

Abstract

N-(5-Bromo-3-methoxypyrazin-2-yl)-5-chlorothiophene-2-sulfonamide 1 was identified as a hit in a CCR4 receptor antagonist high-throughput screen (HTS) of a subset of the AstraZeneca compound bank. As a hit with a lead-like profile, it was an excellent starting point for a CCR4 receptor antagonist program and enabled the rapid progression through the Lead Identification and Lead Optimization phases resulting in the discovery of two bioavailable CCR4 receptor antagonist candidate drugs.
Keywords: Chemokine receptor 4, MDC, TARC, CCR4, antagonist
The chemokine receptor family was originally identified in the 1990s and has since grown in number, complexity, and range of biological functions. Originally thought to be simple cell chemo attractants, chemokines have since been shown to exhibit a broader range of functions covering involvement in HIV-1 infection to hematopoiesis and control of cell growth.1,2
CCR4 is a G protein-coupled receptor (GPCR) and is activated by the CC chemokines, CCL22 (MDC: macrophage-derived chemokine) and CCL17 (TARC: T-cell and activation-related chemokine),3 leading to cell activation and chemotaxis; it is expressed mainly by Th2 lymphocytes.4,5 In keeping with a role for CCR4 in the orchestrated movement of Th2 cells into the allergic lung, both CCL17 and CCL22 have been shown to be elevated in the human lung following allergen challenge.6 This potential role of CCR4 in driving human allergic lung disease led to many pharmaceutical companies attempting to identify CCR4 receptor antagonists for the treatment of allergic rhinitis and asthma,7−15 but as yet, no drug has been discovered (Figure 1).16,17
Figure 1.

Structures of a selection of recently published CCR4 receptor antagonists.
In this communication, we are disclosing our studies on the optimization of N-(5-bromo-3-methoxypyrazin-2-yl)-5-chlorothiophene-2-sulfonamide 1 to afford two clinical candidates AZD-2098 (47) and AZD-1678 (49). High-throughput screening of the AstraZeneca Mixed Chemokine Receptor Antagonist Compound library (∼29,000 compounds) using a hCCR4 fluorescent microvolume assay technology (FMAT) cell binding assay identified compound 1(18,19) as an inhibitor of labeled CCL22 binding to CCR4 (pIC50 = 7.2).20 Subsequently, functional antagonism of 1 was shown using an hCCR4 fluorescence imaging plate reader (FLIPR) intracellular calcium mobilization assay (pIC50 = 7.0) and a human primary Th2 cell chemotaxis assay (pIC50 = 6.7) (Table 1).
Table 1. Hit Profile of Pyrazine Sulfonamide 1.
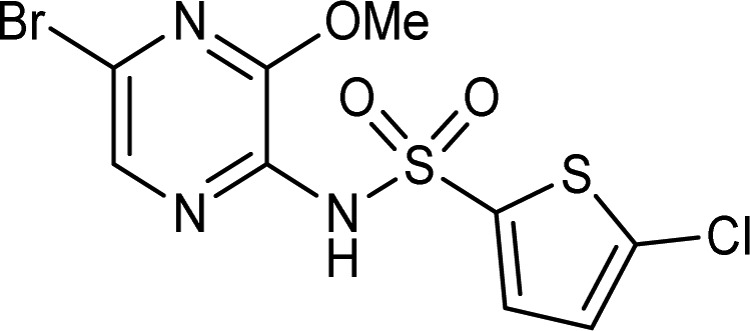
| biological assay/test procedure | compound 1 |
|---|---|
| hCCR4 binding (pIC50) | 7.2 ± 0.2 |
| hPPB (%) | >99.9 |
| calcd WBP (pIC50)a | <4.2 |
| logD | 1.1 |
| solubility (mg/mL) | 0.34 |
| human hepatocytes (μL/mL/106 cells) | <0.2 |
| rat/mouse in vivo PK | |
| Cl (mL/min/kg) | 0.1/0.1 |
| Vss (L/kg) | 0.1/0.1 |
| T1/2 (h) | 16/10 |
| bioavailability (%) | 45/86 |
| rat/mouse CCR4 binding (pIC50) | 8.3/8.3 |
| rat/mouse PPB (%) | 99.85/96.7 |
Calculated from the hCCR4 binding and % age PPB.
