

Abstract
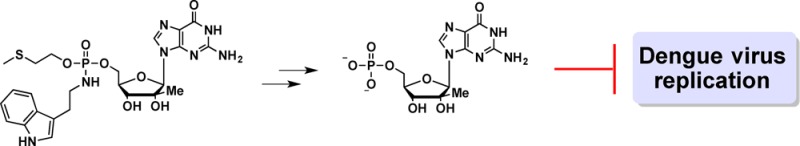
This work describes the synthesis and biological evaluation of an anchimerically activated proTide of 2′-C-β-methylguanosine as an inhibitor of dengue virus 2 (DENV-2). The proTide incorporates a chemically cleavable 2-(methylthio)ethyl moiety and a HINT1 hydrolyzable tryptamine phosphoramidate. Inhibition of DENV-2 replication by proTide 6 was 5-fold greater than the parent nucleoside while displaying no apparent cytotoxicity. Furthermore, we demonstrate with a HINT1 inhibitor that the anti DENV-2 activity of the proTide correlates with the activity of HINT1. Taken together, these results demonstrate that a phosphoramidate based pronucleotide that undergoes an initial nonenzymatic activation step based on anchimeric assistance followed by P–N bond cleavage by HINT1 can be prepared.
Keywords: Phosphoramidate, pronucleotide, Dengue virus, antiviral
Nucleoside/nucleotide analogues have over the years emerged as powerful tools for combating viral infections.1 Specifically, nucleoside analogues require conversion by cellular kinases to their active nucleotide metabolites, which can directly inhibit intracellular enzymes or get incorporated into newly synthesized DNA or RNA.1,2 The tight substrate specificity of human kinases compared to viral kinases provides a basis for selectivity of nucleoside/nucleotide analogues as antiviral therapies.2,3 Incorporation of a nucleoside analogue triphosphate during viral DNA/RNA synthesis may lead to termination of chain elongation, accumulation of mutations during viral replication, or initiation of apoptosis by viral infected cells.1,4
As a well-studied drug target, significant efforts have been made at developing nucleoside analogues that target viral polymerases. Particularly, viral RNA-dependent RNA polymerase (RdRp) has emerged as an attractive drug target because of its pivotal role in the synthesis of the viral genome of RNA viruses.1,4 Several studies have established nucleoside analogues bearing a 2′-C-modification as a common pharmacophore in nucleoside analogue inhibitors of viral polymerases. Specifically, the 2′-C-β-methyl (2′-C-β-Me) modification has emerged as the preferred pharmacophore in viral nucleoside analogue inhibitors. These nucleoside inhibitors, previously developed for hepatitis C virus (HCV), have been shown to be potent inhibitors of DENV due to the phylogenetic similarity between HCV and DENV RdRps.5,6 The most potent 2′-C-β-Me inhibitors of a serotype of DENV (DENV-2) were 2′-C-β-methyladenosine (2′-C-β-Me-A) and 2′-C-β-methylguanosine (2′-C-β-Me-G), with antiviral EC50 values of 4 and 13.6 μM, respectively (Figure 1A).7 In addition, 1′-cyano-4-aza-7,9-dideazaadenosine C-nucleotide was shown to have antiviral activity against DENV-2 (EC50 = 9.46 μM).8
Figure 1.
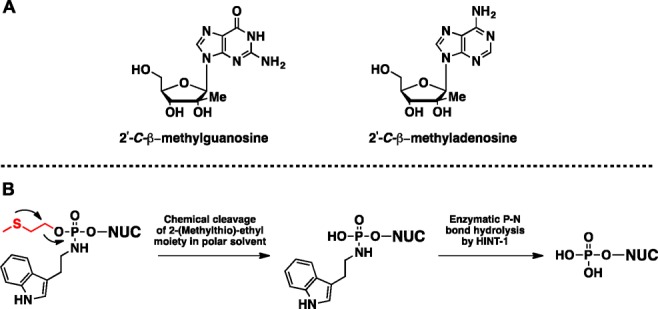
Anti-Dengue nucleosides (A) and activation mechanism of 2-(methylthio)ethyl phosphoramidate diester prodrug (B).
