

Abstract
Background
Glucagon-like peptide-1 (GLP-1) has been reported to exert some beneficial effects on the central nervous system (CNS). However, the effect of GLP-1 on cognitive impairment associated with type 2 diabetes is not well known. This study investigated the effect of GLP-1 on ameliorating memory deficits in type 2 diabetic rats.
Material/Methods
Type 2 diabetic rats were induced by a high-sugar, high-fat diet, followed by streptozotocin (STZ) injection and then tested in the Morris Water Maze (MWM) 1 week after the induction of diabetes. The mRNA expression of Arc, APP, BACE1, and PS1 were determined by real-time quantitative PCR, and the Arc protein was analyzed by immunoblotting and immunohistochemistry.
Results
Type 2 diabetic rats exhibited a significant decline in learning and memory in the MWM tests, but GLP-1 treatment was able to protect this decline and significantly improved learning ability and memory. The mRNA expression assays showed that GLP-1 treatment markedly reduced Arc, APP, BACE1, and PS1 expressions, which were elevated in the diabetic rats. Immunoblotting and immunohistochemistry results also confirmed that Arc protein increased in the hippocampus of diabetic rats, but was reduced after GLP-1 treatment.
Conclusions
Our findings suggest that GLP-1 treatment improves learning and memory deficits in type 2 diabetic rats, and this effect is likely through the reduction of Arc expression in the hippocampus.
MeSH Keywords: Diabetes Mellitus, Type 2; Glucagon-Like Peptide 1; Mild Cognitive Impairment
Background
Type 2 diabetes mellitus (T2DM) has a prevalence of 11.6% in the general adult population in China [1]. This disease results in various complications, including diabetic nephropathy, retinopathy, vasculopathy, and peripheral neuropathy [2], as well as diabetic encephalopathy, which is a complication of DM in the CNS, characterized by neuropathology and mild cognitive deficits [3–5].
As the most common form of dementia, Alzheimer disease (AD) is characterized by progressive decline in cognitive function. Emerging evidence indicates that diabetes is tightly associated with the progression from mild cognitive impairment (MCI) to AD, and increases the risk of dementia [6–8]. In addition, impaired insulin signaling and insulin resistance play key roles in AD pathogenesis [9]. Examination of brain tissue reveals that amyloid beta (Aβ) plaques are crucial in developing AD. Aβ is an abnormal peptide reported to be toxic to the CNS [10]. AD and T2DM share some common pathological features, including metabolic disorders, high cholesterol levels, insulin resistance, Aβ aggregation, and aging-related processes [11–16].
New strategies have been developed to slow the progression of AD, including the application of the incretin hormone glucagon-like peptide-1 (GLP-1) to increase insulin secretion. Besides its protective effect on blood vessels and β cells [17,18], several studies demonstrated that GLP-1 has neuroprotective properties [19]. GLP-1 analog can decrease the levels of Aβ in the brain, protect neurons from glutamate toxicity in vitro, and reduce hyperphosphorylation of tau protein [20–22]. GLP-1 can prevent cognitive decline in models of AD by improving the learning ability of mice [19].
Arc (also known as Arg3.1) is an immediate early gene product and a key factor for memory consolidation, and is required for activity-dependent generation of Aβ [23,24]. Arc physically associates with presenilin1 (PS1) to regulate γ-secretase trafficking and confer activity dependence. Arc-endosome traffics the amyloid precursor protein (APP) and β-secretase (BACE1). Genetic deletion of the Arc gene decreased Aβ load in a transgenic mouse model of AD [24]. These results suggest that Arc is essential for the regulation of Aβ levels by synaptic activity [23]. However, there have been few reports on an alteration of Arc in T2D rats induced by a high-sugar, high-fat diet and STZ injection.
Here, for the first time, we report the effects of GLP-1 treatment on spatial learning and memory deficits in T2D rats in the Morris Water Maze (MWM) test. We also reveal the possible mechanism of Arc involved in GLP-1 treatment against spatial cognitive impairments in type 2 diabetic rats.
