

Abstract
Chronic liver diseases are one of the major public health issues in United States, and there are substantial racial disparities in liver cancer-related mortality. We previously identified racially distinct alterations in the expression of transcripts and proteins of hepatitis C (HCV)-induced hepatocellular carcinoma (HCC) between Caucasian (CA) and African American (AA) subgroups. Here, we performed a comparative genome-wide analysis of normal vs. HCV+ (cirrhotic state), and normal adjacent tissues (HCCN) vs. HCV+HCC (tumor state) of CA at the gene and alternative splicing levels using Affymetrix Human Transcriptome Array (HTA2.0). Many genes and splice variants were abnormally expressed in HCV+ more than in HCV+HCC state compared with normal tissues. Known biological pathways related to cell cycle regulations were altered in HCV+HCC, whereas acute phase reactants were deregulated in HCV+ state. We confirmed by quantitative RT-PCR that SAA1, PCNA-AS1, DAB2, and IFI30 are differentially deregulated, especially in AA compared with CA samples. Likewise, IHC staining analysis revealed altered expression patterns of SAA1 and HNF4α isoforms in HCV+ liver samples of AA compared with CA. These results demonstrate that several splice variants are primarily deregulated in normal vs. HCV+ stage, which is certainly in line with the recent observations showing that the pre-mRNA splicing machinery may be profoundly remodeled during disease progression, and may, therefore, play a major role in HCV racial disparity. The confirmation that certain genes are deregulated in AA compared to CA tissues also suggests that there is a biological basis for the observed racial disparities.
Keywords: hepatitis C, racial disparity, genomic variants, hepatocellular carcinoma, alternative splicing
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the few malignancies in which the incidence is on the rise worldwide, especially in the US [1]. The increasing incidence of HCC in the US is associated with the rise in Hepatitis C virus (HCV) infection [2]. It is estimated that 3.2 million people in the US are infected with HCV, a blood-borne disease linked to 12,000 US deaths a year [3]. Even with the availability of new oral direct acting antiviral drugs [4], it is anticipated that 320,000 patients will die from HCV, 157,000 will develop HCC, and 203,000 will develop cirrhosis in the next 35 years [5]. Inequalities in disease prevalence, treatment, and outcome make HCC an important health problem among minority groups [6]. First, there are disparities in the prevalence of HCV infection with African Americans (AA) being twice as likely to have been infected compared with Caucasian Americans (CA) [7]. Second, there are significant racial/ethnic disparities in access to HCV care [8]. Third, African Americans are also less likely to respond to the new anti-HCV therapy than Caucasian Americans, possibly due to a lower rate of sustained virologic response (SVR) [9], and have considerably lower likelihood of receiving liver transplantation [10]. While much of the existing literature so far has focused on noting the presence of these disparities, little is known about specific biological or genetic factors that are involved. Therefore, there is clear need for molecular/biological approaches to understand the molecular basis for HCV health and racial disparities. Ultimately positive outcomes would allow for the development of novel, affordable and much needed next generation therapeutic care management based on HCV disease state and the racial/ethnic background of patients [11]. We recently reported that racially distinct alterations in the expression of transcripts and proteins exist between CA and AA individuals infected with HCV, as measured by proteomics-based analysis [12]. For example, we showed that the mRNA levels of transferrin (TF), Apolipoprotein A1 (APOA1) and hepatocyte nuclear factor 4-alpha (HNF4α) were significantly altered in AA liver (cirrhotic) and tumor samples compared to CA. It is known that AA with chronic HCV commonly have elevated levels of serum markers of iron stores and altered cholesterol & triglyceride levels [13, 14]. The expression of TF & APOA1 (both involved in iron homeostasis and lipid metabolic processes, respectively) is transcriptionally regulated by HNF4α [15, 16]. Furthermore, HNF4α is also known to be involved in the pathogenesis of HCC [17, 18]. To the best of our knowledge, that was the first study to demonstrate possible link between deregulation of the expression of specific transcripts & proteins and HCV racial disparity between AA and CA subgroups. This finding prompted us to further investigate whether alternative splicing (AS) of genes could be involved in the transcriptome diversity seen between these two ethnic populations. Alternative splicing (AS) is a post-transcriptional event whereby exons are joined by different combinations generating various isoforms from a single gene [19–21]. It has been shown that most genes have at least 2 alternative isoforms [22, 23] contributing to both transcriptome and proteome diversities in various pathophysiological situations including HCV infection and HCC [24, 25].
In this study, we have performed a genome-wide transcriptomic analysis at the gene and splice variants levels in liver and tumor tissue samples of HCV infected individuals using the Affymetrix GeneChip Human Transcriptome array (HTA2.0). The array is especially designed to allow for expression profiling of transcript splice variants. It contains >6.0 million probes covering coding transcripts (70%) and exon-exon splice junctions and non-coding transcripts (30%). Herein, we describe our methods for expression microarray analysis at the genes and splice variants levels using Transcriptome Analysis Console (TAC2.0) software coupled by validation studies to confirm disease-specific splice variants of genes that could be involved in the racial disparity of HCV-induced HCC by real-time qRT-PCR and immunohistochemistry using sixty liver and tumor tissue samples.
RESULTS
Clinical characteristics of tissue samples
A total of 36 snapped frozen liver and tumor samples from CA and AA populations were used in this study. The clinicopathologic characteristics of samples are presented in Supplementary Table 2. As reported in our previous study [12], there were no significant differences of age and sex between samples in the two groups. However, the cirrhotic HCV+ liver samples of AA group had statistically significant laboratory results for aspartate aminotransferase (AST), and alanine aminotransferase (ALT) (p<0.05) compared to CA group. There were no significance differences in the laboratory values for albumin, total albumin and hemoglobin between samples in the two groups.
Identification of differentially expressed genes and splice variants based on diseased states of Caucasian American (CA) population