Although nucleoside/nucleotide analogues are an established antiviral therapy, their development as effective therapeutics is faced with several challenges. Since nucleoside analogues are modified derivatives of naturally occurring nucleosides, the efficiency of conversion to sufficient amounts of their respective 5′-triphosphates, by host kinases, can be problematic. In particular, the first phosphorylation event for several nucleosides has been found to be rate limiting. Consequently, several phosphorylation bypass strategies have been developed for the intracellular delivery of monophosphorylated nucleoside analogues.9−11 Such prodrug strategies have been successfully utilized, as evident in the development of the anti-HCV drug sofosbusvir.12 Sofosbusvir incorporates a proTide phosphoramidate strategy, which utilizes a combination of initial esterase hydrolysis, followed by mixed anhydride chemical hydrolysis, and finally phosphoramidate hydrolysis by hHINT1.13,14
Over the last three decades, a variety of proTide approaches have been successfully explored, such as bis-POM,15 SATE,16 cyclophosphamide,17 and cyclo-Sal,18 as well as several others.9,19 We and others have investigated the utility of phosphoramidate-based proTides for the delivery of antiviral and anticancer nucleoside monophosphates.11,14,20,21
Upon investigation of the metabolism of phosphoramidate proTides, we established that P–N bond hydrolysis by HINT1 is responsible for the intracellular release of the nucleoside monophosphate.22 For example, recently we have shown that 4Ei-1, a monoester phosphoramidate pronucleotide of the eIF4E antagonist N7-benzyl guanosine monophosphate, can serve as a selective inhibitor of the epithelial–mesenchymal transition (EMT).23 Building upon our success at developing monoester phosphoramidates, we have focused our efforts at developing phosphoramidate diester prodrugs because of the potential to engender increased lipophilicity and cell permeability compared to monoester phosphoramidates. Nevertheless, to date, the conversion of most diester phosphoramidates pronucleotides is typically initiated by esterase-dependent hydrolysis of one of the phosphate protecting moieties.19 Consequently, preclinical studies in rodents (rats and mice) have proven to be problematic since the plasma of mice and rats contains considerably more carboxylesterase activity than in humans.24−26 In addition, due to the high carboxylesterase activity of the liver and high liver first pass metabolism, accumulation of nucleoside monophosphate after treatment with a diester phosphoramidate proTide has been reported.27 Thus, high hepatic extraction can limit the application of these proTides for systemic delivery of nucleotides after oral administration. In order to address some of the drawbacks of currently used proTides, we propose a diester phosphoramidate strategy that utilizes an initial chemical activation step to liberate the monoester phosphoramidate, which can be hydrolyzed by HINT1 to release the nucleotide.
Cieślak and co-workers had previously reported the use of 2-(methylthio)ethyl protection group for oligonucleotide synthesis.28 Unmasking of the phosphate oxygen (in a model 4-methylthio-1-butyl system) was shown to be dependent on anchimeric assistance (by sulfur atom) and release of a cyclic sulfonium ion. While the methylthio alkyl protecting groups are stable in organic solvents, intramolecular nucleophilic attack in polar aqueous media has been found to be temperature dependent with a half-life of approximately 2–4 min at 55 °C and at pH 7.0.28 Therefore, we hypothesized that 2-(methylthio)ethyl phosphoramidates may have a longer half-life under physiological conditions (37 °C, pH = 7.4) and thus afford a membrane permeable diester phosphoramidate proTide that is able to undergo intracellular release of the monoester phosphoramidate, followed by HINT1 P–N bond cleavage to deliver the nucleoside monophosphate (Figure 1B). We chose to investigate our hypothesis by preparing a 2-(methylthio)ethyl phosphoramidate diester prodrug of the antiviral nucleoside, 2′-C-β-Me-guanosine, followed by characterization of its in vitro stability and anti-Dengue biological activity.
