

Abstract

The discovery of novel tetrahydropyrrolo[1,2-c]pyrimidines derivatives from Bay41_4109 as hepatitis B virus (HBV) inhibitors is herein reported. The structure–activity relationship optimization led to one highly efficacious compound 28a (IC50 = 10 nM) with good PK profiles and the favorite L/P ratio. The hydrodynamic injection model in mice clearly demonstrated the efficacy of 28a against HBV replication.
Keywords: Hepatitis B virus (HBV); tetrahydropyrrolo[1,2-c]pyrimidines; capsid assembly inhibitor
Hepatitis B is a potentially life-threatening liver disease caused by the hepatitis B virus (HBV) infection. Over 350 million people have chronic HBV infection.1 Chronic HBV infection can lead to liver cirrhosis and hepatocellular carcinoma (HCC).2 Currently, there are two types of anti-HBV agents in the market: immune modulators (IFN-α) and nucleosi(t)des as reverse transcriptase inhibitors (Lamivudine, Entecavir, Telbivudine, Adefovir and Tenofovir).3 However, the usage of IFN-α is limited due to its low efficacy, high cost, and serious side effects; while nucleos(t)ides often result in drug-resistance after long-term treatment.4,5 Moreover, both IFN-α and nucleos(t)ide analogues cannot eradicate chronic HBV infection. Therefore, it is crucial to develop safer and more effective anti-HBV agents with novel mechanisms.6−8
Heteroaryldihydropyrimidines (HAPs), discovered by a cell culture-based screening, were identified as a novel class of HBV inhibitors targeting capsid assembly and RNA packaging.9 It was reported that HAPs could enhance the rate of assembly and cause the formation of aberrant capsid, thus leading to decreased production of virions.10 However, HAPs were hydrophobic due to their special chemical structures.11,12 The poor water solubility significantly affected the drug candidate’s oral PK properties and eventually limited the chance of being developed into a pharmaceutical product.
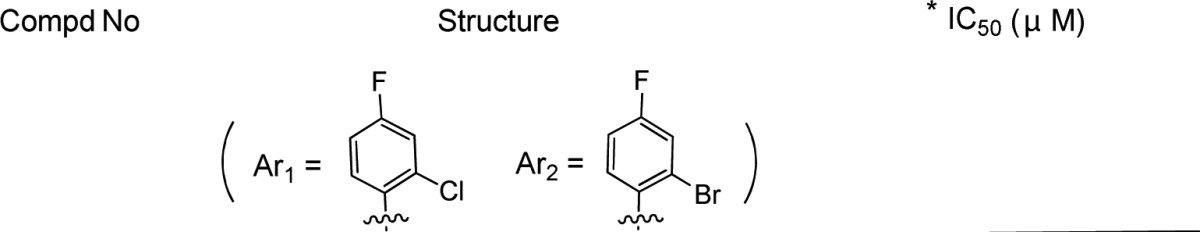
GLS4 (Scheme 1), derived from BAY41–4109 (Scheme 1), is currently in a phase II clinical trial as an anti-HBV agent, which also targets the HBV capsid formation.13−16GLS4 was as potent as the prototype-BAY41–4109. Moreover, it can inhibit both wild-type and Adefovir resistant strains of HBV in cell culture. However, the poor oral bioavailability of GLS4 might become a hurdle for further development.
Scheme 1. Chemical Structures of BAY41-4109 and GLS4.

We herein report the approaches for the design, synthesis, and biological evaluation of a novel series of 3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidine derivatives as HBV capsid assembly inhibitors.
The derivatives (Scheme 2) were designed based on HAPs. Although it was reported that blocking the hydrogen on pyrrimidine with alkyl group resulted in loss of antiviral activity,17,18 we discovered that the fused ring analogues could maintain moderate to excellent potency.
Scheme 2. Chemical Structures of the Derivatives.

The analogues in Table 1 were evaluated by the cell-based anti-HBV assay. The HepG2.2.15 cells were seeded at 4 × 104 cells/well in a 96-well plate (100 μL/well) in seeding medium and incubated overnight at 37 °C in a 5% CO2 humid atmosphere. The next day, compounds were added to the cell culture. Three days later, cells were replenished with fresh medium. After another three-day incubation, cell culture supernatants were collected and stored at −80 °C until HBV DNA was used. HBV DNA was extracted from supernatants with QIAamp 96 DNA Blood Kit and quantified by qPCR.
Table 1. Anti-HBV Activity of the Fused Ring Analogues in HepG 2.2.15 Cells.
