo system to assess CR3 phagocytosis and observed rapid F-actin production (tactin: 7.14 minutes) and bead acidification (tacid: 2.20 minutes) in WT cells (Figures 4D–G, Figure S3B, Movie S2). Correlative immunostaining confirmed that C3 was deposited on pHrodo beads and that ~80% of beads bound by Arpc2−/− macrophages remained external after 40 minutes (Figure 4D, Figures S3C, S3D). Arpc2−/− macrophages required 25.9 minutes to internalize the small fraction of beads that eventually acidified, corresponding to tactin = 16.8 minutes and tacid = 9.1 minutes (Figures 4E, 4F, Figure S3B). However, it was much more common to see C3 beads stalled on the surface of Arpc2−/− cells (Figure 4E, white arrowheads in Arpc2−/− panel; Movie S2). Stalled beads did not generate an F-actin peak (representative data plotted as LA-GFP (no peak) in Figure S3B). These data are also in line with bulk phagocytosis data indicating that CR3 phagocytosis is more dramatically impacted by Arp2/3 complex loss than FcR phagocytosis.
Finally, we sought to understand how the pathway upstream of F-actin assembly was affected by loss of Arp2/3 complex. FcR and SFK levels remained relatively steady at IgG beads throughout the time course in WT cells, while Arpc2−/− cells steadily increased FcR and SFKs at beads over the course of the experiment (Figures S3E, S4). Arpc2−/− F-actin levels again showed a characteristic lag, while WT F-actin levels spiked early and tapered off (Figures S3E, S4). Conversely, CR3 signaling was depressed (although not entirely absent) in Arpc2−/− macrophages, as evidenced by reduced αM, vinculin and F-actin accumulation at C3 beads that did not change over time (Figures S3E, S4). In addition to driving protrusion, Arp2/3-branced actin generation at phagocytic targets could reinforce or amplify the initial outside-in signalling and facilitate the processive advancement of a phagocytic cup.
Arpc2−/− macrophage motility is enhanced by and is reliant on NMIIA
We next turned our attention to cell motility, another important physiological process that requires actin-containing cellular protrusions. Previous studies showed that Arp2/3-deficient fibroblast migration is significantly slower than control cells in a substrate-dependent manner (Rotty et al., 2015; Wu et al., 2012). Interestingly, Arpc2−/− macrophage motility is enhanced compared to WT cells plated on a variety of uniform ECM conditions (Figure 5A). This surprising finding prompted us to understand how Arpc2−/− cells could still polarize, protrude and migrate. Closer examination revealed that WT macrophages moved forward through coordinated lamellipodial protrusion at the front and retraction at the rear (Figure 5B, Movie S3). Arpc2−/− macrophages also maintain the capacity to polarize into protruding and retracting halves as large blebs form at the leading edge and fill in the gaps between adherent filopodia (Figure 5B, Movie S3). At the same time, the rear of the cell is retracted as the cell front protrudes forward (Figure 5B, Movie S3). Since Nonmuscle Myosin II (NMII) is a critical effector of both blebs (Coleman et al., 2001) and tail retraction (Jay et al., 1995), we sought to test the role of NMII in Arpc2−/− macrophage motility.
Figure 5. Enhanced Arpc2−/− macrophage motility requires myosin II.

A) Random migration speed of WT (black bars) or Arpc2−/− macrophages on a range of fibronectin concentrations (KO, grey bars) plotted as mean ± SEM. ****p < 0.0001, ***p = 0.0007. N = at least 89 cells, from 3 separate experiments. B) WT (top) and Arpc2−/− macrophages (bottom) migrating randomly on 1 µg/mL fibronectin. Scale = 10 µm. C) Random migration speed of macrophages (WT and KO, black bars) and fibroblasts (WT and KO, grey bars) in the presence or absence of myosin II-disrupting drugs (15 µM Blebbistatin and 10 µM Y-27632) on 1 µg/mL fibronectin. DMSO = negative control. ***p < 0.0001, **P = 0.0075. N = at least 114 cells, pooled from three separate experiments. D) WT (top) and Arpc2−/− macrophages (bottom) migrating randomly on 1 µg/mL fibronectin in the presence of 15 µM Blebbistatin. Scale = 10 µm. E) Scanning electron micrographs of WT and Arpc2−/− macrophages treated with 15 µM Blebbistatin. Scale = 2 µm. F) Phagocytic index (internalized beads/total beads) of WT (black bars) or Arpc2−/− macrophages (KO, grey bars) in the absence (−) or presence (+) of 15 µM Blebbistatin, after 60 minutes with 2, 6, or 10 micron IgG-opsonized beads. Data is plotted as mean ± SEM. *p = 0.0494. Means represent compiled data from three separate experiments. G) Confocal images, with insets, of F-actin (left) or myosin IIA (right) in WT (top) or Arpc2−/− macrophages (bottom) plated on 1 µg/mL fibronectin. Scale = 10 µm. See also Figure S5, Movie S3.
We targeted NMII function in WT and Arpc2−/− cells with the myosin II inhibitor blebbistatin (Kovács et al., 2004) (Bleb.) or the ROCK inhibitor Y-27632, (Ishizaki et al., 2000) which inhibits one of the major activating kinases upstream of myosin II. Both drugs significantly decrease Arpc2−/− macrophage cell speed (Figure 5C). We compared these results to the effect of inhibiting NMII function in fibroblasts. Arp2/3 complex disruption in fibroblasts strongly reduced migration speed (Figure 5C). Unlike in macrophages, both bleb and Y compound increased WT and Arpc2−/− fibroblast cell speed compared to their DMSO-treated controls (Figure 5C). Blebbistatin-treated WT macrophages show a mild tail retraction defect but can still achieve front/rear polarity, as suggested by our motility data (Figure 5D, Movie S3). Consistent with their reduced cell speed, blebbistatin-treated Arpc2−/− macrophages are unable to define a front and rear edge (Figure 5D, Movie S3). Scanning EM confirmed that blebbistatin treatment blocked bleb formation in Arpc2−/− macrophages, while WT cells showed increased dorsal ruffle formation (Figure 5E). To our surprise, blebbistatin treatment had no significant effect on FcR-mediated phagocytosis in WT or Arpc2−/− macrophages (Figure 5F), suggesting a specific role for NMII in Arpc2−/− macrophage motility rather than a general cellular defect. While fibroblasts express both myosin IIA and IIB, macrophages only express myosin IIA (Figure 5G, Figures S5A, S5B). WT macrophages show exclusion of myosin IIA from lamellipodial structures (Figure 5G, inset). Arpc2−/− macrophages also generate large, F-actin containing protrusions at the leading edge (Figure 5G, inset). The majority of myosin IIA is concentrated in the cell body behind the leading edge, though some puncta in the front do colocalize with actin filaments (Figure 5G, inset). These observations suggest that NMIIA can be similarly distributed with or without the Arp2/3 complex in macrophages.
The Arp2/3 complex is dispensable for chemotaxis to both RTK and GPCR ligands
In vivo, macrophages must migrate toward directional cues in their environment to execute physiological functions (Shi and Pamer, 2011). To address whether the Arp2/3 complex is required in macrophages to migrate directionally to specific extracellular stimuli, we used a microfluidic system (Wu et al., 2012) that can generate stable, long-lasting gradients of both soluble chemotactic cues (i.e. growth factors, cytokines) and substrate-bound haptotactic cues (i.e. ECM). Directionality measurements are calculated by directly observing and tracking cell migration in these gradients (Figure S6A). Forward Migration Index (FMI) is used to quantify directional migration and takes into account how well a cell migrates relative to the external gradient (COSθ) and how well a cell moves persistently in any given direction (d/T). To determine whether the Arp2/3 complex is required for macrophage chemotaxis we used two physiologically relevant chemotactic cues: M-CSF (a ligand of CSFR, a receptor tyrosine kinase (RTK)) or CX3CL1 (a ligand of CX3CR1, a G-protein coupled receptor (GPCR)). Both WT and Arpc2−/− macrophages chemotax with a similar FMI to M-CSF (Figures 6A, 6B, Movie S4) and CX3CL1 (Figures 6C, 6D, Movie S4, Figure S6B), with a positive result defined as a positive average FMI value where 95% confidence intervals do not contain zero (the X-axis). Consistent with this positive functional result, WT and Arpc2−/− macrophages responded similarly to M-CSF and CX3CL1 by phosphorylating Erk1/2, which is a downstream effector of both signaling pathways (Cambien, 2001), (Büscher et al., 1995) (Figure S6C). As with random motility assays, Arpc2−/− macrophages were significantly faster than their WT counterparts in both M-CSF and CX3CL1 chemotaxis experiments (Figures 6B, 6D), underscoring the ability of Arpc2−/− macrophages to respond rapidly and directionally to physiologically relevant soluble migratory cues.