The synthesis of 2′-C-β-Me-G (1) was performed according to a published procedure.29 Treatment of 1 with DMTrCl afforded a mixture of 5′-OH mono-protected nucleoside (2a) as well as a 5′-OH and N2-amino bis-DMTr-protected nucleoside (2b). Subsequent treatment of the mixture with Ac2O afforded a mix of 5′-O-DMTr triacetyl-protected nucleoside (3a) and a 5′-O-N2 bis-DMTr diacetyl-protected nucleoside (3b). Subsequent detritylation of the mixture of 3a and 3b afforded the triacetyl-protected nucleoside 4. The H-phosphonate monoester intermediate 7 was coupled to nucleoside 4 in the presence of pivaloyl chloride to give the corresponding H-phosphonate diester intermediate, which was treated with sat. aq. NaHCO3 and extracted with CH2Cl2 to remove excess pivaloyl chloride. Due to the sensitivity of the nucleoside H-phosphonate monoester intermediate to hydrolysis during silica gel chromatography, the crude organic extract was subjected to oxidative coupling, to install tryptamine, under standard Atherton–Todd reaction conditions. Deacetylation of the resulting phosphoramidate diester 5, with methanolic ammonia yielded 6, as a diastereomeric mixture (7:3).
In addition to preparing the diester phosphoramidate prodrug of 2′-C-β-Me-G, we also prepared the tryptamine monoester derivative of the nucleoside prodrug. Preparation of the monoester phosphoramidate could be achieved via two routes. Route A involved treatment of nucleoside 1 with POCl3 to give the 5′-O-phosphate product 8, which was subsequently used in an EDC-HCl mediated phosphoramidate-forming reaction to give the desired tryptamine phosphoramidate nucleoside prodrug 9. Alternatively, phosphoramidate 9 could be accessed via deprotection of the methylthio ethyl moiety in 10 mM HEPES buffer at 37 °C (route B) (Scheme 1).
Scheme 1.
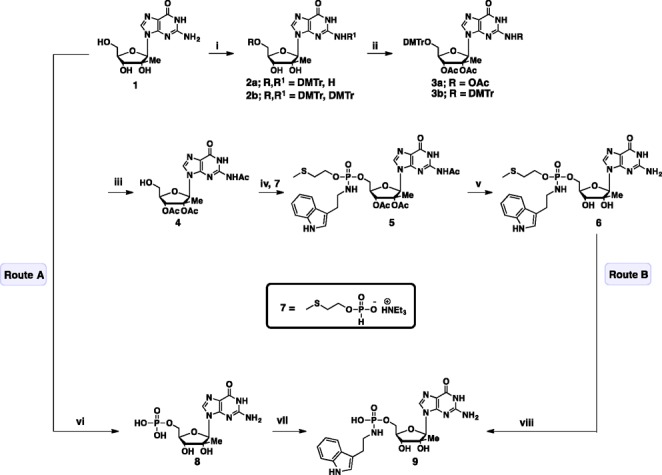
Reagents and conditions: (i) DMTrCl, DMSO, pyridine, rt, overnight; (ii) MeCN, DMAP, Ac2O, pyridine, rt, overnight; (iii) 80% AcOH, 55 °C, 1.5 h, 27% over 3 steps; (iv) 7, PivCl, pyridine, rt, 8 h; then tryptamine, CCl4, NEt3, pyridine, rt, 30 min, 65%; (v) 7 N NH3/MeOH, rt, 4 h, 64%; (vi) (EtO)3PO/POCl3 (10:1), 0 °C, 0.75 h, Dowex-50, 74%; (vii) tryptamine, EDC-HCl, H2O, pH 7, 55 °C, 6 h, Dowex-50WX8, 23%; (viii) 10 mM HEPES, pH 7, 37 °C, 30 h, 84%.
In order to be effective as therapeutic agents, prodrugs in general should be chemically stable without premature decomposition/release of the active metabolite. Consequently, we evaluated the aqueous stability of the nucleoside phosphoramidate diester 6.