Compound 1 exhibited a good lead-like profile21 and was shown to be selective for the CCR4 receptor, through screening against an internal panel of chemokine receptors (e.g., CXCR1 and CXCR2, CCR1, CCR2b, CCR5, CCR7, and CCR8; inactive at 10 μM). When screened against a large panel of receptors and enzymes (∼120 screens at MDS-Pharma), weak or no activity was observed and therefore 1 was chosen as a starting point for the CCR4 antagonist program. Compound 1 had good solubility (0.34 mg/mL), rat bioavailability (F = 45%), and half-life (T1/2 = 16 h) when dosed in rat and a similar profile when dosed in vivo in mouse. This long half-life was accounted for by a very low clearance (0.1 mL/min/kg), which counteracts the small volume of distribution (Vss = 0.1 L/kg).
Compound 1 had very good crossover with rat and mouse CCR4 receptors (both pIC50 = 8.3). Subsequently, this species crossover was shown to be a general feature of the sulfonamide series (see Table 5), with the CCR4 receptor from all three species displaying very similar structure–activity relationships (SAR). When corrected for plasma protein binding, the predicted rat whole blood potency of 1 was pIC50 = 5.5 (rat ppb = 99.85%) and for mouse pIC50 = 6.8 (mouse ppb = 96.7%), giving 1 a suitable profile for use as a target validation tool for in vivo hypothesis testing.
Table 5. Compound Profiles of 47 and 49.
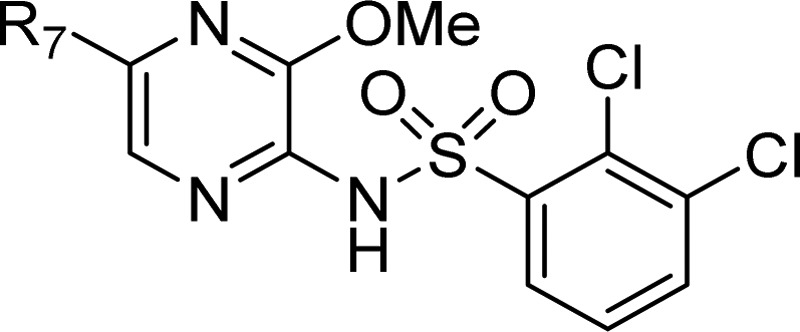
| screen | 47 (R7 = H) | 49 (R7 = F) |
|---|---|---|
| hCCR4 pIC50 | 7.8 ± 0.05 (n = 11) | 8.6 ± 0.1 (n = 10) |
| hPPB (%) | 98.9 | 99.4 |
| calcd WBP (pIC50) | 5.9 | 6.4 |
| logD | 0.35 | 0.6 |
| solubility (mg/mL) | 1.5 | 1.3 |
| human hepatocytes (μL/mL/106 cells) | <0.2 | <1.0 |
| Species Cross Over | ||
| rat CCR4 pIC50 | 8.0 ± 0.27 (n = 3) | 9.0 ± 0.1 (n = 3) |
| mouse CCR4 pIC50 | 8.0 ± 0.04 (n = 3) | 9.0 ± 0.1 (n = 3) |
| dog CCR4 pIC50 | 7.6 ± 0.06 (n = 7) | 8.5 ± 0.2 (n = 3) |
| Rat/Dog in Vivo PKa | ||
| Cl (mL/min/kg) | 2.0/1.5 | 2.3/3.7 |
| Vss (L/kg) | 0.2/0.1 | 0.2/0.2 |
| T1/2 (h) | 1.7/1.0 | 2.5/0.5 |
| F (%) | 100/100 | 100/100 |
| dose-to-man prediction (mg/kg) UID | 1 | 0.1 |
PK studies conducted using 1% sodium bicarbonate solution at 9.0 μmol/kg (p.o.) or 3.0 μmol/kg (i.v.).
However, the calculated human whole blood potency (WBP) was very poor with predicted activity (pIC50) < 4.2 (calculated from hCCR4 pIC50 = 7.2 and hPPB > 99.9%).
With a candidate drug requiring a WBP pIC50 ≈ 6.0, the key issue was to increase WBP by a combination of increasing CCR4 receptor affinity and lowering plasma protein binding,22 while maintaining all the other good features inherent in 1. The high plasma protein binding was attributed to the acidic sulfonamide-NH (measured pKa = 4.1) and the lipophilic nature of the molecule (measured logP = 4.4).