Material and Methods
Materials
GLP-1 was purchased from Shanghai Biotech Company (Shanghai, China). Streptozotocin (STZ) was from Sigma (St. Louis, MO, USA), and dissolved in citrate buffer to a final concentration of 10 mg/ml when used. Antibodies were from various companies including: anti-Abeta42 from Cell Signaling Technology (Boston, MA, USA), anti-Arc from Santa Cruz (CA, USA), anti-GAPDH from Boster Company (Wuhan, China), and the secondary antibody conjugated with horseradish peroxidase (HRP) was from Fude Company (Hangzhou, China). All other chemicals were of analytical grade and commercially available.
Animals
Thirty Sprague-Dawley (SD) rats (6–8 weeks old, weighing 160–180 g) were purchased from the Experiment Animal Center of Southern Medical University (Guangzhou, China). All animal procedures were performed in accordance with the Guidelines of Institutional Animal Care under the supervision of the Animal Usage Committee of China. The temperature of animal housing rooms was maintained constantly at 25°C. Ten rats were assigned to the control (Normal) group and fed with the standard rodent chow until sacrificed. The other 20 rats were fed a high-sugar, high-fat diet for 4 weeks (20% sucrose, 10% lard, 2.5% cholesterol, 1% cholic acid, and 66.5% normal diet), and then received intraperitoneal injection of STZ solution once at a dose of 40 mg/kg [25]. Animals were determined as diabetic when blood glucose levels were higher than16.7 mM for 3 consecutive days (reference range, 5–8 mM). After the type 2 diabetic rats were established experimentally, rats with similar degrees of hyperglycemia were randomly divided into 2 groups: a DM group (STZ plus vehicle) and a GLP-1 group (STZ plus GLP-1). Rats in the GLP-1 group were infused with GLP-1 (30 pmol/kg/min) [26] at the interscapular region by an Alzet Micro-osmotic pump, while age-matched normal rats (Normal), fed with the standard rodent chow, were injected with the vehicle citrate buffer instead. After 1 week of GLP-1 or vehicle treatment, the rats were submitted to the Morris Water Maze (MWM) test for 6 days, after which all rats were sacrificed and the brain tissues were taken out for real-time PCR, immunoblotting, and immunohistochemistry analysis.
Morris Water Maze Test
The Morris Water Maze test was performed as previously described. It consisted of a black circular pool (160 cm in diameter) with water at a temperature of 25±2°C. The pool was divided into 4 quadrants (quadrants 1 to 4). A hidden platform, which was placed in a constant location in the middle ring of quadrant 3, was submerged 1.0 cm beneath the water. In each trial, the animal was given 60 s to locate the submerged platform, and was allowed to rest on the platform for 15 s. If a rat failed to find the platform within 60 s, it was guided to the platform. The swimming path of animals during each trial was recorded. Each rat underwent 4 trials per day for 5 consecutive days. On the sixth day, the percent time spent in the target quadrant and the numbers of crossing were tested with the platform removed.
Immunoblot analysis
The hippocampus of each rat was collected and homogenized in RIPA buffer with PMSF. After centrifugation at 12 000 g for 15 min, the supernatant was collected and the protein concentrations were determined using the BCA (bicinchoninic acid) method [27]. Thirty micrograms of total cell lysate was applied to an SDS-PAGE (10%) for separation and then transferred to a PVDF membrane. The membranes were then probed with primary antibodies (1: 1000 dilution) at 4°C overnight, followed with a secondary antibody (1: 5000 dilution) conjugated with the horseradish peroxidase (HRP) for 1 h at room temperature, and then visualized by enhanced chemiluminescence reagents (ECL; Pierce, Rockford, USA). Bands of interest were quantified by densitometry using Gel-Pro analysis software. The protein expression levels of rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as the housekeeping control.
Real-time quantitative PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The RNA was then reversely transcribed to cDNA using a reverse transcription reagents kit (Takara RR047A). The qPCR was performed on an ABI 7500 instrument (Life Technology, Grand Island, NY 14072) using a standard procedure. β-actin was selected as the housekeeping control gene. An amount of 100 ng cDNA was used for the qPCR kit (Takara RR820A). The relative mRNA abundance was measured by the difference in threshold values between the target gene and the housekeeping control gene, β-actin. The resulting threshold values represented an average of triplicates [28]. The primers used in the qPCR are shown (Table 1).
Table 1.