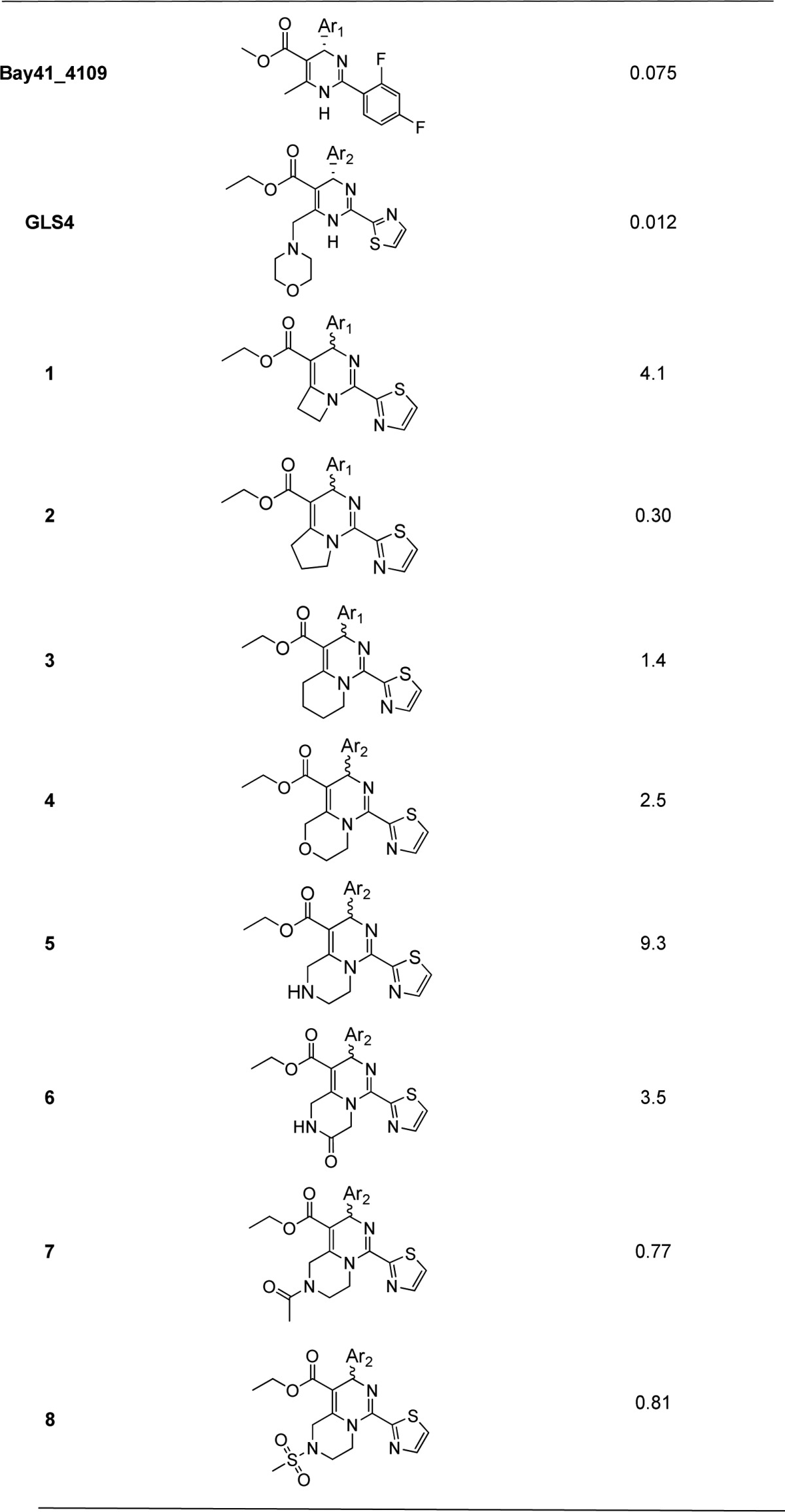
Mean value of three experiments.
The IC50 data in Table 1 showed the fused ring analogues can keep moderate activity. Among them, compound 2 possessed the best potency against HBV.
A general synthetic route for compound 2 is illustrated in Scheme 3. The THP protected ethyl 3-oxoheptanoate was reacted with amidine 10 and the substituted benzaldehyde to produce the cyclized compound 12 under the Biginelli condition.19 Compound 12 was deprotected with p-TsOH to afford compound 13, followed by cyclization in the presence of MsCl/Et3N to generate the racemic compound 2.
Scheme 3. Synthetic Route of 2.

Utilizing the most potent compound 2 as a starting point, we first investigated the effects of different R1 groups as shown in Table 2. The structure–activity relationship (SAR) analysis indicated that the thiazole group is the best moiety in this region.
Table 2. Anti-HBV Activity in HepG2.2.15 Cells via R1 Modification.
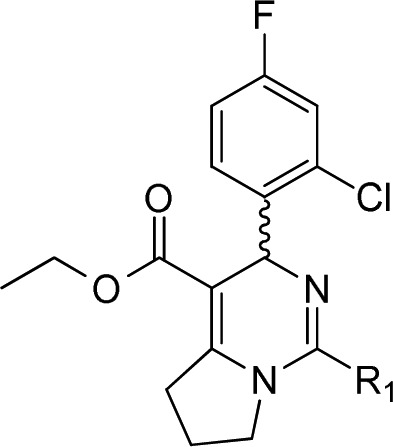
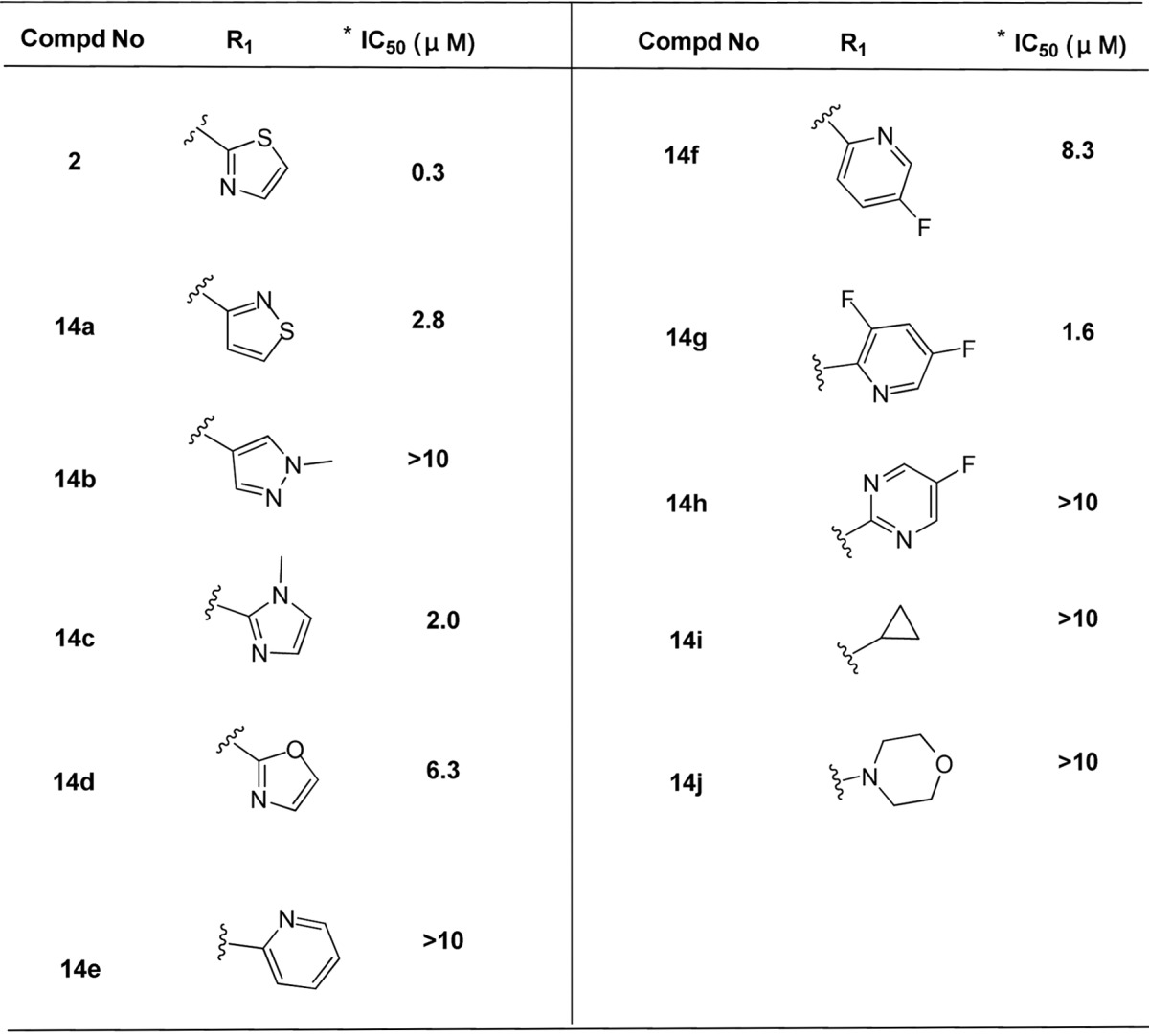
Mean value of three experiments.