Figure 6. The Arp2/3 complex is dispensable for chemotaxis to CSF and CX3CL1, but is required for fibronectin haptotaxis.

A, C, E) Wind rose plots generated from WT or Arpc2−/− macrophages migrating in a CSF gradient (A), CX3CL1 gradient (C) or FN gradient (E). Each plot is from one representative experiment of mixed WT and Arpc2−/− cells. 0°on the rose plot is the direction of the gradient. B, D, F) Forward Migration Index (FMI) of WT and Arpc2−/− macrophages plotted as mean ± 95% CI in CSF (B), CX3CL1 (D), or FN gradients (F). Data tables relate mean ± SEM of cell speed (V), FMI, d/T and total cells tracked (N). Quantitative measurements were pooled from multiple experiments using identical chamber conditions. G) WT and Arpc2−/− macrophages plated on 1 µg/mL collagen stained for αM integrin, vinculin and p-Tyr (4G10), β1 integrin, WAVE2 and F-actin at bound 6 micron Cy5-fibronectin (Cy5-FN) beads after 15 minute incubation. Arrowheads denote bound beads on Arpc2−/− macrophages. Scale = 10 µm. H) Localization of β1 integrin, p-Tyr and F-actin to Cy5-FN beads 5, 15 or 30 minutes after bead binding by WT or Arpc2−/− macrophages plotted as mean integrated pixel density (A.U.) ± SEM. **p = 0.0011, ***p < 0.0001. I) Schematic depiction of integrin-Arp2/3 complex coordination. Upon ligation integrin initiates an outside-in signal (black arrows) that induces the Arp2/3 complex to nucleate a branched actin network (an inside-out response). The branched actin network then reinforces the ‘upstream’ pathway (red arrows), perhaps by coordinating or concentrating these factors at new sites of integrin engagement. Without the Arp2/3 complex, macrophages still produce an outside-in signal but lack the ability to reinforce the initial signal, leading to loss of ECM sensing and inhibition of CR3 phagocytosis. See also Figure S6, Movies S4 and S5.
The Arp2/3 complex is required for macrophage haptotaxis on fibronectin
Fibronectin is upregulated in vivo at wounds and tumors (Barkan et al., 2010; Grinnell et al., 1981) and is known to stimulate adhesion and activate intracellular signaling in macrophages (Proctor, 1987). Furthermore, it has recently been suggested that a cell’s ability to sense and respond appropriately to ECM proteins may be a critical component of physiological processes that may be compromised or activated in pathological settings (Chan et al., 2014; Oudin et al., 2016). ‘Haptotaxis’ refers to a cell’s ability to differentially sense ECM across the length of the cell and to respond by moving directionally toward higher levels of the surface-bound cue. Though conceptually similar to chemotaxis, Arpc2−/− macrophages (which do chemotax) are unable to haptotax on fibronectin gradients like WT controls can (Figures 6E, 6F, Movie S5). Once again, Arpc2−/− macrophages demonstrate increased cell speed compared to WT, despite their inability to migrate directionally (measured by FMI) and their reduced persistence (measured by d/T) in this setting (Figure 6F). Arpc2−/− macrophages adhere to fibronectin-coated substrates and generate vinculin-containing structures (Figure 1C). Serum starved Arpc2−/− macrophages are also able to initiate adhesion-associated signaling, as evidenced by their ability to activate both Src-family kinase signaling and p-Tyr when plated on fibronectin (Figure S6D).
We used Cy5-fibronectin coated beads to assess whether Arpc2−/− cells were able to respond in a spatially defined manner to integrin engagement. We detected integrin enrichment, robust p-Tyr staining and vinculin localization at these beads in WT cells, faithfully recapitulating key events involved in integrin-based adhesion during motility (Figure 6G). WAVE2, Arp2/3 complex and F-actin were also readily apparent at Cy5-FN beads, suggesting a coupling between integrin-based adhesion and Arp2/3-based protrusion in WT cells (Figure 6G, Figure S6E). Indeed, Arp2/3 complex levels rise during the time course and peak 15 minutes after bead binding, coinciding with increasing integrin, p-Tyr and F-actin levels in WT cells during the same timeframe (Figure 6H, Figures S6F, S6G). Arpc2−/− macrophages bind FN-coated beads and localize integrin, signaling proteins, adhesion-associated proteins and F-actin (Figure 6G), though the level of these proteins at beads is low and remains flat throughout the time course (Figure 6H, Figure S6G) similar to our finding with CR3 phagocytosis. These results indicate that while Arpc2−/− macrophages establish efficient cellular protrusions and can bind ECM, the Arp2/3 complex is required to a) generate ECM-responsive protrusions and b) positively reinforce the ‘upstream’ signaling pathway, perhaps by utilizing branched actin networks to concentrate key signaling molecules (Figure 6I).
The Arp2/3 complex is not required for directional motility in vivo
Our in vitro motility experiments motivated us to develop an intravital two-photon microscopy assay to analyze monocyte 3D directional motility in vivo. We generated a mouse line harboring a collection of alleles that simultaneously deletes Arpc2 and induces expression of tdTomato (tdTom) fluorescent protein in CX3CR1-expressing cells in response to tamoxifen treatment (Figure 7A). Primary tdTom+ blood monocytes harvested from these mice lack Arpc2 (Figure 7B). We combined this genetic system with a full-thickness cutaneous wound assay to induce a physiologically-relevant directional cue in mouse ears (Figure 7C). Intravital two-photon imaging revealed that tdTom+ monocytes were robustly recruited to the wound edge one day after wounding (Movie S6, Figure S7A). Individual cells can be identified migrating toward the wound edge in both control (Arpc2+/−) and Arpc2−/− mice (Figure 7D), making it possible to quantitatively analyze motility. Both populations demonstrate robust directional migration in response to wounding (Figure 7E). Arpc2−/− monocytes were less persistent than control monocytes, but moved at a high rate of speed similar to control monocytes, and both populations had strongly positive FMI values, indicating robust directional motility (Figure 7F).
Figure 7. Monocyte motility and directionality in vivo do not require the Arp2/3 complex.

A) Allele combination used for in vivo experimentation. CX3CR1-Cre-ERT2 produced from the endogenous CX3CR1 promoter. Lox-Stop-Lox tdTomato expressed from the endogenous Rosa26 promoter. The Arpc2 allele as previously described. Tamoxifen induces LoxP recombination. B) Control (Arpc2+/−) or Arpc2−/− blood monocytes harvested after tamoxifen treatment. Scale = 10 µm. C) 0.3 mm diameter wounds were induced on day 0. tdTom+ monocytes were imaged the next day as they were recruited to the wound edge (arrow). D) Representative fields demonstrating tdTom+ Control and Arpc2−/− monocytes moving in situ toward a one day old wound over 15 minutes. Arrow denotes the direction of the wound. Individual cells are highlighted across the image series with colored arrows. Scale = 20 µm. E) Wind rose plots generated from endogenous Control (Arpc2+/−) or Arpc2−/− monocytes migrating toward one day old wounds in vivo. Data is from one representative experiment. 0°on the rose plot is the direction of the gradient. F) Forward Migration Index (FMI) of Control (Arpc2+/−) and Arpc2−/− monocytes in vivo plotted as mean ± 95% CI. Data table relates mean ± SEM of cell speed (V), FMI, d/T and total cells tracked (N). Quantitative measurements were pooled from 5 Control or 6 Arpc2−/− adult mice imaged one day after wounding. G) Velocity distribution of Control and Arpc2−/− monocytes from in vivo migration tracks (N = 234 or 382, respectively). Each point on the x-axis corresponds to a range of 50 microns per hour (0–50, 51–100, etc.). Y-axis: fraction of Control (cyan) or Arpc2−/− monocytes (red) migrating within given range. X-axis labels correspond to the upper bound of the 50 micron per hour range. H) FMI distribution of Control and Arpc2−/− monocytes (N = 234 or 382, respectively) from in vivo migration tracks. Each x-axis division corresponds to a range of 0.14, while the y-axis is the fraction of each population that falls within given range. The dotted vertical line divides monocytes with a positive FMI (to the right) that are capable of migrating directionally from those to the left (FMI ≤ 0) that do not. See also Figure S7, Movie S6.
