

Abstract
Genomic instability is one of the primary models of carcinogenesis and a feature of almost all cancers. Homologous recombination (HR) repair protects against genomic instability by maintaining high genomic fidelity during the repair of DNA double strand breaks. The defining step of HR repair is the formation of the Rad51 nucleofilament, which facilitates the search for a homologous sequence and invasion of the template DNA strand. Particulate hexavalent chromium (Cr(VI)), a human lung carcinogen, induces DNA double strand breaks and chromosome instability. Since the loss of HR repair increases Cr(VI)-induced chromosome instability, we investigated the effect of extended Cr(VI) exposure on HR repair. We show acute (24 h) Cr(VI) exposure induces a normal HR repair response. In contrast, prolonged (120 h) exposure to particulate Cr(VI) inhibited HR repair and Rad51 nucleofilament formation. Prolonged Cr(VI) exposure had a profound effect on Rad51, evidenced by reduced protein levels and Rad51 mislocalization to the cytoplasm. The response of proteins involved in Rad51 nuclear import and nucleofilament formation displayed varying responses to prolonged Cr(VI) exposure. BRCA2 formed nuclear foci after prolonged Cr(VI) exposure, while Rad51C foci formation was suppressed. These results suggest that particulate Cr(VI), a major chemical carcinogen, inhibits HR repair by targeting Rad51, causing DNA double strand breaks to be repaired by a low fidelity, Rad51-independent repair pathway. These results further enhance our understanding of the underlying mechanism of Cr(VI)-induced chromosome instability and thus, carcinogenesis.
Keywords: homologous recombination repair, Rad51 nucleofilament, particulate chromium (VI), chromosome instability
Genomic instability is one of the primary models of carcinogenesis and a feature of almost all cancers. One subtype, chromosome instability, can arise due to the misrepair of DNA damage (Pikor et al., 2013). The cell depends upon DNA repair pathways that maintain genomic fidelity, such as homologous recombination (HR), to prevent such errors from occurring. During HR repair, the ends of a DNA double strand break are resected to form a single-stranded DNA overhang. Rad51 is loaded onto the single-stranded DNA, creating a helical nucleoprotein filament responsible for identifying a homologous sequence and strand invasion. This is the defining biochemical step of HR (Baumann et al., 1996; Sung and Robberson, 1995). After the strand successfully invades the donor duplex, repair synthesis occurs using the donor duplex as a template and Holliday junctions are resolved, resulting in high fidelity repair.
Rad51 nucleofilament formation suppresses lower fidelity repair pathways such as single-strand annealing and alternative nonhomologous end joining (alt-NHEJ) (Bennardo et al., 2008; Stark et al., 2004), making it key to maintaining high genomic fidelity. As such, it is important to understand how chemical carcinogens affect Rad51 nucleofilament formation and thus, HR repair. Particulate chromium VI (Cr(VI)) is a known human lung carcinogen. Exposure to particulate Cr(VI) occurs both occupationally and environmentally, making it a major human health hazard.
Acute (24 h) and prolonged (>72 h) Cr(VI) exposure induces DNA damage, chromosome damage and chromosome instability in cultured human lung cells (Holmes et al., 2010; Qin et al., 2014). Additionally, loss of HR repair increases Cr(VI)-induced chromosome instability in Chinese hamster cells (Stackpole et al., 2007). Although proteins early in the HR signaling pathway respond after Cr(VI) exposure, Rad51 foci formation is inhibited after prolonged exposure (Qin et al., 2014), suggesting Rad51 is a target of Cr(VI). Understanding the effect of prolonged Cr(VI) exposure on HR repair is paramount in determining the mechanism of Cr(VI)-induced carcinogenesis. Thus, the goal of this study is to determine the effect of prolonged particulate Cr(VI) exposure on Rad51 localization, its functional unit, the nucleofilament, and the proteins involved in Rad51 nucleofilament formation. Additionally, we directly investigate the effect of prolonged Cr(VI) exposure on homology directed repair with the use of a direct repeat green fluorescent protein (DR-GFP) reporter construct.
MATERIALS AND METHODS
Chemicals and reagents
DMEM and Ham’s F12 50:50 mixture and GlutaGRO (L-alanyl-L-glutamine solution) were purchaed from Mediatech Inc (Herndon, Virginia). Cosmic Calf Serum was purchased from Hyclone (Logan, Utah). Dulbecco’s phosphate buffered saline (PBS), penicillin/streptomycin, sodium pyruvate, trypsin/EDTA, goat serum, HEPES, Prolong Gold Antifade Reagent with DAPI and Alexa Fluor secondary antibodies were purchased from Life Technologies (Grand Island, New York). All plasticware was purchased from BD Biosciences (Franklin Lakes, New Jersey). Zinc chromate (CAS no. 13530-65-9) was purchased from Alfa Aesar (A18178, Ward Hill, MA) and Pfaltz and Bauer (Z00277, Waterbury, Connecticut). 4% paraformaldehyde in PBS was purchased by Alfa Aesar (Ward Hill, Massachusetts). Triton X-100 and Igepal were purchased from Sigma-Aldrich (St Louis, Missouri). Nunc Lab Tek II glass chamber slides were purchased from Thermo Fisher Scientific Inc. (Waltham, Massachusetts). FNC coating mix was purchased from AthenaES (Baltimore, Maryland). MgCl2, MnCl2 and EDTA were purchased from JT Baker (Center Valley, Pennsylvania). PVDF membrane and Bradford assay kit were purchased from BioRad (Hercules, California).