We explored the further SAR-based optimization of compound 2 at the R2 position (Table 3). 2-Bromo-4-fluorophenyl (15a) provided a comparable activity (IC50 = 0.21 μM). Replacement of the phenyl at 2, 3, or 4 position can retain the potency, such as 2-chloro-3-fluorophenyl (15b, IC50 = 0.31 μM). However, it led to less potency if substituted at 2, 5 or 2, 6 positions, for example, 2-chloro-5-fluorophenyl (15c) and 2-chloro-6-fluorophenyl (15d). 2,4-Difluorophenyl (15e, IC50 = 0.51 μM) and 3,4-difluorophenyl (15f, IC50 = 0.43 μM) analogues maintained similar potency. No significant potency improvement was observed by introducing a methyl group at the 2-position of phenyl (15g, IC50 = 0.45 μM). The replacement of 2-bromo-4-fluorophen with 1-methyl-1H-pyrazolor thiazol, resulted in the loss of potency, such as compound 15m or 15k. Besides, introduction of pyridine analogues (15i and 15j) at R2 position also led to the loss of potency.
Table 3. Anti-HBV Activity in HepG2.2.15 Cells via R2 Modification.
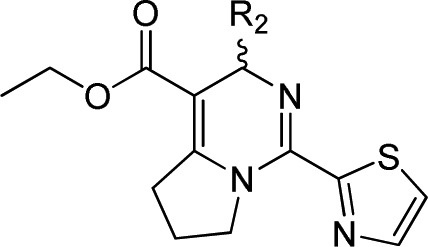
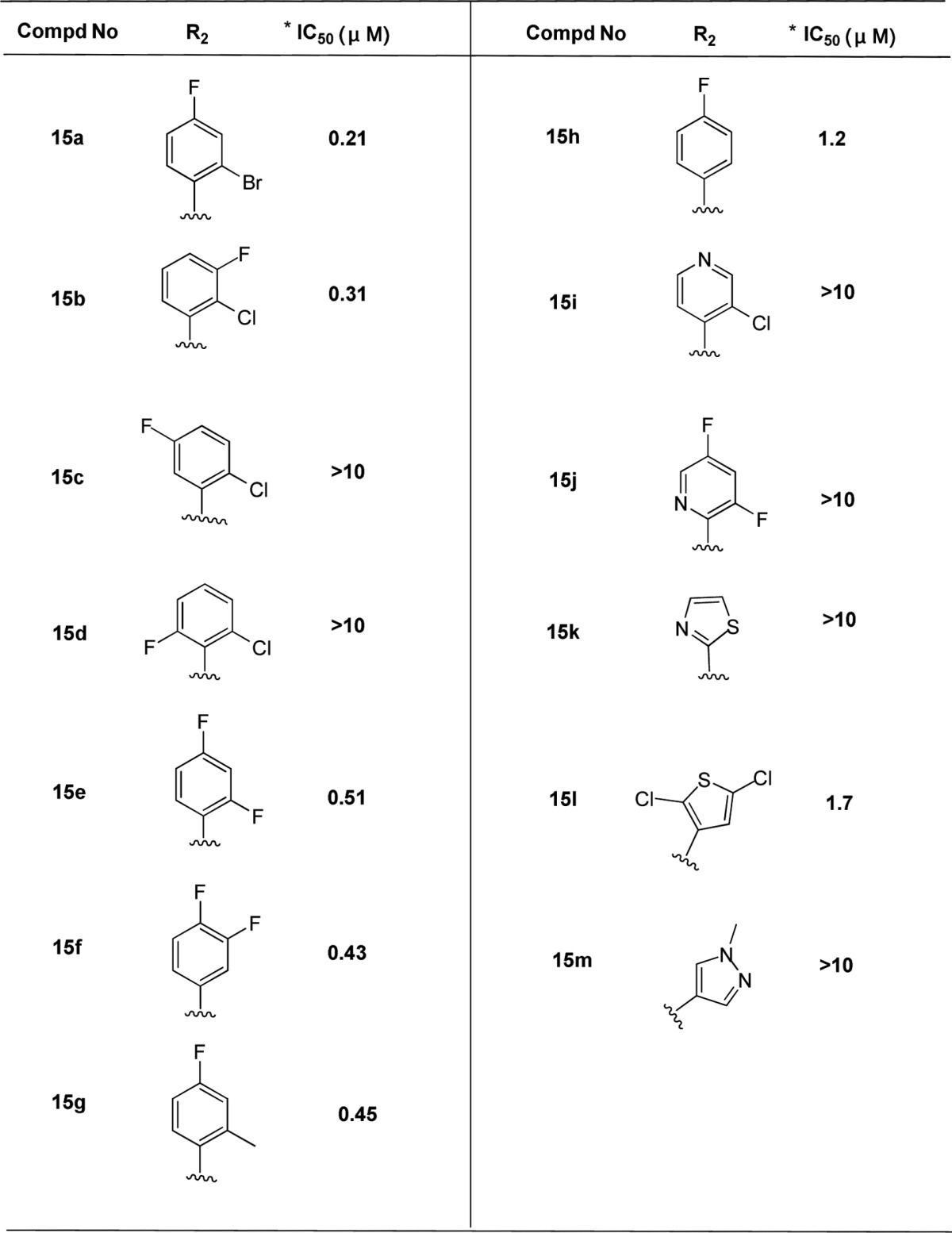
Mean value of three experiments.