We utilized whole ear immunostaining to confirm the completeness of our in vivo knockout system. We found that Arpc2−/−; tdTom+; CD11b (αM integrin)+ cells localize to the wound edge (Figures S7B, S7C) and that ~67% of tdTom+ cells at the wound edge lack p34 staining (9,567 total cells scored for p34-/tdTom+ staining). We further analyzed the distribution of our datasets to ensure that our Arpc2−/− population is not composed of two subpopulations that average out to a positive FMI. The distribution of cell speeds falls along a nearly identical range for control and Arpc2−/− monocytes, in line with the statistically identical values reflected in the mean comparison (Figure 7G). Plotting FMI in a similar fashion reveals that the distribution of Arpc2−/− values is nearly identical to control and is centered firmly at a positive FMI (Figure 7H). Furthermore, the fraction of tracked cells moving with a positive FMI is similar in each population (~0.80), arguing against a non-directional subpopulation of monocytes at Arpc2−/− wounds. Together, these experiments demonstrate that the Arp2/3 complex is not required for monocyte motility or directionality in vivo.
DISCUSSION
The work reported here yields surprising, important insights into how the branched actin-polymerizing Arp2/3 complex contributes to macrophage function in vivo and in vitro. Conditional knockout of Arpc2 in macrophages completely ablates Arp2/3 complex function, causing a number of striking phenotypes, including: loss of actin-containing rosettes and lamellipodia in favor of filopodia and bleb-based protrusion, leading to decreased spread cell area and altered cell shape. Arpc2−/− macrophages generate less F-actin than WT and organize filaments into parallel arrays rather than into puncta characteristic of normal macrophages. Despite these major defects in protrusion and steady state F-actin generation, Arpc2−/− macrophages competently phagocytose IgG-opsonized beads via F-actin rich phagocytic structures. Remarkably, Arpc2−/− macrophages successfully establish front/rear polarity, chemotax to both CSF and CX3CL1, and demonstrate enhanced motility compared to WT cells in both random and directional motility assays. Enhanced Arpc2−/− macrophage motility is dependent upon myosin II function and a combination of weakened adhesion and enhanced contractility likely drives this behavior. Both CR3-mediated phagocytosis and fibronectin haptotaxis are disrupted in Arpc2−/− macrophages, with null cells demonstrating impaired F-actin generation in response to both complement and fibronectin. Direct observation of monocyte motility in vivo yielded the surprising finding that Arpc2−/− monocytes maintain both directionality and cell speeds similar to control. These experiments help refine our understanding of Arp2/3 complex function in phagocytosis, chemotaxis and in vivo directional motility, and advance the notion that the Arp2/3 complex is fundamentally important for integrin-dependent functions in macrophages.
The Arp2/3 complex has long been viewed as the major actin effector of phagocytosis. Our data calls this generality into question, given that Arpc2−/− macrophages and control macrophages treated with CK-666 can competently phagocytose IgG-opsonized beads. It is worth considering how this assumption became so firmly established. Some of the earliest papers on this topic utilized fibroblastic cells transfected with phagocytic receptors, dominant-negative GTPases (Caron and Hall, 1998) and dominant-interfering SCAR/WAVE constructs (Machesky et al., 2000) to establish the central role of the Arp2/3 complex in phagocytosis. While these tools were state-of-the-art at the time, we now realize that many of these reagents have potential off-target effects (Boulter et al., 2010), interpretational limitations (i.e. multiple VCA binding sites on Arp2/3 complex) (Padrick et al., 2011; Ti et al., 2011) or have been supplanted by far more specific approaches. Elegant recent experiments underscore the fundamental importance of F-actin not only as a mediator of protrusion, but also as a means of spatially organizing signaling at the phagocytic cup (Freeman et al., 2016). Our findings suggest that pathways, like FcR phagocytosis, that have an Arp2/3-independent route to actin polymerization can compensate for its loss. In fact, these ‘compensatory’ pathways may be a major underlying driver of FcR phagocytosis revealed only when the Arp2/3 complex is disrupted. However, our data unquestionably demonstrates that the Arp2/3 complex accelerates FcR phagocytosis.
A large body of literature suggests that the Arp2/3 complex should be required for macrophage chemotaxis (Rougerie et al., 2013). The NPFs WASP and WAVE2 have been implicated in CSF and CX3CL1 chemotaxis (Kheir et al., 2005; Park and Cox, 2011; Zicha et al., 1998). We initially hypothesized that Arpc2−/− cells would also fail to chemotax, since WASP and WAVE2 directly activate the Arp2/3 complex (Machesky and Insall, 1998). We were surprised to find the exact opposite: Arpc2−/− macrophages chemotax. It is possible that disruption of a single NPF may cause dysregulation of the Arp2/3 complex (Ishihara et al., 2012), leading to altered lamellipodial protrusion/retraction cycles and defects in migration persistence that deregulate the real drivers of chemotaxis. Another alternative is that NPFs like WASP and WAVE2 have significant Arp2/3 complex-independent functions (as has been proposed for N-WASP (Kovacs et al., 2011) that are required for chemotactic sensing. The emerging consensus that the Arp2/3 complex is dispensable for chemotaxis (Asokan et al., 2014; Collins et al., 2015; Vargas et al., 2015; Wu et al., 2012) meshes well with studies demonstrating that the critical Arp2/3 complex activators WAVE2 and Rac1/2 are not required for leukocyte chemotaxis either, but do lead to morphological changes and migratory behaviors similar to our Arpc2−/− macrophages (Leithner et al., 2016; Wheeler et al., 2006). This body of work can be viewed alongside previous studies on NPF function in macrophages, and the apparent discrepancy should stimulate further investigation. We should not assume that NPF and Arp2/3 complex disruption will lead to identical phenotypes.
Integrin-dependent processes are strongly compromised in Arpc2−/− cells. Early studies utilizing the COS/SCAR-WA system indicated the Arp2/3 complex’s involvement in CR3 phagocytosis (Machesky et al., 2000), but this data is complicated by other studies that found that Rac1, Cdc42 and WASP were all dispensable (Caron and Hall, 1998; Park and Cox, 2009; Tzircotis et al., 2011). More recent studies indicate that Rac1 may in fact be more important than previously thought, but Rho GTPases have received far more attention as drivers of CR3 phagocytosis (Caron and Hall, 1998; Hall et al., 2006; Olazabal et al., 2002; Tzircotis et al., 2011). We initially suspected that the defect would be due to impaired integrin trafficking in Arpc2−/− macrophages since WASH knockout cells clearly demonstrate the importance of this NPF for integrin recycling (Piotrowski et al., 2013; Zech et al., 2011). Surprisingly, the CR3 defect in Arpc2−/− macrophages is not related to lower surface integrin levels, in line with recent findings suggesting that α5β1 integrin recycling does not require Arp2/3 complex (Paul et al., 2015). WASH is known to interact with many other proteins besides the Arp2/3 complex including microtubules, and endosomal and retromer components (Seaman, 2012), indicating once more that NPF phenotypes may not be perfectly recapitulated by loss of Arp2/3 complex function. WT macrophages make prominent dorsal waves that contain the CR3 integrins, F-actin and the Arp2/3 complex. These structures seem to be instrumental in binding C3bi-opsonized beads (Patel and Harrison, 2008) and are not present on the dorsal surfaces of Arpc2−/− macrophages. The prevailing view is that complement-opsonized targets sink directly into the cytoplasm through a process similar to endocytosis (Allen and Aderem, 1996), but our data and others (Patel and Harrison, 2008) indicate that large protrusions are also required. CR3 phagocytosis likely proceeds through a combination of protrusion and vesicular membrane trafficking and is clearly different than FcR phagocytosis. Though Arpc2−/− cells are capable of binding C3bi-opsonized beads, their ability to respond to bead binding is severely compromised.
Arpc2−/− macrophages do not migrate directionally (‘haptotax’) on fibronectin, which differentiates this integrin-dependent directional motility paradigm from chemotaxis. It is clear that Arpc2−/− cells adhere to fibronectin, generate vinculin-containing adhesive structures, establish front/rear polarity and migrate efficiently, but are incapable of generating ECM-responsive actin-based protrusions. Previous work established interactions between Arp2/3 and vinculin (DeMali et al., 2002), as well as FAK (Serrels et al., 2007). These interactions have been implicated in coupling nascent adhesions to nascent lamellipodia (Swaminathan et al., 2016), and the overall importance of lamellipodia for haptotaxis is established (King et al., 2016). WAVE2-Arp2/3 complex activity is known to recruit vinculin and talin to the immune synapse (Nolz et al., 2007). The privileged relationship between Arp2/3 and integrins revealed by our work suggests that positive feedback between integrin-ECM adhesion signaling (outside-in initiation) and spatially directed actin polymerization initiated by the Arp2/3 complex (inside-out response) are required to complete integrin-dependent processes. Arpc2−/− macrophages execute the initial outside-in integrin signaling but cannot generate a protrusive response. Failure to generate an actin based protrusion then deregulates the upstream pathway and halts the further accumulation of key signaling and adhesion molecules (Figure 6I). It is also possible that the force generated by branched actin polymerization positively reinforces the ligand-integrin catch bond (Kong et al., 2009, 2013), resulting in a sustained, spatially directed inside-out response coincident with cellular protrusion. The positive reinforcement provided by the Arp2/3 complex may be especially important for generating and coordinating large integrin-responsive protrusions during haptotaxis or engulfment of large C3-opsonized particles.
