

ABSTRACT
Despite being potentially highly useful for characterizing the biodiversity of phages, metagenomic studies are currently not available for dairy bacteriophages, partly due to the lack of a standard procedure for phage extraction. We optimized an extraction method that allows the removal of the bulk protein from whey and milk samples with losses of less than 50% of spiked phages. The protocol was applied to extract phages from whey in order to test the notion that members of Lactococcus lactis 936 (now Sk1virus), P335, c2 (now C2virus) and Leuconostoc phage groups are the most frequently encountered in the dairy environment. The relative abundance and diversity of phages in eight and four whey mixtures from dairies using undefined mesophilic mixed-strain cultures containing Lactococcus lactis subsp. lactis biovar diacetylactis and Leuconostoc species (i.e., DL starter cultures) and defined cultures, respectively, were assessed. Results obtained from transmission electron microscopy and high-throughput sequence analyses revealed the dominance of Lc. lactis 936 phages (order Caudovirales, family Siphoviridae) in dairies using undefined DL starter cultures and Lc. lactis c2 phages (order Caudovirales, family Siphoviridae) in dairies using defined cultures. The 936 and Leuconostoc phages demonstrated limited diversity. Possible coinduction of temperate P335 prophages and satellite phages in one of the whey mixtures was also observed.
IMPORTANCE The method optimized in this study could provide an important basis for understanding the dynamics of the phage community (abundance, development, diversity, evolution, etc.) in dairies with different sizes, locations, and production strategies. It may also enable the discovery of previously unknown phages, which is crucial for the development of rapid molecular biology-based methods for phage burden surveillance systems. The dominance of only a few phage groups in the dairy environment signifies the depth of knowledge gained over the past decades, which served as the basis for designing current phage control strategies. The presence of a correlation between phages and the type of starter cultures being used in dairies might help to improve the selection and/or design of suitable, custom, and cost-efficient phage control strategies.
KEYWORDS: abundance, bacteriophages, Caudovirales, dairy, diversity, Lactococcus lactis, Leuconostoc, mesophilic, metavirome, Siphoviridae, starter culture, whey
INTRODUCTION
Mesophilic cheese production relies on the activity of specific strains of lactic acid bacteria (LAB), employed as so-called starter cultures, to carry out the acidification of milk and produce the desired flavor. Starter cultures may contain one or more strains of Lactococcus lactis subsp. lactis, Lc. lactis subsp. cremoris, Lc. lactis subsp. lactis biovar diacetylactis, Leuconostoc mesenteroides subsp. cremoris or Le. pseudomesenteroides (1–4). Depending on the starter culture type, the number of strains can be single, multiple (up to six strains), or mixed (usually >50 different undefined strains). Single- and multiple-strain starters are normally defined-strain starters (DSS), unlike mixed-strain starters (MSS), whose strain composition is largely unknown (2, 3, 5–7).
Bacteriophages (phages) attacking starter cultures are of serious economic concern due to their negative impact on fermentations (8). In dairies, phages attacking Lc. lactis are the most frequent causes of delayed or arrested fermentations. Currently, Lc. lactis phages are divided into 10 taxonomic groups, with members of 3 groups being the most frequently encountered in dairies (9). These are the virulent 936 (now designated the Sk1virus genus) and c2 (now designated the C2virus genus) and the heterogeneous P335 quasispecies comprising both virulent and temperate members. The remaining Lc. lactis phage groups (1358, Q54, P087, 949, 1706, P034, and KSY1) have been isolated much less frequently and more from raw milk than from failed fermentations (9).
Phages attacking Leuconostoc species can affect the taste and appearance (e.g., eye formation) of the final product but do not normally interfere with the acidification process. They are grouped primarily into two major groups, i.e., group I (phages attacking Le. mesenteroides) and group II (phages attacking Le. pseudomesenteroides) (10, 11).
The development of an early warning system for the detection of phage attacks requires understanding of the development, diversity, and evolution of phages in the dairy environment, which in turn necessitates the integration of culture-dependent and culture-independent approaches. Traditionally, plaque assays, morphological characterization, restriction enzyme analysis, DNA-DNA hybridization, and PCR-based approaches and sequencing have been primarily employed to characterize emerging phage isolates (6, 9, 12–21). The recent developments of high-throughput sequencing and metagenomics have intensified knowledge on the dynamics and distribution of phages in complex ecosystems (8, 22–26). Although it is possible to generate next-generation sequencing (NGS) libraries from ∼1 ng DNA (27), dairy phages have so far not been studied by using these approaches. This is partly due to the lack of a defined method for extracting the phage DNA from dairy samples.
An established method for the characterization of individual phage isolates is based on a combination of polyethylene glycol (PEG) precipitation (28, 29) and CsCl gradient ultracentrifugation (30). Prior enrichment of the phages using susceptible host strains and subsequent purification provides highly concentrated phage stocks. CsCl gradients, particularly designed for phages propagated over susceptible host strains, enable the preparation of ultrapure phage lysates for morphological and genomic analyses. Adaptation of this method for metagenomic analysis of dairy phage communities requires several optimizations, as has previously been shown for studies of phage communities in the human gut (22). Important considerations include the need for adequate sample clarification prior to PEG precipitation and the maintenance of the diversity of the phages during subsequent purification steps, including CsCl gradient ultracentrifugation. Whey samples from fermentation delays or breaks due to phage attacks of starter strains can, for instance, contain a large amount of casein, which is known to interfere with the phage (DNA) extraction process. Thus, the properties of dairy samples (presence of bacteria, cell debris, various proteins, contaminating nucleic acids, pH variations, etc.) would be the primary challenges for the design of a reliable method.
In the present study, we developed a method for dairy phage metagenomics and investigated the notion that the Lc. lactis 936, P335, c2, and Leuconostoc phage groups are the most frequently encountered in the dairy environment. We assessed whey samples obtained from whey factories in Denmark (D) and Ireland (I) and from a dairy in Germany (G) at three time points. The wheys from Denmark and Germany were produced with undefined mesophilic mixed-strain cultures containing Lactococcus lactis subsp. lactis biovar diacetylactis and Leuconostoc species (DL starter cultures, or simply DL starters), a subgroup of MSS, whereas those from Ireland were produced with DSS. We analyzed the relative distribution of different Lc. lactis and Leuconostoc phages and assessed the overall taxonomic composition. Furthermore, we estimated the diversity of 936 and Leuconostoc phages based on analysis of the distribution of homologous receptor binding protein (RBP) sequences.
RESULTS
Sample clarity and recovery of phages.
High-speed centrifugation (with and without prior adjustment of pH [method 1 and method 3, respectively]) and membrane filtration (method 2) were assessed in terms of two performance parameters: (i) the removal of bulk protein (as examined visually) and (ii) the recovery of spiked phages. In terms of removal of bulk proteins, method 3 appeared to be the most efficient, followed by method 2, while method 1 was the least efficient, indicating that pH adjustment is a simple yet very useful technique to precipitate proteins in milk-based samples. In terms of spiked phages, methods 1 and 3 were found to be more efficient than method 2, retaining >50% of both φ29 and T4 compared to a retention of ∼50% using the last technique. Thus, we considered that high-speed centrifugation was the most efficient method when coupled with prior pH adjustment.
The use of 10% PEG 6000 often yielded precipitation of >90% of the phages in the clarified supernatant, and hence, we did not investigate an alternative phage precipitation technique.
Various CsCl density gradients were assessed in terms of their effectiveness to remove leftover whey proteins as well as to recover adequate amounts of spiked phages. Notably, multiple-layer gradients appeared to be generally less applicable than a modified two-layer CsCl gradient (Fig. 1). The use of multiple-layer gradients failed to provide the intended clarity and recovery due to obstruction of the flow of phages by a band of proteins formed above the 1.3-g ml−1 CsCl layer. The modified gradient, on the other hand, essentially diminished this band by relocating the majority of the proteins to the surface of the gradient. The fraction of spiked phages recovered in the final phage extract was also greater (>20%) when the latter gradient was used, compared to a retention of only <20% with multiple-layer gradients. All in all, adequate sample clarity and recovery (37% of φ29 and ∼24% of T4) could be achieved by combining method 3 with the modified two-layer CsCl gradient.
FIG 1.

Dairy phage metagenomic analysis workflow: overview of method optimization and analysis. Different techniques were assessed in terms of ability to separate bulk proteins and to recover spiked Bacillus subtilis φ29 and Escherichia coli T4 phages from whey samples. Broken-line boxes represent pathways with inadequate protein removal or heavy loss of φ29 and T4. Continuous boldface line boxes represent pathways with removal of a reasonable amount of proteins and/or recovery of a majority of the spiked phages. A continuous faded-line box represents an optional step that depends on the outcome of the previous step. The modified two-layer CsCl gradient was made of a layer of 1.7 g ml−1 CsCl in SM buffer overlaid by a layer of phage suspension (PEG pellet in 1.3 g ml−1 CsCl solution). The method's overall recovery efficiency was assessed following a CsCl equilibrium gradient or the last modified CsCl gradient ultracentrifugation. Local databases containing the genomes of dairy Lc. lactis and Leuconostoc phages and phylogenetically organized receptor-binding protein (RBP) groups were used to perform the respective mapping and Ublast analyses. NMWL, nominal molecular weight limit; PEG, polyethylene glycol; SM, sodium-magnesium (buffer); NGS, next-generation sequencing; GAAS, genome relative abundance and average size.