The SAR generated with respect to modification of the 5-chlorothiophene group is shown in Table 2. Removal of the 5-chloro substituent 2 gave over a 10-fold drop in affinity, as did the isomeric unsubstituted thiophene 3. Addition of a chlorine to the 4-position of the thienyl group also gave a reduction in affinity (compare 4 with 1), while a large substituent (bromine or phenyl) in the 3-position was tolerated (compare 5 and 6 with 1 and 2, respectively). The phenyl analogue 7 was similar to the two unsubstituted thiophenes in potency, and introducing chlorine into the 2-, 3- or 4-positions of the phenyl sulfonamide ring (8, 9 and 10) gave an increase in binding relative to 7. The 6-substituted dichlorophenyl sulfonamides (11–17) had a potency range of almost 3 log units, with the 3,5-dichlorophenylsulfonamide analogue 14 having only weak activity (pIC50 = 5.3) and the 2,3-dichlorophenylsulfonamide 16 being optimal (pIC50 = 8.0), almost a log unit more potent than 1. Similar affinity was achieved with 3,4-dichloro-2-thienyl (compare 18 and 17). A number of other disubstituted phenyl sulfonamides were screened; however, no improvement in binding was seen (19–24 compare with 17). Introduction of a nitrogen into the aromatic ring led to dramatic reduction in potency (25) as did replacement of the aromatic ring with an alkyl group as illustrated with 26.
Table 2. Exploration of the 5-Position of the Pyrazine Ring.
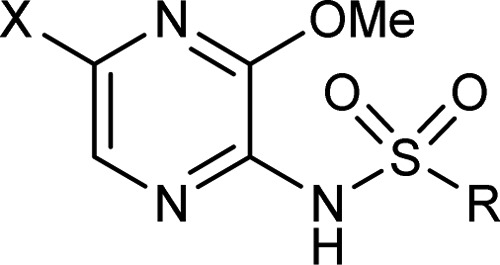
| example | R | X | CCR4 pIC50a |
|---|---|---|---|
| 1 | 5-chloro-2-thienyl | Br | 7.2 ± 0.2 |
| 2 | 2-thienyl | Br | 6.2 ± 0.2 |
| 3 | 3-thienyl | Br | 5.9 ± 0.1 |
| 4 | 4,5-dichloro-2-thienyl | Br | 6.0 ± 0.1 |
| 5 | 3-bromo-5-chloro-2-thienyl | Br | 7.4 ± 0.2 |
| 6 | 3-phenyl-2-thienyl | Br | 6.0 ± 0.1 |
| 7 | phenyl | Br | 6.1 ± 0.1 |
| 8 | 2-chlorophenyl | Br | 6.8 ± 0.2 |
| 9 | 3-chlorophenyl | Br | 7.0 ± 0.2 |
| 10 | 4-chlorophenyl | Br | 6.9 ± 0.2 |
| 11 | 2,6-dichlorophenyl | Br | 6.2 ± 0.2 |
| 12 | 2,5-dichlorophenyl | Br | 5.5 ± 0.1 |
| 13 | 2,4-dichlorophenyl | Br | 6.9 ± 0.2 |
| 14 | 3,5-dichlorophenyl | Br | 5.3 ± 0.1 |
| 15 | 3,4-dichlorophenyl | Br | 6.7 ± 0.2 |
| 16 | 2,3-dichlorophenyl | Br | 8.0 ± 0.2 |
| 17 | 2,3-dichlorophenyl | Cl | 7.9 ± 0.2 |
| 18 | 3,4-dichloro-2-thienyl | Cl | 8.1 ± 0.2 |
| 19 | 2-chloro-3-fluorophenyl | Cl | 7.8 ± 0.2 |
| 20 | 3-chloro-2-fluorophenyl | Cl | 7.1 ± 0.2 |
| 21 | 3-chloro-2-methylphenyl | Cl | 7.7 ± 0.2 |
| 22 | 2-chloro-3-(trifluoromethyl)phenyl | Cl | 7.2 ± 0.1 |
| 23 | 3-chloro-2-cyanophenyl | Cl | 7.2 ± 0.2 |
| 24 | 3-chloro-2-methylthiophenyl | Cl | 7.1 ± 0.2 |
| 25 | 2,3-dichloro-4-pyridyl | Cl | 5.0 ± 0.1 |
| 26 | butyl | Cl | 5.5 ± 0.2 |
Potency is given as pIC50 values with n = ≥ 2 replicates.