Cell culture
For these studies we used an hTERT immortalized clonal cell line derived from human bronchial fibroblasts (WTHBF-6) (Wise et al., 2004). This cell line has normal growth parameters, a normal stable karyotype and a cytotoxic and clastogenic response to metals similar to primary cells (Wise et al., 2004). Cells were cultured as adherent monolayers in DMEM/F12 50:50 mixture, supplemented with 15% cosmic calf serum, 1% L-alanyl-L-glutamine, 1% penicillin/streptomycin, and 0.1 mM sodium pyruvate. Cells were maintained in a 5% CO2-humidified environment at 37 °C.
Treatment with particulate Cr(VI) compound
Zinc chromate was administered as a suspension of particles in cold, sterile water as previously described (Xie et al., 2009). Cells were seeded and allowed 48 h to reenter logarithmic growth before treatment. Cells were treated for 24, 72, and 120 h with concentrations of 0.1–0.3 μg/cm2 zinc chromate. The cytotoxicity of these doses at the various exposure times are previously described (Holmes et al. 2010). Although zinc chromate induces a time and concentration dependent increase in cytotoxicity, plenty of cells survive the 120 h exposures (Holmes et al. 2010). Additionally, annexin V/propidium iodide staining demonstrated 71% of cells are alive after 120 h exposure to our highest dose, 0.3 μg/cm2 zinc chromate (data not shown).
Western blot
Logarithmically growing cells were plated into 100 mm dishes and treated with zinc chromate. After treatment, whole cell extracts were obtained by washing, collecting and incubating cells in 500 μl of extraction buffer (50 mM HEPES, 5 mM MnCl2, 10 mM MgCl2, 2 mM EDTA, 100 mM NaCl, 5 mM KCl, and 0.1% Igepal) with protease and phosphatase inhibitors for 30 min on ice. Cells were sheared with an 18-G needle, followed by a 27-G needle and centrifuged at 14 000 rpm for 10 mins. Supernatant was collected and stored at −80 °C. Nuclear extracts were prepared according to our published methods (Qin et al., 2014). Protein concentration was determined with a Bradford assay and 10 or 15 μg protein were resolved on 12% Bis-Tris SDS-PAGE gels and transferred to PVDF membranes. Immunoblots were probed with anti-Rad51 (Santa Cruz sc-8349; 1:1000) and anti-Rad51C (abcam ab55728; 1:250). Equal loading was confirmed by Alpha-tubulin (Cell Signaling 11H10; 1:500) or H3 (Cell Signaling 9715; 1:500). Immunoblots were incubated with Alexa680 or Alexa800 (1:5000) secondary antibodies and fluorescence detected using an Odyssey Imager (LiCor, Lincoln, Nebraska).
Immunofluorescence
Immunofluorescence staining was conducted as previously described with minor alterations (Xie et al., 2005). Briefly, cells were seeded on glass chamber slides coated with FNC and treated with zinc chromate. At harvest, cells were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 5 min and blocked with 10% goat serum and 5% BSA in PBS for 1 h. For BRCA2 foci, cells were incubated with 0.5% Triton X-100 for 5 mins, fixed with 4% paraformaldehyde and 0.5% Triton X-100 for 15 mins and blocked with 2.5% BSA. Cells were then incubated with anti-RAD51 (Santa Cruz sc-8349; 1:200), anti-RAD51C (abcam ab72063; 1:500) or anti-BRCA2 (Genetex GTX70121; 1:50) antibodies at 4 °C overnight, washed with PBS and incubated with Alexa Fluor 488 (1:3000) for 1 h. Cells were washed with PBS and coverslips were mounted with DAPI. Nuclear foci were scored in 100 cells per concentration/timepoint using fluorescence microscopy. Results were expressed as the percentage of cells with >20 or >40 foci based on background levels such that negative controls had 5% or less of cells with this level. Images of 50 cells per concentration/timepoint were obtained with an Olympus confocal microscope and cytoplasmic and nuclear intensities were measured as integrated intensity using Image J.
Affinity grid capture
Affinity grid specimens were prepared as previously described (Tanner et al., 2012). Briefly, cells were seeded into 100 mm dishes and treated with zinc chromate. After 24 or 120 h treatment, cells were washed twice with PBS and cell pellets were flash frozen in liquid nitrogen. Cytoplasmic and nuclear extracts were prepared using the NE-PER kit (Life Technologies, Grand Island, New York) with phosphatase and protease inhibitors. Lipid monolayers (20% Ni-NTA lipids and 10% DDMA) were incubated for 60 min on ice and applied to the carbon EM grids, followed by His Protein A and anti-RAD51 antibody (Santa Cruz sc-8349; 1:1000). Excess liquid was removed and nuclear or cytoplasmic extracts applied to the prepared EM grids. Specimens were prepared for negative staining with 1% uranyl formate and imaged in a FEI Spirit BioTwin Electron Microscope equipped with a tungsten filament. Rad51 filaments were counted in 1 grid square (37 μm2) and images were recorded on a FEI Eagle 2k HS CCD camera with a pixel size of 30 μm at a nominal magnification of 23 000×.