Phage losses were attributed mainly to pH shock, entrapment in the discarded pellet, and dispersion within the CsCl gradient. In the most efficient method (Fig. 1), the three factors contributed to respective losses of up to 6%, 20%, and 34% of φ29 and 14%, 35%, and 26% of T4.
Phage morphologies.
Transmission electron microscopy (TEM) analysis was conducted in order to examine the morphologies of the most predominant phages. Phage particles were observed in only six samples. Phages could not be observed in the remaining samples due to subthreshold phage titers (the limit of detection for TEM is ∼105 to 106 phages ml−1) (31, 32). Representative TEM micrographs are presented in Fig. 2. Notably, except for one sample (I4), all the other samples contained phages with isometric capsids and long noncontractile tails. The majority of the isometric-headed phages exhibited tail lengths of 141 nm (D3) (Fig. 2, phage D3 on the right) to 151 nm (phage D4 on the left), but shorter tail lengths were also measured, ranging from 120 nm (D5, phage on the right) to 139 nm (D2, phage on the right). In sample I4, a prolate-headed phage was observed with a tail length of 100 nm. The phages did not demonstrate distinct baseplates but rather exhibited slightly enlarged tail terminal ends. Six of the phages demonstrated neck passage structures (NPS) at the head-tail junction. The overall dimensions and morphologies of the isometric-headed phages are generally similar to those documented for previously isolated phages of the 936 group (15, 33–36).
FIG 2.

Representative transmission electron micrographs of phages extracted from whey mixtures. All the micrographs except that of I4 show 936-like phages (I4 is a c2-like phage). Arrowheads indicate neck passage structures (NPS). Numbers below the micrographs are the head dimensions and tail lengths of the phage particles, respectively. Micrographs are shown at identical magnifications.
Optimum parameters.
Mock communities were mapped to db-P or db-RBP databases (in-house databases; see Materials and Methods) in order to optimize the parameters for analysis of sample metaviromes. Reads were mapped to db-P as expected when the length and similarity thresholds were adjusted to 50% and 80%, respectively (see Fig. S1 in the supplemental material). Some reads from the 936 phages were mapped to the genomes of P335 and 949 phages due to relatively short regions of DNA sequence homology between the phages (see Fig. S2 in the supplemental material). Such regions correspond to, for instance, orf11 and orf47 (nps) of phage TP901-1 (a P335 species) and gp047, gp062, gt004, the intergenic space between gp089 and gt005 (gp089-gt005), gt005, and gp128 of phage 949 (a 949 species) (Fig. S2).
Ublast analysis of the mock communities against db-RBP yielded the expected profile when a stringent mapping threshold was used, i.e., 95% minimum query length and similarity (Fig. S1B and S1C). Importantly, this analysis also revealed RBP sequences that were not represented in the mock communities, such as the 1727 and Q49 RBP variants (Fig. S1B). Further analysis of these RBP variants indicated that they show considerable sequence similarity to the fd13 (e.g., fd13 and CaseusJM1) (75 to 80%) and the HD6 (e.g., HD6 and jm3) (69 to 82%) RBP clusters, despite being phylogenetically unrelated. Additionally, most of the RBP sequences of the 936 phages exhibit high sequence similarity toward the 5′ half, which could have increased the possibility of detection of sequences that were not included in the mock communities.
Analysis of spiked phages. (i) Reference mapping.
Metavirome reads were mapped to the spiked phage genomes as summarized in Table 1. Nearly the entire φ29 genome was mapped in all cases (99.9%), with significant fluctuation in depth of coverage along the reference genome. At the terminal ends of φ29, regions with no coverage were observed encompassing 11, 18, 6, 8, and 17 bases (D3, D4, D5, G1, and I2, respectively) (see Fig. S3A in the supplemental material). The fraction of reads mapped to φ29 ranged from 0.2% (D4) to 7.9% (D5), which was generally higher than the fraction of reads mapped to T4 (0.2 [D4] to 4.3% [D5]). This was also consistent with the average coverage (Table 1). With regard to T4, although the mapped read fraction and the average coverage were relatively low, the mapping was nearly complete in D5 (99.5%) and G1 (99.4%). The unmapped portion of the genome corresponded to the terminal 837 (D5) and 957 (G1) bases (Fig. S3B). A very high mapping coverage was also seen in D3 and D4 (94.6% and 93.2%, respectively), although multiple gaps were formed along the alignment. I2 provided the least T4 coverage (58%), which is reasonable given a very small metavirome size (Fig. S3B).
TABLE 1.
Summary of the sequencing results for spiked whey samples
| Sample IDa | Total no. of reads |
Avg read length |
% of reads mapped to reference genomes |
Avg coverage |
||||
|---|---|---|---|---|---|---|---|---|
| Before trimming | After trimming | Before trimming | After trimming | φ29 | T4 | φ29 | T4 | |
| D3 | 2,135,308 | 2,135,098 | 251.0 | 152.0 | 0.4 | 0.4 | 78.4 | 9.08 |
| D4 | 2,731,990 | 2,731,728 | 251.0 | 175.9 | 0.2 | 0.2 | 74.0 | 7.53 |
| D5 | 2,699,702 | 2,699,300 | 251.0 | 209.8 | 7.9 | 4.3 | 2,353.14 | 142.26 |
| G1 | 3,673,882 | 3,673,338 | 251.0 | 201.2 | 6.8 | 2.1 | 2,594.51 | 78.29 |
| I2 | 331,230 | 331,166 | 251.0 | 204.3 | 3.5 | 0.4 | 132.54 | 1.78 |
D, Denmark; G, Germany; I, Ireland.
(ii) De novo assembly.
The method's power to yield adequate sequences for full genome characterization was inferred from de novo assembly of the spiked phages. Nearly the entire φ29 genome was assembled, except the proximate terminal regions, where 56, 1, 6, 8, and 17 bases were absent (D3, D4, D5, G1, and I2, respectively). The assembly was further confirmed by a BLAST query to have significant similarity to the φ29 genome (see Table S3 in the supplemental material). Unlike φ29, T4 was assembled into several short contigs (except in D5 [168,032 bases] and G1 [104,664 bases]), which was apparently due to an inadequate amount of reads to cover the very large T4 genome (Table S3). Importantly, many of the BLAST hits corresponding to the T4 contigs were described as enterobacterial phage RB55 or RB59 (see Table S5 in the supplemental material). These phages belong to the T4virus phage group with 99.96% identity to each other and 99.80% similarity to the T4 phage (37).
Analysis of high-throughput sequencing (HTS) metagenome sequences. (i) Input sequence threshold.
To test if variations in the amount of input sequences can affect the estimation of the relative abundance of phage species and RBP diversity, a test sequencing run and preliminary analysis was carried out with D1 prior to the other metaviromes. With deep sequencing, a total of ∼9.5 million paired-end (PE) reads were generated, ∼5% of which were removed by trimming. In order to assess the impact of varying the amount of input sequences on phage relative abundance and diversity estimations, an array of randomly selected sequences were tested (see Table S2 in the supplemental material). Eventually, the minimum requirements for an accurate estimation of the relative abundance and diversity of phages were found to be ∼50,000 and ∼1,000,000 reads, respectively, indicating that the latter demands a large number of input sequences. Accordingly, we generated at least 1 million reads per each of the study samples. Sequencing of 11 samples generated a total of >12.6 million PE reads (see Fig. S4 in the supplemental material), of which 0.04% were discarded by trimming.
(ii) Comparison of phage content between pairs of metaviromes.
In order to assess the overall similarity in phage content between the metaviromes, they were mapped to each other in a pairwise manner as outlined in Table 2, resulting in a matrix of 121 mapping scores. The matrix clearly indicated that DSS and DL starter-derived whey samples vary considerably in terms of overall phage composition. The similarity score from a comparison of samples derived from the same starter culture type (DSS or DL) was generally higher than when DSS starter-derived samples were compared to DL starter-derived samples. This suggests the presence of significant overlap in phage composition among samples corresponding to the same starter culture type. Nonetheless, the overall composition of I2 appeared to be relatively similar to DL starter-derived samples, which was unexpected for a sample that was derived from DSS starters.
TABLE 2.