Based on the above result, further SAR was investigated at the R3 region (Table 4). We found it led to loss of potency by replacing ethyl with 2-methoxyethyl carboxylate (16c) and 2-(dimethylamino)ethyl carboxylate (16d), but no difference if replaced with a methyl group (16a). The decline in potency was also observed when the ethyl group of 16a was replaced with the similar methoxymethyl group (16b) or 2,2-difluoroethyl group (16e). Furthermore, replacement of the ethyl carboxylate with a N-methoxy carboxylate (16f, IC50 = 5.0 μM) did not maintain the potency.
Table 4. Anti-HBV Activity in HepG2.2.15 Cells via R3 Modification.
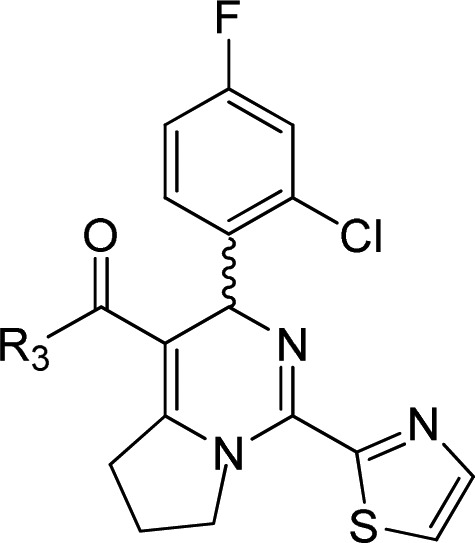
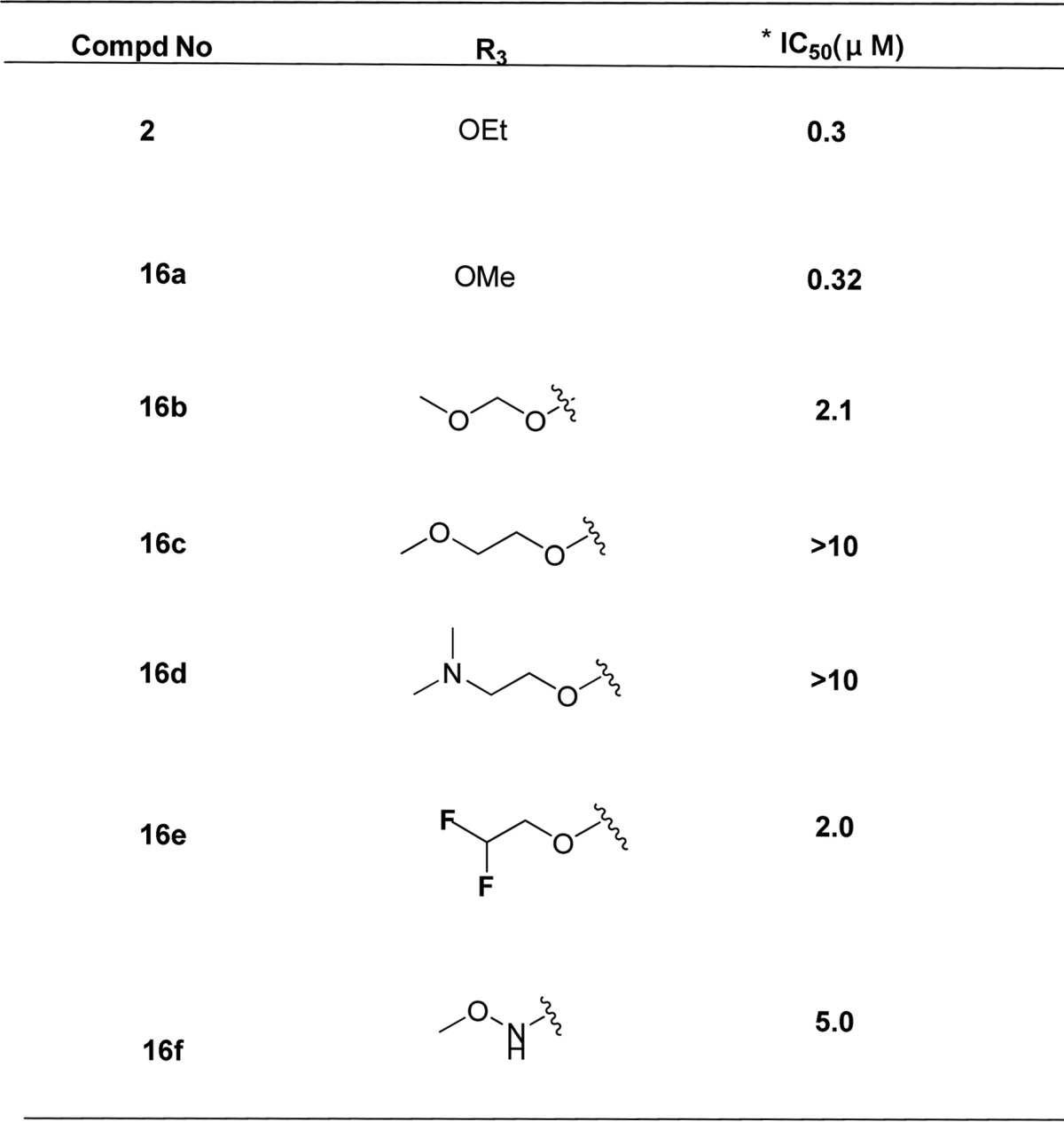
Mean value of three experiments.