Our in vitro experimentation prompted us to test whether the Arp2/3 complex is required for monocyte directional migration in vivo. While full-thickness punch wounds likely elicit numerous directional cues, it is well accepted that most of the monocytes recruited to the wound originate from the peripheral circulation and arrive at the wound edge after extravasation and migration in the dermis across hundreds of microns (Brancato and Albina, 2011; Koh and DiPietro, 2011). Therefore, the directional cue(s) provoking monocyte motility in this setting are likely to act over a long range, much like we would expect for a chemotactic response. Detection of Arpc2−/− monocytes at the wound edge implies that extravasation does not require the Arp2/3 complex. Their ability to migrate robustly through the dermis toward the wound indicates that the Arp2/3 complex is not required for 3D migration, in line with recent observations suggesting that certain formins are the critical drivers of 3D migration in vitro (Paul et al., 2015). Our in vivo data also suggest that lamellipodia are not required for 3D directional motility, given the Arp2/3 complex’s unique ability among actin assembly factors to generate these structures. These data argue that the Arp2/3 complex is dispensable for many important cellular behaviors related to monocyte recruitment in vivo.
An important conclusion arising from these studies is that introducing an identical genetic perturbation into fibroblasts and macrophages can yield divergent phenotypes. Arpc2−/− fibroblast motility is profoundly decreased, while Arpc2−/− macrophage motility is substantially increased compared to respective controls. Additionally, both Blebbistatin and Y-27632 increased fibroblast motility while having a strong inhibitory effect on Arpc2−/− macrophages. These cell type-specific findings argue that Arp2/3 complex and myosin II functions differ, to some extent, according to the functions that they execute in different cell types. We were surprised to find that Arpc2−/− macrophages have reduced F-actin compared to WT macrophages, given our recent finding that Arpc2−/− fibroblasts maintain F-actin levels identical to WT (Rotty et al., 2015). Altering any actin regulatory factor might have context-specific consequences, as we demonstrate here with Arpc2−/− cells. These findings are consistent with the emerging view that actin is a homeostatic system (Davidson and Wood, 2016; Suarez and Kovar, 2016), as important control points might be set or regulated differentially across different cell types as they accomplish specific physiological functions.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources should be directed to and will be fulfilled by the corresponding author, James Bear (jbear@email.unc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse lines and in vivo experimentation
Mouse strains
Mice containing an already described Arpc2 conditional allele (Rotty et al., 2015) were mated with Ink4a/Arf−/− mice and then crossed with a Rosa26 CreERT2 line (C57BL/6J background, from The Jackson Laboratory) until offspring were homozygous for all three alleles in a mixed strain background. These mice were the source of all bone marrow-derived macrophages used in this study. Male and female adult mice were sacrificed from 2–5 months old for bone marrow harvests. All donor mice were immunocompetent and of normal weight. They were not tested, drug treated or subject to any procedures prior to bone marrow harvest. Mice were group housed and given food and water ad libitum. Cage conditions were monitored by the lead author and trained animal facility staff according to institutional animal care and use directives.
Arpc2 conditional mice were crossed with Rosa26 lox-stop-lox tdTom and CX3CR1 CreERt2-IRES-YFP (Parkhurst et al., 2013) mice (C57BL/6J background, The Jackson Laboratory) to yield control mice heterozygous for all three alleles and experimental Arpc2 fl/fl; lsl tdTom/+; CX3CR1 CreERT2/+ mice. These mice were used in intravital microscopy experiments. Male and female adult mice (2–3 months of age at experiment start) were used for longitudinal intravital imaging studies. Experimental mice were compared to littermate controls. All mice were immunocompetent and of normal weight. They were not tested, treated with experimental drugs, or subject to any procedures other than intraperitoneal tamoxifen injection prior to imaging. Mice were group housed and given food and water ad libitum. Cage conditions were monitored by the lead author and trained animal facility staff according to institutional animal care and use directives.
Tamoxifen injection
Arpc2fl/fl; CX3CR1CreERT2-IRES-YFP/+; Rosa26lsl-tdTom/+ mice and control (Arpc2fl/+; CX3CR1CreERT2-IRES-YFP/+; Rosa26lsl-tdTom/+) mice were treated with three doses of 400 µL of a 20 mg/mL Tamoxifen stock solution (dissolved in Sigma corn oil O/N) on Day 1, Day 3 and Day 5 of protocol. On Day 9 mice were transported from our mouse suite to be wounded via ear punch and imaged. Tamoxifen treatment and animal transport were conducted according to IACUC-approved protocols.
Intravital two-photon microscopy and wounding
Full-thickness punch biopsy wounds were induced nine days after the initial tamoxifen treatment. Mice were anesthetized with Isofluorane and 0.3 mm diameter full-thickness punch wounds were made in each ear. Mice were then returned to their cages and maintained overnight in dedicated return space, monitored by trained animal facility staff and the lead author with access to food and water ad libitum. On the first day after wounding mice were again anesthetized and prepped for imaging. Mice were continuously provided with isofluorane/O2 and immobilized on a heating pad on the microscope stage while the animal’s temperature was monitored with a probe connected to the same control as the heating pad. The mouse ear was then spread and immobilized on a custom-designed two-piece aluminum stage with embedded cover glass and thumbscrews. An optical imaging gel (with the same refractive index as water) was deposited on top of ear, coverslip and top of stage were screwed down, and more imaging gel was placed on the coverslip surface. Two-photon imaging was done on an upright FV1000MPE (Olympus) outfitted with an Insight DeepSee IR laser (Spectra-Physics) which we tuned to 1050 nm to excite tdTom fluorescence. An Olympus 25×, 1.05 N.A., 2 mm W.D., water-immersion objective was employed in these experiments. Data was gathered with the Olympus Fluoview software. Wounding and intravital imaging protocols were conducted according to IACUC-approved protocols. Mice were then immediately sacrificed via CO2 followed by cervical dislocation prior to ear harvesting for correlative immunostaining.
The University of North Carolina’s IACUC reviewed and approved all mouse experiments according to national animal use guidelines and recommendations.
Cell harvesting and maintenance
Bone Marrow Isolation
Mice were euthanized via CO2 according to IACUC-approved institutional guidelines, followed by cervical dislocation. Leg bones were harvested and the tops of each bone were removed with a razor blade. Bone marrow was flushed from leg bones harvested from these mice with a 22G needle attached to a 3 mL syringe inserted into the bone. Bone marrow isolates were harvested and cultured in DMEM/10% FBS + 30% L-conditioned media (containing Colony Stimulating Factor, CSF).
Cell Culture (including 4-OHT treatment)
Bone marrow-derived macrophages (BMDMs) were cultured in DMEM (4.5 g/L D-Glucose, L-Glutamate, Sodium Pyruvate) supplemented with 10% FBS, 1% Glutamax, and 30% L media (hereafter referred to as ‘macrophage media’). Dermal fibroblasts (26DF) were cultured in DMEM supplemented with 10% FBS and 1% Glutamax. BMDM and 26DF cells were plated at low density and treated several hours later with 2 µM 4-hydroxy-tamoxifen (4-OHT) to induce recombination of the Arpc2 allele. Two days later fresh media was added with a second dose of 2 µM 4-OHT. On day 5 cells were harvested, lysed or expanded for subsequent experiments.
All macrophages utilized in this study were primary cultures from bone marrow maintained as adherent cells in culture for at least seven days. Dermal Fibroblasts are a stable clonal line established from perinatal mice from the same line as BMDMs. Both male and female mice were used for macrophage bone marrow preparations.
METHOD DETAILS
Reagents
Drugs: CK-666 (Sigma), Cytochalasin D (Sigma), Blebbistatin (Sigma), Y-27632 (Sigma); Antibodies: Arpc2 (Millipore), Arp2 (ECM Bio), Arp3 (clone FMS338, Sigma), GAPDH (clone 6C5, Ambion/Life technologies), actin (Clone C4, Millipore), Vinculin (clone hvin1, Sigma), CD11b/aM integrin (Abcam), b1 integrin (Thermo), b2 integrin (Novus), p-Y418 Src Family Kinase (Cell Signaling), p-Tyr (4G10, EMD Millipore), Myosin IIA (Abcam), Myosin IIB (Cell Signaling), p-Erk 1/2 (Cell Signaling), C3 (Abcam), FcγRII/III (Fisher), Src-Family Kinases (Cell Signaling); Chemicals: 4-hydroxy-tamoxifen (Sigma), Tamoxifen (Sigma), Other: Phalloidin was conjugated to Alexa 488 or Alexa 568 for all experiments (Life Technologies), pHrodo red-avidin (Thermo), biotin-IgG (Rockland), EZ-Link NHS-LC-Biotin (Fisher).