To determine the half-life for deprotection of the 2-(methylthio)ethyl moiety under physiological conditions, we incubated phosphoramidate diester 6 at 37 °C in 20 mM HEPES buffer (pH 7.4). The deprotection reaction was then monitored by reversed phase high-performance liquid chromatography (RP-HPLC). As can be seen in Figure 2, conversion of both diastereomers of 6 to phosphoramidate 9 proceeded smoothly over the course of 24 h with a half-life of 4.50 ± 0.12 h. When the pH of the reaction mixture was lowered to 2.0, the half-life for the cleavage reaction was 3.78 ± 0.32 h. Overall, the results of the stability studies suggest that prodrug 6 should be sufficiently stable in a biological environment and lipophilic enough to cross cell membranes.
Figure 2.

Typical time-course deprotection of 2-(methylthio) ethyl moiety of compound 6 and subsequent formation of compound 9 as monitored by RP-HPLC.
The cytotoxicity and antiviral activities of 1, 6, and 9 were assessed to determine the impact of our prodrug design on cellular uptake and monophosphate delivery. First, compounds 1, 6, and 9 were found to be noncytotoxic (CC50 > 200 μM) to the host Vero cells (Figure 3). Second, as can been seen in Table 1 and Supporting Information Figure 1, the ability of diester phosphoramidate 6 to inhibit viral proliferation was 5-fold greater than the parent nucleoside 1 and over 4-fold greater than monoester phosphoramidate, 9. Furthermore, nucleoside 1 and monoester phosphoramidate 9 showed no significance difference in their ability to inhibit DENV-2 proliferation. These results suggest that from a biological standpoint, the conversion of 2′-C-β-Me-G (1) to the monophosphate by cellular kinases occurs with similar efficiency as the conversion of monoester phosphoramidate 9 into the nucleotide by HINT1. Most importantly, our findings further suggest that diester phosphoramidate 6 is able to penetrate the cell membrane and deliver 2′-C-β-Me-G monophosphate more efficiently than 9, despite the additional achimeric activation step before HINT1 P–N bond cleavage. The necessary role of HINT1 in the activation of phosphoramidates 6 and 9 was confirmed by assessing the antiviral activity of each compound in the presence of HINT1 inhibitor, TrpGc.30
Figure 3.

Cell viability of Vero cells treated with compounds 1, 6, and 9. Vero cells were incubated in the presence of different concentrations of compounds 1, 6, and 9 for 72 h. Cell viability was assayed by Neutral Red dye uptake. Results are from six replicates.
Table 1. In Vitro Biological Activity of Nucleoside 1 and Its Phosphoramidates 6 and 9.
| compd | IC50 (μM)a | observed toxicity (μM) |
|---|---|---|
| 1 | 8.14 ± 0.66 | >200 |
| 6 | 1.59 ± 0.91 | >200 |
| 9 | 6.84 ± 2.36 | >200 |
Experiments were performed in triplicates.
Treatment of DENV-2 infected Vero cells with either compound 6 or 9 in the presence of TpGc resulted in reduced inhibition of viral replication when compared to virus infected cells that were treated with only compound 6 or 9 (Figure 4). In contrast, DENV-2 infected cells that were treated with nucleoside 1 alone, or in combination with TrpGc, did not show a significant difference in antiviral response. This result is expected since the conversion of nucleoside 1 to its monophosphate does not require HINT1. Taken altogether, these results suggest that the phosphoramidase activity of HINT1 is essential for activation of prodrug 6 and 9 to 2′-C-β-Me-G monophosphate.
Figure 4.