DR-GFP homology directed reporter
The hygro-DR-GFP construct developed by Dr Maria Jasin was linearized with SacI and KpnI restriction enzymes and run on an agarose gel to confirm proper digestion. The SacI/KpnI fragment of the hygro-DR-GFP construct was ethanol-precipitated, dried and resuspended in ultra-pure H2O. For stable transfection, WTHBF-6 cells were electroporated at 1200v/30w with 70 μg of the SacI/KpnI hygro-DR-GFP fragment using a Neon transfection system (Life Technologies, Grand Island, New York). Cells underwent selection with 150 μg/ml hygromycin B and hygromycin resistant colonies were selected and expanded individually in selection media. Integration of the hygro-DR-GFP construct was confirmed with Southern blotting.
To measure the repair of an I-SceI generated double strand break after Cr(VI) treatment, cells were plated in 100 mm dishes and treated with 0 and 0.2 μg/cm2 zinc chromate for 24 or 120 h. After treatment, the cells were washed, collected, resuspended in Neon resuspension buffer, mixed with 5 μg of either the I-SceI expression vector pCBASce or a pNZE-CAG control vector and pulsed once at 1200v/30w. A subset of cells from each treatment group was not transfected with a vector as a measurement of background fluorescence. To determine the amount of homology-directed repair, the percentage of GFP-positive cells in 50 000 cells was measured by flow cytometry 48 h after electroporation. Background fluorescence values were subtracted from the raw +I-SceI and +NZE values to calculate the final percentage of GFP-positive cells.
Statistics
Results were expressed as the mean ± SEM (standard error of the mean) of 3 independent experiments. Differences among means were evaluated using a Student’s t-test when comparing 2 variables and a 2-way ANOVA was used when comparing different time points. A 95% confidence interval for the difference in means of each pair of concentrations was constructed based on the Student’s t distribution. All analyses were conducted using GraphPad.
RESULTS
Prolonged Exposure to Particulate Cr(VI) Inhibits the Mediator of HR, Rad51
Rad51 is a central protein of the HR repair pathway. Upon the induction of DNA double strand breaks, Rad51 localizes to the nucleus (Essers et al., 2002). Particulate Cr(VI) induces DNA double strand breaks in a concentration-dependent manner after acute and prolonged exposure (Qin et al., 2014). To begin to understand the impact of Cr(VI) on DNA repair, we examined the response of Rad51 after these exposures. We found particulate chromate significantly reduced whole cell Rad51 protein levels in a time and dose-dependent manner (Figs. 1A and B). For example, whole cell Rad51 levels decreased to 70, 30, and 20% of controls after 24, 72, and 120 h exposure to 0.2 μg/cm2 zinc chromate, respectively. Nuclear Rad51 levels, however, showed a different pattern. After 24 h zinc chromate exposure, nuclear Rad51 protein levels increased initially and then decreased in a dose-dependent maner, but only dropped below control level after the highest dose. However, after 72 and 120 h exposure, a dose-dependent decrease in nuclear Rad51 occurred (Figs. 1A and C). For example, nuclear protein levels were 100% of control after 24 h exposure to 0.2 μg/cm2 zinc chromate, but decreased to 34 and 37% of controls after 72 and 120 h exposure, respectively.
FIG. 1.

Prolonged exposure to zinc chromate decreases whole cell and nuclear Rad51 protein expression in a time and dose dependent manner. A, This figure shows zinc chromate induced a dose-dependent decrease in whole cell Rad51 at all time points. Zinc chromate exposure caused an increase in nuclear Rad51 protein at 24h exposure, but a dose-dependent decrease in nuclear Rad51 protein at 72 and 120 h exposure. Whole cell protein data represent an average of 3 experiments. Error bars = SEM. All doses except for 0.1 μg/cm2 at 72 h exposure were significantly different from controls (*P < .05; # P < .005; ## P < .001). Nuclear protein data represent an average of 3 experiments. Nuclear protein levels were significantly lower than controls after 0.2 μg/cm2 zinc chromate at 72 and 120 h and 0.3 μg/cm2 at all time points (P < .01). B, Representative images of whole cell Rad51 western blots. C, Representative images of nuclear Rad51 western blots.
Due to our observed decrease in nuclear Rad51 and previous reports Rad51 can aggregate in the cytoplasm (Plo et al., 2008; Qin et al., 2014), we tested whether Rad51 accumulates in the cytoplasm after prolonged Cr(VI) exposure. Using immunofluorescence intensity, we determined the average ratio of cytoplasmic/nuclear Rad51 and the percent of cells with cytoplasmic accumulation of Rad51. We found prolonged exposure to zinc chromate increased cytoplasmic Rad51 localization (Figure 2A). The average ratio of cytoplasmic/nuclear Rad51 fluorescence did not change after 24 h exposure to zinc chromate. However, the cytoplasmic/nuclear ratio increased in a dose-dependent manner after 72 and 120 h zinc chromate exposure (Figure 2B). For example, 24 h exposure to 0 and 0.3 μg/cm2 zinc chromate resulted in a slight decrease in the cytoplasmic/nuclear Rad51 ratio from 3.2 to 2.9, respectively, while 120 h exposure resulted in a significant (P < .05) increase in the cytoplasmic/nuclear Rad51 ratio from 3.0 to 5.7, respectively.
FIG. 2.

