

Abstract

We report the synthesis, biological evaluation, and structural study of a series of substituted heteroaryl-pyrazole carboxylic acid derivatives. These compounds have been developed as inhibitors of specific isoforms of carbonic anhydrase (CA), with potential as prototypes of a new class of chemotherapeutics. Both X-ray crystallography and computational modeling provide insights into the CA inhibition mechanism. Results indicate that this chemotype produces an indirect interference with the zinc ion, thus behaving differently from other related nonclassical inhibitors. Among the tested compounds, 2c with Ki = 0.21 μM toward hCA XII demonstrated significant antiproliferative activity against hypoxic tumor cell lines. Taken together, the results thus provide the basis of structural determinants for the development of novel anticancer agents.
Keywords: Carbonic anhydrase, hCA XII inhibitors, heteroaryl-pyrazole carboxylic acids, X-ray crystallography, computational docking, hypoxic tumors
Many currently used antitumor drugs demonstrate relatively poor selectivity for cancer cells, causing frequent toxicity and side effects. A current strategy to overcome this impasse relies on the identification of biological targets that are exclusive, or at least highly prevalent, in cancer cells. One of the most suited hallmarks of cancer is metabolic reprogramming, which offers new opportunities for the development of innovative chemotherapeutics.1 In particular, hypoxia, a common condition in tumor microenvironment, promotes consistent metabolic adaptation, such as the switching of energetic metabolism from aerobic to anaerobic glycolysis.2,3 A critical consequence of this phenomenon is the overproduction of weak acid species, such as lactic acid. In order to preserve their acid–base homeostasis, cancer cells adopt a series of adaptive mechanisms through the activation of specific factors (such as the hypoxia-inducible factor 1, HIF-1), which promptly remove/rebalance the H+ excess from the cells, thus lowering the extracellular pH.4,5
As far as other putative biological pathways involved with hypoxic cancer are concerned, carbonic anhydrases (CAs), a family of metalloenzymes that catalyze the biochemical reaction of carbon dioxide hydration to produce a bicarbonate ion and a proton, are emerging as potential biological targets for cancer therapy.6 Previously, two tumor-associated extracellular transmembrane carbonic anhydrase isozymes (hCA IX and hCA XII) were identified, cloned, and sequenced.6−8 These two CA isoforms have been shown to be expressed in a wide variety of malignancies and appear to be tightly regulated by microenvironmental hypoxia.7,8 This phenomenon is particularly evident in highly malignant solid tumors and has been linked to poor blood perfusion that reduces the provision of oxygen and nutrients for cancer cells, thus inducing a series of adaptive responses (i.e., metabolic shift to anaerobiosis and increased amino acids catabolism) that lead to lower extracellular pH, a condition that is not favorable for cell survival.
In this scenario, the extracellular CA isoforms are implicated in rebalancing the acid/base equilibrium of the extracellular medium.6,9,19 Specifically, hCA XII is minimally expressed in a variety of normal tissues including kidney, colon, prostate, pancreas, ovary, testis, lung, and brain, but its expression is up-regulated in cancer cells originating from these tissues.7 Several recent studies proposed hCA XII as a suitable and attractive target for both diagnostic and therapeutic intervention, particularly on the management of hypoxic tumors normally nonresponsive to classical chemo- and radiotherapies.9−12
Primary sulfonamides, the most clinically used CAs inhibitors (CAIs), have been shown to reverse the effect of the extracellular CAs and inhibit the growth of cancer cells in the low nanomolar or micromolar range.13−15 However, one main drawback of these classical sulfonamide CAIs is the lack of selectivity for inhibiting transmembrane CAs over the other cytoplasmic CA isoforms present in humans and in mouse model systems. For this reason, efforts are being initiated to find novel CAIs, in order to explore molecular diversities and discover original pharmacophores and chemotypes.13−16
This study focuses on the structural optimization of a previously identified hit compound, the 3-(1-ethyl-1H-indol-3-yl)-1H-pyrazole-5-carboxylic acid (1a, Chart 1),17 in order to generate and evaluate a new series of indolylpyrazole-5-carboxylic acids/esters with improved inhibition profile toward hCA IX and hCA XII.
Chart 1. Design and General Structure of Title Compounds.