Cell Lysis and Western Blotting
Cells were washed in cold 1X PBS and then harvested via scraping into ice-cold RIPA buffer (50 mM Tris pH 8, 150 mM NaCl, 0.5% Deoxycholate, 0.1% SDS, 1% NP-40, with 1x concentration of protease and phosphatase inhibitors (Roche PhosStop). Cells were rotated at 4 degrees for 15 minutes and spun at 21,000 × g for 10 minutes at 4 degrees. Supernatant was removed and used for subsequent experiments. Precision Red was used to quantify protein level. Equal protein amounts were run on SDS-PAGE gels, followed by transfer to PVDF membranes in methanol-containing transfer buffer for 75–90 minutes, and blocked for 1h in 5% milk (in 1x PBS-Tween). Primary antibodies were diluted in either 5% milk or 5% BSA (both in PBS-Tween) and incubated overnight at 4°C. HRP-conjugated secondary antibodies were diluted 1:10,000 in 5% milk (in 1x PBS-Tween) for 30 minutes, and chemiluminescent detection was employed to image protein levels on a Bio-Rad ChemiDoc MP imaging system.
Fixation and Immunostaining
Acid-washed coverslips were coated with 10 µg/mL fibronectin Human, Corning) (for fibroblasts) or 1 µg/mL fibronectin (for BMDMs). BMDMs were also plated on 1 µg/mL collagen (Type 1, rat tail, Millipore) for certain experiments, as indicated. Cells were plated overnight on coated coverslips in the presence of full medium, and fixed in cold buffer (paraformaldehyde dissolved to 4% w/v in Krebs-S buffer) for 10 minutes at ambient temperature the next day. After three 1x PBS washes, 0.1% Triton X-100 was employed for 5 minutes to permeabilize cells. A 1:1 ratio of 10% BSA: 10% NGS (dissolved in 1x PBS) was used as blocking buffer (30 minutes at room temperature). Primary antibody dilutions varied depending on the antibody, but all dilutions were done in 1% BSA (in 1x PBS) for 1 hour at room temperature. Secondary antibodies and fluorescently-labeled phalloidin were diluted 1:500 in 1% BSA and incubated on coverslips for 30 minutes. Coverslips were washed briefly and mounted in Fluoromount G prior to imaging (Electron Microscopy Sciences).
Phagocytic Uptake
Experiments were performed according to established protocols. FcR phagocytosis: Latex beads were opsonized with mouse IgG (Life Technologies) for 1h at 37°C. Beads were added to BMDMs in macrophage media on collagen-coated coverslips and incubated for 20 minutes and fixed (20 minute time point) or washed to remove excess beads and incubated for a further 40 minutes (60 minute time point) in a tissue culture incubator. Fixed, intact cells were blocked for 30 minutes and then incubated with goat anti-mouse secondary antibody (conjugated to rhodamine Red-X, 1:750 in 1% BSA) for 15 minutes at room temperature, washed with PBS and permeabilized with 0.1% Triton X-100 for 5 minutes. Cells were blocked again, incubated with goat anti-mouse secondary antibody conjugated to Cy5 and Phalloidin-Alexa 488 (both 1:500 in 1% BSA). Rhodamine Red-X/Cy5 double labeled beads were scored as external, while beads labeled with only Cy5 were scored as internalized. CR3 phagocytosis: Latex beads were opsonized with human IgM (Sigma) for 1h at 37°C, washed, and incubated with mouse complement serum (Innovative Research) for 20 minutes at 37°C. Cells were treated with beads in a similar fashion to FcR phagocytosis, except that CR3 uptake experiments were carried out in the presence of 125 ng/mL PMA in 5% L media in serum free DMEM to minimize non-specific opsonization by antibodies in bovine serum. Fixed, intact cells were blocked and then incubated with rat anti-C3 antibody (1:50), washed and incubated with goat anti-rat-Alexa 568 secondary antibody (1:500, for 15 minutes). Cells were then washed with PBS, permeabilized, blocked, and incubated with rat anti-C3 antibody (1:50). Cells were washed again and incubated with goat anti-rat-Alexa 647 secondary antibody and phalloidin-Alexa 488 (1:500, for 30 minutes). Alexa 568/Alexa 647 double labeled beads were scored as external, while beads labeled with only Alexa 647 were scored as internalized.
Live Cell Phagocytic Uptake
FcR: Commercially available biotin-IgG (Rockland) was reacted at a 1:1 molar ratio with pHrodo-avidin (Thermo) for 1h at RT with gentle rocking. pHrodo-IgG was then adhered to 2 micron latex beads according to standard procedure (above). WT or Arpc2−/− macrophages stably expressing Lifeact-GFP (LA-GFP) were plated on 1 µg/mL collagen overnight in macrophage media. Cells were imaged the next morning via confocal microscopy after addition of pHrodo-IgG beads. GFP and pHrodo were imaged every 10s for 40 minutes as cells interacted with beads. CR3: NHS-LC-Biotin (Fisher) was reacted with human IgM for 1 hour at room temperature (Sigma). Biotin-IgM was added to latex beads according to standard opsonization protocol (above). After one hour, pHrodo red-avidin and mouse complement serum were added together to biotin-IgM containing beads. WT or Arpc2−/− LA-GFP macrophages were prepared in the same fashion as reported above for FcR experiments, except that they were plated in 5% L media in serum free DMEM without PMA. GFP and pHrodo were imaged every 10s for 40 minutes via confocal microscopy as cells interacted with beads. Quantification: pHrodo fluorescence and GFP signal at beads were analyzed via integrated pixel density during the time course. Time 0 for each bead was the point in the time course that it became immobilized on the cell surface. We derived three quantitative values from these data: tactin, the length of time after bead binding required to generate an actin peak; tacid, the length of time between tactin and bead acidification; and tint, the internalization time, or tactin + tacid. The actin peak was defined as the highest LA-GFP value at a bead after a consistent rise in signal, which directly preceded acidification. Bead acidification was defined as the point after the actin peak during which red fluorescence rapidly increases due to lowered pH. Though internalization likely occurred before this point, we chose to treat entry into the phagolysosome as an unambiguous, reproducible phagocytic endpoint.
Confocal Microscopy
Z-stacks were obtained on an inverted Fluoview FV1000 scanning confocal microscope (Olympus), using a 100x PlanApo 1.4 N.A. 100x objective (Olympus). Fluoview software was used to control the microscope and acquire images. Raw .oif files were imported into ImageJ for processing.
Live Cell Imaging
Random motility
Cells were plated on 1 µg/mL fibronectin (Human, corning) for both macrophages and fibroblasts) in glass bottomed dishes (MatTek). Each run was imaged over at least 16 hours, with 10 minute intervals. Cells were imaged at 10x magnification on an Olympus VivaView FL incubator microscope outfitted with a Hamamatsu R2 camera. Experiments involving drug treatments required adding drug to cells just prior to imaging. Cells were exposed to drug for the duration of the experimental run. Cell tracks were generated using the Manual Tracking ImageJ plugin. The ImageJ Chemotaxis plugin was used to calculate velocity from these tracks.
Protrusion/retraction analysis
BMDMs were plated on 1 µg/mL fibronectin in glass bottomed Mat-TEK dishes. Cells were imaged for 10 minutes with 2 second intervals. Cells were imaged at 40x on a Nikon Biostation CT microscope outfitted with a cooled CCD camera. Phase contrast images were collected via Biostation imaging software. Experiments involving drug treatment required that cells be exposed to drug for the duration of the experimental run (1–4 hours).
Chemotaxis
The microfluidic chamber system utilized in these experiments has been previously described in detail(Wu et al., 2012). Briefly, microfluidic chambers were coated with 1 µg/mL collagen before seeding cells for chemotaxis experiments. WT BMDMs were incubated with green cell tracker dye to mark them as controls. These cells were washed and dissociated, before being added with Arpc2−/− BMDMs in the chambers. Cells were allowed to adhere and spread before being transferred to our microfluidic microscope. The central ports of the device were plugged, and outflow ports were connected to waste container. The source inlet port contained chemotactic ligand, while the sink inlet port contained macrophage media or diluted L media, depending on the experiment. A dual syringe pump pushed sink and source media at 20 nL/min, thereby eliminating shear stress from the central cell chamber. Gradients were allowed to establish for 30 minutes to an hour before imaging. Cells were then imaged via DIC at 20x every 10 minutes for at least 16 hours. Fluorescent images were gathered at the beginning and end of the run to differentiate between control and Arpc2−/− macrophages. CSF chemotaxis: 150 ng/mL M-CSF (Mouse, Peprotech) diluted in 0.1x macrophage media was used as a source, while 0.1x macrophage media was used as the sink. CX3CL1 chemotaxis: 300 ng/mL CX3CL1 (Mouse, BioLegend) diluted in macrophage media was used as source, while macrophage media alone was used as the sink. Cell tracks were generated using the Manual Tracking ImageJ plugin. The Image J Chemotaxis plugin was used to calculate velocity, FMI and d/T from these tracks.