Interestingly, when we investigated the substituents at the five-membered ring, we found compounds 26 and 27 with a Boc group also showed good potency with IC50 = 50 and 30 nM, respectively (Table 5). So we did further exploration at this region and identified that methylsulfonyl analogues such as 28a, 29a, and 30 possessed excellent activities. The absolute stereochemistry of 30 was determined by the single crystal X-ray study (see Supporting Information). Compared to (3R,6R) isomers such as 28b (IC50 = 60 nM) and 29b (IC50 = 70 nM), the (3R,6S) isomers (28a and 29a) were 6–7-fold more potent. Based on the efficacy of 28a and 29a, there was no difference if methyl ester replaced by ethyl ester. We kept the ethyl ester moiety and conducted the optimization on the pyrrolo ring, but hydroxyl (31), N,N-dimethyl (32), and methyl (33) analogues did not keep potency.
Table 5. Anti-HBV Activity in HepG2.2.15 Cells.
Mean value of three experiments.
To gain deeper insights into the mechanisms of our compounds, we did the evaluation activity of 28a, Bay41_4109, and GLS4 against HBV capsid assembly in the capsid quenching assay. Compounds 28a, Bay41_4109, and GLS4 had IC50 values of 0.8, 0.43, and 1.2 μM, respectively, which proved the similar caspid assembly inhibition mechanism.
A general synthetic route for compound 28a is illustrated in Scheme 4. The compound 10 reacted with 2-Cl-4-F-benzaldehyde and ethyl 3-oxobutanoate (17) to generate the cyclized compound 18 under the Biginelli condition.1 The enantiopure 20 was obtained through SFC chiral separation of the stereomixtures eluting with a mixed solvent of 85% supercritical CO2/15% EtOH at 100 mL/min rate. The absolute stereochemistry of (−)-enantiomer 20 was determined by its rotation value as reported.21 Bromination of compound 20 generated 21, which was reacted with 22 via three steps to give compound 23. The subsequent reduction of 23 with NaBH4 afforded compound 24. Compound 25 was generated by cyclization of 24 in the presence of MsCl and Et3N. The compounds 26 and 27 were separated by the of SFC separation with higher polarity of 27. Compound 27 reacted with HCl/EtOAc to give the amine intermediate, then coupled with MsCl to generate compound 28a.
Scheme 4. Synthetic Route of 28a.

Computer Modeling on Interaction of Compound 28a with HBV Capsid Protein
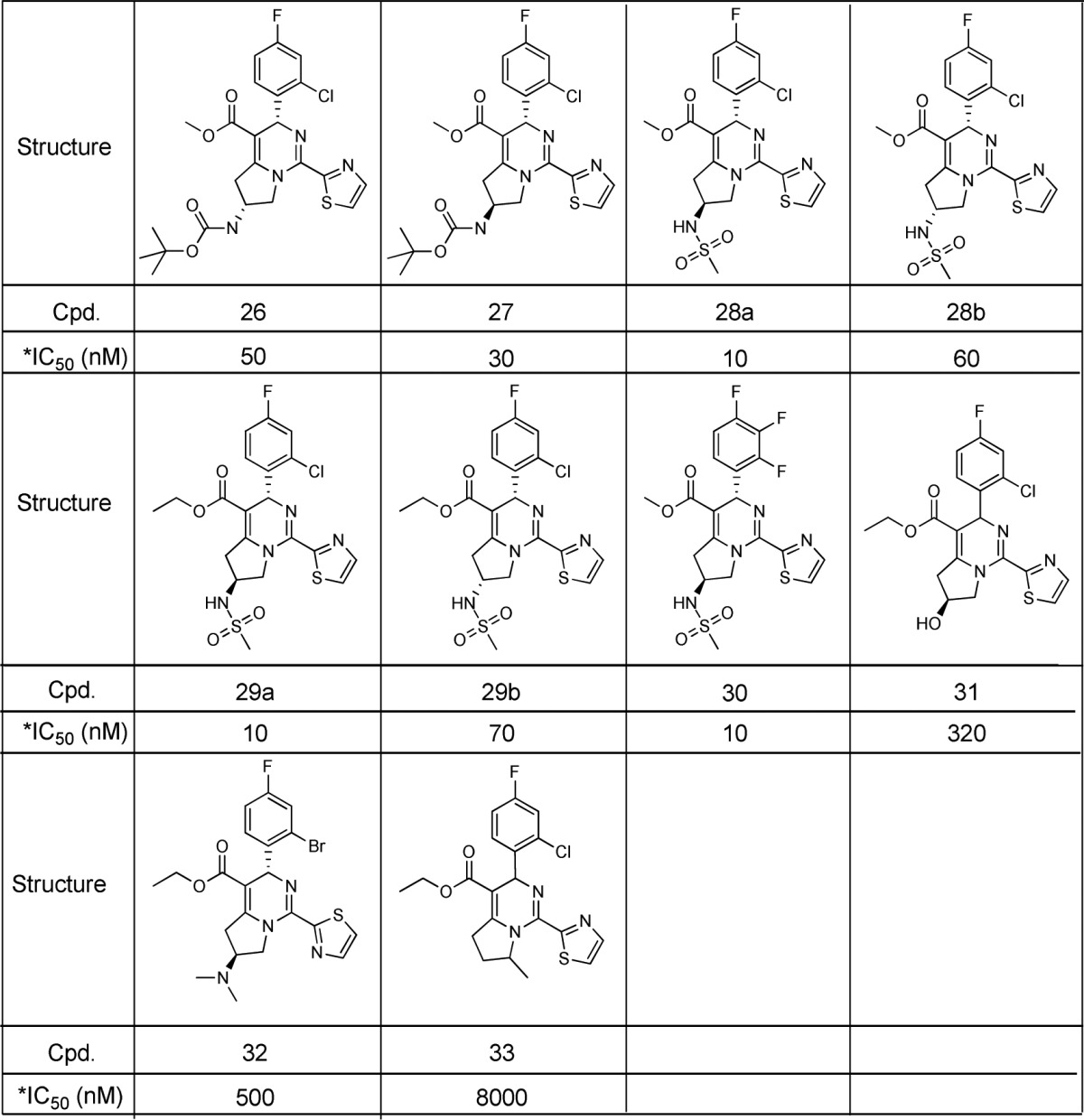
The dihydropyrimidine compounds can potentially bind to the interface of HBV core proteins, disrupting the formation of HBV capsid, therefore blocking the lifecycle of HBV.22−24 To better understand the SAR, some of the analogues in this work were docked into the crystal structure of HBV capsid (PDB code 5GMZ).