Pairwise comparison between metaviromes as carried out by reads-to-contigs mappinga
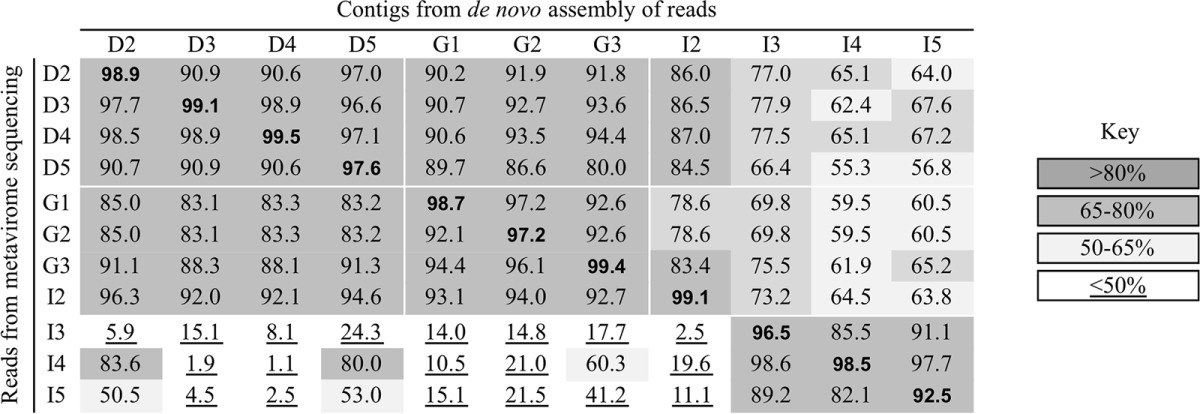
The overall similarity in phage population between whey samples was assessed by pairwise mapping of the metavirome sequences, i.e., by mapping trimmed reads (rows) to contigs from de novo assembly (columns). The mapping threshold was set to a minimum of 50% query length and 80% identity. Entries are shown in bold to indicate that reads and contigs are cognate, i.e., contigs used as references were assembled from the same metavirome, which yielded an expected high average score of ∼97.8 ± 1.9%. All the other entries represent the similarity between unrelated (noncognate) metaviromes. The key next to the table highlights the grouping of the metaviromes according to overall similarity. D, Denmark; G, Germany; I, Ireland.
(iii) BLAST comparison of selected contigs.
To test whether certain phage strains were responsible for the observed sequence similarity among samples derived by the same type of starter, a set of contigs selected for being the highest in read coverage in the respective metavirome assemblies were analyzed by a BLAST query against the NCBI nr database. Notably, the contigs selected from 10 of the 11 metaviromes displayed the greatest similarity to the lactococcal phage sequences in the database (see Table S4 in the supplemental material). Accordingly, the dominant sequence entities in the Danish and German metaviromes appeared to be those of Lc. lactis 936 phages, whereas in the Irish metaviromes sequences of Lc. lactis 936 phages (I2), bacteria (particularly Streptococcus pyogenes) (I3), or Lc. lactis c2 phages (I4 and I5) were dominant. Contigs with no sequence similarity to available sequences were also represented in G3 and I3 (Table S4).
As the majority of the selected contigs were rather short, we tested the universality of the observed finding by analyzing a set of contigs selected for being the longest. To this end, sequences of lactococcal phages (in D2 to D5 and I2), bacteria (in G2, G3, and I3 to I5), or a mixture of phages and bacteria (in G1) appeared to represent the dominant sequence entities in the metaviromes (Table S5). Contigs with no sequence similarity to available sequences were also represented in G1 and G3, possibly indicating sequences of novel phages or viruses or of contaminating host cells.
(iv) Mapping reads to reference genomes.
To estimate the fraction of phages associated with mesophilic cheese production, namely, Lc. lactis and Leuconostoc phages, in the metaviromes, reads were mapped to db-P (in-house database of reference genomes). A total of 1 million randomly selected trimmed reads were used, except for I2, which was mapped entirely due to its small size (∼0.33 million reads). Of the 11 metaviromes, 9 presented a very high proportion of mapped reads, with the average being ∼74% (Fig. S4). The two exceptional metaviromes presenting a relatively low proportion of reads mapping to the database, namely, I3 and I5 (∼9.6% and ∼51.5%, respectively), were both from the Irish whey factories (Fig. S4).
Of the 11 metaviromes, 8 (encompassing all the Danish and German samples and the Irish sample I2) demonstrated the dominance of sequences of the 936 phage group (Table 3). The fraction of mapped reads ranged from 90.0% to 91.5% (in the Danish samples), 85.0 to 93.0% (in the German samples), and 0.6% to 95.1% (in the Irish samples). In the three Irish metaviromes, where 936 phages were a minority, namely, I3, I4, and I5, sequences of the c2 phage group appeared to be the most frequent. The fraction of mapped reads ranged from 0.2% (I2) to 97.8% (I4) and averaged ∼20.8%. The mapping fraction for I3, I4, and I5 alone was significantly higher than for the rest of the samples, averaging ∼80.6%. Therefore, phages of the 936 group were the most frequent in DL starter-derived samples, while phages of the c2 group represented the vast majority in DSS starter-derived samples.
TABLE 3.
Relative abundance of Lc. lactis and Leuconostoc phages in whey mixture metavirome sequences
| Reference genome | Mapped reads (%)a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D2 | D3 | D4 | D5 | G1 | G2 | G3 | I2 | I3 | I4 | I5 | |
| 936 | 90.4 | 91.4 | 91.5 | 90.0 | 85.0 | 92.2 | 93.0 | 95.1 | 2.1 | 0.7 | 0.6 |
| P335 | 5.9 | 4.1 | 4.5 | 5.1 | 6.8 | 3.9 | 3.7 | 2.3 | 36.1 | 0.9 | 1.9 |
| c2 | 2.2 | 0.9 | 0.8 | 1.2 | 1.0 | 0.8 | 0.4 | 0.2 | 47.6 | 97.8 | 96.3 |
| 1358 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| P034 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 |
| 1706 | 0.0 | 0.0 | 0.0 | 0.0 | 0.6 | 0.0 | 0.0 | 0.0 | 0.3 | 0.0 | 0.1 |
| Q54 | 0.3 | 1.4 | 0.9 | 0.7 | 0.6 | 0.7 | 0.3 | 0.9 | 0.2 | 0.0 | 0.0 |
| KSY1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.6 | 0.0 | 0.0 | 0.6 | 0.1 | 0.1 |
| P087 | 0.1 | 0.6 | 0.6 | 0.1 | 0.0 | 0.0 | 0.2 | 0.0 | 0.2 | 0.0 | 0.1 |
| 949 | 1.0 | 1.5 | 1.4 | 1.7 | 1.7 | 1.6 | 1.2 | 1.4 | 0.6 | 0.1 | 0.1 |
| Leuconostoc | 0.1 | 0.1 | 0.1 | 1.0 | 4.1 | 0.1 | 1.3 | 0.0 | 0.5 | 0.1 | 0.1 |
| Satellites | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 11.6 | 0.3 | 0.7 |
Values (scores) represent the percentages of reads mapped to reference genomes and were obtained from statistics generated from mapping of reads to the genome sequences of Lc. lactis and Leuconostoc phages. The mapping threshold was set to a minimum of 50% query length and 80% identity. D, Denmark; G, Germany; I, Ireland.
Compared to the 936 and c2 phages, sequences of the P335 phage group demonstrated a more consistent frequency. The fraction of mapped reads was in the range of 0.9% (I4) to 6.8% (G1), except in I3 (36.1), and averaged ∼6.7%. Contrary to the finding observed in the mock communities (Fig. S2), the vast majority of the P335 genomes were covered by reads, although numerous gaps were also present (see Fig. S5 in the supplemental material). This is suggestive of mapping of primarily P335 sequences, although it is impossible to rule out nonspecific mapping of sequences from the 936 phages. Furthermore, reads that mapped to the 949 genomes averaged ∼1%: however, many of these reads were concentrated around regions corresponding to gp047, gp062, gt004, the gp089-gt005 intergenic space, gt005, gt006, and gp128 of phage 949 (Fig. S5). This might indicate nonspecific mapping of reads potentially originating from 936 phages. The remaining phages were found to be insignificant, except Leuconostoc and satellite phages, which represented 4.1% and 11.6% of the phage populations in G1 and I3, respectively. These results generally indicate that the 936 species is highly dominant in dairies using DL starters, as is the c2 species in most dairies using DSS.
The presence of a large fraction of unclassified reads in some of the metaviromes, such as D3 and I5, prompted us to perform a BLAST analysis of selected de novo-assembled contigs. The results indicated lactococcal phage sequences to be the most frequent hits for a set of contigs selected for being the highest in read coverage (see Table S6 in the supplemental material). Of these, the sequences of 936 phages appeared to be the majority, except for the sequence of c2 phages, which was occasionally encountered in I4 and I5. A set of contigs selected for being the longest presented the greatest similarity to phage or viral sequences. Many of these corresponded to sequences of spiked phages (see Table S7 in the supplemental material), while the fraction of lactococcal phages appeared to be rather low (∼5%). BLAST also revealed bacterial sequences (likely from contamination) in the metavirome extracts, mainly in the German and Irish samples (Table S7).