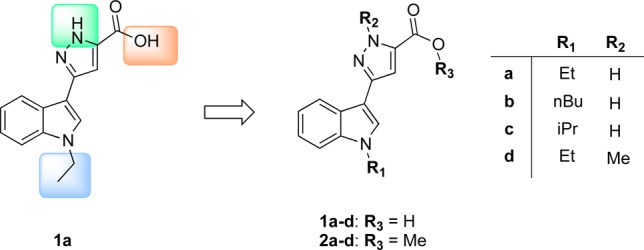
Preliminary major modifications of 1a have been made by varying the substituents at the nitrogen atoms both on the indole and pyrazole rings and by esterification of the carboxylic functionality to generate the series 1a–d and 2a–d (Chart 1). Specifically, N-alkylation of the indole was first explored to modulate lipophilicity; second, N-methylation of the pyrazole was introduced to abolish N–H interaction; finally, on the basis of the assumption that the carboxylic moiety is a necessary feature for the inhibitory activity, we examined the effect of esterification on the activity.
Compounds 1a–d and 2a–d were synthesized starting from the 3-acetyl indole 3 by alkylation reaction with the appropriate alkyl bromide to obtain the N-alkyl-3-acetyl-indole derivatives 4a–c (Scheme 1).
Scheme 1. Synthetic Route for the Preparation of Compounds 1a–d and 2a–d.

These intermediates were reacted with diethyl oxalate in the presence of sodium methoxide in MeOH, to provide the respective β-diketoesters 5a–c. The latter were found to exist in the keto-enol form in DMSO since 1H NMR experiments showed a singlet centered at 6.87 ppm (for 5a,b) and at 6.89 (for 5c) attributable to the enolic CH of the hydroxy-keto motif. Next, the compounds 5a–c were treated with hydrazine monohydrate to generate the pyrazole derivatives 2a–c in appreciable yields (46–93%). Then, the acids 1a–c were obtained by alkaline hydrolysis of the esters 2a–c. Finally, the compound 2d, containing a methyl group on the nitrogen of pyrazole ring, was synthesized by alkylation of intermediate 2a with iodomethane in the presence of NaH. The corresponding acid 1d was obtained from 2d by alkaline hydrolysis with NaOH in ethanol at reflux.
Compounds 1a–d and 2a–d were tested for their ability to inhibit a panel of representative isoforms of human CAs (I, II, IX, and XII, Table 1). Catalytic activities were measured by a stopped-flow technique, as previously described (see Supporting Information).
Table 1. Inhibition of hCA Isoforms I, II, IX and XII with Compounds 1a–d and 2a–d.
|
Ki (μM)a |
||||
|---|---|---|---|---|
| compd | hCA Ib | hCA IIb | hCA IXc | hCA XIIc |
| 1a | 0.042 ± 0.0001 | 1820 ± 9 | 7.79 ± 0.06 | 7.78 ± 0.04 |
| 1b | 5.33 ± 0.04 | 4.70 ± 0.01 | 4.51 ± 0.03 | 0.34 ± 0.006 |
| 1c | 4.83 ± 0.07 | 0.70 ± 0.008 | 18.9 ± 0.21 | 0.28 ± 0.01 |
| 1d | 6.61 ± 0.02 | 0.78 ± 0.009 | 2.91 ± 0.04 | 0.44 ± 0.005 |
| 2a | 4.31 ± 0.06 | 6.95 ± 0.08 | 0.47 ± 0.003 | 0.57 ± 0.008 |
| 2b | 5.93 ± 0.06 | 0.53 ± 0.003 | 7.90 ± 0.08 | 0.35 ± 0.002 |
| 2c | 6.45 ± 0.03 | 0.76 ± 0.05 | 7.36 ± 0.04 | 0.21 ± 0.01 |
| 2d | 0.62 ± 0.005 | 0.41 ± 0.007 | 3.02 ± 0.008 | 0.31 ± 0.01 |
| AAZ | 0.25 ± 0.01 | 0.012 ± 0.001 | 0.025 ± 0.002 | 0.006 ± 0.0005 |
| MZA | 0.78 ± 0.04 | 0.014 ± 0.001 | 0.027 ± 0.008 | 0.034 ± 0.002 |
| DCP | 1.2 ± 0.1 | 0.038 ± 0.003 | 0.050 ± 0.005 | 0.050 ± 0.004 |
Errors in the range of ±5–10% of the reported value from three different determinations.