Gene level differential expression profiles of 12 CA tissues samples (3 normal liver, 3 HCV+ livers, 3 HCV+/HCC+ tumors and 3 HCCN) were determined using HTA2.0 GeneChip Arrays (Affymetrix®) that contain 70,523 detectable transcripts using TAC2.0 software (for filtering criteria see Materials and methods). For normal vs. HCV+, 636 genes were differentially expressed: 350 genes were up-regulated in HCV+ compared to normal (coding 235; non-coding 103; other 12) as shown in Table 1A, whereas 286 genes were down-regulated in HCV+ compared to normal (coding 209; non-coding 73; other 4), Table 1B. For HCCN vs. HCV+HCC, only 61 genes were differentially expressed, as shown in Table 2, using the same algorithm options and filter criteria (see Materials and methods): 47 genes were up-regulated in HCV+HCC compared to HCCN (coding 23; non-coding 6; other 18) and 14 genes were down-regulated in HCV+HCC compared to HCCN (coding 5; non-coding 1; other 8). These results suggest that tumor-adjacent tissue (HCCN) shares biology of the tumors themselves, and only 61 genes are differentially expressed in this case. Figure 1 shows the scatter plot (log 2 scale of expression values) for differentially expressed genes (DEGs) in normal vs. HCV+ state (Figure 1A) and HCCN vs. HCV+HCC state (Figure 1B), respectively. In both cases, most of the genes run along the diagonal axis and can be considered as common genes, expressed similarly in either diseased state, whereas differentially expressed genes with values <-2.0 or <+2.0 are scattered outside the diagonal axis. Examples of these scattered genes (arrows) are shown in Figure 1A (insert 1 C) and Figure 1B (insert 1 D). No overlap of genes (marked) was detected between the two disease stages, which suggest that these genes are differentially expressed based on disease state (normal vs. HCV+ cirrhotic livers; HCCN vs. HCV+/HCC cirrhotic tumors).
Table 1A. The results of differentially expressed genes (DEGs) in normal vs. HCV+ tissue samples.
| Accession Number | Fold Change | Fold Direction | p value | Gene Symbol | Group |
|---|---|---|---|---|---|
| NM_000706 | 13.8 | N UP vs. HCV | 0.01640 | AVPR1A | Coding |
| NM_030754 | 12.05 | N UP vs. HCV | 0.00282 | SAA2 | Coding |
| NM_005949 | 9.48 | N UP vs. HCV | 0.04235 | MT1F | Coding |
| NM_030787 | 6.01 | N UP vs. HCV | 0.00645 | CFHR5 | Coding |
| NM_014926 | 5.96 | N UP vs. HCV | 0.01872 | SLITRK3 | Coding |
| NM_001144904 | 5.79 | N UP vs. HCV | 0.03399 | CLEC4M | Coding |
| NM_000331 | 5.13 | N UP vs. HCV | 0.01927 | SAA1 | Coding |
| NM_001166624 | 5.08 | N UP vs. HCV | 0.01142 | CFHR3 | Coding |
| NM_001201550 | 4.99 | N UP vs. HCV | 0.02009 | CFHR4 | Coding |
| NM_176870 | 4.45 | N UP vs. HCV | 0.02094 | MT1M | Coding |
| NM_001308 | 3.97 | N UP vs. HCV | 0.03343 | CPN1 | Coding |
| NM_001146726 | 3.93 | N UP vs. HCV | 0.00794 | TIMD4 | Coding |
| NM_145290 | 3.68 | N UP vs. HCV | 0.00611 | GPR125 | Coding |
| NM_031900 | 3.62 | N UP vs. HCV | 0.01828 | AGXT2 | Coding |
| NM_020459 | 3.54 | N UP vs. HCV | 0.02778 | PAIP2B | Coding |
| NM_032649 | 3.52 | N UP vs. HCV | 0.00289 | CNDP1 | Coding |
| NM_001159 | 3.45 | N UP vs. HCV | 0.02937 | AOX1 | Coding |
| NM_001361 | 3.31 | N UP vs. HCV | 0.01586 | DHODH | Coding |
| NM_006419 | 3.3 | N UP vs. HCV | 0.00101 | CXCL13 | Coding |
| NM_001039199 | 3.29 | N UP vs. HCV | 0.00756 | TTPAL | Coding |
| NM_001127708 | 3.29 | N UP vs. HCV | 0.03135 | PRG4 | Coding |
| NM_001193646 | 3.28 | N UP vs. HCV | 0.04037 | ATF5 | Coding |
| NM_001143838 | 3.27 | N UP vs. HCV | 0.04855 | SLC13A5 | Coding |
| NM_052972 | 3.25 | N UP vs. HCV | 0.00249 | LRG1 | Coding |
| NM_000028 | 3.2 | N UP vs. HCV | 0.00334 | AGL | Coding |
| NM_000055 | 3.11 | N UP vs. HCV | 0.01262 | BCHE | Coding |
| NM_175737 | 3.09 | N UP vs. HCV | 0.02281 | KLB | Coding |
| NM_000902 | 2.99 | N UP vs. HCV | 0.00453 | MME | Coding |
| NM_016371 | 2.97 | N UP vs. HCV | 0.04476 | HSD17B7 | Coding |
| NM_018078 | 2.95 | N UP vs. HCV | 0.04017 | LARP1B | Coding |
| NM_000133 | 2.93 | N UP vs. HCV | 0.04671 | F9 | Coding |
| NM_001170701 | 2.9 | N UP vs. HCV | 0.00523 | MBLN3 | Coding |
| NM_004944 | 2.89 | N UP vs. HCV | 0.03243 | DNASE1L3 | Coding |
| NM_006691 | 2.81 | N UP vs. HCV | 0.00779 | LYVE1 | Coding |
| NM_014465 | 2.79 | N UP vs. HCV | 0.00251 | SULT1B1 | Coding |
| NM_001161429 | 2.7 | N UP vs. HCV | 0.00854 | RANBP3L | Coding |
| NM_006770 | 2.69 | N UP vs. HCV | 0.01995 | MARCO | Coding |
| NM_001174152 | 2.68 | N UP vs. HCV | 0.00824 | RABEPK | Coding |
| NM_001130991 | 2.62 | N UP vs. HCV | 0.00355 | HYOU1 | Coding |
| NM_033058 | 2.59 | N UP vs. HCV | 0.04228 | TRIM55 | Coding |
| NM_001123 | 2.54 | N UP vs. HCV | 0.02600 | ADK | Coding |
| NM_004169 | 2.52 | N UP vs. HCV | 0.00361 | SHMT1 | Coding |
| NM_005907 | 2.5 | N UP vs. HCV | 0.00967 | MAN1A1 | Coding |
| NM_001128431 | 2.5 | N UP vs. HCV | 0.01099 | SLC39A14 | Coding |
| NM_001128227 | 2.5 | N UP vs. HCV | 0.01359 | GNE | Coding |
| NM_001737 | 2.49 | N UP vs. HCV | 0.01724 | C9 | Coding |
| NM_004911 | 2.47 | N UP vs. HCV | 0.00481 | PDIA4 | Coding |
| NM_000019 | 2.47 | N UP vs. HCV | 0.00874 | ACAT1 | Coding |
| NM_005768 | 2.47 | N UP vs. HCV | 0.03440 | LPCAT3 | Coding |
| NM_000066 | 2.47 | N UP vs. HCV | 0.04159 | C8B | Coding |
| NM_000478 | 2.46 | N UP vs. HCV | 0.00447 | ALPL | Coding |
| NM_145715 | 2.44 | N UP vs. HCV | 0.01064 | TIGD2 | Coding |
| NM_004481 | 2.43 | N UP vs. HCV | 0.03059 | GALNT2 | Coding |
| NM_000236 | 2.43 | N UP vs. HCV | 0.03763 | LIPC | Coding |
| NM_004475 | 2.39 | N UP vs. HCV | 0.00135 | FLOT2 | Coding |
| NM_014730 | 2.38 | N UP vs. HCV | 0.00073 | MLEC | Coding |
| NM_138326 | 2.38 | N UP vs. HCV | 0.03850 | ACMSD | Coding |
| NM_015541 | 2.37 | N UP vs. HCV | 0.04555 | LRIG1 | Coding |
| NM_003658 | 2.36 | N UP vs. HCV | 0.02789 | MT1DP | Coding |
| NM_004108 | 2.34 | N UP vs. HCV | 0.01438 | FCN2 | Coding |
| NM_001242332 | 2.32 | N UP vs. HCV | 0.00197 | USP17L6P | Coding |
| NM_000715 | 2.32 | N UP vs. HCV | 0.02707 | C4BPA | Coding |
| NM_001199758 | 2.31 | N UP vs. HCV | 0.00640 | MTHF5 | Coding |
| NM_001144978 | 2.31 | N UP vs. HCV | 0.00910 | MTHFD2L | Coding |
| NM_181536 | 2.31 | N UP vs. HCV | 0.02866 | PKD1L3 | Coding |
| NM_004388 | 2.3 | N UP vs. HCV | 0.00628 | CTBS | Coding |
| NM_005570 | 2.3 | N UP vs. HCV | 0.01109 | LMAN1 | Coding |
| NM_002168 | 2.29 | N UP vs. HCV | 0.00779 | IDH2 | Coding |
| NM_000348 | 2.27 | N UP vs. HCV | 0.01335 | SRD5A2 | Coding |
| NM_000240 | 2.27 | N UP vs. HCV | 0.02094 | MAO2 | Coding |
| NM_001859 | 2.27 | N UP vs. HCV | 0.03664 | SLC31A1 | Coding |
| NM_005691 | 2.26 | N UP vs. HCV | 0.00742 | ABCC9 | Coding |
| NM_001005375 | 2.26 | N UP vs. HCV | 0.03061 | DAZ4 | Coding |
| NM_000562 | 2.25 | N UP vs. HCV | 0.04361 | C8A | Coding |
| NM_000065 | 2.23 | N UP vs. HCV | 0.04204 | C6 | Coding |
| NM_000608 | 2.22 | N UP vs. HCV | 0.01256 | ORM2 | Coding |
| NM_039654 | 2.22 | N UP vs. HCV | 0.02000 | MIR4450 | Coding |
| NM_005794 | 2.21 | N UP vs. HCV | 0.00033 | DHRS2 | Coding |
| NM_022132 | 2.19 | N UP vs. HCV | 0.01297 | MCCC2 | Coding |
| NM_030782 | 2.18 | N UP vs. HCV | 0.00912 | CLPTM1L | Coding |
| NM_182758 | 2.18 | N UP vs. HCV | 0.01132 | WDR72 | Coding |
| NM_001014797 | 2.16 | N UP vs. HCV | 0.00922 | KCNMA1 | Coding |
| NM_006741 | 2.16 | N UP vs. HCV | 0.01382 | PPP1R1A | Coding |
| NM_181900 | 2.16 | N UP vs. HCV | 0.03056 | STARD5 | Coding |
| NM_005013 | 2.14 | N UP vs. HCV | 0.02120 | NUCB2 | Coding |
| NM_001918 | 2.13 | N UP vs. HCV | 0.03126 | DBT | Coding |
| NM_001161504 | 2.11 | N UP vs. HCV | 0.02578 | ALDH4A1 | Coding |
| NM_001015880 | 2.1 | N UP vs. HCV | 0.00207 | PAPSS2 | Coding |
| NM_001100607 | 2.1 | N UP vs. HCV | 0.01792 | SERPINA10 | Coding |
| NM_001145368 | 2.08 | N UP vs. HCV | 0.00871 | PTPN3 | Coding |
| NM_005045 | 2.07 | N UP vs. HCV | 0.00942 | RELN | Coding |
| NM_138493 | 2.06 | N UP vs. HCV | 0.00822 | CCDC167 | Coding |
| NR_029524 | 2.06 | N UP vs. HCV | 0.01216 | MIR107 | Coding |
| NM_001113239 | 2.02 | N UP vs. HCV | 0.00036 | HIPK2 | Coding |
| NM_003878 | 2.02 | N UP vs. HCV | 0.00058 | GGH | Coding |
| NM_001872 | 2.01 | N UP vs. HCV | 0.04171 | CPB2 | Coding |
| NM_021800 | 2.01 | N UP vs. HCV | 0.04931 | DNAJC12 | Coding |
Table 1B. The results of differentially expressed genes (DEGs) in HCV+ vs. Normal tissue samples.
| Accession Number | Fold Change | Fold Direction | p value | Gene Symbol | Group |