(v) Taxonomic composition.
To estimate the taxonomic composition of the metaviromes, we executed a BLAST comparison with the RefSeq complete viral genomes proteins. Taxonomic affiliations were deduced from the best BLAST hit (threshold of 50 on the BLAST score). The ratio of taxonomic affiliation was very high in the majority of the samples, although it ranged from 12.5% (I3) to 91.8% (D2), with an average affiliation of 63.8%. Notably, no obvious correlation between the degree of taxonomic affiliation and the origin of the samples was observed.
To estimate the proportion of each virotype in the initial sample in terms of the number of viral particles, the taxonomic composition normalized by the genome length of the virotypes was determined, computed via the GAAS tool (38). Notably, certain virotypes appeared to dominate in the metaviromes (Table 4). Double-stranded DNA (dsDNA) viruses belonging to the order Caudovirales and the family Siphoviridae accounted for the majority of the viruses identified. At lower taxonomic ranks, lactococcal phages were found to be highly abundant in all the samples. In all the Danish and German samples and in the Irish sample I2, Lc. lactis 936 phages P008, jm2, P680, jm3, biL170, 340, phage 7, jj50, biBB29, 712, and sk1 appeared to be evenly distributed, whereas Lc. lactis c2 phages bIL67 and c2 dominated in the remaining samples, which were all from Ireland (Table 4). P335 phages generally exhibited low abundance, except in I3 (∼7%). Sequences corresponding to phages TP901-1, BK5-T, Tuc2009, ul36, bIL286, and φLC3 were relatively abundant in I3 (Table 4). In this sample, satellite phages represented ∼17% of the viruses (bIL310, ∼12%; bIL311, ∼1%; bIL312, ∼4%). These phages, however, appeared to be insignificant in the other metaviromes (Table 4).
TABLE 4.
Relative abundance of sequences of different phages in whey mixture metavirome sequencesa
| BLAST hit (bacterial genus and phage name) | Sequence similarity (%) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| D2 | D3 | D4 | D5 | G1 | G2 | G3 | I2 | I3 | I4 | I5 | |
| Lactococcus | |||||||||||
| 936 group | |||||||||||
| P008 | 15.4 | 24.2 | 23.0 | 15.4 | 11.3 | 21.8 | 16.2 | 8.4 | 0.0 | 0.0 | 0.0 |
| jm2 | 15.6 | 11.0 | 11.3 | 14.0 | 9.8 | 14.3 | 18.3 | 46.6 | 0.1 | 0.0 | 0.0 |
| P680 | 10.7 | 8.5 | 9.2 | 7.3 | 8.5 | 7.2 | 8.1 | 3.3 | 0.0 | 0.0 | 0.0 |
| jm3 | 11.5 | 9.5 | 9.3 | 8.9 | 18.1 | 9.1 | 7.5 | 4.0 | 0.0 | 0.0 | 0.0 |
| bIL170 | 8.4 | 8.3 | 9.6 | 7.0 | 7.6 | 9.2 | 9.2 | 6.9 | 0.0 | 0.0 | 0.0 |
| 340 | 7.7 | 9.0 | 8.3 | 5.8 | 4.6 | 8.2 | 5.8 | 2.4 | 0.0 | 0.0 | 0.0 |
| φ7 | 8.3 | 7.6 | 8.4 | 7.3 | 7.4 | 9.9 | 10.1 | 2.3 | 0.1 | 0.2 | 0.1 |
| jj50 | 5.9 | 7.6 | 6.4 | 5.9 | 4.6 | 7.1 | 8.6 | 4.5 | 0.0 | 0.0 | 0.0 |
| bIBB29 | 4.9 | 5.7 | 6.3 | 3.4 | 2.1 | 3.3 | 5.2 | 0.8 | 0.0 | 0.0 | 0.0 |
| 712 | 3.6 | 3.8 | 3.6 | 2.6 | 2.4 | 3.8 | 3.2 | 1.8 | 0.1 | 0.0 | 0.0 |
| sk1 | 2.4 | 2.3 | 2.2 | 2.4 | 1.5 | 3.5 | 2.2 | 0.0 | 0.1 | 0.0 | 0.0 |
| Q54 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 |
| SL4 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 5.6 | 0.0 | 0.0 | 0.0 |
| ASCC191 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.9 | 0.0 | 0.0 | 0.0 |
| sk1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.4 | 0.0 | 0.0 | 0.0 |
| P335 group | |||||||||||
| TP901-1 | 0.3 | 0.2 | 0.3 | 0.2 | 1.2 | 0.4 | 0.1 | 0.3 | 2.0 | 0.0 | 0.1 |
| BK5-T | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 1.7 | 0.0 | 0.2 |
| Tuc2009 | 0.3 | 0.1 | 0.1 | 0.3 | 0.1 | 0.4 | 0.0 | 0.1 | 1.6 | 0.0 | 0.0 |
| φLC3 | 0.4 | 0.2 | 0.4 | 0.4 | 1.1 | 0.3 | 0.1 | 0.1 | 0.8 | 0.0 | 0.0 |
| bIL286 | 0.3 | 0.1 | 0.1 | 0.3 | 0.3 | 0.0 | 0.1 | 0.1 | 1.3 | 0.2 | 0.2 |
| r1t | 0.5 | 0.1 | 0.1 | 0.4 | 0.0 | 0.1 | 1.2 | 0.1 | 0.1 | 0.0 | 0.0 |
| ul36 | 0.1 | 0.1 | 0.1 | 0.3 | 0.2 | 0.4 | 1.0 | 0.0 | 1.5 | 0.0 | 0.1 |
| BM13 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.1 | 0.0 | 0.0 | 0.3 | 0.0 | 0.0 |
| P335 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.6 | 0.1 | 0.1 |
| bIL285 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.6 | 0.0 | 0.0 |
| bIL309 | 0.0 | 0.2 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.4 | 0.0 | 0.0 |
| c2 group | |||||||||||
| c2 | 0.6 | 0.1 | 0.1 | 0.3 | 0.0 | 0.0 | 0.1 | 0.0 | 27.6 | 66.9 | 52.0 |
| bIL67 | 1.6 | 0.0 | 0.0 | 0.5 | 0.5 | 0.0 | 0.0 | 0.0 | 21.2 | 31.6 | 44.9 |
| Satellite phages | |||||||||||
| bIL310 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 12.7 | 0.3 | 0.6 |
| bIL311 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.8 | 0.0 | 0.2 |
| bIL312 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 4.3 | 0.1 | 0.2 |
| Leuconostoc | |||||||||||
| φLN25 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 |
| P793 | 0.0 | 0.0 | 0.0 | 0.2 | 1.2 | 0.0 | 0.4 | 0.0 | 0.0 | 0.0 | 0.0 |
| φLN04 | 0.0 | 0.0 | 0.0 | 0.2 | 1.4 | 0.0 | 0.5 | 0.0 | 0.0 | 0.0 | 0.0 |
| Lmd1 | 0.0 | 0.0 | 0.0 | 0.4 | 0.8 | 0.0 | 0.3 | 0.0 | 0.0 | 0.0 | 0.0 |
| φLN03 | 0.0 | 0.0 | 0.0 | 0.1 | 0.7 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 |
| φLN6B | 0.0 | 0.0 | 0.0 | 0.2 | 0.3 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| φLN12 | 0.0 | 0.0 | 0.0 | 0.0 | 0.3 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 |
| Lactobacillus | |||||||||||
| Ld25A | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 |
| Lc-Nu | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 2.0 | 0.0 | 0.1 |
| φAQ113 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.5 | 0.0 | 0.0 |
| Bacillus | |||||||||||
| φ29 | 0.0 | 0.7 | 0.5 | 14.0 | 12.7 | 0.0 | 0.0 | 5.8 | 0.0 | 0.0 | 0.0 |
| Shigella | |||||||||||
| SfIV | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.9 | 0.0 | 0.2 |
| Escherichia | |||||||||||
| fiAA91-ss | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 3.2 | 0.0 | 0.0 |
| Others | 0.3 | 0.1 | 0.1 | 1.2 | 0.5 | 0.4 | 0.2 | 0.7 | 6.1 | 0.1 | 0.3 |
Relative abundance was considered as the fraction of reads affiliated to NCBI's RefSeq database (threshold of 50 on the BLAST score) accessed via the Metavir analysis pipeline. All hits with an abundance level of <0.25% in all metaviromes were excluded for brevity. D, Denmark; G, Germany; I, Ireland.
(vi) Diversity of 936 and Leuconostoc phages.