Effect of TrpGc on antiviral activity in DENV-2 infected Vero cells treated with compounds 1, 6, and 9. Inhibition of HINT1 reverts the protective effect against DENV-2 replication. Vero cells were infected with DENV-2 (moi = 0.2) and treated with 100 μM compounds 1, 6, and 9 alone or in combination with 100 μM TrpGc. After 72 h, supernatants were processed for RNA extraction and production of viral progeny was measured by qPCR specific for DENV-2. Results represent the percentage of replication to the untreated DENV-2 infected control. Results are represented as mean of four different experiments. *DENV-2 + (6) versus DENV-2 + (6) + TrpGc (p = 0.0229). #DENV-2 + (9) versus DENV-2 + (9) + TrpGc (p = 0.0126). ***DENV-2 versus DENV-2 + (6), (9), and (1) (p = 0.0305, 0.0208, and 0.0002, respectively).
Overall, our results demonstrate that the 2-(methylthio)ethyl phosphate masking moiety can be incorporated into a phosphoramidate proTide of an antiviral ribonucleoside, which enhances the antiviral potency of the parental nucleoside. This approach circumvents the need for an initiating enzymatic activation step while being dependent on the intracellular liberation of the nucleoside monophosphate from the monoester phosphoramidate metabolite by HINT1. The results of studies addressing the potential for this approach to enhance the oral and systemic delivery of nucleoside monophosphates will be reported in due course.
Acknowledgments
Funding from the University of Minnesota Foundation and support of A.O. by NIH training grant T32 GM008700 is gratefully acknowledged.
Glossary
ABBREVIATIONS
- DMTrCl
4,4′-dimethoxytrityl chloride
- HINT1
histidine triad nucleotide-binding protein 1
- POCl3
phosphorus(V) oxychloride
- EDC-HCl
1-(3-(dimethylamino)propyl)-3-ethylcarbodiimide hydrochloride
- DMSO
dimethyl sulfoxide
- DMAP
4-dimethylaminopyridine
- eIF4E
eukaryotic translation initiation factor 4E
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00277.
Full synthetic procedure for all test compounds, 1HNMR, 13CNMR, 31PNMR, and HPLC characterization for all test compounds, and full procedure for biological evaluation of test compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12, 447–64. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Chen Y. L.; Yokokawa F.; Shi P. Y. The search for nucleoside/nucleotide analog inhibitors of dengue virus. Antiviral Res. 2015, 122, 12–9. 10.1016/j.antiviral.2015.07.010. [DOI] [PubMed] [Google Scholar]
- Yeo K. L.; Chen Y. L.; Xu H. Y.; Dong H.; Wang Q. Y.; Yokokawa F.; Shi P. Y. Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob. Agents Chemother. 2015, 59, 2086–93. 10.1128/AAC.04779-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen D. B.; Eldrup A. B.; Bartholomew L.; Bhat B.; Bosserman M. R.; Ceccacci A.; Colwell L. F.; Fay J. F.; Flores O. A.; Getty K. L.; Grobler J. A.; LaFemina R. L.; Markel E. J.; Migliaccio G.; Prhavc M.; Stahlhut M. W.; Tomassini J. E.; MacCoss M.; Hazuda D. J.; Carroll S. S. A 7-deaza-adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob. Agents Chemother. 2004, 48, 3944–53. 10.1128/AAC.48.10.3944-3953.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V.; Dolja V. V. Evolution and taxonomy of positive-strand RNA viruses: implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. 10.3109/10409239309078440. [DOI] [PubMed] [Google Scholar]