While an increase in affinity was achieved with compounds 16 and 17, changing the 5-chlorothiophene group for 2,3-dichlorophenyl did not significantly change the lipophilicity. Consequently, the hPPB figures for these two compounds was also high (>99.9%).
The exploration of the 3-position is shown in Table 3. Initially the atom linking the 3-substituent to the pyrazine ring was investigated. Changing methoxy for methylthio, methylamino, or ethyl (28, 29, and 30) gave a large drop in potency, establishing oxygen as the optimal atom with which to attach the 3-substituent to the pyrazine ring. Next, a small number of 3-alkoxy analogues were synthesized (27, 31, and 32). In each case, there was a substantial drop in potency relative to the parent methoxy-substituted compound (1 or 17). However, the 3-allyloxy analogue (33) maintained activity and with the 3-propargyloxy (34) analogue affording a slight increase in activity. Further increases in activity were obtained with 3-OCH2Aryl and 3-OCH2heteroaryl (35, 36, 37, and 38), where substitution gave substantial increase in activity (pIC50 up to 9.5). Although this SAR demonstrated how to improve on CCR4 affinity, the increase in lipophilicity associated with these changes did not address the plasma protein binding issue with hPPB > 99.9% in each case.
Table 3. Exploration of the 3-Position of the Pyrazine Ring.
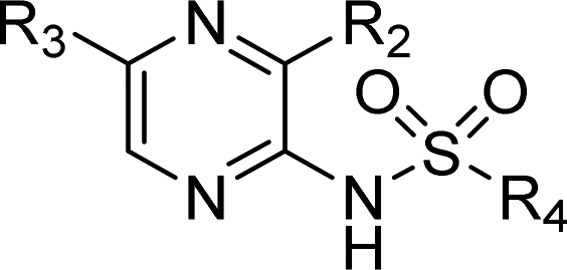

Potency is given as pIC50 values with n = ≥2 replicates. *44% inhibition at 10 μM.
In contrast to the 3-position, the exploration of the 5- and 6-positions on the pyrazine ring allowed for the discovery of compounds with increased activity combined with lower lipophilicity relative to 17 (Table 4).
Table 4. Exploration of the 5- and 6-Position of the Pyrazine Ring.
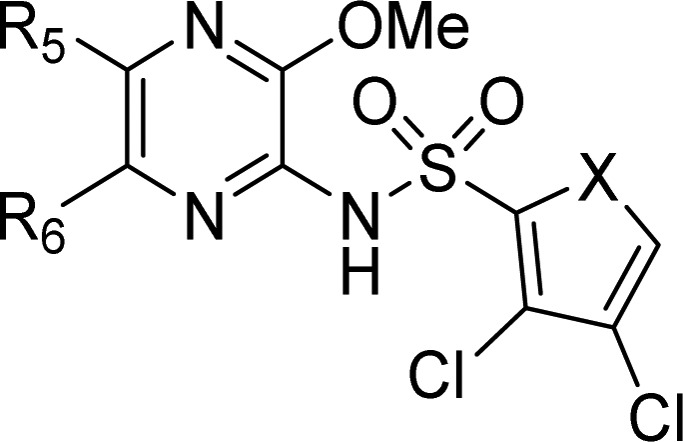
| example | X | R5 | R6 | CCR4 receptor antagonism (pIC50)a | LogP | hPPB (%) | calcd WBP (pIC50) |
|---|---|---|---|---|---|---|---|
| 17 | CH=CH | Cl | H | 7.9 ± 0.2 | 4.1 | >99.9 | |
| 39 | CH=CH | H | Cl | 8.1 ± 0.3 | 4.5 | >99.9 | |
| 40 | CH=CH | OEt | H | 7.9 ± 0.2 | 4.1 | >99.9 | |
| 41 | CH=CH | SO2Me | H | 5.3 ± 0.2 | |||
| 42 | CH=CH | Me | H | 7.4 ± 0.2 | 3.0 | 99.65 | 4.9 |
| 43 | CH=CH | H | Me | 7.3 ± 0.3 | >99.8 | ||
| 44 | CH=CH | CH2OH | H | 7.6 ± 0.2 | 2.1 | 96.2 | 6.2 |
| 45 | CH=CH | CH2OH | Cl | 8.4 ± 0.2 | 3.2 | 99.6 | 6.0 |
| 46 | CH=CH | CH2OH | Me | 7.4 ± 0.1 | 2.4 | 98.7 | 5.5 |
| 47 | CH=CH | H | H | 7.8 ± 0.05 | 2.5 | 98.9 | 5.8 |
| 48 | S | H | H | 7.7 ± 0.2 | 2.8 | 97.5 | 6.1 |
| 49 | CH=CH | F | H | 8.6 ± 0.1 | 3.1 | 99.4 | 6.4 |
| 50 | CH=CH | H | F | 8.4 ± 0.2 | 3.3 | >99.8 | |
| 51 | CH=CH | F | F | 9.0 ± 0.3 | >99.8 |
Potency is given as pIC50 values with n = ≥2 replicates.