Prolonged Cr(VI) exposure induces the mislocalization of Rad51 in response to DNA damage. This figure shows prolonged exposure to zinc chromate caused an increase in cytoplasmic Rad51 localization. Data represent an average of at least 3 experiments. Error bars = standard error of the mean. A, Representative images of Rad51 subcellular localization after 24 and 120 h exposure to 0.2 μg/cm2 zinc chromate. B, Rad51 cytoplasmic to nuclear ratio determined by fluorescent intensity. The cytoplasmic/nuclear ratio after exposure to 0.2 and 0.3 μg/cm2 zinc chromate at 120 h were significantly higher than control (P < .05). C, Percent of cells with cytoplasmic accumulation of Rad51 and cytoplasmic Rad51 aggregates. The percent of cells with cytoplasmic accumulation of Rad51 after 0.1 μg/cm2 zinc chromate at 72 and 120 h and 0.3 μg/cm2 zinc chromate at 120 h were significantly higher than control (P <.05). The percent of cells with cytoplasmic Rad51 accumulation and cytoplasmic Rad51 aggregate formation correlate.
We then calculated the percent of cells with cytoplasmic accumulation of Rad51 and cytoplasmic Rad51 aggregates (Figure 2C). Cells with a cytoplasmic intensity >95% of control cells were considered positive for cytoplasmic accumulation. Cytoplasmic aggregates were defined as clusters of Rad51 concentrated in the cytoplasm. There was no significant increase in the percent of cells with cytoplasmic Rad51 accumulation and no cytoplasmic aggregates detected at 24 h zinc chromate exposure. The percent of cells with cytoplasmic Rad51 accumulation significantly (P < 0.05) increased in a dose-dependent manner after 72 and 120 h zinc chromate exposure. Cytoplasmic Rad51 aggregates were first detected at 72 h exposure and increased in a time- and dose-dependent manner. The percent of cells with cytoplasmic Rad51 accumulation and cytoplasmic Rad51 aggregate formation showed a positive correlation.
Rad51 forms a nucleofilament on the single-stranded DNA overhang during HR repair, facilitating the search for a homologous sequence and subsequent strand invasion. The Rad51 nucleofilament is the functional unit but it is unclear how much Rad51 is necessary for proper filament formation. To determine whether the effects of prolonged Cr(VI) expsosure on Rad51 result in the inhibition of nucleofilament formation, we used affinity grid capture and transmission electron microscopy (TEM) to visualize Rad51 nucleofilaments after 24 and 120 h exposure to zinc chromate (Figure 3A). No protein complexes were found on negative control grids coated with the Ni-NTA lipids, His-Protein A and the anti-Rad51 antibody (Supplementary Figure 1A). Specificity of the Rad51 antibody was confirmed by visualization of a single band (approximately 37 kDa) by western blotting (Supplementary Figure 1B). However, it is important to note that any proteins interacting with Rad51 may be bound to the grid as part of the Rad51 nucleofilament structure. The Rad51 nucleofilament structures visualized consisted of either one or numerous filaments attached to darker ‘nodules’. As it was difficult to determine whether the more complex structures consisted of 1 Rad51 nucleofilament wrapped around itself or several nucleofilaments twisted together, we counted each Rad51 nucleofilament structure only once to be conservative. For example, each image shown in Figure 3A was counted as 1 Rad51 nucleofilament. 24 h exposure to 0.2 μg/cm2 zinc chromate induced a 2.7-fold increase in Rad51 nucleofilaments over the control while 120 h exposure inhibited Rad51 nucleofilament formation, resulting one-fourth of control values (Figure 3B).
FIG. 3.

Prolonged Cr(VI) exposure inhibits Rad51 nucleofilament formation. This figure shows prolonged exposure to zinc chromate decreased the number of Rad51 nucleofilaments and resulted in the formation of primarily simple Rad51 filament structures. Data represent an average of 3 experiments. A, Representative TEM images of Rad51 nucleofilaments. B, Number of Rad51 nucleofilaments in 37 μm2. The number of Rad51 nucleofilaments significantly increased (P < .05) after 24 h 0.2 μg/cm2 zinc chromate exposure and significantly decreased (P < .05) after 120 h exposure to the same concentration. C, Quantification of ‘simple’ or ‘complex’ Rad51 filament structures. 120 h to 0.2 μg/cm2 zinc chromate significantly shifted the filament composition towards simple filaments (P < .05).
The structure of the Rad51 nucleofilaments was distinctly different following 24 and 120 h exposure to zinc chromate. Rad51 nucleofilaments were categorized as ‘simple’ or ‘complex’ filament structures and quantified. ‘Simple’ filaments were defined as linear structures with less than 2 extended branching points and no cross-over of filaments. ‘Complex’ filaments were defined as structures with 2 or more extended branching points and/or at least 1 cross-over of filaments. Untreated controls contained nucleofilaments of both categories, varying between 40 and 60%. After 24 h exposure to 0.2 μg/cm2 zinc chromate, the composition was similar to the controls, with 65% complex and 35% simple filaments. Prolonged exposure of 120 h to the same concentration significantly shifted the composition towards simple filaments, with only 23% complex and 77% simple filaments (Figure 3C).
Prolonged Exposure to Particulate Cr(VI) Suppresses Rad51 Mediator Proteins
Having determined prolonged exposures to particulate Cr(VI) inhibit the functional unit of Rad51 during HR repair, we then asked what effect these exposures have on the proteins involved in forming the Rad51 nucleofilament. Normally, Rad51 is transported into the nucleus in response to DNA damage and a multitude of proteins cooperate to load Rad51 onto the ssDNA overhang and stabilize the Rad51 nucleofilament. Both BRCA2 and Rad51C have been shown to transport Rad51 into the nucleus in response to DNA damage (Gildemeister et al., 2009; Jeyasekharan et al., 2013). Additionally, BRCA2 is responsible for loading Rad51 onto the ssDNA (Jensen et al., 2010) while Rad51C plays a role in stabilizing the Rad51 filament (Amunugama et al., 2013). We chose to investigate the effect of prolonged Cr(VI) exposure on BRCA2 and Rad51C foci formation due to their key roles in forming and maintaining the Rad51 nucleofilament.