The primers used in the qPCR.
| Name | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| Arc | ctgagatgctggagcacgta | gccttgatggacttcttcca |
| APP | gtgatctacgagcgcatgaa | agaaggcatgagagcatcgt |
| BACE1 | gctgcagtcaagtccatcaa | attgctgaggaaggatggtg |
| PS1 | caagagctgctgtccaggaa | tgaaaatggcgagcaggagt |
| β-actin | caacacagtgctgtctggtg | gatccacatctgctggaag |
Immunohistochemistry
Animals were sacrificed and the brains were fixed in 4% paraformaldehyde solution for 24 h at 4°C. Brains were paraffin-embedded and cut into 3-μm-thick sections. After rehydration through xylene and graded ethanol, sections were treated with 3% (v/v) hydrogen peroxide and blocked with 10% (v/v) normal goat serum for 45 min at room temperature, followed by incubation with rabbit anti-Arc antibody (Cell Signal Technology, 1: 200) or anti-Aβ42 antibody (Santa Cruz, 1: 200) for 24 h at 4°C. The immunostaining was detected using the appropriate HRP-conjugated secondary antibodies (DAKO, Glostrup, Denmark) and diaminobenzidine tetrahydrochloride (DAB). The images were captured under an Olympus microscope (Tokyo, Japan). At least 5 random fields of each section were examined, and the semi-quantitative evaluations were analyzed with Image-Pro Plus software version 6.0.
Statistical analysis
All analyses were carried out with GraphPad Prism 4.0 software (GraphPad Software, San Diego, CA, USA). Data are expressed as the mean ±SEM and then analyzed using one-way ANOVA followed by Tukey’s multiple comparison test. A p value <0.05 was considered statistically significant.
Results
GLP-1 ameliorates hyperglycemia of type 2 diabetic rats
As shown in Table 2, blood glucose levels of all rats were normal before the diabetic model was established. After that, the blood glucose levels of diabetic rats were significantly higher than that of the normal group (23.8±2.1 mmol/L vs. 6.6±0.4 mmol/L; p<0.01), and the blood glucose levels of rats in the DM+GLP-1 group were also significantly higher than in the normal group (23.7±2.3 mmol/L vs. 6.6±0.4 mmol/L; p<0.01).
Table 2.
Blood glucose levels of rats in different experimental groups.
| Group | n | Blood glucose (mmol/L) | ||
|---|---|---|---|---|
| Before model establishment | Before treatment | After treatment | ||
| Normal | 10 | 6.6±0.3 | 6.6±0.4 | 6.9±0.8 |
| DM | 10 | 6.9±0.6 | 23.8±2.1** | 25.1±1.3** |
| DM±GLP-1 | 10 | 6.7±0.5 | 23.7±2.3** | 18.2±1.1## |
Compared with normal group before treatment,
P<0.01; compared with DM group after treatment,
P<0.01.
After GLP-1 treatment for 1 week, GLP-1 significantly reduced the blood glucose levels of the rats in the experimental group compared with the DM group (18.2±1.1 mmol/L vs. 25.1±1.3 mmol/L; p<0.01).
These results indicate that the type 2 diabetic rat model was established successfully and GLP-1 administration was effective in controlling glucose.
GLP-1 treatment improves learning and memory deficits in type 2 diabetic rats
After 1 week of GLP-1 or vehicle treatment, all the rats were subjected to Morris Water Maze testing to assess spatial cognitive abilities. The latency to find the platform (escape latency) is shown in Figure 1A. The escape latency of the normal group decreased gradually during the 5 days of training. However, the DM group rats took a significantly longer time to find the platform (33.417±5.115s, n=12) compared to normal group rats (19.825±2.530s, n=11) on the 3rd day of training (p<0.05). The GLP-1 group took a significantly shorter time to find the platform (11.0±2.9s, n=8) compared to rats in the DM group (33.8±4.5s, n=10) (p<0.05).
Figure 1.

Treatment with GLP-1 improved learning and memory deficits in diabetic rats. (A) The average escape latencies of rats in searching for the hidden platform in MWM; (B) for the memory test, the swimming tracks; (C) the percentage of time spent in the target quadrant in probe trial; (D) times of crossing platform in the target quadrant. Data are presented as means ±SEM. Statistical analyses were performed by one-way ANOVA. # P<0.05 versus the normal group; * P<0.05 versus the DM group.