A wide variety of substituents were tolerated in the 5- and 6-positions, and it was discovered that the percentage plasma protein binding was reduced by lowering the lipophilicity of the compounds, affording plasma protein binding below 99% for compounds of modest lipophilicity (logP < 3.0). For 44, 45, 47, 48, and 49, the calculated WBP (pIC50) of 5.8–6.4 was achieved. These met the candidate drug target profile set of WBP pIC50 ≈ 6.0. Of these four compounds, 47 and 49 had the best overall profile for progression (Table 5). Compounds 47 and 49 demonstrated good selectivity for the CCR4 receptor when screened in-house against a range of other chemokine receptors (CXCR1 and CXCR2, CCR1, CCR2b, CCR5, CCR7, and CCR8; all inactive at 10 μM) and little or no activity when screened against a large panel of receptors and enzymes (∼120 screens at MDS-Pharma). A pharmacokinetic profile commensurate with once-daily oral administration enabled both compounds to progress as candidate drugs.23
The antagonist potency of 47 and 49 was assessed in a number of cell systems in which a response mediated by the human CCR4 receptor can be evoked by MDC or TARC and quantified using changes in intracellular calcium concentration or chemotaxis. The cell systems used were a cell line (CHO) transfected with the human CCR4 receptor and human CD4+, CD45RA+ Th2 cells in 0.3% HSA. Findings are summarized in Table 6. The reduced potency in the presence of added protein is consistent with the drop-off predicted from plasma protein binding measurements. Both 47 and 49 demonstrated no agonist activity at concentrations up to 10 μM.
Table 6. Potency of 47 and 49 for Inhibition of Cellular Responses Mediated by the Human CCR4 Receptor.
| cell system | assay readout | 47 (pIC50 mean ± SEM) | 49 (pIC50 mean ± SEM) |
|---|---|---|---|
| inhibition of CCL22 Ca2+ response | MDC-induced Ca2+ flux | 7.5 ± 0.04 (n = 3) (CHO cells expressing hCCR4) | 8.4 ± 0.1 (n = 4) (HEK cells expressing hCCR4) |
| inhibition of Th2 cell CCL22 driven chemotaxis in 0.3% HSA | MDC-induced chemotaxis | 6.3 ± 0.2 (n = 3) | 6.8 ± 0.2 (n = 3) |
| inhibition of Th2 cell CCL17 driven chemotaxis in 0.3% HSA | TARC-induced chemotaxis | 6.3 ± 0.1 (n = 3) | 6.5 (n = 1) |
The anti-inflammatory effects of 47 were investigated in Brown–Norway rats sensitized to ovalbumin. Sensitized rats were dosed orally (BID) with 47 at 0.22, 0.75, 2.2, 3.0, 7.5, and 15 μmol/kg 1 h prior to antigen challenge and every 12 h thereafter prior to termination 96 h post-challenge. Histopathological examination of the lung tissue showed a marked, dose-dependent reduction in a range of histological correlates with reduced alveolitis and leukocyte trafficking in the microvasculature being the most diagnostic features of efficacy. The changes were first visible at a dose of 0.22 μmol/kg and maximal at 7.5 μmol/kg. Plasma samples were taken 12 h after the last dose of compound for measurement of terminal trough concentrations of 47 to demonstrate a strong PKPD correlation (Table 7).