|---|---|---|---|---|---|
| NM_020299 | -30.81 | HCV UP vs. N | 0.00242 | AKR1B10 | Coding |
| NM_001130080 | -14.86 | HCV UP vs. N | 0.02019 | IFI27 | Coding |
| NM_000584 | -8.33 | HCV UP vs. N | 0.03313 | IL8 | Coding |
| NR_026703 | -7.05 | HCV UP vs. N | 0.02314 | VTRNA1-1 | Coding |
| NM_000582 | -6.02 | HCV UP vs. N | 0.03381 | SPP1 | Coding |
| NM_004864 | -5.65 | HCV UP vs. N | 0.00097 | GDF15 | Coding |
| NM_033049 | -5.46 | HCV UP vs. N | 0.03079 | MUC13 | Coding |
| NM_001040092 | -4.93 | HCV UP vs. N | 0.00379 | ENPP2 | Coding |
| NM_001565 | -4.79 | HCV UP vs. N | 0.00803 | CXCL10 | Coding |
| NM_006149 | -3.89 | HCV UP vs. N | 0.00061 | LGALS4 | Coding |
| NM_001046 | -3.84 | HCV UP vs. N | 0.02276 | SLC12A2 | Coding |
| NR_002921 | -3.83 | HCV UP vs. N | 0.00306 | SNORA75 | Coding |
| NM_006398 | -3.77 | HCV UP vs. N | 0.04837 | UBD | Coding |
| NM_025130 | -3.66 | HCV UP vs. N | 0.02106 | HKDC1 | Coding |
| NM_000492 | -3.61 | HCV UP vs. N | 0.00914 | CFTR | Coding |
| NM_000552 | -3.59 | HCV UP vs. N | 0.00285 | VWF | Coding |
| NR_002953 | -3.45 | HCV UP vs. N | 0.00506 | SNORA11 | Coding |
| NM_001128175 | -3.39 | HCV UP vs. N | 0.00364 | DTNA | Coding |
| NM_031310 | -3.38 | HCV UP vs. N | 0.00235 | PLVAP | Coding |
| AF533910 | -3.33 | HCV UP vs. N | 0.04893 | HLA-DQA1 | Coding |
| NR_002915 | -3.3 | HCV UP vs. N | 0.00041 | SNORA74A | Coding |
| NM_001166395 | -3.29 | HCV UP vs. N | 0.00387 | CHST4 | Coding |
| AF287958 | -3.29 | HCV UP vs. N | 0.01057 | HLA-A | Coding |
| NM_016591 | -3.26 | HCV UP vs. N | 0.03060 | BICC1 | Coding |
| NM_005245 | -3.21 | HCV UP vs. N | 0.01618 | FAT1 | Coding |
| NM_144975 | -3.2 | HCV UP vs. N | 0.01512 | SLFN5 | Coding |
| NM_021983 | -3.11 | HCV UP vs. N | 0.01176 | HLA-DRB4 | Coding |
| NR_003016 | -3.09 | HCV UP vs. N | 0.02789 | SNORA26 | Coding |
| NM_005567 | -3.05 | HCV UP vs. N | 0.00582 | LGALS3BP | Coding |
| NM_020638 | -3.03 | HCV UP vs. N | 0.02594 | FGF23 | Coding |
| NM_006274 | -2.95 | HCV UP vs. N | 0.00198 | CCL19 | Coding |
| NM_001901 | -2.87 | HCV UP vs. N | 0.04083 | CTGF | Coding |
| NM_001144964 | -2.84 | HCV UP vs. N | 0.00177 | NEDD4L | Coding |
| NM_001003954 | -2.81 | HCV UP vs. N | 0.00160 | ANXA13 | Coding |
| NM_017533 | -2.81 | HCV UP vs. N | 0.02032 | MYH4 | Coding |
| NM_005961 | -2.73 | HCV UP vs. N | 0.00874 | MUC6 | Coding |
| NM_002345 | -2.72 | HCV UP vs. N | 0.02683 | LUM | Coding |
| NM_001164617 | -2.71 | HCV UP vs. N | 0.03061 | GPC3 | Coding |
| NM_138694 | -2.68 | HCV UP vs. N | 0.00081 | PKHD1 | Coding |
| NM_001206567 | -2.68 | HCV UP vs. N | 0.00272 | IFI16 | Coding |
| NM_001242758 | -2.68 | HCV UP vs. N | 0.00823 | HLA-A | Coding |
| NM_002354 | -2.68 | HCV UP vs. N | 0.02366 | EPCAM | Coding |
| NM_005218 | -2.59 | HCV UP vs. N | 0.03577 | DEFB1 | Coding |
| NM_001781 | -2.58 | HCV UP vs. N | 0.03613 | CD69 | Coding |
| NM_016548 | -2.57 | HCV UP vs. N | 0.00153 | GOLM1 | Coding |
| NM_000587 | -2.52 | HCV UP vs. N | 0.01468 | C7 | Coding |
| NM_002867 | -2.47 | HCV UP vs. N | 0.03684 | RAB3B | Coding |
| NM_001546 | -2.46 | HCV UP vs. N | 0.00355 | ID4 | Coding |
| NM_005233 | -2.45 | HCV UP vs. N | 0.01517 | EPHA3 | Coding |
| NM_005261 | -2.43 | HCV UP vs. N | 0.01036 | GEM | Coding |
| NM_002989 | -2.42 | HCV UP vs. N | 0.00164 | CCL21 | Coding |
| NM_002416 | -2.37 | HCV UP vs. N | 0.02732 | CXCL9 | Coding |
| NM_005556 | -2.37 | HCV UP vs. N | 0.02828 | KRT7 | Coding |
| NM_138788 | -2.34 | HCV UP vs. N | 0.00009 | TMEM45B | Coding |
| NM_015529 | -2.34 | HCV UP vs. N | 0.03311 | MOXD1 | Coding |
| NM_032211 | -2.28 | HCV UP vs. N | 0.00438 | LOXL4 | Coding |
| NM_000346 | -2.28 | HCV UP vs. N | 0.00737 | SOX9 | Coding |
| NM_173648 | -2.25 | HCV UP vs. N | 0.00153 | CCDC141 | Coding |
| NM_003319 | -2.25 | HCV UP vs. N | 0.00285 | TTN | Coding |
| NM_003246 | -2.23 | HCV UP vs. N | 0.03008 | THBS1 | Coding |
| NM_000366 | -2.23 | HCV UP vs. N | 0.04147 | TPM1 | Coding |
| NM_001198695 | -2.17 | HCV UP vs. N | 0.00717 | MFAP4 | Coding |
| NM_001128310 | -2.17 | HCV UP vs. N | 0.01904 | SPARCL1 | Coding |
| NM_001105549 | -2.16 | HCV UP vs. N | 0.00629 | ZNF83 | Coding |
| NM_003897 | -2.15 | HCV UP vs. N | 0.01088 | IER3 | Coding |
| NM_004791 | -2.15 | HCV UP vs. N | 0.04359 | ITGBL1 | Coding |
| NM_001005180 | -2.14 | HCV UP vs. N | 0.00085 | TRIM22 | Coding |
| NM_018420 | -2.14 | HCV UP vs. N | 0.01240 | SLC22A15 | Coding |
| NM_005841 | -2.14 | HCV UP vs. N | 0.01787 | SPRY1 | Coding |
| NM_182832 | -2.14 | HCV UP vs. N | 0.04488 | PLAC4 | Coding |
| NM_002392 | -2.13 | HCV UP vs. N | 0.00520 | MDM2 | Coding |
| NM_001080538 | -2.13 | HCV UP vs. N | 0.01548 | AKR1B15 | Coding |
| NM_014314 | -2.13 | HCV UP vs. N | 0.02827 | DDX58 | Coding |
| NM_000141 | -2.09 | HCV UP vs. N | 0.00133 | FGFR2 | Coding |
| NM_006291 | -2.09 | HCV UP vs. N | 0.03200 | TNFAIP2 | Coding |
| NM_001129 | -2.07 | HCV UP vs. N | 0.04471 | AEBP1 | Coding |
| NM_001005473 | -2.06 | HCV UP vs. N | 0.02827 | PLCXD3 | Coding |
| NM_014256 | -2.06 | HCV UP vs. N | 0.04406 | B3GNT3 | Coding |
| NM_144682 | -2.05 | HCV UP vs. N | 0.00055 | SLFN13 | Coding |
| NM_198281 | -2.05 | HCV UP vs. N | 0.01338 | GPRIN3 | Coding |
| NM_001098484 | -2.02 | HCV UP vs. N | 0.01968 | SLC4A4 | Coding |
| NM_001253835 | -2.01 | HCV UP vs. N | 0.03487 | IGFBP7 | Coding |
Table 2. The results of differentially expressed genes (DEGs) in HCC vs. HCCN samples.
| Accession Number | Fold Change | Fold Direction | p value | Gene Symbol | Group |
|---|---|---|---|---|---|
| NR_028370 | 3.53 | HCC UP vs. HCCN | 0.04806 | PCNA-AS1 | Coding |
| NM_080593 | 2.35 | HCC UP vs. HCCN | 0.04926 | HIST1H2BK | Coding |
| NM_006332 | 2.21 | HCC UP vs. HCCN | 0.04400 | IFI30 | Coding |
| NM_001145845 | 2.2 | HCC UP vs. HCCN | 0.03077 | ROBO1 | Coding |
| NM_001244871 | 2.11 | HCC UP vs. HCCN | 0.04974 | DAB2 | Coding |
| NR_039890 | 2.01 | HCC UP vs. HCCN | 0.03285 | MIR4737 | Coding |
| NR_004398 | -2.20 | HCC UP vs. HCCN | 0.01972 | SNORD82 | Coding |
Figure 1. Global gene expression profiling data of hepatitis C tissue samples.
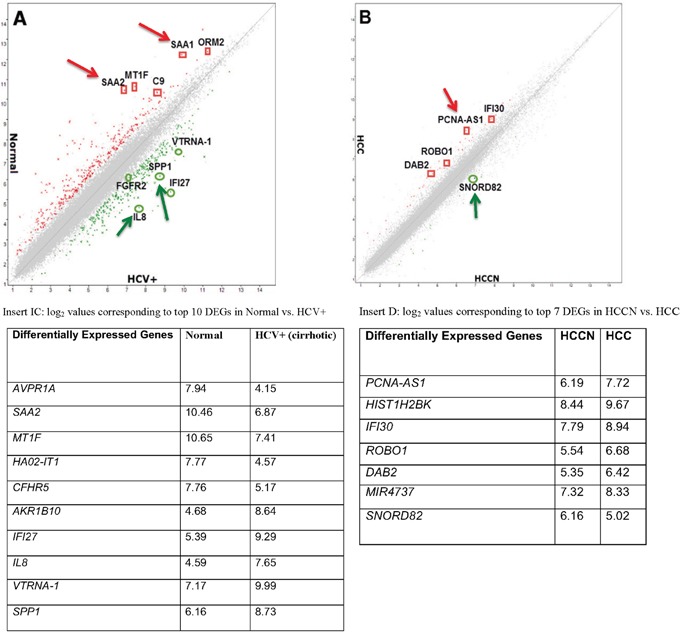
(A): Scatter plot presenting the values of log2 for each gene in the normal (Y-axis) vs. HCV+ cirrhotic samples (X-axis). (B): Scatter plot presenting the values of log2 for each gene in the HCCN (X-axis) vs. HCV+HCC tumor samples (Y-axis). Insert (C): Table indicating the log2 values corresponding to top 10 DEGs in normal vs. HCV+ samples. Insert (D): Table indicating the log2 values corresponding to top 7 DEGs in HCCN vs. HCV+ HCC samples.
For alternative splicing analysis, based on the algorithm options and filter criteria stated in the materials and methods, we were able to detect splice variant events only in normal vs. HCV+ stage (cirrhotic) and not in HCCN vs. HCV+HCC stage (tumor). This could be due to the low numbers of DEGs detected in the tumor state (61 genes) and/or the cut off and filter criteria. However, in normal vs. HCV+ stage about 12,650 genes were expressed in both conditions (coding). Only 15% of genes have at least one PSR or junction with SI (linear) <-2.0 or >+2.0 to indicate alternative splicing. For non-coding, about 2,943 of genes were expressed in both conditions. Only 2.7% of genes were found to have at least one PSR or junction with SI (linear) <-2.0 or >+2.0 to indicate alternative splicing. Table 3 shows various alternative splicing events (coding) for the top 30 genes identified in normal vs. HCV+ livers.
Table 3. The results of alternative splicing (AS) events in Normal vs. HCV+ tissue samples using Affymetrix Human Transcriptomic Array 2.0 (HTA 2.0).