To further investigate the spatial memory ability of rats in the MWM test, probe trials without the platform were tested on day 6. The representative swimming traces of rats in probe trials were shown in Figure 1B. The percentage of time in the target quadrant for the DM group was 17.79±3.99%, which was significantly smaller (29.66±7.38%) (p<0.05) than that of the normal group (Figure 1C). The percentage of time in the target quadrant for the GLP-1 group (30.13±3.50%) was more than that of the DM group (p<0.05). The crossing times in the target quadrant of each group were calculated (Figure 1D). In the DM group, crossing time in the target quadrant was 1.250±0.50, which was significantly shorter than that of the normal group (4.25±1.26) (p<0.05). The crossing time in the target quadrant of the GLP-1 group (3.5±0.57) was significantly longer than for the DM group (p<0.05).
Our results show that diabetic rats exhibited spatial memory and cognitive dysfunction, and GLP-1 treatment can efficiently reverse these deficits.
GLP-1 treatment inhibits the mRNA expression of PS1, APP, BACE1, and Arc in the hippocampus of diabetic rats
To elucidate the possible molecular mechanism underlying the protection of GLP-1 against STZ-mediated deficit in spatial cognition, we assessed the changes in 4 factors involved in the pathway of Aβ accumulation (Figure 2). Because APP, BACE1, PS1, and Arc are important proteins associated with Aβ deposition [23,24], we first investigated the levels of these factors in the hippocampus of rats using real-time PCR after the MWM test. Compared with the normal group, the DM group rats had a significant increase in mRNA expression of APP, BACE1, and PS1 (p<0.01), and these increases were reversed by GLP-1 treatment (P <0.01). Arc was significantly increased in the DM group compared with the normal group (p<0.01), and was dramatically reduced in the GLP-1 group (P <0.01). These results indicate that GLP-1 acts as a neuroprotective factor, possibly by modulating the expression of APP, BACE1, PS1, and Arc.
Figure 2.

Expression of the ARC, APP, PS1, and BACE1 in the hippocampus. All of these mRNA levels were notably increased in the diabetic rats and decreased in the GLP-1-treated ones. Data are presented as means ±SEM. Statistical analyses were performed by one-way ANOVA. ### P<0.01 versus the normal group; *** P<0.01 versus the DM group.
GLP-1 treatment attenuates the levels of Arc and Aβ accumulation in the hippocampus of diabetic rats
The levels of Arc protein in the rat hippocampus were determined by immunoblotting (Figure 3). Compared with the normal group, the DM group had a significant increase in expression of Arc protein (p<0.01), and this increase was reversed by the GLP-1 treatment (p<0.05). Similar results were further confirmed in the rat brains by immunohistochemistry. Little positive staining of Arc was observed in the sections from the control and GLP-1-treated rats (Figure 4), while positive staining increased in the hippocampus of the diabetic rats, which decreased when GLP-1 was administrated.
Figure 3.

Immunoblotting analysis of hippocampal Arc expression. Arc levels were dramatically increased in the diabetic rats and reduced in the GLP-1-treated ones. Data are presented as means ±SEM. Statistical analyses were performed by one-way ANOVA. ** P<0.01 versus the normal group; # P<0.05 versus the DM group.
Figure 4.

Immunostaining of Arc and Aβ protein in hippocampal regions of rats. Immunohistochemical localization of Arc and Aβ were examined in the hippocampal region. Little positive staining for Arc and Aβ was observed in sections from control and GLP-1 groups. Markedly increased Arc and Aβ staining was observed in the hippocampus of type 2 diabetic rats (DM). DAPI counterstaining indicates nuclear localization (blue). Data are presented as means ±SEM. Statistical analyses were performed by one-way ANOVA. ** P<0.01 versus the normal group; ## P<0.01 versus the DM group. Scale bar: 50 μm.
Aβ accumulation is the hallmark characteristic in AD and AD-related brain diseases. In the present study, Aβ accumulation was also analyzed. As shown in Figure 4, immunohistochemistry results indicated that the Aβ42 immunostaining was increased in the hippocampus of diabetic rats compared with those in the control group (p<0.01), but was reduced after GLP-1 treatment (p<0.01).