Table 7. Terminal Plasma Concentrations of 47.
| dose (μmol/kg) | total plasma concentration (μM) | free plasma concentration/pIC50 |
|---|---|---|
| 0.22 | <LOQa | |
| 0.75 | <LOQa | |
| 2.2 | <LOQa | |
| 3.0 | 0.033 ± 0.018 | 0.6 |
| 7.5 | 0.14 ± 0.069 | 3 |
| 15 | 0.49 ± 0.18 | 9 |
LOQ was 0.03 μM for the 0.22, 0.75, and 2.2 μmol/kg doses.
The synthesis of 47 and 49 is shown (Scheme 1). Dibromination of 2-aminopyrazine followed by selective displacement of the bromine in the 3-position with sodium methoxide gave 2-amino-5-bromo-3-methoxypyrazine. The bromine in the 5-position was then removed by hydrogenation and coupling of the 2,3-dichlorophenylsulphonyl chloride with 2-amino-3-methoxypyrazine afforded 47. A highly selective 5-position nitration of 47 followed by reduction of the nitro group 52 gave an amino-pyrazine 53 that was diazotized in the presence of hydrofluoric acid to give 49.
Scheme 1.

Reagents and conditions: (i) Br2, dichloromethane, 2,6-lutidine (85-95%); (ii) NaOMe (3 equiv), MeOH, reflux (90-95%); (iii) H2, Pd/C, ethanol, rt (95%) (iv) ArSO2Cl, KOtBu, 0–25°C, THF (80–95%); (v) HNO3, AcOH, 70–85 °C (75-82%); (vi) H2, Pd/C, AcOH, 60 °C (75-92%); (vii) HBF4, CH3CN, 0–5 °C, NaNO2 (40%) or HF/pyridine/NaNO2 (85%). Range of percentage yields across multiple reactions and scale (∼100 mg to >50 g).
In summary, a new series of potent and bioavailable amino-pyrazine sulfonamide CCR4 receptor antagonists have been developed from a lead optimization program initiated from a high-throughput screening/synthesis campaign. The initial hit series proved to have reasonable activity but was hindered by having very high human plasma protein binding resulting in a low free fraction of compound. Through chemical modification, both the potency of the series and the free fraction were enhanced to ultimately deliver two clinical candidates. Further studies on these compounds will be reported in due course.
Acknowledgments
The author’s thank Drs Michael Bernstein, Sara Duncan, and Eva Lenz for NMR spectroscopy support and Mrs. Hemma Pancholi for detailed analysis work.
Glossary
ABBREVIATIONS
- CCR4
CC chemokine receptor 4
- CCL12
CC chemokine ligand 12
- F
bioavailability
- FLIPR
fluorescence imaging plate reader
- FMAT
fluorescent microvolume assay technology
- GPCR
G protein-coupled receptor
- MDC
macrophage-derived chemokine
- PPB
plasma protein binding
- TARC
T-cell and activation-related chemokine
- UID
once-a-day
- Vss
volume of distribution at steady state
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00315.
Representative synthetic procedures and analytical data for 47 and 49. Description of biological assay (CCR4 receptor antagonism using FMAT whole cell binding) (PDF)
Author Present Address
‡ School of Pharmacy, Centre for Biomolecular Sciences, The University of Nottingham, University Park, Nottingham, NG7 2RD, U.K.
Author Present Address
§ Discovery Sciences, Innovative Medicines & Early Development, AstraZeneca, Darwin Building, Cambridge Science Park, Cambridge, CB4 0FZ, U.K.
Author Present Address
∥ IMED Oncology, Innovative Medicines & Early Development, AstraZeneca, Chesterford Research Park, Cambridge, CB10 1XL, U.K.