Figure 4 demonstrates BRCA2 nuclear foci formation increased in a concentration-dependent manner after 24, 72, and 120 h exposure. For example, the percent of cells with BRCA2 foci increased from 4.5 to 16.8, after 24 h exposure to 0 or 0.2 μg/cm2 zinc chromate, respectively; and further increased to 29% after a 120 h exposure to 0.2 μg/cm2 zinc chromate (Figs. 4A and B).
FIG. 4.

Prolonged Cr(VI) exposure induces BRCA2 foci formation. This figure shows both acute and prolonged zinc chromate exposure induced BRCA2 foci formation. Data represent an average of 4 experiments. Error bars = SEM. A, Percent of cells with BRCA2 foci. The percent of cells with BRCA2 foci increased in a concentration-dependent manner (*P < .05; # P < .005). B, Representative images of BRCA2 foci.
Interestingly, prolonged zinc chromate exposure did inhibit Rad51C foci formation. Although the percent of cells with Rad51C foci significantly (P < .05) increased in a concentration-dependent manner after 24 h exposure, Rad51C foci formation was inhibited after 72 h and significantly (P < .05) suppressed after 120 h (Figs. 5A and B). For example, 0.2 μg/cm2 zinc chromate-induced Rad51C foci formation increased 3.6 fold after 24 h exposure but decreased to 0.69% of control after 120 h exposure. Data are represented as percent of control to normalize for a time-dependent increase in Rad51C foci in the untreated controls (data not shown). To determine if the lack of Rad51C foci was due to a decrease in protein expression, whole cell Rad51C protein was measured by western blot (Figs. 5C and D). No temporal or concentration-related changes in Rad51C protein levels were observed.
FIG. 5.

Prolonged Cr(VI) exposure inhibits Rad51C localization. This figure shows prolonged exposure to zinc chromate inhibited Rad51C foci formation but not RAD51C protein levels. A, The percent of cells with Rad51C foci (relative to control). The percent of cells with Rad51C foci was significantly higher than control after 24 h exposure to 0.2 and 0.3 μg/cm2 zinc chromate. However, the percent of cells with Rad51C foci decreased in a concentration and time-dependent manner after 72 and 120 h exposure and was significantly lower than control after 120 h exposure to 0.3 μg/cm2 zinc chromate (*P < .05). B, Representative images of Rad51C foci. C, Rad51C whole cell protein levels. No time or concentration related change in Rad51C protein levels were observed after zinc chromate exposure. D, Representative images of whole cell Rad51C western blots.
Prolonged Exposure to Particulate Cr(VI) Inhibits Homology Directed DNA Repair
Having established prolonged exposure to Cr(VI) inhibits Rad51 nucleofilament formation, we hypothesized that prolonged Cr(VI) exposure inhibits homology directed repair in human lung fibroblasts. To test this hypothesis, we stably transfected a recombination repair construct into WTHBF-6 cells (Figure 6A). The repair construct consists of a DR-GFP reporter that assays for gene conversion events, resulting in the expression of functional GFP which is detectable by flow cytometry (Pierce et al. 1999). The DR-GFP reporter contains 2 mutated GFP genes organized as direct repeats. One of the mutated GFP sequences contains a recognition site for the Sce-I endonuclease (SceGFP) and will obtain a double strand break when I-SceI is expressed in vitro. The downstream mutated GFP gene (iGFP) has the base-pairs of the Sce-I recognition site incorporated in place of the wild-type GFP sequences and 2 stop codons that inactivate the gene. The iGFP sequence acts as the donor template for the broken SceGFP gene during HR, resulting in a functional GFP that provides a fluorescent signal.
FIG. 6.