The binding pose of sulphonamide compound 28a in the core protein is shown in Figure 1. The F- or Cl-phenyl occupies a compact and hydrophobic pocket, which consists of Thr33, Ile105, Leu30, Pro25, and Trp102 in one of the core proteins (chain B) and Val124, Arg127 and Thr128 in the other core protein (chain C). This pocket is very tight and hydrophobic, where the fluorine, chlorine, and bromine analogues are potent, but polar and bulky groups are generally not well tolerated (Table 3). The dihydropyrimidine can catch a key hydrogen bond to Trp102, as well as have hydrophobic interactions with Leu140 and Val124. However, the dihydropyrimidine core has a unique conformation, which facilitates the F-, Cl-phenyl binding to the tight pocket with an optimal dihedral angle. The thiazole can form hydrophobic interactions with Pro25 and Thr128, as well as π–π stacking with Phe23 and Tyr118. Some heterocycles with high polarity, such as pyrrozole and pyridine, can reduce activity. Moreover, saturated rings may even significantly drop in activity (Table 2). Most of these interactions lie in the deep pocket, stabilizing the conformation of 28a in the interface of HBV core proteins.
Figure 1.
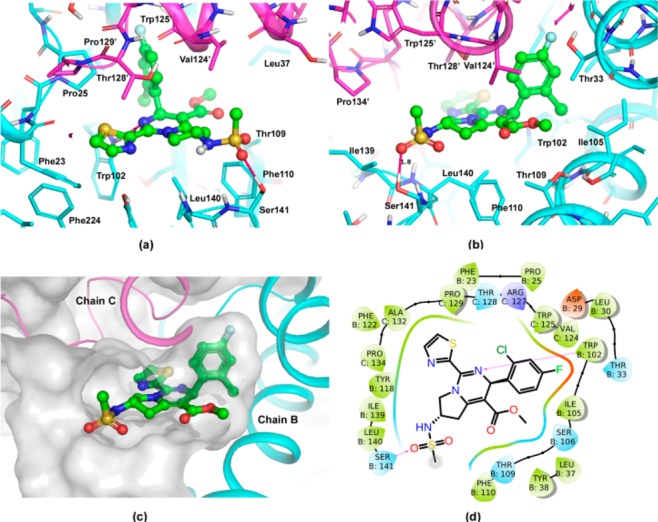
Putative binding pose of compound 28a in HBV capsid. The core proteins, chains B and C, are shown in magenta and cyan cartoon. The ligand is shown as ball-and stick, where carbon atoms are in green. (a) View from the left-hand side of the entrance to binding site. (b) View from the right-hand side. (c) Binding pocket of the interface of core proteins. (d) Two-dimensional plot of compound 28a in HBV capsid. The figures were prepared by Pymol25 and Maestro26 packages.
The fused pyrrolidine can form hydrophobic interactions with Leu140, Phe110, and Val124, moreover, it acts as a rigid linker, which can deliver a hydrophilic tail to a region the near solvent. Given the high lipophilicity of lead compound 2, the free space around this region makes it possible to introduce hydrophilic groups to improve the solubility and pharmacokinetics properties. The vicinity of this hydrophilic region has Leu140 and Ser141, where two amides on main chain and a hydroxyl on side chain point to the inner pocket, forming a hydrogen bond donor rich region. The sulphonamide potentially can catch a hydrogen bond to Ser141, stabilizing the conformation of compound 28a in the binding pocket (Figure 1). For the potent analogue 26, its carbamate tail can also catch a hydrogen bond to Ser141, and its t-Bu can have additional hydrophobic interactions with Pro134 and Ile139. However, those compounds with less hydrogen bond acceptors on the tail (31, see Table 5) and even basic compounds (32) may lose the hydrogen bond at this region and thus become less potent. For the flexibility of pyrrolidine, both R and S enantiomers are tolerated in the pocket, but generally the compounds in S configuration can catch hydrogen bonds and fit binding pocket better. Substitutions at position 7 of tetrahydropyrrolopyrimidine are very close to the thiazole ring, which can change its conformation and affect protein binding. For example, the addition of methyl in 33 significantly drops in activity.
Physical Properties, ADME/PK Profiles of GLS4 and 28a
Compound 28a showed desirable in vitro potency and physicochemical properties (Table 6). In the in vitro DDI assessment, compound 28a displayed limited risk on CYP inhibition in a panel of five enzymes (CYP1A2, 2C9, 2C19, 2D6, and 3A4). Compound 28a exhibited moderate plasma clearance (Cl) (21 mL min–1 kg–1) with good oral bioavailability of 54% in mice, while the oral bioavailability of GLS4 was only 14%.