To estimate the diversity of phages, we analyzed reads by Ublast against db-RBP (in-house database of RBP sequences). The result is summarized in Table S8 in the supplemental material. Of 19 RBP variants representing 936 phages, a maximum of 4 appeared to be predominant per sample. Two variants, namely, SCH and phage 7, were found to be widely distributed, whereas others, including 645, ASCC406, 1727, and ΦL.6, seemed to be associated with many of the samples. Notably, SCH was almost the sole variant detected in G1, while phage 7 was the major variant in D5, G3, and I2. In the Irish samples I3, I4, and I5, generally very small quantities of RBPs were detected, which is consistent with the low prevalence of 936 phages in these samples (Table S8).
Sequences corresponding to the RBP sequences of Leuconostoc phages were detected in just three samples (D5, G1, and G3) (see Table S9 in the supplemental material), with the LN6B variant being more frequent than the others. A very small number of sequences corresponding to the P793 variant were also detected in G1 and G3 (Table S9). The LN6B variant, together with the P793 and φLN23 variants, represents Le. pseudomesenteroides phages, possibly suggesting the dominance of these phages over Le. mesenteroides phages in the dairies involved.
DISCUSSION
One of the primary challenges with the extraction of phages from different dairy metagenomic samples is the difficulty involved in separating various residues (proteins, bacterial cells, and cell debris) before concentrating the phages. The pH variation among different dairy samples (bulk starter, milk, whey, etc.) partly explains the processing challenges. The extraction method described in this study included a step whereby samples were adjusted to pH ∼4.6, which has been shown to precipitate ∼80% of the total nitrogen in dairy samples (39). Incorporation of this step facilitated the removal of a significant fraction of residues by low-speed centrifugation. However, there was unexpected loss of phages during this initial sample processing stage, which was attributed mainly to pH shock (loss of infectivity) and entrapment of phages in the pellet. The former loss seemed reasonable, as the spiked phages were not adapted to the dairy environment and hence could have been relatively sensitive to low pH. Conversely, dairy phages tolerate low pH, and hence minimal (if any) losses may be expected.
Upon PEG precipitation of phages, smaller proteins likely pose the greatest challenge, as they precipitate concurrently with the phages. These proteins are impossible to separate by standard CsCl gradient ultracentrifugation (40), as they tend to aggregate at the interface between the phage suspension and the CsCl layer, preventing the flow of phages to their buoyant density (isopycnic point). Thus, a modified two-layer CsCl gradient was optimized to lower the blocking effect of proteins on the flow of the phages. It consists of an upper layer of phage suspension (prepared by resuspending the PEG-precipitated pellet with 1.3 g ml−1 CsCl in SM buffer, consisting of 18 mM MgSO4·7H2O, 0.1 M NaCl, 0.05 M Tris-HCl, pH 8.0) and a lower layer of 1.7 g ml−1 CsCl. The usefulness of this gradient lies in its potential to alter the direction of flow of proteins (density, <1.3 g ml−1) without affecting the direction of flow of (most) phages (>1.3 g ml−1), which was proved by the aggregation of proteins on the top of the gradient. The higher density of the phage suspension increases the physical distance between the phages and smaller proteins within the gradient, which ultimately increases the sample clarity. The absence of intermediate CsCl layers (1.45 and/or 1.5 g ml−1) increases the phage diversity being extracted as it promotes the gathering of the phages just above the 1.7 g ml−1 CsCl layer.
Overall, the extraction method enables progressive removal of bulk proteins while retaining an adequate fraction of phages for metagenomic studies. The recovery of adequate φ29 and T4 phages for assembly of almost the entire genomes, as demonstrated by reference mapping (both phages) and de novo assembly (only φ29), demonstrates this. Of note, the observed drop in sequence coverage toward the terminal ends of the genomes is attributed to the Nextera XT transposome technology (41). The protocol's ability to recover T4, which has a very large genome (>8 times the size of φ29) (42, 43), proves its suitability for metagenomic characterization of a wide variety of phages, including dairy phages. Yet, the low success rate with de novo assembly of the T4 genome suggests its strength toward phages of smaller genome sizes. Besides, the relatively low efficiency of recovery would not allow the protocol to be combined with, for instance, TEM examination due to a detection threshold of 105 to 106 phages ml−1 (31, 32). Thus, further optimizations are needed to improve the efficiency of recovery, thereby to extend the method's applicability.
The influence of starter culture type on the composition of phages is evident from the predominance of 936 phages in whey from DL starters and of primarily c2 phages in whey from DSS. This, combined with the relatively conserved nature of the genomes of 936 and c2 phages, could have contributed to the observed overall similarity among the different metaviromes. The situation might have been different if the heterogeneous P335 phages were dominant, since these phages show much less overall genome conservation (44, 45). Nevertheless, the dominance of 936 phages in Danish and German whey appears to be due to the employment of undefined cultures, whereas the dominance of c2 phages in most Irish whey is likely due to the application of strains sensitive to specific phages rather than to the starter type itself.
The dominance of 936 phages in the present metavirome study is consistent with most previous lactococcal phage isolation studies (6, 18, 36, 46–53). Detection of mostly SCH and phage 7 and occasionally 645, ASCC406, 1727, and ΦL.6 RBP variants may suggest the presence of a limited diversity of these phages in the dairies. Phages within these RBP variants infect mostly strains of Lc. lactis subsp. cremoris, while the 645 RBP variant infects strains of both Lc. lactis subsp. lactis and Lc. lactis subsp. cremoris (35). Furthermore, the presence of NPS in the majority of phages is consistent with the recent increase in the frequency of phages displaying this structure (18, 36, 52, 54). This highlights the view that NPS might enhance host range and adaptation (33), though the NPS of the temperate TP901-1 phage does not appear to be important for infection, assembly, and stability (55). Thus, the function of NPS for 936 phages is yet to be elucidated.
Probably, the most striking finding of the analysis of the c2 phages is their dominance in the majority of samples derived from defined cultures, which is in stark contrast to the notion that the frequency of isolation of members of c2 species has dropped lately due to the adoption of antiphage strategies (14). As far as we are aware, no previous study has published similar findings in Ireland. Elsewhere, c2 phages have occasionally been reported to be more frequent than other lactococcal phages (56, 57). The finding may indicate that the c2 phages in Irish dairies are relatively tolerant to cheese milk thermal treatments, which is in accordance with a recent study that reported that many c2 phages survive traditional cheese milk heat treatments (58). Phages of the c2 group require the host receptor phage infection protein (PIP) in order to attack sensitive strains (59, 60). Strains that carry mutations in pip acquire complete resistance against c2 phages (61). It appears that starter culture manufacturers in Ireland do not generally select for pip mutants or that other genes such as yjaE (62) could replace pip during infection.
The relatively low abundance of P335 phages appears to be in accordance with the reported decrease in the frequency of isolation of P335 phages due to the adoption of antiphage strategies (14). It is possible that the increased frequency of P335 phages in I3 was primarily due to induction of temperate P335 prophages during cheese production. The increased frequency of satellite phages in the same whey mixture might thus be due to coinduction of P335 and satellite phages. Chopin et al. suggested that satellite phages of Lc. lactis possibly rely on phages from the P335 group for multiplication (44). It is therefore possible that the satellite phages in I3 have acquired certain modules from P335 phages prior to induction, which may indicate the presence of coevolution of these phages in the corresponding dairy.
Lc. lactis phages other than 936, P335, and c2 were generally detected much less frequently. Although the 949 phage group was detected in all whey mixtures, further evaluation of individual mappings revealed mostly localized mapping of reads at the regions corresponding to gp047, gp062, gp089 to gt005, and gp128 in 949, likely suggesting nonspecific mapping of reads originating potentially from 936 phages such as CB13 (63). This is consistent with the taxonomic composition analysis, which revealed just a small fraction of 949 phages. Furthermore, Leuconostoc phages were represented mostly by the LN6B RBP variant, indicating that Le. pseudomesenteroides phages were relatively more common. The exceptional abundance of Leuconostoc phages in G1 was reflected also by the detection of a large number of Leuconostoc phage RBP in this metavirome.
In conclusion, the method described in this study allows metagenomic studies of dairy phages. Particularly, the CsCl gradient enables the isolation of phages with a wide range of genome sizes. The findings from this study support the previous notion that Lc. lactis phages of the 936, P335, and c2 species are the most frequently encountered in the dairy environment. The composition of the phage population was somehow linked to the starter culture propagation regime, as shown from the dominance of 936 phages in whey from DL starters and that of c2 phages in (most) whey from DSS. Concurrent increases in the frequency of P335 and satellite phages may indicate coinduction and evolution of these phages. Future work would possibly describe the mechanisms whereby starter culture types influence the composition of the phage population.
MATERIALS AND METHODS
Optimization of isolation of phage communities from whey.