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Pease J.; Horuk R. Chemokine Receptor Antagonists. J. Med. Chem. 2012, 55 (22), 9363–9392. 10.1021/jm300682j. [DOI] [PubMed] [Google Scholar]
- Charo I. F.; Ransohoff R. M. The Many Roles of Chemokines and Chemokine Receptors in Inflammation. N. Engl. J. Med. 2006, 354 (6), 610–621. 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- Proudfoot A. E. I. Chemokine receptors: multifaceted therapeutic targets. Nat. Rev. Immunol. 2002, 2 (2), 106–115. 10.1038/nri722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonecchi R.; Bianchi G.; Bordignon P. P.; D’Ambrosio D.; Lang R.; Borsatti A.; Sozzani S.; Allavena P.; Gray P. A.; Mantovani A.; Sinigaglia F. Differential Expression of Chemokine Receptors and Chemotactic Responsiveness of Type 1 T Helper Cells (Th1s) and Th2s. J. Exp. Med. 1998, 187 (1), 129–134. 10.1084/jem.187.1.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochner B. S.; Hudson S. A.; Xiao H. Q.; Liu M. C. Release of Both CCR4-Active and CXCR3-Active Chemokines during Human Allergic Pulmonary Late-Phase Reactions. J. Allergy Clin. Immunol. 2003, 112 (5), 930–934. 10.1016/j.jaci.2003.08.012. [DOI] [PubMed] [Google Scholar]
- Pilette C.; Francis J. N.; Till S. J.; Durham S. R. CCR4 Ligands Are up-Regulated in the Airways of Atopic Asthmatics after Segmental Allergen Challenge. Eur. Respir. J. 2004, 23 (6), 876–884. 10.1183/09031936.04.00102504. [DOI] [PubMed] [Google Scholar]
- Hall D.; Ford A.; Hodgson S.. Therapeutic Potential of CCR4 Antagonistste. In New Drugs and Targets for Asthma and COPD; Karger Medical and Scientific Publishers, 2010; Vol. 39, pp 161–165. [Google Scholar]
- Newhouse B.; Allen S.; Fauber B.; Anderson A. S.; Eary C. T.; Hansen J. D.; Schiro J.; Gaudino J. J.; Laird E.; Chantry D.; Eberhardt C.; Burgess L. E. Racemic and Chiral Lactams as Potent, Selective and Functionally Active CCR4 Antagonists. Bioorg. Med. Chem. Lett. 2004, 14 (22), 5537–5542. 10.1016/j.bmcl.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Yokoyama K.; Ishikawa N.; Igarashi S.; Kawano N.; Masuda N.; Hattori K.; Miyazaki T.; Ogino S.; Orita M.; Matsumoto Y.; Takeuchi M.; Ohta M. Potent CCR4 Antagonists: Synthesis, Evaluation, and Docking Study of 2,4-Diaminoquinazolines. Bioorg. Med. Chem. 2008, 16 (17), 7968–7974. 10.1016/j.bmc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- Yokoyama K.; Ishikawa N.; Igarashi S.; Kawano N.; Hattori K.; Miyazaki T.; Ogino S.; Matsumoto Y.; Takeuchi M.; Ohta M. Discovery of Potent CCR4 Antagonists: Synthesis and Structure–activity Relationship Study of 2,4-Diaminoquinazolines. Bioorg. Med. Chem. 2008, 16 (14), 7021–7032. 10.1016/j.bmc.2008.05.036. [DOI] [PubMed] [Google Scholar]
- Burdi D. F.; Chi S.; Mattia K.; Harrington C.; Shi Z.; Chen S.; Jacutin-Porte S.; Bennett R.; Carson K.; Yin W.; Kansra V.; Gonzalo J.-A.; Coyle A.; Jaffee B.; Ocain T.; Hodge M.; LaRosa G.; Harriman G. Small Molecule Antagonists of the CC Chemokine Receptor 4 (CCR4). Bioorg. Med. Chem. Lett. 2007, 17 (11), 3141–3145. 10.1016/j.bmcl.2007.03.030. [DOI] [PubMed] [Google Scholar]
- Purandare A. V.; Wan H.; Somerville J. E.; Burke C.; Vaccaro W.; Yang X.; McIntyre K. W.; Poss M. A. Core Exploration in Optimization of Chemokine Receptor CCR4 Antagonists. Bioorg. Med. Chem. Lett. 2007, 17 (3), 679–682. 10.1016/j.bmcl.2006.10.091. [DOI] [PubMed] [Google Scholar]