Table 6. Physical Properties, ADME/PK Profiles.
| compound ID | GLS4 | 28a | |
|---|---|---|---|
| MW/tPSA | 508/76 | 484/100 | |
| CLogP/LogD | 4.7/4.4 | 2.7/2.8 | |
| KS (μM) | 14 | 22 | |
| liver/plasma ratio of AUC0–4h (μM·h) | 9 | 10 | |
| CYP (μM) 1A2/2C9/2C19/2D6/3A4 | >50/16/4/52/2 | >50/28/39/>50/11 | |
| IV (1 mpk) | T1/2 (h) | 1.5 ± 0.1 | 1.1 ± 0.1 |
| Vdss (L/kg) | 4.1 ± 0.6 | 2.0 ± 0.2 | |
| Cl (mL/min/kg) | 56 ± 7 | 21 ± 3 | |
| AUC0-last (μM·h) | 0.6 ± 0.1 | 1.7 ± 0.2 | |
| POa,b (10 mpk) | Cmax (μM) | 0.7 ± 0.1 | 4.7 ± 0.3 |
| AUC0-last (μM·h) | 0.8 ± 0.1 | 9.1 ± 1.5 | |
| F% | 14 | 54 | |
The single-dose pharmacokinetics of GLS4 and 28a was carried out in female Balb/c mice (n = 3) according to standard procedures.
Vehicle: 1.00 mg/mL in 0.5% HEC.
The in vivo antiviral activity of 28a was evaluated in a hydrodynamic injection (HDI) HBV mouse model.27 In consideration of higher oral exposure of 28a than GLS4, 28a was dosed at 50 mpk, BID versus GLS4 at 100 mpk, BID. As shown in Figure 2, 28a demonstrated a statistically significant reduction (p < 0.01) of HBV DNA in both mouse plasma and liver. In comparison with the vehicle group, 50 mpk (BID) of compound 28a treatment achieved 2.05 and 2.57 log viral DNA reduction in mouse plasma on days 3 and 5, respectively (Figure 2A), while 100 mpk (BID) of reference compound GLS4 treatment achieved 0.35 and 1.56 log viral DNA reduction in mouse plasma on days 3 and 5, respectively. Treatment of the mice with 28a and GLS4 reduced 0.68 and 0.91 log viral load of DNA in mouse liver on day 7, respectively (Figure 2B).
Figure 2.
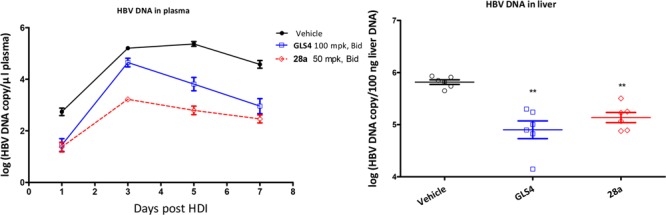
Effect of treatment of GLS4 and 28a on inhibition of HBV replication in HDI mice. Mice (n = 6 per group) were orally dosed with vehicle (0.5% HEC), 28a (50 mpk, BID), or GLS4 (100 mpk, BID). The levels of HBV DNA in plasma (A) and liver (B) were determined by qPCR. Error bars represent standard error. Statistical analysis was performed by Student’s t test. ** p < 0.01.
Conclusion
In summary, we have described the successful structural optimization of HAPs to 3,5,6,7-tetrahydropyrrolo[1,2-c]pyrimidines as a novel class of HBV capsid assembly inhibitors. Some of these newly developed derivatives demonstrated highly potent in vitro activities against HBV replication. Structural optimization resulted in the identification of one lead compound 28a with an IC50 value of 10 nM in HepG2.2.15 cells. Compound 28a also showed good oral bioavailability with the favorite L/P ratio (10:1) in mice. The HBV HDI mouse model demonstrated that 28a significantly reduced HBV DNA in both mouse plasma and liver. These results demonstrate that 28a is a promising lead compound for further optimization.
Glossary
ABBREVIATIONS
- KS
kinetic solubility
- PK
pharmacokinetics
- L/P ratio
liver/plasma ratio of AUC
- SAR
structure–activity relationship
- CYP
cytochrome P450
- hERG
human ether-a-go-go related gene
- IV
intravenous
- PO
per oral
- AUC
area under curve
- mpk
mg/kg
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00288.