Discussion
Much research attention has recently focussed on the effects of GLP-1 on improving learning and memory deficits, although with limited reports and no consistency in animal models and observation criteria. Han et al. injected Aβ into the hippocampus of rat brains (4 nmol in 4 μl) to induce brain damage and then injected the GLP-1 analog, liraglutide, to invert the effects; the results suggested that pretreatment with liraglutide effectively protects against the Aβ25–35-induced impairment of spatial memory and deficit of L-LTP [29]. In addition, this protective effect revealed a dose-dependent pattern. However, this experimental model was significantly different from the natural process of T2DM. Thus, we preferred to induce diabetic rats with a high-glucose, high-fat diet and STZ injection in our experiment. This experimental model of type 2 diabetes replicates the natural history and metabolic characteristics of the syndrome and is widely accepted in diabetes studies [25,30]. A high-sugar, high-fat diet represents the eating habits of many people who prone to obesity and diabetes. Low-dose STZ induced partial islet injury, and also mimics partial islet function impairment in type 2 diabetes mellitus. We also used the classic Morris Water Maze test to assess spatial memory and cognitive deficit.
In our animal model, the time point of learning and memory deficits appeared at Week 4 after T2DM was established, which was much earlier than the 4 months reported by Wang [31]. This is the first report that GLP-1 is able to reverse the learning and memory deficits in the diabetic rats induced by a high-glucose, high-fat diet and injection of STZ. Our results are very similar to those of Bomfim et al., who used the analog of GLP-1, Exendin-4 in APP/PSI mutant rats [32]. These results further confirmed that GLP-1 can improve learning and memory abilities.
Emerging evidence shows that type 2 diabetes increases the risk of AD [10,12]. It has been connected to AD as a form of brain insulin resistance [33]. We confirmed that the combination of a high-sugar, high-fat diet with injection of STZ resulted in significant impairment of spatial learning and memory in freely moving rats. Then, the neuroprotective effects of GLP-1 were investigated. The escape latency and the times crossing the platform in the STZ plus GLP-1 group were similar to the those in the normal control group. We, for the first time, found that GLP-1 significantly protected against cognitive impairment in diabetic rats induced by a high-glucose, high-fat diet and injection of STZ.
The mechanisms linking diabetes to cognitive deficit are still unclear. Mounting evidence shows that Aβ deposition in the brain is involved in diabetes-associated cognitive deficit [13,15]. It is clear that Aβ is a cleavage product derived from APP, and on sequential cleavage by β-secretase and γ-secretase. APP is processed to generate various peptide species, including the more toxic form Aβ42, which is prone to oligomerization, leading to neurotoxicity [34].
Several studies indicated that GLP-1 and its analogs can slow the progression of AD by reducing Aβ deposition. Hunter et al. confirmed that GLP-1 and its analog can be injected peripherally and be taken up into the brain [35]. Wang et al. demonstrated that Val(8) GLP-1 enhanced synaptic plasticity in short- and long-term applications and preserved synaptic functionality in the brains of a mouse model of AD [36]. In McClean’s study, liraglutide reduced plaque formation and protected memory and synaptic plasticity in the brains of a mouse model of AD [37]. In line with our study, immunohistochemistry results revealed that Aβ42 immunostaining was increased in the hippocampus of diabetic rats, but reduced after GLP-1 treatment. Our results suggest that GLP-1 treatment can reduce Aβ deposition.
To determine the possible molecular mechanism of GLP-1 effects on the reduction of Aβ deposition [31], we further investigated the genes involved. As there are reports on the features of AD disease, such as Aβ aggregation, hippocampal atrophy, and synaptic loss in STZ induced diabetic rats, we looked at the signal pathway of Arc activation, which has been reported to elevate Aβ level [32]. In this experiment, we studied the key gene transcript levels of APP, BACE1, PS1, and Arc in the diabetic rats and found they were higher than those in the normal controls (P<0.05).
Previous studies demonstrated that Arc plays a vital role in the activity-dependent generation of Aβ. Arc is also a postsynaptic protein that recruits dynamin and endophilin 2/3 to early recycling endosomes that traffic AMPA receptors to decrease synaptic strength of plasticity. In addition, Arc-endosome traffics BACE1 and APP, and Arc physically associates with presenilin1 (PS1) to regulate γ-secretase trafficking and confer activity dependence [24]. Moreover, Arc is required for synaptic activity-regulated Aβ generation [23]. Although recently progress has been made, the mechanisms by which diabetes develops into AD remain unknown. Our study found that the level of Arc was dramatically increased in type 2 diabetic rats and was remarkably reduced after GLP-1 treatment. These results suggest that Arc may be a critical factor linking diabetes and cognition impairment, and GLP-1 may exert its neuroprotective effect through modulating the levels of Arc.