Haptotaxis
The microfluidic chamber system has been previously adapted for haptotaxis experiments and has been described previously in detail(Wu et al., 2012). Briefly, source chambers were filled with 200 µg/mL Cy5-conjugated fibronectin, which was used to directly assess gradient formation in the central cell chamber. Chambers were then washed several times with PBS and cells were loaded as a mixed population with WT BMDMs labeled green with cell tracker dye. Cells were allowed to spread in full macrophage media and then imaged over at least 16 hours, with 10 minute intervals. Cells were imaged at 10x magnification on an Olympus VivaView FL incubator microscope outfitted with a Hamamatsu R2 camera. Cell tracks were generated using the Manual Tracking ImageJ plugin. The Image J Chemotaxis plugin was used to calculate velocity, FMI and d/T from these tracks.
Scanning Electron Microscopy
Sample preparation was done as previously described. Briefly, cells were plated on 12mm round coverslips and fixed with 2.5% glutaraldehyde/0.15M sodium phosphate buffer (pH 7.4) at room temperature. Cells were post-fixed with 1% osmium tetroxide with 2% tannic acid in water for 10 minutes and 1% osmium tetroxide in water for 10 minutes. Coverslips were then rehydrated in ethanol and critical point dried using CO2 as transitional solvent (Samdri-795 critical point dryer, Tousimis Research Cop., Rockville, MD). Coverslips were then mounted and coated with 10 nm of gold-palladium alloy (60Au:40Pd, Hummer X Sputter Coater, Anatech USA, Union City, C). A Zeiss Supra 25 FESEM operating at 5kV, with 5mm working distance and 10 µm aperture was used to gather images (Carl Zeiss SMT Inc., Peabody, MA).
Superresolution Microscopy
Fixed WT and Arpc2−/− macrophages were imaged on a Zeiss 880 LSM confocal equipped with an Airyscan detector. Samples were imaged with the 63× 1.4 NA Plan-Apo objective compatible with the Airyscan detector. Images of phalloidin staining at phagocytic cups were collected and processed using Zeiss software to assemble individual z-slices with 1.4x better resolution compared to standard confocal microscopy. These individual slices were assembled into maximum intensity z-projections in ImageJ.
Intravital live cell imaging and analysis
Multiple z-positions were imaged during each run with 3 micron intervals between slices. Each z-stack was imaged for 40 minutes at one minute intervals to analyze monocyte recruitment to the wound edge. Image series were compiled in the Fluoview software, with each frame containing a maximum intensity projection of the z-stack at each time point. Image series were then imported into ImageJ and xy drift was corrected with the StackReg plugin(Thevenaz et al., 1998). Individual cell tracks were then generated with the Manual Tracking plugin. The Chemotaxis plugin was used to calculate velocity, FMI and d/T from these cell tracks.
Whole ear immunostaining
Mice were sacrificed via CO2 immediately following intravital imaging, according to IACUC-approved methods. The dorsal and ventral surfaces of the ear were gently peeled apart using forceps and fixed for 10 minutes in 1% PFA. Ears were then washed 3×10 minutes in 0.3% Triton (in PBS), blocked for 30 minutes in 0.3% Triton-containing blocking buffer (5% BSA/5% NGS in PBS) and incubated for one hour at room temperature with primary antibody (in 1% BSA, no Triton). Ears were then washed and incubated with secondary antibodies (1:500 in 1% BSA) for 30 minutes at RT, washed briefly and mounted. Confocal microscopy of these samples was conducted according to protocol above.
Flow cytometry analysis
WT and Arpc2−/− macrophages were dissociated and treated with Fc blocking reagent (Innovex Biosciences) for 30 minutes on ice. Half of the sample was used as a secondary only negative control. The other half was incubated for 30 minutes on ice with integrin-binding antibodies diluted in FACS buffer (2% FBS in PBS), washed, and labeled with Alexa 488-conjugated secondary antibody (1:500 in FACS buffer) for 30 minutes followed by flow cytometry analysis. All data were collected using a Bio-Rad S3 Cell Sorter. Analysis was done using FlowJo software (FlowJo LLC).
Fibronectin/IgG/C3 bead quantification
WT or Arpc2−/− macrophages were plated separately on 1 µg/mL collagen-coated coverslips overnight at 37°C. Cy5-FN was diluted to 5 µg/mL in sterile PBS and incubated with 30 µL 6 micron latex beds for 1h at 37°C with gentle rocking. Opsonized beads were spun down and washed several times with cold PBS. Fresh 6 micron Cy5-FN opsonized beads were added to BMDMs for 5 minutes and then immediately fixed, or washed thoroughly to remove excess beads and allowed to continue interacting with beads for 15 or 30 minutes before these samples were also fixed. All staining was done according to the immunofluorescence protocol outlined above. Samples were imaged via confocal microscopy, also outlined above. Quantification strategy is reported below under Image Analysis section. Unlabeled IgG or C3 beads were also added to cells in separate experiments for 5, 15, or 30 minutes followed by fixation, staining, imaging and quantification, as reported in Image Analysis section.
QUANTIFICATION AND STATISTICAL ANALYSIS
F-actin, cell size and cell shape analysis
After staining with fluorescent phalloidin, images were taken on an Olympus IX81 microscope controlled by Metamorph software (Molecular Devices) using a 0.30 N.A. 10x objective. Phalloidin images were imported to ImageJ and background subtracted. Cells were outlined by hand and integrated pixel density (i.e. F-actin staining), cell area and cell perimeter were measured on a per cell basis. Integrated densities are reported in arbitrary units (A.U.). Spread cell area was directly reported based on these measurements. Cell shape was determined quantitatively with the following equation: (4π*cell area)/(cell perimeter2). A circle would have a value of 1, while a straight line would have a value of 0.
Image Analysis
Integrated density of FcR/CR3/FN pathway components at beads
Levels were analyzed via confocal microscopy of fixed macrophages bound to IgG, complement or fibronectin-opsonized beads. These z-series were imported into ImageJ. The z-slice corresponding to the midpoint (i.e. widest diameter) of the bead was chosen and staining intensity around the bead was analyzed by drawing a region of interest at the bead’s widest diameter in the z-stack. The measurements are reported as integrated pixel densities (A.U.).
Surface integrin levels in spread cells
Cells plated on collagen-coated coverslips were fixed but not permeabilized and subsequently incubated with integrin antibodies directed against extracellular domains. Samples were then prepared according to our standard IF protocol (above) and imaged on an Olympus IX81 microscope controlled by Metamorph software Molecular Devices) using a 0.30 N.A. 10x objective. Cells were outlined in ImageJ and integrated pixel density is reported (in A.U.).
Directionality definitions
Persistence (d/T) measures how well a cell moves in any given direction, as d is the shortest path between the cell’s position at t0 and tfinal and T is the total distance traveled between t0 and tfinal. A value of 1 means that the cell moves directly from point a to point c, while a value <<1 means that the cell meanders much more along the path from a to c. It should be noted that persistence is not a measure of directional migration on its own, as it has no external frame of reference (i.e. movement toward a chemotactic source). Forward Migration Index (FMI) is our main measure of directionality as it takes d/T into account as well as an external frame of reference. FMI = Cosθ*(d/T), where θ is the angle formed between an ideal directionally migrating cell and the cell’s actual endpoint, both compared to the cell’s starting position. An FMI value of 1 is the idealized chemotactic value, indicating perfect directional migration. An FMI value of 0 denotes random migration. We employ 95% confidence intervals when we plot FMI to determine whether a cell is migrating directionally. If these intervals encompass 0, then the population is not moving directionally. Otherwise, we interpret the population’s motility as directional.
Velocity distribution
We plotted intravital monocyte velocity as a distribution of values ranging from 0 to 800 microns/h. Each point on the x-axis corresponds to a range of 50 microns per hour (0–50, 51–100, etc.), with the y-axis corresponding to the fraction of each population that falls within that range. The x-axis labels actually correspond to the upper bound of a 50 micron per hour range for that point distribution.
FMI distribution
We plotted FMI measurements from intravital monocyte cell tracks as a distribution of values ranging from −1 to 1. Each bar on the x-axis corresponds to a range of 0.14, with the y-axis corresponding to the fraction of each population that falls within that range.