Full length.
Catalytic domain. AAZ, acetazolamide; MZA, methazolamide; DCP, dichlorophenamide.
Although structural modifications of the lead compound 1a reduced the potency and selectivity against hCA I isoform (work is in progress to clarify the behavior of this outlier compound), these new compounds showed an interesting selectivity toward hCA XII, with Ki values ranging from 0.21 to 0.57 μM (Table 1). Moreover, with the exception of 1b and 2a, the tested compounds effectively interfere with hCA II catalytic activities in submicromolar/micromolar concentration range (Ki values of 0.41–6.95 μM, Table 1). On the contrary, no significant inhibition or slightly weaker inhibitory action toward hCA I and hCA IX (with the exception of 2d and 2a, with Ki values of 0.62 and 0.47 μM, for hCA I and hCA IX, respectively), were revealed.
Overall, the biological profile appeared to be substantially independent from the nature of substituents on both the indole and the pyrazole rings. However, although a proper structure–activity relationship (SAR) for hCA XII inhibition is not feasible at this stage, it is plausible that structural determinants of this chemotype (i.e., indole ring system, lypophilic substituents at the nitrogen indole, and carboxylic functionality) appear to be an optimal combination to effectively inhibit the hCA XII (with a certain mutual interference with cytosolic hCA II catalytic activity). Again, no relevant differences could be observed between acids and esters on the inhibition profile. Nevertheless, among the tested compounds, compound 2c (Ki = 0.21 μM) proved to be the most active compound in inhibiting hCA XII, thus demonstrating to be one of the most effective nonsulfonamide-based inhibitors toward this CA isoform.
Since the indolylpyrazole-5-carboxylate backbone appeared to be an original chemotype for CA inhibition, the interaction of representative compound 1d with hCA II was studied by X-ray crystallography and determined to 1.2 Å resolution (statistics summarized in Table S1). Structural analysis of the hCA II/1d complex indicates that the compound does not bind directly to the active site zinc(II) ion, making it dissimilar to other potent CA inhibitors.18 For example, apart from the classical (i.e., sulfonamides and their isosteres) zinc binders and nonclassical (e.g., carboxylates, hydroxamates, dithiocarbamates, and phosphonates) CAIs, other classes of compounds were found to act through another mechanism of action by (a) anchoring to the metal ion coordinated nucleophile binding in a more distant part of the active site cavity compared with the zinc binders (phenols, polyamines, esters, sulfocoumarins), (b) occluding the entrance of the active site cavity (coumarins), and (c) binding outside the active site (benzoic acids).18
Instead, the compound 1d utilizes a carboxylic acid moiety to substitute the typical location of a zinc-binding group (ZBG).19,20
As such, the compound resides buried in the hCA II active site and interacts with adjacent residues to the catalytic zinc. This mode of binding results in the observation of a disordered water molecule coordinated to the active site zinc (Figure 1). However, compound 1d displaces the ordered water network in the active site that has been shown to be important for catalysis. Key interactions are observed between the carbonyl oxygen of compound 1d and the backbone NH of both Thr199 and Thr200 (2.3 and 2.4 Å, respectively). The carbonyl hydroxyl also forms a hydrogen bond with a coordinated solvent molecule within the active site (bond distance = 3.2 Å), which is then stabilized by a hydrogen bond with the amide of Gln92 (bond distance = 2.3 Å). Compound 1d also displays extensive interactions with the hydrophobic pocket of the hCA II active site. Specifically, van der Waals interactions with residues Val121, Phe131, and Pro202 of the enzymes hydrophobic pocket are predicted to stabilize the “tail” region of compound 1d and are therefore suggested to greatly stabilize the enzyme/inhibitor complex (Figure 2).
Figure 1.