There have been few studies published on the alteration of Arc in type 2 diabetic rats induced by a diet of high glucose, high fat, and injection of STZ. The molecular mechanisms underlying the neuroprotective effects of GLP-1 and its analogs remain unclear. Some studies showed that phosphorylation of ERK1/2, PKC, and PKA might be increased in the diabetic rats compared with age-matched control rats [38,39]. Other studies revealed that Arc exerted its functions on the activation of PKA or ERK1/2 [40,41]. Thus, the pathway ERK1/2, PKC, PKA→Arc→Aβ (Figure 5) may be the link between diabetes and AD, and the cognitive impairment is able to be reversed by the GLP-1 treatment at the early stage of AD.
Figure 5.
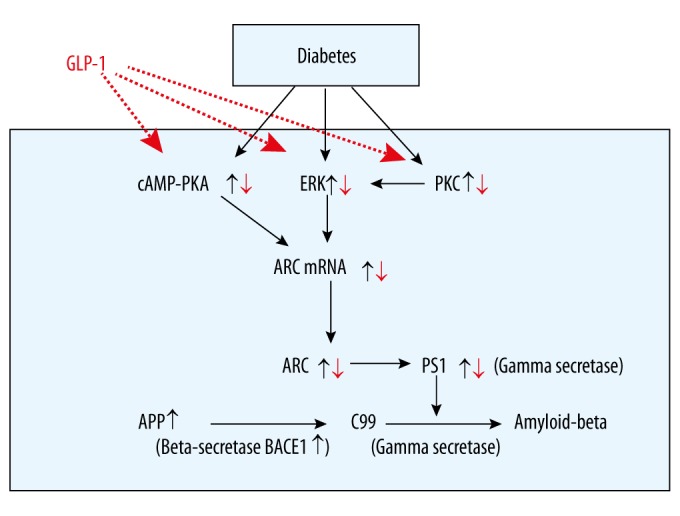
The possible pathway of GLP-1 treatment in rescuing cognitive impairments in diabetic rats. ARC – activity-regulated cytoskeleton-associated protein; APP – amyloid precursor protein; PKA – protein kinase A; PKC – protein kinase C; BACE1(β-secretase) – β-site APP-cleaving anzyme1; PS1 – presenilin 1; C99 – b-secretase-generated C-terminal APP fragment.
Conclusions
In conclusion, the present study confirmed the protective effects of administration of GLP-1 against spatial memory and cognitive dysfunction in type 2 diabetic rats, which were induced by a high-sugar, high-fat diet and injection of STZ. The effective neuroprotective action of GLP-1 may have resulted from reducing the Arc level. Arc is likely the potential target for treatment of diabetes-associated memory deficits in patients.