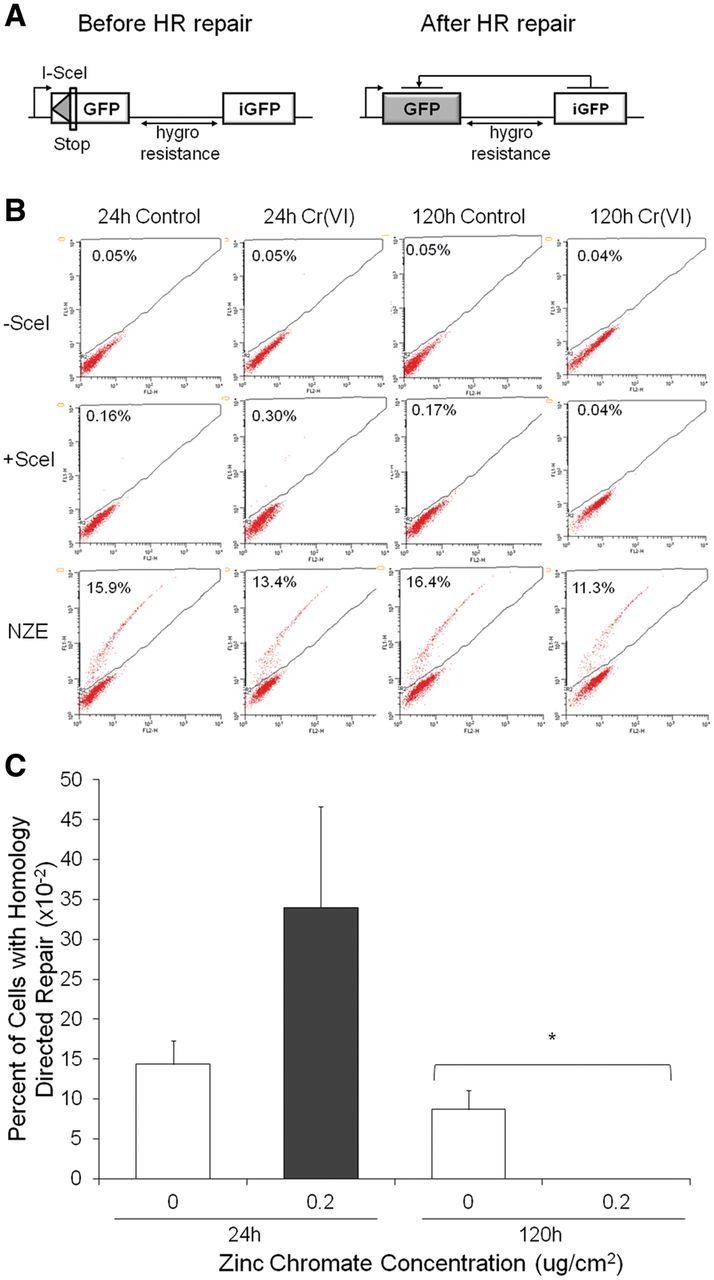
Prolonged Cr(VI) exposure inhibits homology directed repair in human lung cells. This figure shows prolonged exposure to zinc chromate inhibited homology directed repair. Data represent an average of 3 experiments. Error bars = SEM. A, hygro-DR-GFP reporter construct. The construct was stably inserted into WTHBF-6 cells. Cleavage of the I-SceI site in vitro and repair by homology directed repair using the downstream iGFP repeat results in GFP+ cells. B, Flow cytometry analysis of Cr(VI)-treated cells after integration of the I-SceI expression vector. The control vector (NZE) demonstrates equal transfection efficiency between conditions. C, Percent of cells with homology directed repair after zinc chromate exposure and integration of the I-SceI expression vector. The percent of cells with homology directed repair after 120 h exposure to 0.2 μg/cm2 zinc chromate was significantly less then control (P < .05), while there was no significant difference after 24 h exposure.
Cells treated with 0.2 μg/cm2 zinc chromate for 24 or 120 h were transfected with the pCBASce vector to transiently express the I-SceI endonuclease (Figure 6B). I-SceI expression in non-treated control cells led to an average of 11.5% of cells being GFP+ (Figure 6C). Cells treated with zinc chromate for 24 h showed an increase in homology directed repair, with 34% of cells being GFP+. However, 120 h exposure to zinc chromate completely inhibited homology directed repair with 0% of cells being GFP+. Transfection with a control pNZE-CAG construct, which expresses a wild-type GFP protein from the same control elements as the pCBASce construct, demonstrated the differences in GFP signal could not be explained by differences in tranfection efficiency (data not shown).
DISCUSSION
HR repair is a high fidelity DNA repair pathway that is crucial to maintaining genomic stability. The inhibition of the defining step in the HR repair pathway, formation of the Rad51 nucleofilament, results in the use of error-prone repair pathways such as single strand annealing and alternative NHEJ (Baumann et al., 1996; Bennardo et al., 2008; Stark et al., 2004). Although the involvement of HR in repairing heavy metal-induced DNA damage and protecting against chromosome instability has been demonstrated (Bryant et al., 2006; Helleday et al., 2000; Qin et al., 2014; Stackpole et al., 2007), little data are available determining the effect of heavy metal exposure on HR repair.
This study revealed significant differences between the effects of acute and prolonged particulate Cr(VI) exposure on DNA repair signaling. Acute exposure, represented by a 24 h particulate Cr(VI) treatment, induced a normal DNA repair response. Using the DR-GFP construct, we found HR repair was active and even slightly enhanced, although not significantly. This observation is in agreement with previously reported induction of HR repair after 24 h Cr(VI) exposure evidenced by the induction of recombination in the hprt gene of the SPD8 cell line (Helleday et al., 2000). In contrast, our data show a different response following prolonged (>72 h) particulate Cr(VI) exposure. When cells were exposed to prolonged Cr(VI), we observed a loss of HR repair.
This report is the first to show Cr(VI)-induced inhibition of HR repair. Other documentation of metal-induced inhibition of HR repair involved sodium arsenite exposure (Zhang et al., 2014). Interestingly, sodium arsenite exposure decreased HR repair after 24 h while Cr(VI)-induced inhibition of HR only occurs after prolonged exposure. There was also a difference in the extent of HR inhibition. Sodium arsenite decreased the percent of cells with homology directed repair 3-fold while Cr(VI) induced a complete suppression of HR repair. These differences suggest arsenite and chromate exhibit different mechanisms of HR repair inhibition.
Our data show Rad51 responded appropriately to acute Cr(VI) exposure, evidenced by its localization to the nucleus, an increase in the number and complexity of Rad51 nucleofilaments and no increase in Rad51 cytoplasmic accumulation. The observed increase in Rad51 monofilaments agrees with a previous report of Rad51 nuclear foci formation following acute Cr(VI) exposure (Qin et al., 2014).