| Accession Number | Fold Change (FC) | Gene Symbol | Group | Splicing Index (SI)* | Splicing Events |
|---|---|---|---|---|---|
| NM_005950 | 10.24 | MT1G | Coding | -2.14 | Cassette Exon |
| NM_176870 | 9.94 | MT1M | Coding | -2.37 | Cassette Exon |
| NM_005949 | 7.44 | MT1F | Coding | -2.84 | |
| NM_017460 | 6.68 | CYP3A4 | Coding | 3.18 | |
| NM_017460 | 6.68 | CYP3A4 | Coding | 2.19 | |
| NM_017460 | 6.68 | CYP3A4 | Coding | -2.03 | |
| NM_017460 | 6.68 | CYP3A4 | Coding | -2.22 | Cassette Exon |
| NM_017460 | 6.68 | CYP3A4 | Coding | -4.27 | Alternative 5' Donor Site |
| NM_017460 | 6.68 | CYP3A4 | Coding | -4.36 | |
| NM_030787 | 6.44 | CFHR5 | Coding | 2.03 | |
| NM_000669 | 5.58 | ADH1C | Coding | 2.08 | Alternative 5' Donor Site |
| NM_000669 | 5.58 | ADH1C | Coding | -4.86 | Cassette Exon |
| NM_001881 | 4.81 | CRHBP | Coding | 2.15 | Alternative 5' Donor Site |
| NM_001881 | 4.81 | CRHBP | Coding | -4.8 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -2.3 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -2.31 | |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -2.36 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -2.46 | |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -2.76 | Alternative 3' Acceptor Site |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -3.72 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -4.19 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -4.4 | Cassette Exon |
| NM_019844 | 4.74 | SLCO1B3 | Coding | -4.84 | |
| NM_003708 | 4.49 | RDH16 | Coding | -3.3 | Alternative 5' Donor Site |
| NM_177550 | 4.47 | SLC13A5 | Coding | 2.66 | |
| NM_177550 | 4.47 | SLC13A5 | Coding | -2.54 | |
| NM_177550 | 4.47 | SLC13A5 | Coding | -5.52 | Alternative 3' Acceptor Site |
| NM_003645 | 4.42 | SLC27A2 | Coding | -3.63 | |
| NM_001308 | 4.37 | CPN1 | Coding | -2.86 | Alternative 3' Acceptor Site |
| NM_006100 | 4.36 | ST3GAL6 | Coding | 2.41 | |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -2.1 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -2.19 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -2.21 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -2.66 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -2.8 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -3.23 | |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -3.57 | Alternative 5' Donor Site |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -3.81 | Cassette Exon |
| NM_006100 | 4.36 | ST3GAL6 | Coding | -6.23 | Alternative 3' Acceptor Site |
| NM_004944 | 4.33 | DNASE1L3 | Coding | -3.74 | Intron Retention |
| NM_004944 | 4.33 | DNASE1L3 | Coding | -5.49 | Alternative 5' Donor Site |
| NM_004944 | 4.33 | DNASE1L3 | Coding | -6.67 | |
| NM_018388 | 4.22 | MBNL3 | Coding | -2.13 | |
| NM_018388 | 4.22 | MBNL3 | Coding | -4.34 | Cassette Exon |
| NM_012068 | 3.8 | ATF5 | Coding | -2.2 | Alternative 5' Donor Site |
| NM_012068 | 3.8 | ATF5 | Coding | -3.13 | Cassette Exon |
| NM_012068 | 3.8 | ATF5 | Coding | -3.2 | |
| NM_030754 | 3.69 | SAA2 | Coding | 22.12 | |
| NM_030754 | 3.69 | SAA2 | Coding | 12.96 | |
| NM_030754 | 3.69 | SAA2 | Coding | 10.89 | |
| NM_030754 | 3.69 | SAA2 | Coding | 8.47 | |
| NM_030754 | 3.69 | SAA2 | Coding | 8.4 | Intron Retention |
| NM_030754 | 3.69 | SAA2 | Coding | 6.78 | Cassette Exon |
| NM_030754 | 3.69 | SAA2 | Coding | 5.96 | |
| NM_030754 | 3.69 | SAA2 | Coding | 5.25 | Cassette Exon |
| NM_030754 | 3.69 | SAA2 | Coding | 5.11 | Cassette Exon |
| NM_030754 | 3.69 | SAA2 | Coding | 5.01 | |
| NM_030754 | 3.69 | SAA2 | Coding | 3.97 | Cassette Exon |
| NM_030754 | 3.69 | SAA2 | Coding | 2.52 | |
| NM_030754 | 3.69 | SAA2 | Coding | -2.63 | Alternative 5' Donor Site |
| NM_024039 | 3.65 | MIS12 | Coding | -2.1 | Cassette Exon |
| NM_024039 | 3.65 | MIS12 | Coding | -2.79 | |
| NM_024039 | 3.65 | MIS12 | Coding | -3.67 | Alternative 5' Donor Site |
| NM_005952 | 3.65 | MT1X | Coding | -10.33 | Alternative 3' Acceptor Site |
| NM_005952 | 3.61 | MT1X | Coding | -3.98 | Cassette Exon |
| NM_005952 | 3.6 | MT1X | Coding | -4.2 | |
| NM_005952 | 3.59 | MT1X | Coding | -2.23 | Cassette Exon |
| NM_024331 | 3.59 | TTPAL | Coding | -2.96 | |
| NM_001361 | 3.54 | DHODH | Coding | -2.03 | |
| NM_000236 | 3.54 | LIPC | Coding | -4.04 | Alternative 5' Donor Site |
| NM_000236 | 3.48 | LIPC | Coding | -2.27 | Cassette Exon |
| NM_031900 | 3.48 | AGXT2 | Coding | -4.04 | |
| NM_052972 | 3.41 | LRG1 | Coding | -3.18 | Alternative 5' Donor Site |
| NM_032565 | 3.39 | EBPL | Coding | -2.06 | |
| NM_032565 | 3.39 | EBPL | Coding | -2.11 | Cassette Exon |
| NM_024641 | 3.39 | MANEA | Coding | -2.2 | Cassette Exon |
| NM_020988 | 3.39 | GNAO1 | Coding | -2.2 | Cassette Exon |
| NM_020988 | 3.39 | GNAO1 | Coding | -2.97 | Cassette Exon |
| NM_020988 | 3.37 | GNAO1 | Coding | -3.68 | |
| NM_020988 | 3.37 | GNAO1 | Coding | -2.04 | Cassette Exon |
| NM_020988 | 3.37 | GNAO1 | Coding | -2.19 | |
| NM_020988 | 3.37 | GNAO1 | Coding | -2.46 | |
| NM_000028 | 3.37 | AGL | Coding | -2.74 | Cassette Exon |
| NM_000028 | 3.36 | AGL | Coding | -2.96 | |
| NM_000028 | 3.36 | AGL | Coding | 26.12 | |
| NM_000028 | 3.36 | AGL | Coding | 12.96 | |
| NM_000028 | 3.36 | AGL | Coding | 8.08 | Intron Retention |
| NM_000331 | 3.36 | SAA1 | Coding | 4.49 | |
| NM_000331 | 3.27 | SAA1 | Coding | 2.21 | |
| NM_000331 | 3.27 | SAA1 | Coding | -2.05 | Cassette Exon |
| NM_000331 | 3.27 | SAA1 | Coding | -2.66 | Alternative 5' Donor Site |
| NM_000331 | 3.27 | SAA1 | Coding | -3.06 | Alternative 3' Acceptor Site |
| NM_001159 | 3.27 | AOX1 | Coding | -3.42 | |
| NM_015506 | 3.27 | MMACHC | Coding | -3.86 | Alternative 5' Donor Site |
Results were obtained following data normalization using Affymetrix Transcriptome Analysis Console 2.0 (TAC 2.0) software, which determines the Splicing Index (SI) of a gene and q-value <0.05 FC as criteria for selection.
*SI = The ratio of the exon intensities in Normal vs. HCV+ livers after normalization to their respective gene intensities in each sample. SI = (0) value indicates that the Probeset Selection Region (PSR) is present at equal levels in both Normal and HCV+ livers. SI = (+) value implies elevated inclusion, and (-) value suggests increased PSR skipping in Normal vs. HCV+ livers.
Differentially expressed genes are involved in a number of pathways and networks associated with disease state
To gain insights into the molecular pathways involving the identified differentially expressed genes, Ingenuity Pathway Analysis (IPA) of experimental data was performed by Ingenuity software as we previously reported [12]. Using the list of 636 genes involved in normal vs. HCV+ (cirrhotic) events and 61 genes involved in HCCN vs. HCV+HCC (tumor) events, IPA identified several pathways and function that might be relevant for each disease stage as shown in Tables 4A and 4B, respectively. Top associated network functions for differentially expressed genes in HCV+ cirrhotic state (Table 4A) were: 1) Hepatic fibrosis/hepatic stellate cell activation, 2) Antigen presentation pathway, 3) Graft-versus-host disease signaling, 4) Inhibition of matrix metalloproteases, and 5) T-helper cell differentiation. These data suggest that acute inflammatory phase is involved in HCV+ cirrhotic state as a result of HCV-induced oxidative stress. Genes such as SAA1, SAA2 and LGALS4 known to be involved in acute inflammatory phase were detected in this disease state (Tables 1A and 1B; Figure 1A). For HCCN vs. HCV+HCC (tumor stage), top associated network functions for differentially expressed genes (Table 4B) were: 1) GADD 45 signaling, 2) Cell cycle control of chromosomal replication, 3) Estrogen-mediated S-phase entry, 4) Cell cycle: G2/M DNA damage checkpoint regulation, 5) Cyclins and cell cycle regulation. These data suggest that cell cycle signaling pathways are certainly involved in HCV-induced HCC (tumor phase). Genes such as PCNA-AS1 and HIST1H2BK known to be involved in cell cycle regulation pathways were detected in this disease stage (Table 2; Figure 1B).
Table 4A. Functional analysis of 636 differentially expressed genes (DEGs) between Normal vs. HCV+ tissue samples.
| Top Canonical Pathways | ||
|---|---|---|
| Name | p-value | ratio |
| Hepatic Fibrosis/Hepatic Stellate Cell Activation | 4.25E-04 | 28/127 (0.22) |
| Antigen Presentation Pathway | 4.34E-04 | 8/18 (0.44) |
| Graft-versus-Host Disease Signaling | 1.48E-03 | 8/21 (0.381) |
| Inhibition of Matrix Metalloproteases | 2.89E-03 | 8/23 (0.348) |
| T Helper Cell Differentiation | 3.37E-03 | 11/39 (0.282) |
| Top Toxicity Functions | ||
| Name | p-value | # Molecules |
| Liver Cirrhosis | 4.96E-03 – 4.96E-03 | 5 |
| Liver Necrosis/Cell Death | 1.01E-01 – 1.01E-01 | 4 |
| Liver Adhesion | 1.14E-01 – 1.14E-01 | 1 |
| Liver Fibrosis | 2.16E-01 – 6.22E-01 | 3 |
| Liver Proliferation | 2.16E-01 – 6.22E-01 | 3 |
| Molecular and Cellular Functions | ||
| Name | p-value | # Molecules |
| DNA Replication, Recombination, and Repair | 2.29E-02 – 2.29E-02 | 3 |
Table 4B. Functional analysis of 61 differentially expressed genes (DEGs) between HCCN vs. HCC tissue samples.
| Top Canonical Pathways | ||
|---|---|---|
| Name | p-value | ratio |
| GADD45 Signaling | 2.93E-06 | 8/19 (0.421) |
| Cell Cycle Control of Chromosomal Replication | 1.07E-05 | 8/22 (0.364) |
| Estrogen-mediated S-Phase Entry | 2.24E-05 | 8/24 (0.333) |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 2.31E-05 | 11/46 (0.239) |
| Cyclins and Cell Cycle Regulation | 6.44E-05 | 13/69 (0.188) |
| Top Toxicity Functions | ||
| Name | p-value | # Molecules |
| Hepatocellular Carcinoma | 3.50E-03 – 5.87E-01 | 9 |
| Liver Hyperplasia/Hyperproliferation | 3.50E-03 – 5.87E-01 | 31 |
| Glutathione Depletion in Liver | 5.37E-02 – 5.38E-01 | 2 |
| Liver Damage | 5.37E-02 – 3.92E-01 | 7 |
| Liver Degradation | 5.37E-02 – 5.37E-02 | 1 |
| Molecular and Cellular Functions | ||
| Name | p-value | # Molecules |
| Carbohydrate Metabolism | 1.42E-03 – 1.42E-03 | 3 |
| Drug Metabolism | 1.42E-03 – 1.42E-03 | 3 |
| Molecular Transport | 1.42E-03 – 3.73E-02 | 7 |
| Small Molecule Biochemistry | 1.42E-03 – 3.73E-02 | 10 |
| Post-Translational Modification | 2.88E-03 – 2.88E-03 | 2 |
Target validation of gene expression and splice variants in Caucasian and African Americans tissue samples
In order to determine whether the racial disparity seen in HCV associated HCC is partly due to the diversity in gene expression and splice variants events between CA and AA, we selected a representative group of genes for qRT-PCR cross validation analysis. For normal vs. HCV+ (cirrhotic state), we selected the following genes: SAA1, AOX1 and SLC13A5. Representative examples of the amplicon binding sites for the PCR primer sequences are shown in Supplementary Figures 1 and 2. For HCCN vs. HCV+HCC (tumor stage), the following genes were selected: PCNA-AS1, IFI30, DBA2, ROBO1, and SNORD82. The expression of these eight genes was validated by qRT-PCR using an independent test set of 24 liver and tumor tissue samples (12 CA and 12 AA). The qRT-PCR results are shown in Tables 5A and 5B. The data suggest that good concordance of the results is seen using HTA2.0 arrays and qRT-PCR analysis. However, there is a distinct difference in SAA1 expression level between CA & AA samples (Table 5A). The overall fold change (FC) of SAA1 in CA samples has a positive value because the overall gene expression in HCV+ cirrhotic liver is down compared to normal (Table 1A) resulting in a positive fold-change (FC) value. Although the overall FC (qRT-PCR) in AA samples (Table 5A) has a positive value, it is actually lower than CA, because the overall gene expression in HCV+ cirrhotic liver is higher in CA, thus lower value of FC is seen. Similar profile is seen in genes expressed in HCCN vs. HCV+HCC (tumor state): PCNA-AS1, ROBO1, DAB2, and IFI30 (Table 5A, lower part). As shown in Table 5B, SAA1 has an overall SI positive value in both HTA2.0 and qRT-PCR analyses. However, the SI value in AA samples (qRT-PCR) is lower compared to CA. This relates to the overall gene signal being higher in HCV+ cirrhotic liver (Table 5A, upper), thus more sliced out (higher signal) compared to normal. These data suggest that the observed disparity in HCV-induced HCC seen in CA and AA tissue samples could be due, in part, to transcriptome diversity of specific genes like SAA1, PCNA-AS1, IFI30, DBA2, and ROBO1.