- Ng K. K.; Arnold J. J.; Cameron C. E. Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 2008, 320, 137–56. 10.1007/978-3-540-75157-1_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio G.; Tomassini J. E.; Carroll S. S.; Tomei L.; Altamura S.; Bhat B.; Bartholomew L.; Bosserman M. R.; Ceccacci A.; Colwell L. F.; Cortese R.; De Francesco R.; Eldrup A. B.; Getty K. L.; Hou X. S.; LaFemina R. L.; Ludmerer S. W.; MacCoss M.; McMasters D. R.; Stahlhut M. W.; Olsen D. B.; Hazuda D. J.; Flores O. A. Characterization of resistance to non-obligate chain-terminating ribonucleoside analogs that inhibit hepatitis C virus replication in vitro. J. Biol. Chem. 2003, 278, 49164–70. 10.1074/jbc.M305041200. [DOI] [PubMed] [Google Scholar]
- Cho A.; Saunders O. L.; Butler T.; Zhang L.; Xu J.; Vela J. E.; Feng J. Y.; Ray A. S.; Kim C. U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett. 2012, 22, 2705–7. 10.1016/j.bmcl.2012.02.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner C. R.; Iyer V. V.; McIntee E. J. Pronucleotides: toward the in vivo delivery of antiviral and anticancer nucleotides. Med. Res. Rev. 2000, 20, 417–451. . [DOI] [PubMed] [Google Scholar]
- Hecker S. J.; Erion M. D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. 10.1021/jm701260b. [DOI] [PubMed] [Google Scholar]
- Mehellou Y. The ProTides Boom. ChemMedChem 2016, 11, 1114–1116. 10.1002/cmdc.201600156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers B. B.; Beavers J. W. F.; Klibanov O. M. Sofosbuvir, a novel nucleotide analogue inhibitor used for the treatment of hepatitis C virus. J. Pharm. Pharmacol. 2014, 66, 1653–1666. 10.1111/jphp.12294. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Pathirana R. N.; Balzarini J.; De Clercq E. Intracellular delivery of bioactive AZT nucleotides by aryl phosphate derivatives of AZT. J. Med. Chem. 1993, 36, 1048–52. 10.1021/jm00060a013. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Perrone P.; Madela K.; Neyts J. The phosphoramidate ProTide approach greatly enhances the activity of beta-2′-C-methylguanosine against hepatitis C virus. Bioorg. Med. Chem. Lett. 2009, 19, 4316–20. 10.1016/j.bmcl.2009.05.122. [DOI] [PubMed] [Google Scholar]
- Farquhar D.; Chen R.; S K. 5′-[4-(Pivaloyloxy)-1,3,2-dioxaphosphorinan-2-yl]-2 0-deoxy-5-¯uorouridine: A membrane-permeating prodrug of 5-¯uoro-20-deoxyuridylic acid(FdUMP). J. Med. Chem. 1995, 38, 488–495. 10.1021/jm00003a012. [DOI] [PubMed] [Google Scholar]
- Girardet J.-L.; Perigaud C.; Aubertin A. M.; Gosselin G.; Kirn A.; Imbach J.-L. Increase of the anti-HIV activity of d4T in human T-cell culture by the use of the SATE pronucleotide approach. Bioorg. Med. Chem. Lett. 1995, 5, 2981–2984. 10.1016/0960-894X(95)00525-7. [DOI] [Google Scholar]
- Borch R. F.; Canute G. W. Synthesis and antitumor properties of activated cyclophosphamide analogues. J. Med. Chem. 1991, 34, 3044–3052. 10.1021/jm00114a013. [DOI] [PubMed] [Google Scholar]
- Meier C.; Knispel T.; De Clercq E.; Balzarini J. CycloSal-pronucleotides of 2′,3′-dideoxyadenosine and 2′,3′-dideoxy-2′,3′-didehydroadenosine: synthesis and antiviral evaluation of a highly efficient nucleotide delivery system. J. Med. Chem. 1999, 42, 1604–1614. 10.1021/jm981096z. [DOI] [PubMed] [Google Scholar]