Statistical Analysis
Unless otherwise noted, all means are graphed or presented in tables with standard error of the mean. FMI graphs contain error bars representing 95% confidence intervals. Unpaired two-tailed t tests were utilized to assess statistical significance, with p-values < 0.05 being considered significant. GraphPad Prism was used to generate all graphs and run all statistical tests. All statistical information for quantitative datasets can be found in corresponding figure legends, including exact n values and what n represents for each experiment.
Supplementary Material
Highlights.
Arp2/3 complex deletion disrupts actin assembly and cellular protrusion
FcR phagocytosis and chemotaxis do not require Arp2/3 complex
The Arp2/3 complex drives integrin-responsive directed protrusion
Endogenous Arpc2−/− monocytes migrate directionally in response to wounding
Acknowledgments
This work was supported by NIH grants to J.E.B. (GM110155, U01EB018816) and J.P.T. (AI029564). We thank Victoria Madden and Kristen White at the Microscopy Services Core for assistance with SEM sample prep and microscopy. We are grateful to the UNC-Olympus Imaging Research Center and the Hooker Imaging Core, and to Robert Curran’s administrative and technical assistance in both contexts. We thank Stephen Jones and Tao Bo for additional technical assistance, and members of our laboratories for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTION
J.D.R. designed and performed experiments, analyzed data, and wrote the paper; H.E.B. performed experiments; S.L.C. performed experiments and analyzed data; S.B.A. performed experiments and analyzed data; N.C. performed experiments and analyzed data; J.P.T. proposed and directed experiments; J.E.B proposed and designed experiments and wrote the paper. All authors reviewed and made comments on the manuscript prior to publication.
The authors declare no conflicts of interest.
References
- Allen LA, Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor-mediated phagocytosis in macrophages. J. Exp. Med. 1996;184:627–637. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asokan SB, Johnson HE, Rahman A, King SJ, Rotty JD, Lebedeva IP, Haugh JM, Bear JE. Mesenchymal Chemotaxis Requires Selective Inactivation of Myosin II at the Leading Edge via a Noncanonical PLCγ/PKCα Pathway. Dev. Cell. 2014;31:747–760. doi: 10.1016/j.devcel.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Green JE, Chambers AF. Extracellular matrix: a gatekeeper in the transition from dormancy to metastatic growth. Eur. J. Cancer. 2010;46:1181–1188. doi: 10.1016/j.ejca.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boczkowska M, Rebowski G, Kast DJ, Dominguez R. Structural analysis of the transitional state of Arp2/3 complex activation by two actin-bound WCAs. Nat. Commun. 2014;5:3308. doi: 10.1038/ncomms4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat. Cell Biol. 2010;12:477–483. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancato SK, Albina JE. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am. J. Pathol. 2011;178:19–25. doi: 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büscher D, Hipskind RA, Krautwald S, Reimann T, Baccarini M. Rasdependent and -independent pathways target the mitogen-activated protein kinase network in macrophages. Mol. Cell. Biol. 1995;15:466–475. doi: 10.1128/mcb.15.1.466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambien B. Signal transduction pathways involved in soluble fractalkine-induced monocytic cell adhesion. Blood. 2001;97:2031–2037. doi: 10.1182/blood.v97.7.2031. [DOI] [PubMed] [Google Scholar]
- Caron E, Hall A. Identification of two distinct mechanisms of phagocytosis controlled by different Rho GTPases. Science. 1998;282:1717–1721. doi: 10.1126/science.282.5394.1717. [DOI] [PubMed] [Google Scholar]
- Chan KT, Asokan SB, King SJ, Bo T, Dubose ES, Liu W, Berginski ME, Simon JM, Davis IJ, Gomez SM, et al. LKB1 loss in melanoma disrupts directional migration toward extracellular matrix cues. J. Cell Biol. 2014;207:299–315. doi: 10.1083/jcb.201404067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell Biol. 2001;3:339–345. doi: 10.1038/35070009. [DOI] [PubMed] [Google Scholar]
- Collins SR, Yang HW, Bonger KM, Guignet EG, Wandless TJ, Meyer T. Using light to shape chemical gradients for parallel and automated analysis of chemotaxis. Mol. Syst. Biol. 2015;11:804. doi: 10.15252/msb.20156027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson AJ, Wood W. Unravelling the Actin Cytoskeleton: A New Competitive Edge? Trends Cell Biol. 2016;26:569–576. doi: 10.1016/j.tcb.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMali KA, Barlow CA, Burridge K. Recruitment of the Arp2/3 complex to vinculin: coupling membrane protrusion to matrix adhesion. J. Cell Biol. 2002;159:881–891. doi: 10.1083/jcb.200206043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. doi: 10.1016/0092-8674(94)90528-2. [DOI] [PubMed] [Google Scholar]
- Flannagan RS, Jaumouillé V, Grinstein S. The Cell Biology of Phagocytosis. Annu. Rev. Pathol. Mech. Dis. 2012;7:61–98. doi: 10.1146/annurev-pathol-011811-132445. [DOI] [PubMed] [Google Scholar]
- Freeman SA, Grinstein S. Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol. Rev. 2014;262:193–215. doi: 10.1111/imr.12212. [DOI] [PubMed] [Google Scholar]
- Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, Hinz B, van der Merwe PA, Das R, Grinstein S. Integrins Form an Expanding Diffusional Barrier that Coordinates Phagocytosis. Cell. 2016;164:128–140. doi: 10.1016/j.cell.2015.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goley ED, Rodenbusch SE, Martin AC, Welch MD. Critical Conformational Changes in the Arp2/3 Complex Are Induced by Nucleotide and Nucleation Promoting Factor. Mol. Cell. 2004;16:269–279. doi: 10.1016/j.molcel.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Grinnell F, Billingham RE, Burgess L. Distribution of fibronectin during wound healing in vivo. J. Invest. Dermatol. 1981;76:181–189. doi: 10.1111/1523-1747.ep12525694. [DOI] [PubMed] [Google Scholar]
- Hall AB, Gakidis MAM, Glogauer M, Wilsbacher JL, Gao S, Swat W, Brugge JS. Requirements for Vav Guanine Nucleotide Exchange Factors and Rho GTPases in FcγR- and Complement-Mediated Phagocytosis. Immunity. 2006;24:305–316. doi: 10.1016/j.immuni.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Ishihara D, Dovas A, Park H, Isaac BM, Cox D. The Chemotactic Defect in Wiskott-Aldrich Syndrome Macrophages Is Due to the Reduced Persistence of Directional Protrusions. PLoS One. 2012;7:e30033. doi: 10.1371/journal.pone.0030033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological Properties of Y-27632, a Specific Inhibitor of Rho-Associated Kinases. Mol. Pharmacol. 2000;57 [PubMed] [Google Scholar]
- Jay PY, Pham PA, Wong SA, Elson EL. A mechanical function of myosin II in cell motility. J. Cell Sci. 1995;108 doi: 10.1242/jcs.108.1.387. [DOI] [PubMed] [Google Scholar]
- Kheir WA, Gevrey J-C, Yamaguchi H, Isaac B, Cox D. A WAVE2-Abi1 complex mediates CSF-1-induced F-actin-rich membrane protrusions and migration in macrophages. J. Cell Sci. 2005;118:5369–5379. doi: 10.1242/jcs.02638. [DOI] [PubMed] [Google Scholar]
- King SJ, Asokan SB, Haynes EM, Zimmerman SP, Rotty JD, Alb JG, Tagliatela A, Blake DR, Lebedeva IP, Marston D, et al. Lamellipodia are crucial for haptotactic sensing and response. J. Cell Sci. 2016;129:2329–2342. doi: 10.1242/jcs.184507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TJ, DiPietro LA. Inflammation and wound healing: the role of the macrophage. Expert Rev. Mol. Med. 2011;13:e23. doi: 10.1017/S1462399411001943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, García AJ, Mould AP, Humphries MJ, Zhu C. Demonstration of catch bonds between an integrin and its ligand. J. Cell Biol. 2009;185 doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Li Z, Parks WM, Dumbauld DW, García AJ, Mould AP, Humphries MJ, Zhu C. Cyclic mechanical reinforcement of integrin-ligand interactions. Mol. Cell. 2013;49:1060–1068. doi: 10.1016/j.molcel.2013.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs EM, Verma S, Ali RG, Ratheesh A, Hamilton Na, Akhmanova A, Yap AS. N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat. Cell Biol. 2011;13:934–943. doi: 10.1038/ncb2290. [DOI] [PubMed] [Google Scholar]