Table 5A. qRT-PCR validation of 8 selected DEGs.
| Disease Stage | Gene Symbol | Accession Number | Fold Change (FC) | |||
|---|---|---|---|---|---|---|
| HTA 2.0 | qRT-PCR | |||||
| Normal vs. HCV+ | CA | AA | CA | AA | ||
| SAA1 | NM_000331 | 3.36 | NA | 3.12 | 2.0* | |
| AOX1 | NM_001159 | 3.45 | NA | 3.10 | 3.3 | |
| SLC13A5 | NM_001143838 | 3.27 | NA | 3.51 | 3.0 | |
| HCCN vs. HCV+HCC | PCNA-AS1 | NR_028370 | 3.53 | NA | 3.2 | 0.99* |
| ROBO1 | NM_001145845 | 2.20 | NA | 2.9 | 0.20* | |
| DAB2 | NM_001244871 | 2.20 | NA | 3.0 | 0.55* | |
| IFI30 | NM_001244871 | 2.21 | NA | 2.0 | 0.72* | |
| SNORD82 | NR_004398 | -2.20 | NA | -2.0 | -2.0 | |
CA: Caucasian American; AA: African American.
*p<0.05; mean average of 3 biological replicates from each cohort.
Table 5B. qRT-PCR validation of alternative splicing of 3 selected genes.
| Disease Stage | Gene Symbol | Accession Number | Splicing Index (SI) | |||
|---|---|---|---|---|---|---|
| HTA 2.0 | qRT-PCR | |||||
| Normal vs. HCV+ | CA | AA | CA | AA | ||
| SAA1 | NM_000331 | 10.77 | NA | 9.12 | 3.21* | |
| AOX1 | NM_001159 | -2.55 | NA | -2.10 | -1.38 | |
| SLC13A5 | NM_001143838 | -1.37 | NA | -1.61 | -1.12 | |
CA: Caucasian American; AA: African American.
*p<0.05; mean average of 3 biological replicates from each cohort.
Hepatocyte nuclear factor 4α (HNF4α) and serum amyloid A1 (SAA1)-associated protein staining patterns in liver and tumor tissue samples
Since SAA1 is transcriptionally regulated by HNF4α [26], we examined the staining patterns of both proteins in 72 tissues sections for CA and AA using immunohistochemical analysis (Figures 2 and 3). Intense staining for SAA1 and P1/P2-HNF4α was observed in normal liver tissues for both CA (Figure 2Aa, and 2Ad) and AA (2Ba, and 2Bd). In contrast, the staining reactivity for both proteins showed a tendency to decrease in HCV+ cirrhotic livers of AA (Figure 2Bb, and 2Be) compared to CA (2Ab, and 2Ae). As shown in Figure 2C and 2D, the percentage of reactivity for SAA1 and P1/P2-HNF4α are 6.5 and 40 in AA, whereas in CA they are 25 and 50, respectively. Likewise, the staining patterns for both SAA1 and P1/P2-HNF4α in HCC are different in AA compared to CA samples. In AA tumor samples, there was no staining detected for SAA1 (Figure 2Bc), whereas intense staining was detected for P1/P2-HNF4α (Figure 2Bf). For CA tumor samples, staining was detected for both proteins, although less than what is detected in normal tissues (Figure 2Ac, and 2Af). Figure 3A illustrates the staining pattern of P1-HNF4α in tissue samples for both CA and AA. In HCV+ tissues, the percentage reactivity of P1-HNF4α is higher in CA (125%), and lower in AA (50%). There is no clear difference in HCC staining reactivity of P1-HNF4α between CA and AA.
Figure 2. Immunohistochemical staining of SAA1 and P1/P2-HNF4α.
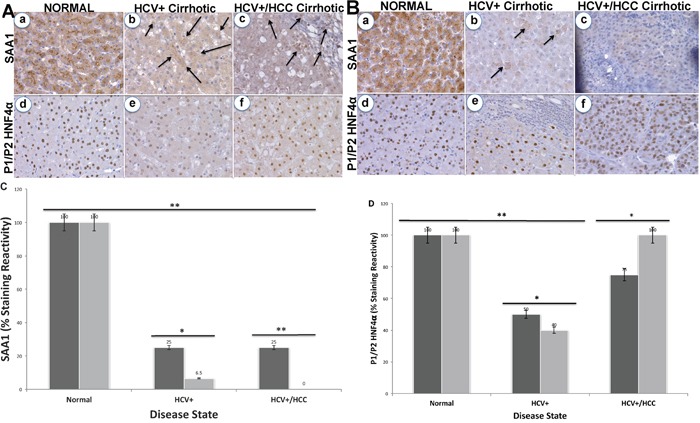
(A) Normal (a and d, respectively), HCV+ cirrhotic (b and e, respectively), and HCV+/HCC cirrhotic (c and f, respectively) in CA. (B) Normal (a and d, respectively), HCV+ cirrhotic (b and e, respectively), and HCV+/HCC cirrhotic (c and f, respectively) in AA. Bar graphs = % staining reactivity (Y-axis) vs. disease state (X-axis) for SAA1 (C) and P1/P2-HNF4α (D). Black bar = CA; Gray bar = AA (n=3 – 4 tissue sections from 24 paraffin embedded tissue blocks ± S.E; *p<0.05; **p<0.001).
Figure 3. Immunohistochemical staining of P1-HNF4α.
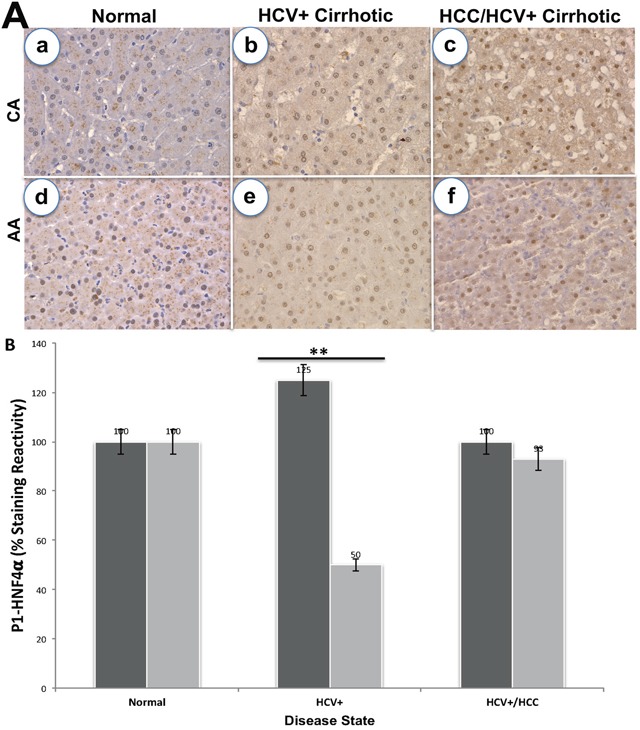
(A) Staining in normal, HCV+ and HCC for CA (a-c) and AA (d-f) tissue samples. (B) Bar graphs = % staining reactivity (Y-axis) vs. disease state (X-axis) for CA, black bar and AA, grey bar (n=3 – 4 tissue sections from 24 paraffin embedded tissue blocks ± S.E; *p<0.05; **p<0.001).
DISCUSSION
We previously showed [12] that there are distinct alterations in the expression of transcripts and proteins exist in CA liver and tumor tissue samples based on HCV disease state. However, the levels of expression were different when the results were cross- validated on tissue samples of AA cohort. The aim of the current study was to follow up on these findings and investigate, at the whole transcriptome level, the extent to which splice variant events may play a role in this genomic diversity of HCV disease state and racial disparity. Alternative splicing of mRNA is a major mechanism that generates diverse mRNA transcript isoforms from a single gene, and subsequently differentiates proteins to have varying cellular processes [19–23]. These variants are targeted as biomarkers in disease diagnosis, prognosis and treatment [27–29].
In the present study, genome-wide analyses of genes and alternative splicing events of human liver and tumor tissues were performed using the newly developed Affymetrix Human Transcriptome 2.0 arrays (HTA 2.0). With a high density of oligonucleotide probes, these arrays cover the exonic regions of human genome as well as junction regions between adjacent exons. Many changes were apparent in HCV+ cirrhotic vs. normal livers, even more so than HCV+HCC vs. HCCN. This may indicate that HCV+ cirrhotic livers, as a type of intermediary lesion in HCV disease progression, already exhibited strong signs of alternations. From the molecular changes evidenced in HCV+ (Figure 1A), it is clear that HCV+ cirrhotic livers are not merely accumulating alterations that will be found in HCV+HCC (Figure 1B). Possibly, the evolution to HCC follows a more strictly clonal expansion, which may select for gene changes important for clonal growth while eliminating less relevant modifications. According to this hypothesis, HCV+ cirrhotic livers may have different outcomes, some evolving toward cancer (HCC), whereas others could be prone to disappearance. In this case, we were able to identify more genes expressed in normal vs. HCV+ (636 DEGs), whereas only 61 DEGs were detected in HCCN vs. HCV+HCC. No overlap of genes was detected between the two disease states.