Whey mixtures used in this study were stored at −60°C until needed to prevent potential inactivation of phages. Test whey samples were thawed (in a water bath, ≤30°C), spiked with ∼106 ml−1 phages of Bacillus subtilis φ29 (42) and Escherichia coli T4 (43), and centrifuged at 300 × g for 5 min. The supernatant was incubated with 1 M NaCl (wt/vol) (Sigma-Aldrich, USA) for 1 h at 4°C, and the mixture was centrifuged at ∼28,000 × g for 15 min (method 1), filtered by 100,000 nominal molecular weight limit (NMWL) Amicon centrifugal filters (Merck Millipore, USA) according to the supplier's recommended procedures (method 2), or adjusted to pH ∼4.6 using 1 M HCl and/or 1 M NaOH and centrifuged at ∼28,000 × g for 15 min (method 3) (Fig. 1). The supernatant from methods 1 and 3 was incubated with 10% PEG 6000 (wt/vol) (Sigma-Aldrich) for 1 h at 4°C, and the phages were pelleted by centrifugation at 15,000 × g for 15 min. The resultant pellet was resuspended by 1 ml SM buffer and incubated overnight at 4°C. The pellet from method 3 was resuspended also by 1 ml SM buffer supplemented with 1.3 g ml−1 CsCl (Sigma-Aldrich) and incubated as above. The phage suspension in SM buffer was purified by CsCl block (106,750 × g for 2.5 h) and equilibrium (175,000 × g for 22 h) gradient ultracentrifugations (both at 15°C), while that in CsCl-containing SM buffer was purified by a modified two-layer CsCl gradient centrifuged at 106,750 × g for 2.5 h at 15°C (Fig. 1). Phages were collected from the portion of the gradient containing visible bands or corresponding to 1.4 to 1.5 g ml−1 density using a needle installed on a syringe (the refractive index was measured using a handheld refractometer [Bellingham + Stanley, UK]). The phage extract was stored at −20°C until needed.
The samples were examined visually for clarity and by the double-agar plaque assay (22, 64) for recovery of spiked phages following the NaCl centrifugation, PEG precipitation, and CsCl gradient ultracentrifugation steps.
Isolation of phage communities and electron microscopy.
Phages were isolated from 20 ml of whey mixture using method 3 combined with the modified two-layer CsCl gradient (Fig. 1). Whey mixtures D3, D4, D5, G1, and I2 were spiked with ∼106 ml−1 of φ29 and T4 phages prior to isolation, whereas D1, D2, G2, G3, I3, I4, and I5 were isolated without spiking. The phages were analyzed by transmission electron microscopy (TEM) to assess their morphotypes. Adsorption of CsCl-purified phage lysates to an ultrathin carbon film floated on a freshly coated mica sheet and negative staining with 2% (wt/vol) uranyl acetate were performed as previously described (65). The film was applied to a 400-mesh copper grid (Agar Scientific, United Kingdom), and images of the phages were taken using a MegaView G2 charge-coupled-device (CCD) camera (Emsis, Germany) installed onto a Tecnai 10 transmission electron microscope (FEI, Netherlands) operated at an acceleration voltage of 80 kV.
Extraction of phage metaviromes.
The phage lysate was dialyzed against dialysis buffer (10 mM NaCl, 50 mM Tris-Cl [pH 8.0], 10 mM MgCl2) essentially as described by Sambrook and Russell (40). The pH of the suspension was adjusted to 7.25 ± 0.25 using 1 M HCl and/or 1 M NaOH, and 0.1 volume of 10× DNase I reaction buffer (10 mM Tris-HCl, 2.5 mM MgCl2, 0.1 mM CaCl2), 50 units ml−1 of DNase I (Sigma-Aldrich), and 1 μl ml−1 of RNase A solution (R6148; Sigma-Aldrich) were added. The mixture was incubated for 30 min at 37°C, and DNase I was inactivated by the addition of 10 mM EDTA (pH 8.0) and 1% SDS (both from Sigma-Aldrich). The mixture was treated with 50 μg ml−1 proteinase K and incubated at 55°C for 1 h. Phage DNA was extracted with the GenElute Bacterial Genomic DNA kit (Sigma-Aldrich) as per the manufacturer's protocol and eluted with 50 μl elution buffer. The DNA was stored at −20°C until required.
High-throughput sequencing and analysis. (i) Phage sequence databases and phylogenetic tree.
The genomes of Lc. lactis (936, P335, c2, 1358, Q54, P087, 949, 1706, P034, KSY1, bIL310, bIL311, and bIL312) and Leuconostoc phages available in GenBank (https://www.ncbi.nlm.nih.gov/genome/viruses/) until 15 June 2016 were downloaded and, together with genomes obtained from in-house sources, were used to construct a phage full-genome sequence database called db-P. Accessible RBP sequences of 936 and Leuconostoc phages were used (i) to construct an in-house RBP database called db-RBP and (ii) to generate a phylogenetic tree (parameters: neighbor-joining method, Jukes-Cantor distance measured with bootstrap analysis with 100 replicates). According to this phylogenetic tree, the RBPs in the db-RBP were separated into 19 variants and were later used for estimation of phage diversity (see below).
(ii) Optimization of analysis parameters.
Three Illumina NGS sequencing-simulated metaviromes (mock communities) were constructed to optimize parameters for phage relative abundance and diversity estimations. Each mock community contained 1 million reads from selected Lc. lactis 936, c2, and Leuconostoc phages (see Table S1 in the supplemental material). The mock communities were mapped to db-P (CLC Genomics Workbench 8.5.1; Qiagen, Denmark) and queried against db-RBP variants (Ublast algorithm [66]) through varying the query length and similarity thresholds until the expected phage distribution was achieved. The outputs from db-RBP BLAST (amount of reads aligned to each RBP) were transformed into reads per kilobase per million mapped reads (RPKM) values (67) through normalization of the RBP size variations. The phage diversity in a given mock community was estimated from the number of unique RBP variants formed in that metavirome following combining of RPKM values corresponding to phylogenetically related RBPs (RBPs of the same variant).
(iii) Test run.
An HTS sequencing library for D1 was prepared using the Nextera XT DNA kit (Illumina, USA) according to the manufacturer's protocol. The library was deep sequenced as 2 × 250 paired-end (PE) reads. Unless indicated otherwise, the analysis of reads was performed on CLC Genomics Workbench 8.5.1. Adapters, low-quality reads (quality limit, <0.05), reads containing >2 ambiguous nucleotides, and short reads (<15 bp) were discarded. The minimum number of reads required for phage relative abundance and diversity estimations was assessed by analysis of an array of randomly selected reads from D1 by (i) mapping to db-P (50% query length and 80% identity thresholds) and (ii) Ublast against db-RBP variants (95% query length and identity thresholds).
(iv) Final run.
Library construction, sequencing, and trimming of the remaining metaviromes were performed as described above except that I3, I4, and I5 were sequenced as 2 × 300 PE reads. The spiked metaviromes (D3, D4, D5, G1, and I2) were mapped to the φ29 and T4 genomes using 50% query length and 80% identity thresholds. Metaviromes were then assembled using the “de novo assembly” tool with default parameters, including analysis of coverage (50% query length and 80% identity thresholds). Contigs were (i) assessed with NCBI's BLAST tool (68) using default parameters and (ii) extracted and employed as references for analyzing the metaviromes using the “map reads to references” tool with 50% query length and 80% identity thresholds. The relative abundance and diversity of different phages in each metavirome were estimated by mapping the trimmed reads to db-P or by Ublast against db-RBP, essentially as described above for D1, using 1 million reads or the entire metavirome as an input, respectively. The output from db-RBP analysis was further treated as described above.
(v) Taxonomic composition.
One million randomly selected trimmed reads were uploaded to Metavir (metavir-meb.univ-bpclermont.fr) for BLASTx comparison against the NCBI Refseq complete viral genome protein sequence database. Taxonomic affiliations were deduced from the best BLAST hit (using a threshold of 50 on the BLAST score) and was normalized by the genome length using the GAAS tool (38).
Data availability.
The metaviromes can be accessed through the European Nucleotide Archive (69) under accession number PRJEB17619.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by FTP (Project number 0602-022170B) and was carried out as part of the MetaPhageLAB project. Musemma K. Muhammed has been the recipient of a Ph.D. stipend from the MetaPhageLAB project. Jennifer Mahony is the recipient of a Starting Investigator Research Grant (SIRG) (Ref. No. 15/SIRG/3430) funded by Science Foundation Ireland (SFI). Josué L. Castro-Mejía was supported by the Counteracting Age-related Loss of Skeletal Muscle Mass (CALM) project (University of Copenhagen Excellence Programme for Interdisciplinary Research). Douwe van Sinderen is supported by a Principal Investigator award (Ref. No. 13/IA/1953) through SFI.
We acknowledge the whey factories for their cooperation and Angela Back (Max-Rubner Institut [MRI], Germany) and Bashir Aideh (University of Copenhagen, Denmark) for technical assistance.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00888-17.