In contrast, we observed prolonged particulate Cr(VI) exposure inhibited Rad51 nucleofilament formation, evidenced by a decrease in both the number of nucleofilaments detected and the complexity of the nucleofilament structures. The observed Cr(VI)-induced inhibition of Rad51 monofilaments is in agreement with previously reported decreases in Rad51 foci levels following prolonged Cr(VI) exposure (Qin et al., 2014). Additionally, we demonstrated that both nuclear and overall Rad51 protein levels significantly decreased in a time and concentration-dependent manner, beginning at 72 h Cr(VI) exposure. Although Zhang et al. (2014) showed arsenite inhibits HR repair, it did not inhibit the response of Rad51 to the site of damage. This difference further supports that the mechanism of HR inhibition is different for these 2 heavy metals.
There are several factors that may be responsible for the particulate Cr(VI)-induced inhibition of Rad51 nucleofilament formation. One explanation is that insufficient Rad51 protein is available for proper nucleofilament formation and stabilization. Candelli et al. (2014) showed Rad51 nucleation on single-stranded DNA and the stability of the resulting foci was strongly dependent on Rad51 concentration. However, the same study demonstrated that as few as 3–6 Rad51 molecules can form stable foci. Although our data show dramatic decreases in Rad51 protein following particulate Cr(VI) exposure, the fact that some Rad51 protein remains available even after prolonged exposure suggests the decrease in protein level is not entirely responsible for the inhibition of Rad51 nucleofilament formation.
Another possible cause of Cr(VI)-induced inhibition of Rad51 nucleofilament formation is the disruption of Rad51 mediator proteins. Rad51 nucleofilament formation is regulated by the cooperation of numerous proteins. We considered BRCA2 and Rad51C, which are responsible for Rad51 nuclear import as well as the formation and stabilization of the Rad51 nucleofilament. BRCA2 foci formation increased in a concentration-dependent manner after acute and prolonged Cr(VI) exposure. This suggests BRCA2, the protein responsible for loading Rad51 onto the ssDNA (Jensen et al., 2010), is functional after Cr(VI) exposure. In contrast, Rad51C foci formation increased after acute Cr(VI) exposure, but was inhibited following prolonged exposure. Cr(VI) exposure had no effect on Rad51C whole cell protein levels, suggesting the lack of Rad51C foci was not due to a decrease in protein but rather a lack of response to the Cr(VI)-induced DNA damage. Rad51C plays a role in the stabilization of the Rad51 nucleofilament, downstream of its formation (Amunugama et al., 2013). Therefore, the observed inhibition of a Rad51C response suggests a defect in Rad51 nucleofilament stabilization.
Since Rad51 nuclear transport is crucial to its response to DNA damage, the mislocalization of Rad51 protein is a third potential explanation for the Cr(VI)-induced inhibition of Rad51 nucleofilament formation. Both BRCA2 and Rad51C have previously been shown to import Rad51 into the nucleus (Gildemeister et al., 2009; Jeyasekharan et al., 2013). Interestingly, we observed prolonged Cr(VI) exposure induced the subcellular mislocalization of Rad51. Rad51 localization to the cytoplasm, represented as the cytoplasmic/nuclear ratio, increased in a dose-dependent manner after prolonged particulate Cr(VI) exposure. This outcome correlated with an increase in the percent of cells with Rad51 cytoplasmic accumulation. These observations are consistent with our previous report of a time- and dose-dependent increase in cytoplasmic Rad51 aggregates (Qin et al., 2014). Our observations of Rad51 mislocalization to the cytoplasm and Rad51C inhibition corresponded temporally, occurring after 72 h of particulate Cr(VI) exposure. Therefore, we hypothesize that the inhibition of Rad51C induces the cytoplasmic accumulation of Rad51. Future work is needed to determine the mechanisms of particulate Cr(VI)-induced Rad51 nucleofilament inhibition and subcellular mislocalization.
In summary, our data show prolonged exposure to particulate Cr(VI) inhibits HR repair through the inhibition of Rad51 nucleofilament formation. As HR repair protects against Cr(VI)-induced chromosome instability, this finding further enhances our understanding of the underlying mechanism of Cr(VI)-induced carcinogenesis. Our results suggest that DNA double strand breaks need to be resolved by a Rad51-independent pathway following prolonged particulate Cr(VI) exposure. The resulting switch to lower fidelity DNA repair pathways following prolonged exposure may be a driving force of Cr(VI)-induced chromosome instability. Future work is aimed at determining the activity of low fidelity repair pathways following prolonged particulate Cr(VI) exposure.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Carly Winton, Vasilea Karageorge, William Dearnaley, and Brian Gilmore of Virginia Tech Carilion Research Institute for their assistance with the affinity capture of the Rad51 filaments and acquiring the TEM images. We thank Geron Corporation for the use of the hTERT materials as well as DrHong Xie, Christy Gianios and Shouping Huang for administrative and technical assistance.
FUNDING
This work was supported by the National Institute of Environmental Health Sciences (ES016893 to J.P.W.); National Aeronautics and Space Administration (NASA) (ACD FSB-2009 to J.P.W.) and The Maine Center for Toxicology and Environmental Health.