- Allen S.; Newhouse B.; Anderson A. S.; Fauber B.; Allen A.; Chantry D.; Eberhardt C.; Odingo J.; Burgess L. E. Discovery and SAR of Trisubstituted Thiazolidinones as CCR4 Antagonists. Bioorg. Med. Chem. Lett. 2004, 14 (7), 1619–1624. 10.1016/j.bmcl.2004.01.072. [DOI] [PubMed] [Google Scholar]
- Liu C.-J.; Yu S.-L.; Liu Y.-P.; Dai X.-J.; Wu Y.; Li R.-J.; Tao J.-C. Synthesis, Cytotoxic Activity Evaluation and HQSAR Study of Novel Isosteviol Derivatives as Potential Anticancer Agents. Eur. J. Med. Chem. 2016, 115, 26–40. 10.1016/j.ejmech.2016.03.009. [DOI] [PubMed] [Google Scholar]
- Procopiou P. A.; Barrett J. W.; Barton N. P.; Begg M.; Clapham D.; Copley R. C. B.; Ford A. J.; Graves R. H.; Hall D. A.; Hancock A. P.; Hill A. P.; Hobbs H.; Hodgson S. T.; Jumeaux C.; Lacroix Y. M. L.; Miah A. H.; Morriss K. M. L.; Needham D.; Sheriff E. B.; Slack R. J.; Smith C. E.; Sollis S. L.; Staton H. Synthesis and Structure–Activity Relationships of Indazole Arylsulfonamides as Allosteric CC-Chemokine Receptor 4 (CCR4) Antagonists. J. Med. Chem. 2013, 56 (5), 1946–1960. 10.1021/jm301572h. [DOI] [PubMed] [Google Scholar]
- Pease J. E.; Horuk R. Chemokine Receptor Antagonists: Part 1. Expert Opin. Ther. Pat. 2009, 19 (1), 39–58. 10.1517/13543770802641346. [DOI] [PubMed] [Google Scholar]
- Cahn A.; Hodgson S.; Wilson R.; Robertson J.; Watson J.; Beerahee M.; Hughes S. C.; Young G.; Graves R.; Hall D.; van Marle S.; Solari R. Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of GSK2239633, a CC-Chemokine Receptor 4 Antagonist, in Healthy Male Subjects: Results from an Open-Label and from a Randomised Study. BMC Pharmacol. Toxicol. 2013, 14 (1), 14. 10.1186/2050-6511-14-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews G.; Jones C.; Wreggett K. A. An Intracellular Allosteric Site for a Specific Class of Antagonists of the CC Chemokine G Protein-Coupled Receptors CCR4 and CCR5. Mol. Pharmacol. 2007, 73 (3), 855–867. 10.1124/mol.107.039321. [DOI] [PubMed] [Google Scholar]
- Slack R. J.; Russell L. J.; Barton N. P.; Weston C.; Nalesso G.; Thompson S.-A.; Allen M.; Chen Y. H.; Barnes A.; Hodgson S. T.; Hall D. A. Antagonism of Human CC-Chemokine Receptor 4 Can Be Achieved through Three Distinct Binding Sites on the Receptor. Pharmacol. Res. Perspect. 2013, 1 (2), e00019. 10.1002/prp2.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter A.; Timothy J.; Kindon N.; Roberts B.; Steele J.; Stocks M.; Nicholas T.. Preparation of N-pyrazinylthiophenesulfonamides as Chemokine Receptor Modulators. WO 2003051870, 2003.
- Davis A. M.; Teague S. J.; Kleywegt G. J. Application and Limitations of X-Ray Crystallographic Data in Structure-Based Ligand and Drug Design. Angew. Chem., Int. Ed. 2003, 42 (24), 2718–2736. 10.1002/anie.200200539. [DOI] [PubMed] [Google Scholar]
- Luker T.; Bonnert R.; Paine S. W.; Schmidt J.; Sargent C.; Cook A. R.; Cook A.; Gardiner P.; Hill S.; Weyman-Jones C.; Patel A.; Thom S.; Thorne P. Zwitterionic CRTh2 Antagonists. J. Med. Chem. 2011, 54 (6), 1779–1788. 10.1021/jm1014549. [DOI] [PubMed] [Google Scholar]
- Grime K. H.; Barton P.; McGinnity D. F. Application of In Silico, In Vitro and Preclinical Pharmacokinetic Data for the Effective and Efficient Prediction of Human Pharmacokinetics. Mol. Pharmaceutics 2013, 10 (4), 1191–1206. 10.1021/mp300476z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.