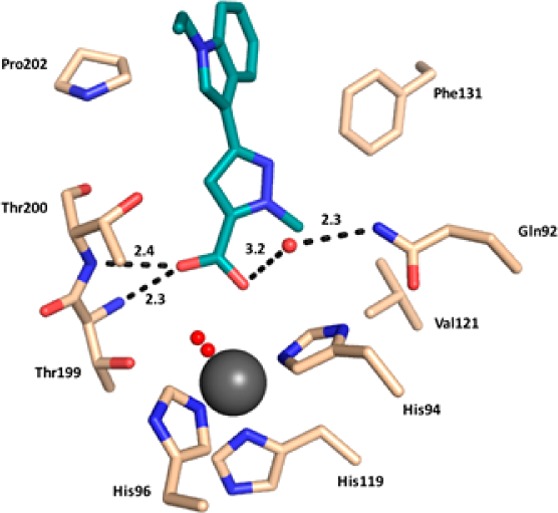
hCA II (light brown) in complex with compound 1d (deep teal). Hydrogen bond interactions are shown with distances (Å) and amino acids labeled. Water molecules are depicted as red spheres. Zinc ion is represented as a gray sphere. This chemotype binds with residues located in proximity of the catalytic site, which displaces the ordered water network, thus definitely producing an indirect interference with the zinc ion.
Figure 2.
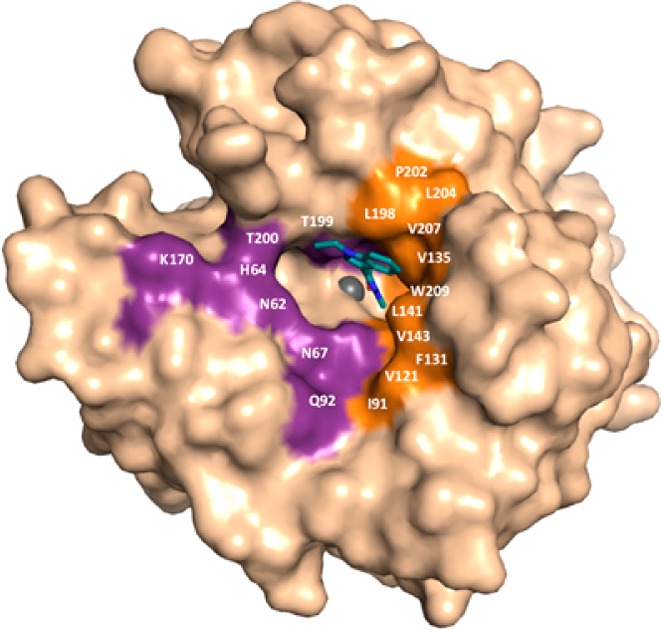
Surface representation showing the location of compound 1d (deep teal) in the hCA II active site (light brown). Hydrophobic (orange) and hydrophilic (purple) residues are highlighted and relative locations labeled.
In order to investigate the interation of the other compounds 1a–c and 2a–d within the catalytic sites of hCA II (new PDB code: 5CJL) and hCA XII (PDB code: IJCZ), a series of docking studies was performed. Preliminarily, we envisaged that in the assembly of ligand–target complex, water molecules could play a crucial role in careful calculations. To be able to accomplish the calculation with manageable computational complexity, the docking procedure recently proposed by Olson and Forli21 was employed. This procedure allows the effect of water molecules located in proximity of ligands to be considered. This method entails the use of a monatomic pseudoatom (W) as a surrogate of the water molecule. First, compound 1d was docked into the X-ray crystal structure, obtaining the same binding mode as in the X-ray crystallography, within the experimental resolution limits (Figure S1A). This established the robustness of the method, and then 1d was also modeled with CA XII (Figure S1B).
After this, the docking settings were optimized, and docking runs were performed for all remaining ligands (i.e., 1a–c and 2a–d) in the active sites of hCA II and XII isoforms. Graphical representations of top-ranking binding modes obtained for these ligands are shown in Figures S2A,B, S3, and S4.
From the structural point of view, hCA II and XII exhibit considerable tertiary structure similarity, with an rmsd value of 1.2 Å; some discrepancies are present on the surface of proteins in concomitance of random coiled regions (Figure S5). Moreover, only three residues exhibited a rmsd greater than 3 Å. Focusing on the catalytic region, the primary sequence appears highly conserved, with the exception of nine residues (i.e., A65S, N67K, E69D, I91T, P131A, G132S, V135S, Q136N, and L204N, indicated as ResCAII-number-ResCAXII). As shown in Table S2, 18 and 22 residues, for hCA II and XII, respectively, stabilize the ligand–protein complex. Moreover, the residues at positions 94, 131, 135, 198, 200, and 202, were also involved in ligand interaction. Furthermore, the key residue Thr199 is predicted to establish an interaction only for hCA II, whereas residues in positions 91, 92, 96, and 141 are predominantly involved in contacts with hCA XII active site. Although differences were found between the binding modes of ligands to the same CA isoform, a similar orientation pattern and disposition within the catalytic site were observed. In the case of the hCA II, all ligands accommodated almost perpendicularly to the plane of the three coordinating histidines with the carboxylate group directed toward the zinc-bound water. In the case of hCA XII, ligands are in a decentered position with the scaffold lying along the protein wall formed by residues Gln92 and His94. This particular arrangement implies that the carboxylate group makes contact with the deep-water from the lateral side. In all cases (with the exception of the less active compound 1a), it allows the formation of a hydrogen bond with the imidazole ring of His96. Regarding the most active compound 2c against hCA XII, docking results showed that the most favorable binding conformation with hCA II is superimposable to that of compound 1d obtained experimentally by X-ray crystallography (Figures S1A and 3A).