Synthetic procedures, analytical data, assay protocol, and inhibitor mechanism of action (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Yang X.; Xu X. A new series of HAPs as anti-HBV agents targeting at capsid assembly. Bioorg. Med. Chem. Lett. 2014, 24, 4247. 10.1016/j.bmcl.2014.07.032. [DOI] [PubMed] [Google Scholar]
- Liang T. J. Hepatitis B: The virus and disease. Hepatology 2009, 49, S13. 10.1002/hep.22881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferir G.; Kaptein S.; Neyts J. Antiviral treatment of chronic hepatitis B virus infections: the past, the present and the future. Rev. Rev. Med. Virol. 2008, 18, 19. 10.1002/rmv.554. [DOI] [PubMed] [Google Scholar]
- Janssen H. L.; Van-Zonneveld M.; Senturk H.; Zeuzem S.; Akarca U. S.; Cakaloglu Y.; Simon C.; So T. M.; Gerken G.; de Man R. A. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet 2005, 365, 123. 10.1016/S0140-6736(05)17701-0. [DOI] [PubMed] [Google Scholar]
- Zoulim F.; Poynard T.; Degos F.; Slama A.; El Hasnaoui A.; Blin P.; Mercier F.; Deny P.; Landais P.; Parvaz P. A prospective study of the evolution of lamivudine resistance mutations in patients with chronic hepatitis B treated with lamivudine. J. Viral Hepat. 2006, 13, 278. 10.1111/j.1365-2893.2005.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish R.; Jia J.; Locarnini S.; Zoulim F. Selection of chronichepatitis B therapy with high barrier to resistance. Lancet Infect. Dis. 2012, 12, 341. 10.1016/S1473-3099(11)70314-0. [DOI] [PubMed] [Google Scholar]
- Kim S. R.; Yang J.; Kudo M.; Hino O. Recent Advances in theManagement of Chronic Hepatitis B. Hepat.Mon. 2011, 11, 601. 10.5812/kowsar.1735143X.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang K. B.; Kim H. J. Management of antiviral drug resistance inchronic hepatitis B. World. J. Gastroenterol. 2014, 20, 11641. 10.3748/wjg.v20.i33.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber O.; Schlemmer K. H.; Hartmann E.; Hagelschuer I.; Paessens A.; Graef E.; Deres K.; Goldmann S.; Niewoehner U.; Stoltefuss J.; Haebich D.; Ruebsamen-Waigmann H.; Wohlfeil S. Inhibition of human hepatitis B virus (HBV) by a novel nonnucleosidiccompound in a transgenic mouse model. Antiviral Res. 2002, 28, 69. 10.1016/S0166-3542(01)00216-9. [DOI] [PubMed] [Google Scholar]
- Deres K.; SchrÖder C. H.; Paessens A.; et al. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science 2003, 299, 893. 10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- Bourne C. R.; Finn M. G.; Zlotnick A. Global Structural Changes in Hepatitis B Virus Capsids Induced by the Assembly Effector HAP1. J. Virol. 2006, 80, 11055. 10.1128/JVI.00933-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stray S. J.; Zlotnick A. BAY 41–4109 has multiple effects on Hepatitis B virus capsid assembly. J. Mol. Recognit. 2006, 19, 542. 10.1002/jmr.801. [DOI] [PubMed] [Google Scholar]
- Wu G.; Liu B.; Zhang Y.; Li J.; Arzumanyan A.; Clayton M. M.; Schinazi R. F.; Wang Z.; Goldmann S.; Ren Q.; Zhang F.; Feitelson M. A. Preclinicalcharacterization of GLS4, an Inhibitorof hepatitis B virus core particle assembly. Antimicrob. Agents Chemother. 2013, 57, 5344. 10.1128/AAC.01091-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wei Z.; Wu G.; Wang J.; Zhang Y.; Li J.; Zhang H.; Xie X.; Wang X.; Wang Z.; Wei L.; Wang Y.; Chen H. In vitroinhibition of HBVreplication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antiviral Ther. 2012, 17, 793. 10.3851/IMP2152. [DOI] [PubMed] [Google Scholar]
- Zhou X.; Gao Z.; Meng J.; Chen X.; Zhong D. Effects of ketoconazoleand rifampicin on the pharmacokinetics of GLS4, a novelanti-hepatitis B virus compound, in dogs. Acta Pharmacol. Sin. 2013, 34, 1420. 10.1038/aps.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wei Z.; Wu G.; et al. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil- resistant HBV mutations. Antiviral Ther. 2012, 17, 793. 10.3851/IMP2152. [DOI] [PubMed] [Google Scholar]
- Hinkle G.; Sepp-Lorenzino L. Hepatitis b virus (hbv) irna compositions and methods of use thereof. WO2016077321A1, 2016.
- Slee H. D.; Chen Y.; Zhang X. 2-Amino-N-pyrimidin-4-ylacetamides as A2A Receptor Antagonists: 1. Structure–Activity Relationships and Optimization of Heterocyclic Substituents. J. Med. Chem. 2008, 51, 1730. 10.1021/jm701187w. [DOI] [PubMed] [Google Scholar]
- Kappe C. O. 100 years of the biginelli dihydropyrimidine synthesis. Tetrahedron 1993, 49, 6937. 10.1016/S0040-4020(01)87971-0. [DOI] [Google Scholar]
- The absolute stereochemistry of (−)-enantiomer 20 was determined by its rotation value as reported in WO201437480.
- Zlotnick A.; Venkatakrishnan B.; Tan Z.; Lewellyn E.; Turner W.; Francis S. Core Protein: A Pleiotropic Keystone in the HBV Lifecycle. Antiviral Res. 2015, 121, 82. 10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z.; Lin X.; Zhou M.; Liu Y.; Zhu W.; Chen W.; Zhang W.; Guo L.; Liu H.; Wu G.; Huang M.; Jiang M.; Xu Z.; Zhou Z.; Qin N.; Ren S.; Qiu H.; Zhong S.; Zhang Y.; Zhang Y.; Wu X.; Shi L.; Shen F.; Mao Y.; Zhou X.; Yang W.; Wu J. Z.; Yang G.; Mayweg A. V.; Shen H. C.; Tang G. Design and Synthesis of Orally Bioavailable 4-Methyl Heteroaryldihydropyrimidine Based Hepatitis B Virus (HBV) Capsid Inhibitors. J. Med. Chem. 2016, 59, 7651. 10.1021/acs.jmedchem.6b00879. [DOI] [PubMed] [Google Scholar]
- Klumpp K.; Lam A. M.; Lukacs C.; Vogel R.; Ren S.; Espiritu C.; Baydo R.; Atkins K.; Abendroth J.; Liao G.; Efimov A.; Hartman G.; Flores O. A. High-Resolution Crystal Structure of a Hepatitis B Virus Replication Inhibitor Bound to the Viral Core Protein. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 15196. 10.1073/pnas.1513803112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.8; Schrödinger, LLC: New York [Google Scholar]
- Schrödinger Release 2017–2: Maestro; Schrödinger, LLC: New York, 2017. [Google Scholar]
- Huang L. R.; Gabel Y. A.; Graf S.; Arzberger S.; Kurts C.; Heikenwalder M.; Knolle P. A.; Protzer U. Transfer of HBV genomes using low doses of adenovirus vectors leads to persistentinfection in immune competent mice. Gastroenterology 2012, 142, 1447. 10.1053/j.gastro.2012.03.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.