REFERENCES
- 1.Atamer Z, Ali Y, Neve H, Heller KJ, Hinrichs J. 2011. Thermal resistance of bacteriophages attacking flavour-producing dairy Leuconostoc starter cultures. Int Dairy J 21:327–334. doi: 10.1016/j.idairyj.2010.11.005. [DOI] [Google Scholar]
- 2.Erkus O, de Jager VCL, Spus M, van Alen-Boerrigter IJ, van Rijswijck IMH, Hazelwood L, Janssen PWM, van Hijum SAFT, Kleerebezem M, Smid EJ. 2013. Multifactorial diversity sustains microbial community stability. ISME J 7:2126–2136. doi: 10.1038/ismej.2013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smid EJ, Erkus O, Spus M, Wolkers-Rooijackers JC, Alexeeva S, Kleerebezem M. 2014. Functional implications of the microbial community structure of undefined mesophilic starter cultures. Microb Cell Fact 13(Suppl 1):S2. doi: 10.1186/1475-2859-13-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tariq MA, Louise F, Everest C, Cowley L, De Soyza A, Holt GS, Bridge H, Perry A, Perry JD, Bourke S, Cummings S, Lanyon CV, Barr JJ, Smith DL. 2015. A metagenomic approach to characterize temperate bacteriophage populations from cystic fibrosis and non-cystic fibrosis bronchiectasis patients. Front Microbiol 6:97. doi: 10.3389/fmicb.2015.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stadhouders J, de Vos WM. 1991. Starter cultures for cheese production, p 77–112. In Currell BC, Dam-Mieras RCE (ed), Biotechnological innovations in food processing, 1st ed Butterworth-Heinemann Ltd., Oxford, England. [Google Scholar]
- 6.Kleppen HP, Bang T, Nes IF, Holo H. 2011. Bacteriophages in milk fermentations: diversity fluctuations of normal and failed fermentations. Int Dairy J 21:592–600. doi: 10.1016/j.idairyj.2011.02.010. [DOI] [Google Scholar]
- 7.Nielsen EW. 1999. Long term use of a cheddar starter and development of phages with homology to its bacteria. Int Dairy J 8:1003–1009. doi: 10.1016/S0958-6946(99)00025-4. [DOI] [Google Scholar]
- 8.Park EJ, Kim KH, Abell GCJ, Kim MS, Roh SW, Bae JW. 2011. Metagenomic analysis of the viral communities in fermented foods. Appl Environ Microbiol 77:1284–1291. doi: 10.1128/AEM.01859-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deveau H, Labrie SJ, Chopin MC, Moineau S. 2006. Biodiversity and classification of lactococcal phages. Appl Environ Microbiol 72:4338–4346. doi: 10.1128/AEM.02517-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali Y, Kot W, Atamer Z, Hinrichs J, Vogensen FK, Heller KJ, Neve H. 2013. Classification of lytic bacteriophages attacking dairy Leuconostoc starter strains. Appl Environ Microbiol 79:3628–3636. doi: 10.1128/AEM.00076-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kot W, Hansen LH, Neve H, Hammer K, Jacobsen S, Pedersen PD, Sørensen SJ, Heller KJ, Vogensen FK. 2014. Sequence and comparative analysis of Leuconostoc dairy bacteriophages. Int J Food Microbiol 176:29–37. doi: 10.1016/j.ijfoodmicro.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 12.Azaïez SRC, Fliss I, Simard RE, Moinau S. 1998. Monoclonal antibodies raised against native major capsid proteins of lactococcal c2-like bacteriophages. Appl Environ Microbiol 64:4251–4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emond E, Holler BJ, Boucher I, Vandenbergh PA, Vedamuthu ER, Kondo JK, Moineau S. 1997. Phenotypic and genetic characterization of the bacteriophage abortive infection mechanism AbiK from Lactococcus lactis. Appl Environ Microbiol 63:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garneau JE, Tremblay DM, Moineau S. 2008. Characterization of 1706, a virulent phage from Lactococcus lactis with similarities to prophages from other Firmicutes. Virology 373:298–309. doi: 10.1016/j.virol.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 15.Jarvis AW. 1984. Differentiation of lactic streptococcal phages into phage species by DNA-DNA homology. Appl Environ Microbiol 47:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvis AW. 1978. Serological studies of a host range mutant of a lactic streptococcal bacteriophage. Appl Environ Microbiol 36:785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moineau S, Pandian S, Klaenhammer TR. 1994. Evolution of a lytic bacteriophage via DNA acquisition from the Lactococcus lactis chromosome. Appl Environ Microbiol 60:1832–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rousseau GM, Moineau S. 2009. Evolution of Lactococcus lactis phages within a cheese factory. Appl Environ Microbiol 75:5336–5344. doi: 10.1128/AEM.00761-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Verreault D, Gendron L, Rousseau GM, Veillette M, Massé D, Lindsley WG, Moineau S, Duchaine C. 2011. Detection of airborne lactococcal bacteriophages in cheese manufacturing plants. Appl Environ Microbiol 77:491–497. doi: 10.1128/AEM.01391-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahony J, van Sinderen D. 2014. Current taxonomy of phages infecting lactic acid bacteria. Front Microbiol 5:7. doi: 10.3389/fmicb.2014.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kot W, Neve H, Heller KJ, Vogensen FK. 2014. Bacteriophages of Leuconostoc, Oenococcus, and Weissella. Front Microbiol 5:186. doi: 10.3389/fmicb.2014.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castro-Mejía JL, Muhammed MK, Kot W, Neve H, Franz CMAP, Hansen LH, Vogensen FK, Nielsen DS. 2015. Optimizing protocols for extraction of bacteriophages prior to metagenomic analyses of phage communities in the human gut. Microbiome 3:1–14. doi: 10.1186/s40168-014-0066-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GGZ, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, Felts B, Dinsdale EA, Mokili JL, Edwards RA. 2014. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 5:1–11. doi: 10.1038/ncomms5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Willner D, Thurber RV, Rohwer F. 2009. Metagenomic signatures of 86 microbial and viral metagenomes. Environ Microbiol 11:1752–1766. doi: 10.1111/j.1462-2920.2009.01901.x. [DOI] [PubMed] [Google Scholar]
- 25.Cottrell MT, Kirchman DL. 2012. Virus genes in Arctic marine bacteria identified by metagenomic analysis. Aquat Microb Ecol 66:107–116. doi: 10.3354/ame01569. [DOI] [Google Scholar]
- 26.Sharon I, Battchikova N, Aro E-M, Giglione C, Meinnel T, Glaser F, Pinter RY, Breitbart M, Rohwer F, Béjà O. 2011. Comparative metagenomics of microbial traits within oceanic viral communities. ISME J 5:1178–1190. doi: 10.1038/ismej.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Illumina, Inc. 2014. Nextera library validation and cluster density optimization. https://www.illumina.com/documents/products/technotes/technote_nextera_library_validation.pdf.