Tables 1A & 1B show specific gene expression alterations in normal vs. HCV+. The signature of 350 probes corresponding to downregulated genes in HCV+ compared to normal is shown in Table 1A. Among the highest down- regulated genes are: AVR1A, SAA2, MT1F, CFHR5, SLITRK3, CLEC4M, SAA1, CPN1, TIMD4, GPR125, and AOX1. Most of these genes have not been described to be associated with HCV+ cirrhotic livers, although several of the changes agreed to previous reports including variations in the expression levels of SAA1, SAA2 or MT1F [30–33]. For example, SAA1 and SAA2 are well-known acute phase reactants, and their serum levels were shown to be down regulated in HBV-associated HCC patients compared to healthy individuals [34]. In our study, both SAA1 and SAA2 are down regulated in HCV+ liver compared to normal (Figure 1A). As tumor suppressor, metallothionein 1F (MT1F) has been shown to be down regulated in several tumors as part of cancer initiation and/or progression [35]. The signature of 286 probes corresponding to upregulated genes in HCV+ compared to normal is shown in Table 1B. Among the highest upregulated genes are: AKR1B10, IFI27, IL8, VTRNA1-1, SPP1, GDF15, CXCL10, IGLC7, and LGALS4. The expression of these genes is known to be strongly associated with HCV-induced liver cirrhosis and/or HCC [36–45]. In Figure 1A, both SPP1 and IL8 are upregulated in HCV+ cirrhotic liver compared to normal.
The signature of 61 probes corresponding to genes showing expression alterations in HCCN vs. HCV+HCC is shown in Table 2. In this disease state, 47 genes (77%) are upregulated, whereas 14 genes (23%) are downregulated. Among the top deregulated probes, PCNA-AS1 has been found to be the most up-regulated probes in HCV+HCC compared to HCCN, whereas SNORD82, among the downregulated probes (Figure 1B). Both genes are considered long non-coding RNAs (lncRNAs) and well recognized to play major regulatory roles in disease development. For example, PCNA-AS1 was shown to act as an upstream regulator in HCC [46], and SNORD82 has been found to be involved in the development of prostate and breast cancers [47, 48]. Ingenuity Pathway Analysis (IPA) was performed using Ingenuity software, as we reported previously [12] to understand the correlation between the canonical biological pathways and the deregulated genes identified in this study. Among the top 5 canonical pathways for normal vs. HCV+ state (Table 5A) was Hepatic Fibrosis/Satellite Cell Activation (p=4.25E-04). In hepatic fibrosis, hepatotoxins like HCV initiate a cascade of stress related pro-inflammatory events, which eventually activate Hepatic Stellate cells (HSCs). Activated HSCs secrete cytokines that perpetuate their activated state. Continued liver injury results in an accumulation of activated HSCs, which in turn synthesize large amount of extracellular matrix (ECM) proteins, leading to severe fibrosis and eventually liver cirrhosis. SAA1 and SAA2 genes are among the molecules activated in this disease state (acute phase reactants), and both are down regulated indicating a possible involvement in disease initiation to HCC. For HCCN vs. HCV+HCC state (Table 5B), GADD45 Signaling was the top pathway identified (p=2.93E-06). It has been implicated in stress signaling response that can result in cell cycle arrest, DNA repair, cell survival, senescence, and apoptosis. This response is mediated via a complex binding to several proteins involved in these processes, including PCNA and thus PCNA-ASI was found to be upregulated in HCC (Figure 1B).
We next validated the expression of 8 DEGs by real-time qRT-PCR using independent samples for CA and AA, as shown in Table 5A. Although it is clearly shown in this table that there is good concordance in results obtained using both platforms, the level of SAA1 in AA samples (normal vs. HCV+ state) is significantly lower than that of CA (p<0.05). Thus, immune response to chronic HCV infection may play a crucial role in HCV racial disparities. Four (PCNA-AS1, ROBO1, DAB2 and IFI30) out 5 transcripts with increased expression in HCCN vs. HCV+HCC state (Table 2) were found to be significantly lower (p<0.05) in AA compared to CA samples. Thus, in addition to the immune response-associated genes, these genes could also play a role in HCV/HCC racial disparities seen between CA and AA samples, and might be valuable markers for early diagnosis of the disease based on racial background of patients.
Since SAA1 (acute response reactant) is transcriptionally regulated by HNF4α [49] we validated the expression of both using immunohistochemical analysis. HNF4α is a member of the superfamily of ligand-dependent transcription factors (TFs) and master regulator of tissue-specific gene expression in the liver [50]. It inhibits progression of HCC in mice [17, 18]. There are two alternative promoters that drive expression of HNF4α gene (P1 and P2) and give rise to HNF4α isoforms that differ by 16-38 amino acids in their terminal region [51]. While the different isoforms have identical DNA and ligand binding domains, there subtle yet significant functional differences between the HNF4α isoforms. Both P1- and P2-driven HNF4α are expressed in the fetal liver but only P1- HNF4α is expressed in the normal adult liver [52], and P1- HNF4α is down regulated in human HCC while P2- HNF4α is upregulated [51]. Furthermore, P1- HNF4α is known to repress the activation of the P2 promoter [51], which could explain the switch between the two isoforms. In this study, we used both H1415 and K9218 monoclonal antibodies to detect P1/P2- and P1-promoter-driven HNF4α, respectively, in the liver and tumor samples to determine how the expression of these two isoforms may play a role in SAA1 expression patterns. Our data in Figure 2 clearly indicate that staining reactivity of SAA1 and P1/P2-HNF4α is altered based on HCV disease state and race. For example, staining reactivity (%) for SAA1 (Figure 2C) in CA is 25% for both HCV+ cirrhotic and HCC states, whereas in AA samples it is only 6.5% and 0.0%, respectively. This indicate that the marker for “acute inflammatory phase” is much lower in HCV+ of AA compared to CA cohort. As shown in Figure 2D, the staining reactivity of P1/P2- HNF4α, which is a measure of both isoforms, is lower in HCV+ for both CA and AA tissue samples. However, it is clearly shown in Figure 3B that the low staining reactivity is related to P1- HNF4α isoform, and mainly in AA tissue samples. These data clearly indicate that the acute inflammatory phase as measured by SAA1 level is severely compromised in AA compared to CA as a result of dysregulation of HNF4α isoforms. Our results also show that changes in splicing profiles in normal vs. HCV+ state could possibly contribute to the observed HCV disease state racial disparity (Table 3). The alternative splicing events of three genes (SAA1, AOX1 and SLC13A5) from the 28-gene set (Table 3) were confirmed by real-time qRT-PCR in normal vs. HCV+ state. Specifically, we validated the expression of SAA1, AOX1, and SLC13A5. For SAA1, the expression of exon 1 to 2 and exon 1 to 3 (Supplementary Figure 1), for AOX1 4 to 5, and the exon 12 to 13, for SLC13A5 exon 10 to 12 (Supplementary Figure 2). We found that the splicing index (SI) of SAA1 is significantly lower (p<0.05) in AA compared to CA (Table 5B). This suggests that splicing events occurred mainly in specific disease state (HCV+ cirrhotic) predominantly in AA cohort. The role played by these alternative splice products in HCV+ will thus require further investigations, together with the other alternative transcripts detected. In sum, our study suggests that altered gene expression, and splice variants are important events in HCV racial disparities between Caucasian and African Americans.
In conclusion, our genomic variants study showed that genes were differentially expressed between HCCN and HCV+HCC but, also, to a large extent, between normal and HCV+ (cirrhotic) state. Many of these genes are involved in biological pathways pertinent to the overall pathophysiological response to HCV infection. The observation that several splice variants were deregulated in normal vs. HCV+ is certainly in line with the recent observations showing that the pre-mRNA splicing machinery may be profoundly remodeled during HCV disease progression, and may, therefore, play a major role in the disease outcome. Target validation analyses showed that some of these genes are significantly deregulated especially in AA compared to CA tissue samples. These observations suggest that socioeconomic factors may not fully explain the differences in HCV racial disparity, but rather biological/genetic factors should also be considered. Further analyses will be required to determine if these gene variants are predictive markers of the pathophysiological evolution in HCV disease progression. It would be of great interest to determine whether our differentially expressed genes and splice variants are under some kind of coordinated control. This certainly will allow for the development of next generation therapeutic care management for HCV disease state based on racial/ethnic backgrounds of patients.
MATERIALS AND METHODS
Sample preparation and data analysis
Total RNA was extracted from 12 tissue samples of Caucasian individuals (3 normal livers, 3 HCV+/HCC- (cirrhotic livers), 3 HCV+/HCC+ (cirrhotic tumors) and 3 normal adjacent tissue matched pairs HCCN) using the RNeasy mini kit (Qiagen, Valencia, CA, USA) and quantified using Nanodrop ND-100 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), as previously reported [12]. RNA samples were then subjected to RNA amplification using the SensationPlus FFPE Amplification and WT Labeling Kit (Affymetrix Inc., Santa Clara, CA, USA), as previously reported [53, 54]. The biotin double-stranded cDNA products were hybridized to Affymetrix HTA 2.0 arrays using an Affymetrix hybridization kit. Hybridized HTA 2.0 arrays were scanned with an Affymetrix GeneChip® 3000 fluorescent scanner. Image generation and feature extraction was performed using Affymetrix GeneChip Command Console Software. The raw data (.*CEL) were analyzed using the Transcriptome Analysis Console (TAC) 2.0 software, which allows for the identification of differentially expressed genes (DEG) & exons and the visualization of alternative splicing events for determining possible transcript isoforms that may exist in samples.
For microarray data analysis, two parallel analyses (gene-level and alternative splicing level) were performed. Data were normalized using quantile normalization, and background noise was detected using Detection Above Background (DABG) algorithm. Only the probesets characterized by a DABG p-value <0.05 in at least 50% of the samples were considered for statistical analysis. We performed an unpaired Student's t-test to compare gene intensities between normal vs. HCV+ and HCCN vs. HCV+HCC. Genes were considered significantly regulated when Fold Change (FC), linear <-2.0 or >+2.0 and ANOVA p-value (condition pair) <0.05. Analysis of the splicing level was also performed using TAC 2.0 software, which determines among other parameters, the Splicing Index (SI) of a gene. The SI corresponds to a comparison of gene-normalized exon-intensity values between the two analyzed experimental conditions [55]. Additional criteria used beside SI: q-value <0.05, a gene is expressed in both conditions (normal vs. HCV+, and HCCN vs. HCV+HCC), a Probset Ratio (PSR)/Junction must be expressed in at least one condition, and a gene must contain at least one PSR value.
Reverse transcription PCR validation
Validation of 8 selected differentially expressed genes (DEGs) and splice variants was performed on 24 independent tissue samples (12 CA, and 12 AA) at various disease state (normal, HCV+ and HCC). mRNA levels were measured using the SYBR-GREEN quantitative RT-PCR (qRT-PCR) method as previously reported [12] by the ABI 7900HT Fast Real Time PCR System (Applied Biosystems). cDNAs were amplified using specific primers indicated in Supplementary Table 1; data results were normalized against alpha-ACTIN (ACTIN1), beta-2-Microglobin (B2M), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Relative RNA levels of genes were calculated using the comparative Ct method 2-ΔΔCt [56]. For splice variants, alt-spliced (A) and constitutive (C) exons were identified in TAC 2.0, and qRT-PCR primer sets were designed using Primer3 (http://www.ncbi.nlm.nih.gov/tools/primer-blast/) as shown in Supplementary Table 1. By designing specific primer pairs for constitutively expressed flanking exons (Supplementary Figure 1 and 2), it is possible to simultaneously amplify isoforms that include or skip the target exon [57]. The identities of variant specific amplicons were simultaneously verified and quantitated by melt curve analysis, and the products were confirmed either present or absent using agarose gel electrophoresis. Splice Index (SI) was calculated for (A) by normalizing fold change (FC) to the average FC of (C) for each splicing event. For amplicon spanning exons 4-5 in AOX1 (Supplementary Table 1), the calculated FC (A)/average FC (C) value is less than 1 (0.47), indicating decreased exon 5 inclusion in Normal vs. HCV+. This is finally reported as -1/0.47 = -2.1, as a negative number (Table 5B). For SAA1, the reported positive SI number (9.12) indicates increased exon 3 inclusion in Normal vs. HCV+. Each sample was measured in triplicate and values were reported as average.