- Pradere U.; Garnier-Amblard E. C.; Coats S. J.; Amblard F.; Schinazi R. F. Synthesis of nucleoside phosphate and phosphonate rodrugs. Chem. Rev. 2014, 114, 9154–9218. 10.1021/cr5002035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham T. W.; Wagner C. R. A phosphoramidite-based synthesis of phosphoramidate amino-acid diesters of antiviral nucleosides. Nucleosides Nucleotides 1994, 13, 1891–1903. 10.1080/15257779408010671. [DOI] [Google Scholar]
- Wagner C. R.; McIntee E. J.; Schinazi R. F.; Abraham T. W. Aromatic amino-acid phosphoramidate diesters and triesters of 3′-azido-3′-deoxythymidine (AZT) are nontoxic inhibitors of HIV-1 replication. Bioorg. Med. Chem. Lett. 1995, 5, 1819–1824. 10.1016/0960-894X(95)00302-A. [DOI] [Google Scholar]
- Chou T. F.; Bieganowski P.; Shilinski K.; Cheng J.; Brenner C.; Wagner C. R. 31P NMR and genetic analysis establish hinT as the only Escherchia coli purine nucleoside phosphoramidase and as essential for growth under high salt conditions. J. Biol. Chem. 2005, 280, 15356–61. 10.1074/jbc.M500434200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith K. A.; Zhou B.; Avdulov S.; Benyumov A.; Peterson M.; Liu Y.; Okon A.; Hergert P.; Braziunas J.; Wagner C. R.; Borok Z.; Bitterman P. B. Transforming Growth Factor-β1 Induced Epithelial Mesenchymal Transition is blocked by a chemical antagonist of translation factor eIF4E. Sci. Rep. 2016, 5, 18233. 10.1038/srep18233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardelli C.; Attenni B.; Donghi M.; Meppen M.; Pacini B.; Harper S.; Di Marco A.; Fiore F.; Giuliano C.; Pucci V.; Laufer R.; Gennari N.; Marcucci I.; Leone J. F.; Olsen D. B.; MacCoss M.; Rowley M.; Narjes F. Phosphoramidate prodrugs of 2′-C-methylcytidine for therapy of hepatitis C virus infection. J. Med. Chem. 2009, 52, 5394–407. 10.1021/jm900447q. [DOI] [PubMed] [Google Scholar]
- Minagawa T.; Kohno Y.; Suwa T.; Tsuji A. Species differences in hydrolysis of isocarbacyclin methyl ester (TEI-9090) by blood esterases. Biochem. Pharmacol. 1995, 49, 1361–5. 10.1016/0006-2952(95)00071-7. [DOI] [PubMed] [Google Scholar]
- Rudakova E. V.; Boltneva N. P.; Makhaeva G. F. Comparative analysis of esterase activities of human, mouse, and rat blood. Bull. Exp. Biol. Med. 2011, 152, 73–5. 10.1007/s10517-011-1457-y. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Gilles A.; Madela K.; Aljarah M.; Holl S.; Jones S.; Vernachio J.; Hutchins J.; Ames B.; Bryant K. D.; Gorovits E.; Ganguly B.; Hunley D.; Hall A.; Kolykhalov A.; Liu Y.; Muhammad J.; Raja N.; Walters R.; Wang J.; Chamberlain S.; Henson G. Phosphoramidate ProTides of 2′-C-methylguanosine as highly potent inhibitors of hepatitis C virus. Study of their in vitro and in vivo properties. J. Med. Chem. 2010, 53, 4949–57. 10.1021/jm1003792. [DOI] [PubMed] [Google Scholar]
- Cieślak J.; Grajkowski A.; Livengood V.; Beaucage S. L. Thermolytic 4-methylthio-1-butyl group for phosphate/thiophosphate protection in solid-phase synthesis of DNA oligonucleotides. J. Org. Chem. 2004, 69, 2509–15. 10.1021/jo035861f. [DOI] [PubMed] [Google Scholar]
- Li N. S.; Piccirilli J. A. Efficient synthesis of 2′-C-beta-methylguanosine. J. Org. Chem. 2006, 71, 4018–20. 10.1021/jo0602165. [DOI] [PubMed] [Google Scholar]
- Garzón J.; Herrero-Labrador R.; Rodríguez-Muñoz M.; Shah R.; Vicente-Sánchez A.; Wagner C. R.; Sánchez-Blázquez P. HINT1 protein: a new therapeutic target to enhance opioid antinociception and block mechanical allodynia. Neuropharmacology 2015, 89, 412–23. 10.1016/j.neuropharm.2014.10.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.