Both compounds shared common residue interactions (Table S2), with slight differences for the absence of a hydrophobic contact with Val121 (for 2c), due to the lacking N-methyl group in position 1 of pyrazole ring and the presence of a methoxy group receptor that is able to establish additional contact with Trp209. It is worth noting that, when docked in hCA XII catalytic site, compound 2c appeared located inside a binding pocket formed by 13 residues (Figure 3B and Table S2), with the presence of three strong hydrogen bonds with His96 and Thr200. An additional observation could be made about the comparison of the inhibition values between 2a and 2d. Although compound 2a is not the most potent compound toward hCA XII, it exhibits a significant selectivity profile trans-membrane vs cytosolic isoforms (i.e., hCA XII/hCA II), which resulted about 10-fold higher (as ratio of selectivity indexes) than that of 2d (KihCA XII/Ki hCA II = 12.2 and 1.3, for 2a and 2d, respectively, Table 1). Considering that the only structural difference of 2a with respect to compound 2d is the N-methylation of the pyrazole ring, this modification might be relevant for selectivity. Analyses of postdocking energies such as estimated free energy of binding and related estimated inhibition constant (E.I.C.) values, calculated for each compound (i.e., E.I.C. = 83.29 and 34.42 μM, against hCA II, and 5.38 and 6.60 μM, against hCA XII, for 2a and 2d, respectively), seem to support this behavior.
Figure 3.
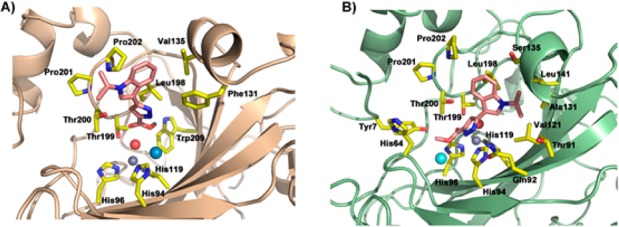
Predicted binding modes for the most promising compound 2c within the catalytic pockets of hCA II (A) and XII (B) isoforms. hCA II and XII are colored as light brown and pale green, respectively; side chain of significant residues are represented as yellow sticks and labeled. Ligand 2a is in pink sticks. Zinc cofactor, zinc-bound water, and pseudoatoms are depicted as spheres and colored in gray, red, and pale blue or cyan, respectively.
The inhibitory effect of the compounds 1a, 2a, 1c, and 2c on CA activity (particularly of hCA IX and/or XII) was evaluated in terms of cell viability of three human cancer cell lines (hormone-independent prostate cells, PC-3; human embryonic kidney, HEK 293 cells; human neuroblastoma cells, SH-SY5Y). First, HEK 293 cells were treated for 24 h with increasing concentrations of 1a, 2a, 1c, and 2c (Figure S6A). No significant antiproliferative effect was detected at the 1, 10, 30, or 100 μM of compound treatment, with the exception of 2c, which exhibited a dose-dependent cell growth inhibition reaching roughly the 70% antiproliferative effect at the highest concentration. Better results were found when the compounds were tested on PC-3 cells at the same conditions (Figure S6B) as well as on SH-SY5Y cell lines after 48 h of treatment (∼85 growth inhibition, Figure S6C).