Immunohistochemistry
Study tissue blocks (24 samples, including 3 normal; 3 HCV+, 3 HCCN and 3 HCV+/HCC for CA and AA, respectively) were selected after histopathologic review by pathologists. Three 4-tissue sections were selected from each block (total = 96 tissue slides). All of the tissue slides were treated to heat induced epitope retrieval (HIER) in a decloaker (BIocare Inc.) using HIER-L solution (citrate buffer, pH 6.0, Thermo Fisher). Detection for serum amyloid A1 protein (SAA1) and hepatocyte nuclear factor 4-alpha (HNF4α) isoforms was performed by incubating slides in a rabbit anti-mouse antibody (SAA1, Clone # 902738, R&D Systems, Cat # MBA30191, dilutions 1:50), (P1/P2-HNF4α, Clone # H1415, R&D Systems, Cat # PP-H1415-00, dilutions 1:100) or (P1-HNF4α, Clone # K9219, Cat # PP-K9218-00, dilutions 1:100) overnight at 4°C followed by incubation in a horseradish peroxide-conjugated anti-rabbit antibody, then developing with 3,3’-diaminobenzidine tetrahydrochloride chromogen. For negative control, the primary antibodies were replaced with PBS. Liver sections were used as positive controls. Staining reactivity for each protein/tissue slide was graded by two pathologists (MMY and SB) as consensus using a semi-quantitative scoring system (0 – 4) as previously reported [58]. The staining reactivity of 3-4 tissue slides was plotted for SAA1, P1/P2- and P1- HNF4α.
Pathways, functional enrichment and interactive network analysis
Gene networks and canonical pathways representing key genes were identified through the use of QIAGEN'S Ingenuity Pathway Analysis software (IPA, QIAGEN Redwood City, www.qiagen.com/ingenuity, content version 18841524, release date 06/26/2014) as previously reported [12]. Briefly, the data sets containing gene identifiers and corresponding fold change and p-values were uploaded into the web-delivered application and each gene identifier was mapped to its corresponding gene object in the IPA software. Fisher's exact test was performed to calculate a P-value assigning probability of enrichment to each biological function and canonical pathway within the IPA library.
Statistical analysis
The data were expressed as mean±SE, and analyzed with the Student's t-test between two groups. Changes were considered statistically significant if the P-value was <0.05.
Ethics statement
Washington State University (WSU) Office of Research Assurances has found that the study is exempt from the need for the Institutional Research Board (IRB) approval. Thirty-six snapped frozen tissue samples (12 included in the original analysis and 24 for target validation study), as well as 25 tissue sections from formalin-fixed paraffin-embedded blocks were obtained from the IRB approved University of Kansas Medical Center Liver Center Tissue Bank. All specimens with anonymized identifiers were histopathologically confirmed by a pathologist.
SUPPLEMENTARY MATERIALS FIGURES AND TABLES
Acknowledgments
The authors would like to thank University of Kansas Medical Center – Liver Center Tissue Bank for providing us with tissue samples. We thank Mr. Ryan Maynard for his assistance with HTA2.0 data analysis and TAC2.0 software. The authors are grateful for the excellent technical assistance of Ms. Zahra Afsharinejad and Ms. Kelly Hudkins. The critical review of the manuscript by Dr. Theo Bammler is greatly appreciated.
Abbreviations
- HCV
Hepatitis C virus
- HCC
Hepatocellular carcinoma
- HTA2.0
Human Transcriptome Array 2.0
- HCV+
HCV positive cirrhotic liver
- HCV+HCC
HCV positive liver tumor
- HCCN
Tumor adjacent normal tissue
- CA
Caucasian American
- AA
African American
- AS
Alternative splicing
- DEGs
Differentially expressed genes
- IPA
Ingenuity pathway analysis
- qRT-PCR
Quantitative real-time-PCR
- FC
Fold Change
- SI
Splicing Index
- PSR
Probeset Ratio
- IHC
immunohistochemistry
Author contributions
Conceived and designed the experiments: SSD, MMY. Performed IHC study: MMY, SB. Contributed reagents/materials: SSD, BR. Analyzed the data: SSD, MMY, SB, ND. Wrote the paper: MMY, SSD. All authors read and approved the final version of the manuscript.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
FUNDING
Not applicable.
REFERENCES
- 1.Yeh MM, Yeung RS, Apisarnthanarax S, Bhattacharya R, Cuevas C, Harris WP, Hon TL, Padia SA, Park JO, Riggle KM, Daoud SS. Multidisciplinary perspective of hepatocellular carcinoma: A Pacific Northwest experience. World J Hepatol. 2015;7:1460–1483. doi: 10.4254/wjh.v7.i11.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edlin BR. Perspective: test and treat this silent killer. Nature. 2011;474:S18–S19. doi: 10.1038/474S18a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobson IM, Davis GL, El-Serag H, Negro F, Trépo C. Prevalence and challenges of liver diseases in patients with chronic hepatitis C virus infection. Clin Gastroenterol Hepatol. 2010;8:924–33. doi: 10.1016/j.cgh.2010.06.032. [DOI] [PubMed] [Google Scholar]
- 4.Gonzalez-Grande R, Jimenez-Perez M, Arjona Gonzalez, Mostazo Terres J. New approaches in the treatment of hepatitis C. World J Gastroenterol. 2016;22:1421–32. doi: 10.3748/wjg.v22.i4.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chhatwal J, Wang X, Ayer T, Kabiri M, Chung RT, Hur C, Donohue JM, Roberts MS, Kanwal F. Hepatitis C disease burden in the United States in the area of oral directing-acting antivirals. Hepatology. 2016;64:1442–14450. doi: 10.1002/hep.28571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.DeSantis C, Naishadham D, Jemal A. Cancer statistics for African Americans, 2013. CA Cancer J Clin. 2013;63:151–166. doi: 10.3322/caac.21173. [DOI] [PubMed] [Google Scholar]
- 7.Saab S, Jackson C, Nieto J, Francois F. Hepatitis C in African Americans. Am J Gastroenterol. 2014;109:1576–84. doi: 10.1038/ajg.2014.243. [DOI] [PubMed] [Google Scholar]
- 8.Trooskin SB, Navarro VJ, Winn RJ, Axelrod DJ, McNeal AS, Velez M, Herrine SK, Rossi S. Hepatitis C risk assessment, testing and referral for treatment in urban primary care: role of race and ethnicity. World J Gastroenterol. 2007;13:1074–8. doi: 10.3748/wjg.v13.i7.1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Su F, Green PK, Berry K, Ioannou GN. The association between race/ethnicity and the effectiveness of direct antiviral agents for hepatitis C virus infection. Hepatol. 2017;65:426–438. doi: 10.1002/hep.28901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Artinyan A, Mailey B, Sanchez-Luege N, Khalili J, Sun CL, Bhatia S, Wagman LD, Nissen N, Colquhoun SD, Kim J. Race, ethnicity, and socioeconomic status influence the survival of patients with hepatocellular carcinoma in the United States. Cancer. 2010;116:1367–77. doi: 10.1002/cncr.24817. [DOI] [PubMed] [Google Scholar]
- 11.Daoud SS. Hepatitis C pharmacogenetics: possible solutions for an existing problem. J Pharmacogenomics Pharmacoproteomics. 2013;4:2. [Google Scholar]
- 12.Dillon ST, Bhasin MK, Feng X, Koh DW, Daoud SS. Quantitative proteomic analysis in HCV-induced HCC reveals sets of proteins with potential significance for racial disparity. J Transl Med. 2013;11:239. doi: 10.1186/1479-5876-11-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ioannou GN, Dominitz JA, Weiss NS, Heagerty PJ, Kowdley KV. Racial differences in the relationship between hepatitis C infection and iron stores. Hepatology. 2003;37:795–801. doi: 10.1053/jhep.2003.50147. [DOI] [PubMed] [Google Scholar]
- 14.Samantray J, Zambare S, Seyoum B, Abou-Samra AB. Glucose control and lipid metabolism in African American patients with type 2 diabetes mellitus and chronic hepatitis C viral infection. Endocr Pract. 2011;17:363–8. doi: 10.4158/EP10175.OR. [DOI] [PubMed] [Google Scholar]
- 15.Mogilenko DA, Dizhe EB, Shavva VS, Lapikov IA, Orlov SV, Perevozchikov AP. The role of the nuclear receptors HNF4 alpha, PPAR alpha, and LXRs in the TNF alpha-mediated inhibition of human Apolipoprotein A-1 gene expression in HepG2 cells. Biochemistry. 2009;48:11950–60. doi: 10.1021/bi9015742. [DOI] [PubMed] [Google Scholar]
- 16.Fang B, Mane-Padros D, Bolotin E, Jiang T, Sladek FM. Identification of a binding motif specific to HNF4 by comparative analysis of multiple nuclear receptors. Nucleic Acids Res. 2012;40:5343–5356. doi: 10.1093/nar/gks190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatziapostolou M, Polytarchou C, Aggelidou E, Drakaki A, Poultsides GA, Jaeger SA, Ogata H, Karin M, Struhl K, Hadzopoulou-Cladaras M, Iliopoulos D. An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular carcinoma. Cell. 2011;147:1233–47. doi: 10.1016/j.cell.2011.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bonzo JA, Ferry CH, Matsubara T, Kim JH, Gonzalez FJ. Suppression of hepatocyte proliferation by hepatocyte nuclear factor 4α in adult mice. J Biol Chem. 2012;287:7345–56. doi: 10.1074/jbc.M111.334599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Venables JP. Aberrant and alternative splicing of cancer. Cancer Res. 2004;64:7647–7654. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 20.Pal S, Gupta R, Davuluri RV. Alternative transcription and alternative splicing in cancer. Pharmacol Therap. 2012;136:283–294. doi: 10.1016/j.pharmthera.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Biamonti G, Catillo M, Pignataro D, Montecucco A, Ghigna C. The alternative splicing side of cancer. Semin Cell Dev Biol. 2014;32:30–36. doi: 10.1016/j.semcdb.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 22.Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008;40:1413–1415. doi: 10.1038/ng.259. [DOI] [PubMed] [Google Scholar]