- 28.Albertsson P, Frick G. 1960. Partition of virus particles in a liquid two-phase system. Biochim Biophys Acta 15:230–237. doi: 10.1016/0006-3002(60)90228-6. [DOI] [PubMed] [Google Scholar]
- 29.Philipson L, Albertsson PÅ, Frick G. 1960. The purification and concentration of viruses by aqueous polymer phase systems. Virology 11:553–571. doi: 10.1016/0042-6822(60)90100-8. [DOI] [PubMed] [Google Scholar]
- 30.Bachrach U, Friedmann A. 1971. Practical procedures for the purification of bacterial viruses. Appl Microbiol 22:706–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laue M, Bannert N. 2010. Detection limit of negative staining electron microscopy for the diagnosis of bioterrorism-related micro-organisms. J Appl Microbiol 109:1159–1168. doi: 10.1111/j.1365-2672.2010.04737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ackermann H-W. 2009. Basic phage electron microscopy, p 113–126. In Clokie MRJ, Kropinski A (ed), Bacteriophages: methods and protocols, vol 1 Isolation, characterization and interaction. Humana Press, New York, NY. [Google Scholar]
- 33.Crutz-Le Coq A-M, Cantele F, Lanzavecchia S, Marco S. 2006. Insights into structural proteins of 936-type virulent lactococcal bacteriophages. Arch Virol 151:1039–1053. doi: 10.1007/s00705-005-0709-4. [DOI] [PubMed] [Google Scholar]
- 34.Dupont K, Vogensen FK, Neve H, Bresciani J, Josephsen J. 2004. Identification of the receptor-binding protein in 936-species lactococcal bacteriophages. Appl Environ Microbiol 70:5818–5824. doi: 10.1128/AEM.70.10.5818-5824.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mahony J, Kot W, Murphy J, Ainsworth S, Neve H, Hansen LH, Heller KJ, Sørensen SJ, Hammer K, Cambillau C, Vogensen FK, Van Sinderen D. 2013. Investigation of the relationship between lactococcal host cell wall polysaccharide genotype and 936 phage receptor binding protein phylogeny. Appl Environ Microbiol 79:4385–4392. doi: 10.1128/AEM.00653-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Murphy J, Bottacini F, Mahony J, Kelleher P, Neve H, Zomer A, Nauta A, van Sinderen D. 2016. Comparative genomics and functional analysis of the 936 group of lactococcal Siphoviridae phages. Sci Rep 6:1–13. doi: 10.1038/s41598-016-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yaung SJ, Esvelt KM, Church GM. 2015. Complete genome sequences of T4-like bacteriophages RB3, RB5, RB6, RB7, RB9, RB10, RB27, RB33, RB55, RB59, and RB68. Genome Announc 3:e01122-14. doi: 10.1128/genomeA.01122-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Angly FE, Willner D, Prieto-Davo A, Edwards RA, Schmieder R, Vega-Thurber R, Antonopoulos DA, Barott K, Cottrell MT, Desnues C, Dinsdale EA, Furlan M, Haynes M, Henn MR, Hu Y, Kirchman DL, McDole T, McPherson JD, Meyer F, Miller RM, Mundt E, Naviaux RK, Rodriguez-Mueller B, Stevens R, Wegley L, Zhang L, Zhu B, Rohwer F. 2009. The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput Biol 5:1–13. doi: 10.1371/journal.pcbi.1000593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mulvihill DM, Ennis MP. 2003. Functional milk proteins: production and utilization, p 1175–1228. In Fox PF, McSweeney PLH (ed), Advanced dairy chemistry, 3rd ed, vol 1 Proteins. Springer US, Boston, MA. [Google Scholar]
- 40.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 41.Kot W, Vogensen FK, Sørensen SJ, Hansen LH. 2014. DPS-A rapid method for genome sequencing of DNA-containing bacteriophages directly from a single plaque. J Virol Methods 196:152–156. doi: 10.1016/j.jviromet.2013.10.040. [DOI] [PubMed] [Google Scholar]
- 42.Meijer WJJ, Horcajadas JA, Salas M. 2001. phi29 family of phages. Microbiol Mol Biol Rev 65:261–287. doi: 10.1128/MMBR.65.2.261-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller ES, Kutter E, Mosig G, Kunisawa T, Rüger W, Arisaka F, Ru W. 2003. Bacteriophage T4 genome. Microbiol Mol Biol Rev 67:86–156. doi: 10.1128/MMBR.67.1.86-156.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chopin A, Bolotin A, Sorokin A, Ehrlich SD, Chopin M. 2001. Analysis of six prophages in Lactococcus lactis IL1403: different genetic structure of temperate and virulent phage populations. Nucleic Acids Res 29:644–651. doi: 10.1093/nar/29.3.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mahony J, Martel B, Tremblay DM, Neve H, Heller KJ, Moineau S, Van Sinderen D. 2013. Identification of a new P335 subgroup through molecular analysis of lactococcal phages Q33 and BM13. Appl Environ Microbiol 79:4401–4409. doi: 10.1128/AEM.00832-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atamer Z, Dietrich J, Müller-Merbach M, Neve H, Heller KJ, Hinrichs J. 2009. Screening for and characterization of Lactococcus lactis bacteriophages with high thermal resistance. Int Dairy J 19:228–235. doi: 10.1016/j.idairyj.2008.10.012. [DOI] [Google Scholar]
- 47.Bissonnette F, Labrie S, Deveau H, Lamoureux M, Moineau S. 2000. Characterization of mesophilic mixed starter cultures used for the manufacture of aged cheddar cheese. J Dairy Sci 83:620–627. doi: 10.3168/jds.S0022-0302(00)74921-6. [DOI] [PubMed] [Google Scholar]
- 48.Casey CN, Morgan E, Daly C, Fitzgerald GF. 1993. Characterization and classification of virulent lactococcal bacteriophages isolated from a Cheddar cheese plant. J Appl Bacteriol 74:268–275. doi: 10.1111/j.1365-2672.1993.tb03025.x. [DOI] [Google Scholar]
- 49.Mahony J, Murphy J, van Sinderen D. 2012. Lactococcal 936-type phages and dairy fermentation problems: from detection to evolution and prevention. Front Microbiol 3:335. doi: 10.3389/fmicb.2012.00335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miklic A, Rogelj I. 2003. Characterization of lactococcal bacteriophages isolated from Slovenian dairies. Int J Food Sci Technol 38:305–311. doi: 10.1046/j.1365-2621.2003.00676.x. [DOI] [Google Scholar]
- 51.Moineau S, Borkaev M, Holler BJ, Walker SA, Kondo JK, Vedamuthu ER, Vandenbergh PA. 1996. Isolation and characterization of lactococcal bacteriophages from cultured buttermilk plants in the United States. J Dairy Sci 79:2104–2111. doi: 10.3168/jds.S0022-0302(96)76584-0. [DOI] [Google Scholar]
- 52.Murphy J, Royer B, Mahony J, Hoyles L, Heller K, Neve H, Bonestroo M, Nauta A, van Sinderen D. 2013. Biodiversity of lactococcal bacteriophages isolated from 3 Gouda-type cheese-producing plants. J Dairy Sci 96:4945–4957. doi: 10.3168/jds.2013-6748. [DOI] [PubMed] [Google Scholar]
- 53.Szczepańska AK, Hejnowicz MS, Kołakowski P, Bardowski J. 2007. Biodiversity of Lactococcus lactis bacteriophages in Polish dairy environment. Acta Biochim Pol 54:151–158. [PubMed] [Google Scholar]
- 54.Castro-Nallar E, Chen H, Gladman S, Moore SC, Seemann T, Powell IB, Hillier A, Crandall KA, Chandr PS. 2012. Population genomics and phylogeography of an Australian dairy factory derived lytic bacteriophage. Genome Biol Evol 4:382–393. doi: 10.1093/gbe/evs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vegge CS, Neve H, Brøndsted L, Heller KJ, Vogensen FK. 2006. Analysis of the collar-whisker structure of temperate lactococcal bacteriophage TP901-1. Appl Environ Microbiol 72:6815–6818. doi: 10.1128/AEM.01033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moineau S, Fortier J, Ackermann HW, Pandian S. 1992. Characterization of lactococcal bacteriophages from Quebec cheese plants. Can J Microbiol 38:875–882. doi: 10.1139/m92-143. [DOI] [Google Scholar]
- 57.Raiski A, Belyasova N. 2009. Biodiversity of Lactococcus lactis bacteriophages in the Republic of Belarus. Int J Food Microbiol 130:1–5. doi: 10.1016/j.ijfoodmicro.2008.12.024. [DOI] [PubMed] [Google Scholar]
- 58.Marvig CL, Aideh B, Neve H, Heller KJ, Knøchel S, Vogensen FK. 2011. Heat tolerance of dairy lactococcal c2 phages. Int Dairy J 21:556–560. doi: 10.1016/j.idairyj.2011.03.004. [DOI] [Google Scholar]
- 59.Geller BL, Ivey RG, Trempy JE, Hettinger-Smith B. 1993. Cloning of a chromosomal gene required for phage infection of Lactococcus lactis subsp. lactis C2. J Bacteriol 175:5510–5519. doi: 10.1128/jb.175.17.5510-5519.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Valyasevi R, Sandine WE, Geller BL. 1991. A membrane protein is required for bacteriophage c2 infection of Lactococcus lactis subsp. lactis C2. J Bacteriol 173:6095–6100. doi: 10.1128/jb.173.19.6095-6100.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Babu KS, Spence WS, Monteville MR, Geller BL. 1995. Characterization of a cloned gene (pip) from Lactococcus lactis required for phage infection. Dev Biol Stand 85:569–575. [PubMed] [Google Scholar]
- 62.Stuer-Lauridsen B, Janzen T. July. 2006. Bacteriophage resistant lactic acid bacteria. Patent WO 2006/072631 A1. World Intellectual Property Organization, Geneva, Switzerland. [Google Scholar]
- 63.Samson JE, Moineau S. 2010. Characterization of Lactococcus lactis phage 949 and comparison with other lactococcal phages. Appl Environ Microbiol 76:6843–6852. doi: 10.1128/AEM.00796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lillehaug D. 1997. An improved plaque assay for poor plaque-producing temperate lactococcal bacteriophages. J Appl Microbiol 83:85–90. doi: 10.1046/j.1365-2672.1997.00193.x. [DOI] [PubMed] [Google Scholar]
- 65.Vegge CS, Vogensen FK, Mc Grath S, Neve H, van Sinderen D, Brøndsted L. 2006. Identification of the lower baseplate protein as the antireceptor of the temperate lactococcal bacteriophages TP901-1 and Tuc2009. J Bacteriol 188:55–63. doi: 10.1128/JB.188.1.55-63.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Edgar RC, Bateman A. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 67.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. 2008. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 68.Altschup SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 69.EMBL-EBI 2017. European Nucleotide Archive. EMBL-EBI, Hinxton, UK: http://www.ebi.ac.uk/ena. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The metaviromes can be accessed through the European Nucleotide Archive (69) under accession number PRJEB17619.