Since hypoxia-induced upregulation of hCA IX and XII are key components of the complex response of cancer cells to the evolving low oxygen environment, compound 2c was further investigated for its antiproliferative potency on neuroblastoma SH-SY5Y cells in normal and in simulated hypoxic conditions using cobalt(II) chloride (Figure 4).22 As expected, the addition of cobalt(II) chloride, a known chemical inducer of hypoxia-inducible factors, significantly increased the susceptibility of the cells to the 2c treatment, showing a 40% reduction of cell viability at an inhibitor concentration of 30 μM (p < 0.05 vs untreated cells).
Figure 4.

Dose-dependent antiproliferative activity of compound 2c on human neuronal SH-SY5Y cells (measured by percentage of cell viability after 48 h of treatment) assessed in normal and simulated hypoxic conditions.
A new series of compounds bearing a 3-(1H-indol-3-yl)pyrazole-5-carboxylic acid scaffold have been synthesized and evaluated for their selective inhibition of hCA XII. Among the tested compounds, 2c (Ki = 0.21 μM) resulted the most potent hCA XII inhibitor and exhibited cytotoxicity toward three different cancer cell lines. Interestingly, this chemotype behaves differently from other related nonclassical inhibitors. In fact, it does not directly bind to the metal ion or to the zinc-bound water/hydroxide ion: the zinc ion–carboxylate distances in our model are ∼3.78 and ∼4.62 Å (for contact with each oxygen atom), too much longer to support a favorable zinc ion coordination to carboxylate functionality of the ligand. Otherwise, this compound seems to establish contacts with residues located in proximity of the catalytic site, which displaces the ordered water network (shorter zinc ion–water distance, i.e., 2.26 and 1.94 Å, for zinc–O1 and zinc–O2, was revealed), thus definitively producing an indirect interference with the zinc ion. However, 2c displayed improved antiproliferative effects when tested in simulated hypoxia conditions, considering the putative expected involvement of hCA XII inhibition. These results could provide structural determinants for the discovery of novel anticancer agents with original mechanism of action, warranting further development.
Acknowledgments
This research was financed in part the National Institutes of Health, project CA165284. C.L. is supported by the National Center for Advancing Translational Sciences of the National Institutes of Health under University of Florida Clinical and Translational Science Awards TL1TR001428 and UL1TR001427. We would also like to acknowledge the staff at Cornell High Energy Synchrotron Source (CHESS) for assisting with X-ray data collection.
Glossary
ABBREVIATIONS
- hCA
human carbonic anhydrase
- CAIs
carbonic anhydrase inhibitors
- Ki
inhibition constant
- ZBG
zinc-binding group
- PC-3
hormone-independent prostate cells
- HEK 293
human embryonic kidney cells
- SH-SY5Y
human neuroblastoma cells
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00229.
Experimental details for chemistry, X-ray crystallography, and molecular modeling. 1H NMR spectra for title compounds 1b–d and 2a–d, and their intermediates 4a–c and 5a–c. Mass spectra for 1b–d, 2a–d, and 5a–c. X-ray crystallography statistics for data processing and refinement of 4d in complex with hCA II. Details of experimental biology procedure. Additional molecular modeling data. Antiproliferative activity of compounds 1a, 2a, 1c, and 2c (PDF)
Author Contributions
∇ These authors contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Notes
PDB ID 5CJL: 1d in complex with hCA II (Table S1).