- Kovács M, Tóth J, Hetényi C, Málnási-Csizmadia A, Sellers JR. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004;279:35557–35563. doi: 10.1074/jbc.M405319200. [DOI] [PubMed] [Google Scholar]
- Leithner A, Eichner A, Müller J, Reversat A, Brown M, Schwarz J, Merrin J, de Gorter DJ, Schur F, Bayerl J, et al. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nat. Cell Biol. 2016;18:1253–1259. doi: 10.1038/ncb3426. [DOI] [PubMed] [Google Scholar]
- Lemière J, Valentino F, Campillo C, Sykes C. How cellular membrane properties are affected by the actin cytoskeleton. Biochimie. 2016;130:33–40. doi: 10.1016/j.biochi.2016.09.019. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Insall RH. Scar1 and the related Wiskott–Aldrich syndrome protein, WASP, regulate the actin cytoskeleton through the Arp2/3 complex. Curr. Biol. 1998;8:1347–1356. doi: 10.1016/s0960-9822(98)00015-3. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME, Pollard TD. Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. U. S. A. 1999;96:3739–3744. doi: 10.1073/pnas.96.7.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machesky LM, May RC, Caron E, Hall A. Involvement of the Arp2/3 complex in phagocytosis mediated by FcgammaR or CR3. Nat. Cell Biol. 2000;2:246–248. doi: 10.1038/35008673. [DOI] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell a, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolz JC, Medeiros RB, Mitchell JS, Zhu P, Freedman BD, Shimizu Y, Billadeau DD. WAVE2 Regulates High-Affinity Integrin Binding by Recruiting Vinculin and Talin to the Immunological Synapse. Mol. Cell. Biol. 2007;27:5986–6000. doi: 10.1128/MCB.00136-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olazabal IM, Caron E, May RC, Schilling K, Knecht DA, Machesky LM. Rho-kinase and myosin-II control phagocytic cup formation during CR, but not FcgammaR, phagocytosis. Curr. Biol. 2002;12:1413–1418. doi: 10.1016/s0960-9822(02)01069-2. [DOI] [PubMed] [Google Scholar]
- Oudin MJ, Jonas O, Kosciuk T, Broye LC, Guido BC, Wyckoff J, Riquelme D, Lamar JM, Asokan SB, Whittaker C, et al. Tumor Cell-Driven Extracellular Matrix Remodeling Drives Haptotaxis during Metastatic Progression. Cancer Discov. 2016;6:516–531. doi: 10.1158/2159-8290.CD-15-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padrick SB, Doolittle LK, Brautigam Ca, King DS, Rosen MK. Arp2/3 complex is bound and activated by two WASP proteins. Proc. Natl. Acad. Sci. U. S. A. 2011;108:E472–E479. doi: 10.1073/pnas.1100236108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Cox D. Cdc42 Regulates Fc Receptor-mediated Phagocytosis through the Activation and Phosphorylation of Wiskott-Aldrich Syndrome Protein (WASP) and Neural-WASP. Mol. Biol. Cell. 2009;20:4500–4508. doi: 10.1091/mbc.E09-03-0230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Cox D. Syk regulates multiple signaling pathways leading to CX3CL1 chemotaxis in macrophages. J. Biol. Chem. 2011;286:14762–14769. doi: 10.1074/jbc.M110.185181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR, Lafaille JJ, Hempstead BL, Littman DR, Gan W-B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell. 2013;155:1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PC, Harrison RE. Membrane Ruffles Capture C3bi-opsonized Particles in Activated Macrophages. Mol. Biol. Cell. 2008;19:4628–4639. doi: 10.1091/mbc.E08-02-0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul NR, Allen JL, Chapman A, Morlan-Mairal M, Zindy E, Jacquemet G, Fernandez del Ama L, Ferizovic N, Green DM, Howe JD, et al. α5β1 integrin recycling promotes Arp2/3-independent cancer cell invasion via the formin FHOD3. J. Cell Biol. 2015;210:1013–1031. doi: 10.1083/jcb.201502040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piotrowski JT, Gomez TS, Schoon RA, Mangalam AK, Billadeau DD. WASH Knockout T Cells Demonstrate Defective Receptor Trafficking, Proliferation, and Effector Function. Mol. Cell. Biol. 2013;33:958–973. doi: 10.1128/MCB.01288-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD, Marchand J-B, Kaiser DA, Higgs HN. Interaction of WASP/Scar proteins with actin and vertebrate Arp2/3 complex. Nat. Cell Biol. 2001;3:76–82. doi: 10.1038/35050590. [DOI] [PubMed] [Google Scholar]
- Price J, Vance R. The Macrophage Paradox. Immunity. 2014;41:685–693. doi: 10.1016/j.immuni.2014.10.015. [DOI] [PubMed] [Google Scholar]
- Proctor RA. Fibronectin: An Enhancer of Phagocyte Function. Clin. Infect. Dis. 1987;9:S412–S419. doi: 10.1093/clinids/9.supplement_4.s412. [DOI] [PubMed] [Google Scholar]
- Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, Johnson HE, Haugh JM, Kovar DR, Bear JE. Profilin-1 serves as a gatekeeper for actin assembly by Arp2/3-dependent and -independent pathways. Dev. Cell. 2015;32:54–67. doi: 10.1016/j.devcel.2014.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougerie P, Miskolci V, Cox D. Generation of membrane structures during phagocytosis and chemotaxis of macrophages: role and regulation of the actin cytoskeleton. Immunol. Rev. 2013;256:222–239. doi: 10.1111/imr.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman MNJ. The retromer complex - endosomal protein recycling and beyond. J. Cell Sci. 2012;125:4693–4702. doi: 10.1242/jcs.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrels B, Serrels A, Brunton VG, Holt M, McLean GW, Gray CH, Jones GE, Frame MC. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007;9:1046–1056. doi: 10.1038/ncb1626. [DOI] [PubMed] [Google Scholar]
- Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C, Kovar DR. Internetwork competition for monomers governs actin cytoskeleton organization. Nat. Rev. Mol. Cell Biol. 2016;17:799–810. doi: 10.1038/nrm.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM. Ultrastructure of protrusive actin filament arrays. Curr. Opin. Cell Biol. 2013;25:574–581. doi: 10.1016/j.ceb.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svitkina TM, Borisy GG. Arp2/3 Complex and Actin Depolymerizing Factor/Cofilin in Dendritic Organization and Treadmilling of Actin Filament Array in Lamellipodia. J. Cell Biol. 1999;145:1009–1026. doi: 10.1083/jcb.145.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaminathan V, Fischer RS, Waterman CM. The FAK-Arp2/3 interaction promotes leading edge advance and haptosensing by coupling nascent adhesions to lamellipodia actin. Mol. Biol. Cell. 2016;27:1085–1100. doi: 10.1091/mbc.E15-08-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenaz P, Ruttimann UE, Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Thrasher AJ, Burns SO. WASP: a key immunological multitasker. Nat. Rev. Immunol. 2010;10:182–192. doi: 10.1038/nri2724. [DOI] [PubMed] [Google Scholar]
- Ti S-C, Jurgenson CT, Nolen BJ, Pollard TD. Structural and biochemical characterization of two binding sites for nucleation-promoting factor WASp-VCA on Arp2/3 complex. Proc. Natl. Acad. Sci. U. S. A. 2011;108:E463–E471. doi: 10.1073/pnas.1100125108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzircotis G, Braga VMM, Caron E. RhoG is required for both FcγR- and CR3-mediated phagocytosis. J. Cell Sci. 2011;124:2897–2902. doi: 10.1242/jcs.084269. [DOI] [PubMed] [Google Scholar]
- Vargas P, Maiuri P, Bretou M, Sáez PJ, Pierobon P, Maurin M, Chabaud M, Lankar D, Obino D, Terriac E, et al. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nat. Cell Biol. 2015;18:43–53. doi: 10.1038/ncb3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler AP, Wells CM, Smith SD, Vega FM, Henderson RB, Tybulewicz VL, Ridley AJ. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J. Cell Sci. 2006;119:2749–2757. doi: 10.1242/jcs.03024. [DOI] [PubMed] [Google Scholar]
- Wu C, Asokan SB, Berginski ME, Haynes EM, Sharpless NE, Griffith JD, Gomez SM, Bear JE. Arp2/3 is critical for lamellipodia and response to extracellular matrix cues but is dispensable for chemotaxis. Cell. 2012;148:973–987. doi: 10.1016/j.cell.2011.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zech T, Calaminus SDJ, Caswell P, Spence HJ, Carnell M, Insall RH, Norman J, Machesky LM. The Arp2/3 activator WASH regulates 5 1-integrin-mediated invasive migration. J. Cell Sci. 2011;124:3753–3759. doi: 10.1242/jcs.080986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zicha D, Allen WE, Brickell PM, Kinnon C, Dunn GA, Jones GE, Thrasher AJ. Chemotaxis of macrophages is abolished in the Wiskott-Aldrich syndrome. Br. J. Haematol. 1998;101:659–665. doi: 10.1046/j.1365-2141.1998.00767.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.