Footnotes
Source of support: This work was supported by grants from the National 973 Projects (no. 2011CB504006), the Songshan Lake Science and Technology Fund (no. 2010B025 and no. 2010B026), the Guangdong Science and Technology Fund (no. 2010B090400041), and the Doctoral Program of Higher Education of the Ministry of Education (no. 20104433110014)
References
- 1.Xu Y, Wang L, He J, Bi Y, et al. Prevalence and control of diabetes in Chinese adults. JAMA. 2013;310:948–59. doi: 10.1001/jama.2013.168118. [DOI] [PubMed] [Google Scholar]
- 2.Rathmann W, Giani G. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:2568–69. doi: 10.2337/diacare.27.10.2568. author reply 2569. [DOI] [PubMed] [Google Scholar]
- 3.Bauduceau B, Doucet J, Bordier L, Garcia C, et al. Hypoglycaemia and dementia in diabetic patients. Diabetes Metab. 2010;36(Suppl 3):S106–11. doi: 10.1016/S1262-3636(10)70476-6. [DOI] [PubMed] [Google Scholar]
- 4.Francis GJ, Martinez JA, Liu WQ, Xu K, et al. Intranasal insulin prevents cognitive decline, cerebral atrophy and white matter changes in murine type I diabetic encephalopathy. Brain. 2008;131:3311–34. doi: 10.1093/brain/awn288. [DOI] [PubMed] [Google Scholar]
- 5.Zhou Y, Luo Y, Dai J. Axonal and dendritic changes are associated with diabetic encephalopathy in rats: An important risk factor for Alzheimer’s disease. J Alzheimers Dis. 2013;34:937–47. doi: 10.3233/JAD-121762. [DOI] [PubMed] [Google Scholar]
- 6.Tun PA, Nathan DM, Perlmuter LC. Cognitive and affective disorders in elderly diabetics. Clin Geriatr Med. 1990;6:731–46. [PubMed] [Google Scholar]
- 7.Ryan CM. Neurobehavioral complications of type I diabetes. Examination of possible risk factors. Diabetes Care. 1988;11:86–93. doi: 10.2337/diacare.11.1.86. [DOI] [PubMed] [Google Scholar]
- 8.Velayudhan L, Poppe M, Archer N, et al. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- 9.Kroner Z. The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Altern Med Rev. 2009;14:373–79. [PubMed] [Google Scholar]
- 10.Park SA. A common pathogenic mechanism linking type-2 diabetes and Alzheimer’s disease: Evidence from animal models. J Clin Neurol. 2011;7:10–18. doi: 10.3988/jcn.2011.7.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strachan MWJ, Reynolds RM, Marioni RE, Price JF. Cognitive function, dementia and type 2 diabetes mellitus in the elderly. Nat Rev Endocrinol. 2011;7:108–14. doi: 10.1038/nrendo.2010.228. [DOI] [PubMed] [Google Scholar]
- 12.Janson J, Laedtke T, Parisi JE, et al. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–81. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 13.Bitel CL, Kasinathan C, Kaswala RH, et al. Amyloid-β and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis. 2012;32:291–305. doi: 10.3233/JAD-2012-120571. [DOI] [PubMed] [Google Scholar]
- 14.Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer’s disease. Eur J Pharmacol. 2004;490:127–33. doi: 10.1016/j.ejphar.2004.02.050. [DOI] [PubMed] [Google Scholar]
- 15.Li ZG, Zhang W, Sima AA. Alzheimer-like changes in rat models of spontaneous diabetes. Diabetes. 2007;56:1817–24. doi: 10.2337/db07-0171. [DOI] [PubMed] [Google Scholar]
- 16.Valente T, Gella A, Fernàndez-Busquets X, Unzeta M, et al. Immunohisto-chemical analysis of human brain suggests pathological synergism of Alzheimer’s disease and diabetes mellitus. Neurobiol Dis. 2010;37:67–76. doi: 10.1016/j.nbd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Yi Z, Sun HL, Hong C, et al. Glucagon-like peptide-1 (GLP-1) protects vascular endothelial cells against advanced glycation end products (AGEs) – induced apoptosis. Med Sci Monit. 2012;18:BR286–91. doi: 10.12659/MSM.883207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bojanowska E. Physiology and pathophysiology of glucagon-like peptide-1 (GLP-1): the role of GLP-1 in the pathogenesis of diabetes mellitus, obesity, and stress. Med Sci Monit. 2005;11:RA271–78. [PubMed] [Google Scholar]
- 19.Holscher C. The incretin hormones glucagonlike peptide 1 and glucose-dependent insulinotropic polypeptide are neuroprotective in mouse models of Alzheimer’s disease. Alzheimers Dement. 2014;10:S47–54. doi: 10.1016/j.jalz.2013.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Gault VA, Holscher C. GLP-1 agonists facilitate hippocampal LTP and reverse the impairment of LTP induced by beta-amyloid. Eur J Pharmacol. 2008;587:112–17. doi: 10.1016/j.ejphar.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 21.Gilman CP, Perry T, Furukawa K, et al. Glucagon-like peptide 1 modulates calcium responses to glutamate and membrane depolarization in hippocampal neurons. J Neurochem. 2003;87:1137–44. doi: 10.1046/j.1471-4159.2003.02073.x. [DOI] [PubMed] [Google Scholar]