SUPPLEMENTARY DATA
Supplementary data are available online at Supplementary Data
REFERENCES
- Amunugama R., Groden J., Fishel R. (2013). The HsRAD51B-HsRAD51C stabilizes the HsRAD51 nucleoprotein filament. DNA Repair 12, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P., Benson F. E., West S. C. (1996). Human Rad51 protein promotes ATP-dependent homologous pairing and strand transfer reactions in vitro. Cell 87, 757–766. [DOI] [PubMed] [Google Scholar]
- Bennardo N., Cheng A., Huang N., Stark J. M. (2008). Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 4, e1000110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant H. E., Ying S., Helleday T. (2006). Homologous recombination is involved in repair of chromium-induced DNA damage in mammalian cells. Mutat. Res. 599, 116–123. [DOI] [PubMed] [Google Scholar]
- Candelli A., Holthausen J. T., Depken M., Brouwer I., Franker M.A.M., Marchetti M., Heller I., Bernard S., Garcin E. B., Modesti M., et al. (2014). Visualization and quantification of nascent Rad41 filament formation at single-moner resolution. Proc. Natl. Acad. Sci U S A 111, 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essers J., Houtsmuller A. B., vanVeelen L., Paulusma C., Nigg A. L., Pastink A., Vermeulin W., Hoeijmakers J. H. J., Kanaar R. (2002). Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. Embo J. 21, 2030–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gildemeister O. S., Sage J. M., Knight K. L. (2009). Cellular redistribution of Rad51 in response to DNA damge: novel role for Rad51C. J. Biol. Chem. 284, 31945–31952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T., Nilsson R., Jenssen D. (2000). Arsenic [III] and heavy metal ions induce intrachromosomal homologous recombination in the hprt gene of V79 Chinese hamster cells. Environ. Mol. Mutagen. 35, 114–122. [DOI] [PubMed] [Google Scholar]
- Holmes A. L., Wise S. S., Pelsue S. C., Aboueissa A. M., Lingle W., Salisbury J., Gallagher J., Wise J. P., Sr (2010). Chronic exposure to zinc chromate induces centrosome amplification and spindle assembly checkpoint bypass in human lung fibroblasts. Chem. Res. Toxicol. 23, 386–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen R. B., Carreira A., Kowalczykowski S. C. (2010). Purified human BRCA2 stimulates RAD51-mediated recombination. Nature 467, 678–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyasekharan A. D., Liu Y., Hattori H., Pisupati V., Jonsdottir A. B., Rajendra E., Lee M., Sundaramoorthy E., Schlachter S., Kaminski C. F., et al. (2013). A cancer-associated BRCA2 mutation reveals masked nuclear export signals controlling localization. Nat. Struct. Mol. Biol. 20, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce A. J., Johnson R. D., Thompson L. H., Jasin M. (1999). XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13, 2633–2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikor L., Thu K., Vucic E., Lam W. (2013). The detection and implication of genome instability in cancer. Cancer Metastasis Rev. 32, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plo I., Laulier C., Gauthier L., Lebrun F., Calvo F., Lopez B. S. (2008). AKT1 inhibits homologous recombination by inducing cytoplasmic retention of BRCA1 and RAD51. Cancer Res. 68, 9404–9412. [DOI] [PubMed] [Google Scholar]
- Qin Q., Wise S. S., Browning C. L., Thompson K. N., Holmes A. L., Wise Sr J. P. (2014). Homologous recombination repair signaling in chemical carcinogenesis: prolonged particulate hexavalent chromium exposure suppresses the Rad51 response in human lung cells. Toxicol. Sci. 142, 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackpole M. M., Wise S. S., Grlickova Duzevik E., Munroe R. C., Thompson W. D., Thacker J., Thompson L. H., Hinz J. M., Wise J. P., Sr (2007). Homologous recombination repair protects against particulate chromate-induced chromosome instability in Chinese hamster cells. Mutat. Res. 625, 145–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark J. M., Pierce A. J., Oh J., Pastink A., Jasin M. (2004). Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 24, 9305–9316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung P., Robberson D. L. (1995). DNA strand exchange mediated by a RAD51-ssDNA nucleoprotein filament with polarity opposite to that of RecA. Cell 82, 453–461. [DOI] [PubMed] [Google Scholar]
- Tanner J. R., Degen K., Gilmore B. L., Kelly D. F. (2012). Capturing RNA-dependent pathways for cryo-EM analysis. Comput. Struct. Biotechnol. J. 1, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise S. S., Elmore L. W., Holt S. E., Little J. E., Antonucci P. G., Bryant B. H., Wise J. P., Sr (2004). Telomerase-mediated lifespan extension of human bronchial cells does not affect hexavalent chromium-induced cytotoxicity or genotoxicity. Mol. Cell. Biochem. 255, 103–111. [DOI] [PubMed] [Google Scholar]
- Xie H., Wise S. S., Holmes A. L., Xu B., Wakeman T. P., Pelsue S. C., Singh N. P., Wise J. P., Sr (2005). Carcinogenic lead chromate induces DNA double strand breaks in human lung cells. Mutat. Res. 586, 160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie H., Holmes A. L., Young J. L., Qin Q., Joyce K., Pelsue S. C., Peng C., Wise S. S., Jeevarajan A. S., Wallace W. T., et al. (2009). Zinc chromate induces chromosome instability and DNA double strand breaks in human lung cells. Toxicol. Appl. Pharmacol. 234, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F., Paramasivam M., Cai Q., Dai X., Wang P., Lin K., Song J., Seidman M. M., Wang Y. (2014). Arsenite binds to the RING finger domains of RNF20-RNF40 histone E3 ubiquitin ligase and inhibits DNA double-strand break repair. J. Am. Chem. Soc. 136, 12884–12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.