Notes
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Gatenby R. A.; Gillies R. J. Why do cancers have high aerobic glycolysis?. Nat. Rev. Cancer 2004, 4, 891–899. 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- Gilkes D. M.; Semenza G. L.; Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. 10.1038/nrc3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza G. L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol.: Mech. Dis. 2014, 9, 47–71. 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- Chiche J.; Brahimi-Horn M. C.; Pouysségur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J. Cell. Mol. Med. 2010, 14, 771–794. 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri D.; Supuran C. T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discovery 2011, 10, 767. 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- Kallio H.; Rodriguez Martinez A.; Hilvo M.; Hyrskyluoto A.; Parkkila S. Cancer-associated carbonic anhydrases IX and XII: effect of growth factors on gene expression in human cancer cell lines. J. Cancer Mol. 2010, 5, 73–78. [Google Scholar]
- Wykoff C. C.; Beasley N. J. P.; Watson P. H.; Turner K. J.; Pastorek J.; Sibtain A.; Wilson G. D.; Turley H.; Talks K. L.; Maxwell P. H.; Pugh C. W.; Ratcliffe P. J.; Harris A. L. Hypoxia-inducible expression of tumor-associated carbonic anhydrases. Cancer Res. 2000, 60, 7075–7083. [PubMed] [Google Scholar]
- Pastorekova S.; Zatovicova M.; Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr. Pharm. Des. 2008, 14, 685–698. 10.2174/138161208783877893. [DOI] [PubMed] [Google Scholar]
- Parkkila S. Significance of pH regulation and carbonic anhydrases in tumour progression and implications for diagnostic and therapeutic approaches. BJU Int. 2008, 101 (Suppl. 4), 16–21. 10.1111/j.1464-410X.2008.07643.x. [DOI] [PubMed] [Google Scholar]
- Chiche J.; Ilc K.; Laferrière J.; Trottier E.; Dayan F.; Mazure N. M.; Brahimi-Horn M. C.; Pouysségur J. Hypoxia-inducible carbonic anhydrase IX and XII promote tumor cell growth by counteracting acidosis through the regulation of the intracellular pH. Cancer Res. 2009, 69, 358–368. 10.1158/0008-5472.CAN-08-2470. [DOI] [PubMed] [Google Scholar]
- Ivanov S.; Liao S. Y.; Ivanova A.; Danilkovitch-Miagkova A.; Tarasova N.; Weirich G.; Merrill M. J.; Proescholdt M. A.; Oldfield E. H.; Lee J.; Zavada J.; Waheed A.; Sly W.; Lerman M. I.; Stanbridge E. J. Expression of hypoxia-inducible cell-surface transmembrane carbonic anhydrases in human cancer. Am. J. Pathol. 2001, 158, 905–919. 10.1016/S0002-9440(10)64038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supuran C. T. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discovery 2008, 7, 168–181. 10.1038/nrd2467. [DOI] [PubMed] [Google Scholar]
- Supuran C. T.Drug Design of Zinc-Enzyme Inhibitors: Functional, Structural, and Disease Applications, Part II; Wiley: Hoboken, NJ, 2009. [Google Scholar]
- Supuran C. T., Scozzafava A., Conway J., Eds. Carbonic Anhydrase: Its Inhibitors and Activators; CRC Press: Boca Raton, FL, 2004. [Google Scholar]
- Alterio V.; Di Fiore A.; D’Ambrosio K.; Supuran C. T.; De Simone G. Multiple binding modes of inhibitors to carbonic anhydrases: how to design specific drugs targeting 15 different isoforms?. Chem. Rev. 2012, 112, 4421–4468. 10.1021/cr200176r. [DOI] [PubMed] [Google Scholar]
- Sechi M.; Innocenti A.; Pala N.; Rogolino D.; Carcelli M.; Scozzafava A.; Supuran C. T. Inhibition of α-class cytosolic human carbonic anhydrases I, II, IX and XII, and β-class fungal enzymes by carboxylic acids and their derivatives: New isoform-I selective nanomolar inhibitors. Bioorg. Med. Chem. Lett. 2012, 22, 5801–5806. 10.1016/j.bmcl.2012.07.094. [DOI] [PubMed] [Google Scholar]
- Supuran C. T. How many carbonic anhydrase inhibition mechanisms exist?. J. Enzyme Inhib. Med. Chem. 2016, 31, 345–360. 10.3109/14756366.2015.1122001. [DOI] [PubMed] [Google Scholar]
- Lomelino C. L.; Supuran C. T.; McKenna R. Non-classical inhibition of carbonic anhydrase. Int. J. Mol. Sci. 2016, 17, 1150. 10.3390/ijms17071150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langella E.; D’Ambrosio K.; D’Ascenzio M.; Carradori S.; Monti S. M.; Supuran C. T.; De Simone G. A combined crystallographic and theoretical study explains the capability of carboxylic acids to adopt multiple binding modes in the active site of carbonic anhydrases. Chem. - Eur. J. 2016, 22, 97–100. 10.1002/chem.201503748. [DOI] [PubMed] [Google Scholar]
- Forli S.; Olson A. J. A force field with discrete displaceable waters and desolvation entropy for hydrated ligand docking. J. Med. Chem. 2012, 55, 623–638. 10.1021/jm2005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Okail M. S. Cobalt chloride, a chemical inducer of hypoxia-inducible factor-1α in U251 human glioblastoma cell line. J. Saudi Chem. Soc. 2010, 14, 197–201. 10.1016/j.jscs.2010.02.005. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.