- 22.Li L, Zhang ZF, Holscher C, et al. (Val(8)) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol. 2012;674:280–86. doi: 10.1016/j.ejphar.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 23.Cheng X, Wu J, Geng M, Xiong J. Role of synaptic activity in the regulation of amyloid beta levels in Alzheimer’s disease. Neurobiol Aging. 2014;35:1217–32. doi: 10.1016/j.neurobiolaging.2013.11.021. [DOI] [PubMed] [Google Scholar]
- 24.Wu J, Petralia RS, Kurushima H, Patel H, et al. Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell. 2011;147:615–28. doi: 10.1016/j.cell.2011.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srinivasan K, Viswanad B, Asrat L, et al. Combination of high-fat diet-fed and low-dose streptozotocin-treated rat: A model for type 2 diabetes and pharmacological screening. Pharmacol Res. 2005;52:313–20. doi: 10.1016/j.phrs.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Owens DR, Monnier L, Bolli GB. Differential effects of GLP-1 receptor agonists on components of dysglycaemia in individuals with type 2 diabetes mellitus. Diabetes Metab. 2013;39:485–96. doi: 10.1016/j.diabet.2013.09.004. [DOI] [PubMed] [Google Scholar]
- 27.Kapoor KN, Barry DT, Rees RC, et al. Estimation of peptide concentration by a modified bicinchoninic acid assay. Analytical Biochem. 2009;393:138–40. doi: 10.1016/j.ab.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 28.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–8. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 29.Han WN, Holscher C, Yuan L, et al. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging. 2013;34:576–88. doi: 10.1016/j.neurobiolaging.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 30.Bhatta A, Sangani R, Kolhe R, et al. Deregulation of arginase induces bone complications in high-fat/high-sucrose diet diabetic mouse model. Mol Cell Endocrinol. 2015;422:211–20. doi: 10.1016/j.mce.2015.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang JQ, Yin J, Song YF, et al. Brain aging and AD-like pathology in streptozotocin-induced diabetic rats. J Diabetes Res. 2014;2014:796840. doi: 10.1155/2014/796840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bomfim TR, Forny-Germano L, Sathler LB, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease – associated Abeta oligomers. J Clin Invest. 2012;122:1339–53. doi: 10.1172/JCI57256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Reports. 2009;42:475–81. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113( Pt 11):1857–70. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- 35.Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. 2012;13:1–6. doi: 10.1186/1471-2202-13-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang XH, Li L, Hölscher C, et al. Val 8 -glucagon-like peptide-1 protects against Aβ1–40-induced impairment of hippocampal late-phase long-term potentiation and spatial learning in rats. Neuroscience. 2010;170:1239–48. doi: 10.1016/j.neuroscience.2010.08.028. [DOI] [PubMed] [Google Scholar]
- 37.Moller JB, Jusko WJ, Gao W, Hansen T, et al. Mechanism-based population modelling for assessment of L-cell function based on total GLP-1 response following an oral glucose tolerance test. J Pharmacokinet Pharmacodyn. 2011;38:713–25. doi: 10.1007/s10928-011-9216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao MH, Xiang YC, Huang JY, et al. The disturbance of hippocampal CaMKII/PKA/PKC phosphorylation in early experimental diabetes mellitus. CNS Neurosci Ther. 2013;19:329–36. doi: 10.1111/cns.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sacai H, Sasaki-Hamada S, Sugiyama A, et al. The impairment in spatial learning and hippocampal LTD induced through the PKA pathway in juvenile-onset diabetes rats are rescued by modulating NMDA receptor function. Neurosci Res. 2014;81–82:55–63. doi: 10.1016/j.neures.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 40.Thompson JL, Shuttleworth TJ. Anchoring protein AKAP79-mediated PKA phosphorylation of STIM1 determines selective activation of the ARC channel, a store-independent Orai channel. J Physiol. 2015;593:559–72. doi: 10.1113/jphysiol.2014.284182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xiu F, Lin L, Cai L, Ji S. NAc Shell Arc/Arg3.1 protein mediates reconsolidation of morphine CPP by increased GluR1 cell surface expression: Activation of ERK-coupled CREB is required. Int J Neuropsychopharmacol. 2015;18(9) doi: 10.1093/ijnp/pyv030. pii: pyv030. [DOI] [PMC free article] [PubMed] [Google Scholar]