- 23.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Klingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hanoun N, Bureau C, Diab T, Gayet O, Dusetti N, Selves J, Vinel JP, Buscail L, Cordelier P, Torrisani J. The SV2 variant of KLF6 is downregulated in hepatocellular carcinoma and displays anti-proliferative and pro-apoptotic functions. J Hepatol. 2010;53:880–8. doi: 10.1016/j.jhep.2010.04.038. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Lv G, Chu Z, Piao L, Liu X, Wang T, Jiang Y, Zhang P. Identification of natural splice variants of SAMHD1 in virus-infected HCC. Oncol Rep. 2014;31:687–92. doi: 10.3892/or.2013.2895. [DOI] [PubMed] [Google Scholar]
- 26.Bauza G, Miller G, Kaseje N, Wang Z, Sherburne A, Agarwal S, Burke PA. Injury-induced changes in liver specific transcription factors HNF-1α and HNF-4α. J Surg Res. 2012;175:298–304. doi: 10.1016/j.jss.2011.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi Q, Tang L. Alternative spliced variants as biomarkers of colorectal cancer. Curr Drug Metab. 2011;12:966–74. doi: 10.2174/138920011798062355. [DOI] [PubMed] [Google Scholar]
- 28.Pavlidou A, Kroupis C, Dimas K. Association of survivin splice variants with prognosis and treatment of breast cancer. World J Clin Oncol. 2014;5:883–94. doi: 10.5306/wjco.v5.i5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun F, Indran IR, Zhang ZW, Tan MH, Li Y, Lim ZL, Hua R, Yang C, Soon FF, Li J, Xu HE, Cheung E, Yong EL. A novel prostate cancer therapeutic strategy using icaritin-activated arylhydrocarbon-receptor to co-target androgen receptor and its splice variants. Carcinogenesis. 2015;36:757–68. doi: 10.1093/carcin/bgv040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji YR, Kim HJ, Bae KB, Lee S, Kim MO, Ryoo ZY. Hepatic serum amyloid A1 aggravates T cell-mediated hepatitis by inducing chemokines via Toll-like receptor 2 in mice. J Biol Chem. 2015;290:12804–11. doi: 10.1074/jbc.M114.635763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olteanu S, Kandel-Kfir M, Shaish A, Almog T, Shemesh S, Barshack I, Apte RN, Harats D, Kamari Y. Lack of Interleukin-1α in Kupffer cells attenuates liver inflammation and expression of inflammatory cytokines in hypercholesterolaemic mice. Dig Liver Dis. 2014;46:433–9. doi: 10.1016/j.dld.2014.01.156. [DOI] [PubMed] [Google Scholar]
- 32.Hansen MT, Forst B, Cremers N, Quagliata L, Ambartsumian N, Grum-Schwensen B, Klingelhöfer J, Abdul-Al A, Hermann P, Osterland M, Stein U, Nielsen GH, Scherer PE, et al. A link between inflammation and metastasis: serum amyloid A1 and A3 induce metastasis, and are targets of metastasis-induced S100A4. Oncogene. 2015;34:424–35. doi: 10.1038/onc.2013.568. [DOI] [PubMed] [Google Scholar]
- 33.Werynska B, Pula B, Muszczynska-Bernhard B, Gomulkiewicz A, Piotrowska A, Prus R, Podhorska-Okolow M, Jankowska R, Dziegiel P. Metallothionein 1F and 2A overexpression predicts poor outcome of non-small cell lung cancer patients. Exp Mol Pathol. 2013;94:301–8. doi: 10.1016/j.yexmp.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 34.He X, Wang Y, Zhang W, Li H, Luo R, Zhou Y, Liao CL, Huang H, Lv X, Xie Z, He M. Screening differential expression of serum proteins in AFP-negative HBV-related hepatocellular carcinoma using iTRAQ-MALDI-MS/MS. Neoplasma. 2014;61:17–26. [PubMed] [Google Scholar]
- 35.Yan DW, Fan JW, Yu ZH, Li MX, Wen YG, Li DW, Zhou CZ, Wang XL, Wang Q, Tang HM, Peng ZH. Downregulation of metallothionein 1F, a putative oncosuppressor, by loss of heterozygosity in colon cancer tissue. Biochim Biophys Acta. 2012;1822:918–26. doi: 10.1016/j.bbadis.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 36.Semmo N, Weber T, Idle JR, Beyoglu D. Metabolomics reveals that aldose reductase activity due to AKR1B10 is upregulated in hepatitis C virus infection. J Viral Hepat. 2015;22:617–24. doi: 10.1111/jvh.12376. [DOI] [PubMed] [Google Scholar]
- 37.Ha SY, Song DH, Lee JJ, Lee HW, Cho SY, Park CK. High expression of aldo-keto-reductase 1B10 is an independent predictor of favorable prognosis in patients with hepatocellular carcinoma. Gut Liver. 2014;8:648–54. doi: 10.5009/gnl13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matkowskyj KA, Bai H, Liao J, Zhang W, Li H, Rao S, Omary R, Yang GY. Aldoketoreductase family 1B10 (AKR1B10) as a biomarker to distinguish hepatocellular carcinoma from benign liver lesions. Hum Pathol. 2014;45:834–43. doi: 10.1016/j.humpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sato S, Genda T, Hirano K, Tsuzura H, Narita Y, Kanemitsu Y, Kikuchi T, Iijima K, Wada R, Ichida T. Up-regulated aldo-keto reductase family 1 member B10 in chronic hepatitis C: association with serum alpha-fetoprotein and hepatocellular carcinoma. Liver Int. 2012;32:1382–90. doi: 10.1111/j.1478-3231.2012.02827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jablonowska E, Wojcik K, Koslinska-Berkan E, Szymanska B, Omulecka A, Piekarska A. Expression of selected genes in liver biopsy specimen in relation to early virological response in patients with chronic hepatitis C with HCV-mono- and HIV/HCV co-infection. Arch Virol. 2014;159:1365–71. doi: 10.1007/s00705-013-1930-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Langhans B, Kramer B, Louis M, Nischalke HD, Huneburg R, Staratschek-Jox A, Odenthal M, Manekeller S, Schepke M, Kalff J, Fischer HP, Schultze JL, Spendgler U. Intrahepatic IL-8 producing Foxp3+CD4+ regulatory T cells and fibrogenesis in chronic hepatitis C. J Hepatol. 2013;59:229–35. doi: 10.1016/j.jhep.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 42.Ali FT, Ali MA, Elgizawy MM, Elsawy AM. Secreted phosphoprotein 1 promoter genetic variants are associated with the response to pegylated interferon α plus ribavirin combination therapy in Egyptian patients with chronic hepatitis C virus infection. Gut Liver. 2015;9:516–10. doi: 10.5009/gnl14162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu X, Chi X, Gong Q, Gao L, Niu Y, Chi X, Cheng M, Si Y, Wang M, Zhong J, Niu J, Yang W. Association of serum level of growth differentiation factor 15 with liver cirrhosis and hepatocellular carcinoma. PLoS One. 2015;10:e0127518. doi: 10.1371/journal.pone.0127518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wandrer F, Falk C, John K, Skawran B, Manns MP, Schulze-Osthoff K, Bantel H. Interferon-mediated cytokine induction determines sustained virus control in chronic HCV infection. J Infect Dis. 2016;213:746–54. doi: 10.1093/infdis/jiv505. [DOI] [PubMed] [Google Scholar]
- 45.Cai Z, Zeng Y, Xu B, Gao Y, Wang S, Zeng J, Chen L, Huang A, Liu X, Liu J. Galectin-4 serves as a prognostic biomarker for the early recurrence/metastasis of hepatocellular carcinoma. Cancer Sci. 2014;105:1510–7. doi: 10.1111/cas.12536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuan SX, Tao QF, Wang J, Yang F, Liu L, Wang LL, Zhang J, Yang Y, Liu H, Wang F, Sun SH, Zhou WP. Antisense long non-coding RNA PCNA-ASI promotes tumor growth by regulating proliferating cell nuclear antigen in hepatocellular carcinoma. Cancer Lett. 2014;349:87–94. doi: 10.1016/j.canlet.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 47.Ho SM, Cheong A, Lam HM, Hu WY, Shi GB, Zhu X, Chen J, Zhang X, Mededovic M, Leung YK, Prins GS. Exposure of human prostaspheres to bisphenol A epigenetically regulates SNORD family noncoding RNAs via histone modification. Endocrinology. 2015;156:3984–95. doi: 10.1210/en.2015-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Askarian-Amiri ME, Crawford J, French JD, Smart CE, Smith MA, Clark MB, Ru K, Mercer TR, Thompson ER, Lakhani SR, Vargas AC, Campbell IG, Brown MA, et al. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA. 2011;17:878–91. doi: 10.1261/rna.2528811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bauza G, Miller G, Kaseje N, Wang Z, Sherburne A, Agarwal S, Burke PA. Injury-induced changes in liver specific transcription factors HNF-1α and HNF4α. J Surg Res. 2012;175:298–304. doi: 10.1016/j.jss.2011.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hwang-Verslues WW, Sladek FM. HNF4a role in drug metabolism and potential drug target? Curr Opin Pharmacol. 2010;10:698–705. doi: 10.1016/j.coph.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tanaka T, Jiang S, Hotta H, Takano K, Iwanari H, Sumi K, Daigo K, Ohashi R, Sugai M, Ikegame C, Umezu H, Hirayama Y, Midorkawa Y, et al. Dysregulated expression of P1 and P2 promoter-deriven hepatocyte nuclear factor 4α in the pathogenesis of human cancer. J Pathol. 2006;208:662–672. doi: 10.1002/path.1928. [DOI] [PubMed] [Google Scholar]
- 52.Briancon N, Baily N, Clotman F, Jacquemin P, Lemaigre FP, Weiss MC. Expression of the alpha7 isoform of hepatocyte nuclear factor (HNF) 4 is activated by HNF6/OC-2 and HNF1 and repressed by HNF4alpha 1 in the liver. J Biol Chem. 2004;279:33398–408. doi: 10.1074/jbc.M405312200. [DOI] [PubMed] [Google Scholar]
- 53.Roberts L, Bowers J, Sensinger K, Lisowski A, Getts R, Anderson MG. Identification of methods for use of formalin-fixed, paraffin-embedded tissue samples in RNA expression profiling. Genomics. 2009;94:341–8. doi: 10.1016/j.ygeno.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 54.Pillai R, Deeter R, Rigl CT, Nystrom JS, Miller MH, Buturovic L, Henner WD. Validation and reproducibility of a microarray-based gene expression test for tumor identification in formalin-fixed, paraffin-embedded specimens. J Mol Diagn. 2011;13:48–56. doi: 10.1016/j.jmoldx.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.de la Grange P, Gratadou L, Delord M, Dutertre M, Aubonuf D. Splicing factor and exon profiling across human tissues. Nucleic Acids Res. 2010;38:2828–38. doi: 10.1093/nar/gkq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:2003–2007. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vandenbroucke II, Vandesompele J, De Paepe A, Messiaen L. Quantification of splice variants using real-time PCR. Nucleic Acid Res. 2001;29:e68–e69. doi: 10.1093/nar/29.13.e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan ES, Yeh MM. The use of immunohistochemistry in liver tumors. Clin Liver Dis. 2010;14:687–703. doi: 10.1016/j.cld.2010.10.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.