

Abstract
Tsetse flies (Diptera: Glossinidae) have medical significance as the obligate vectors of African trypanosomes. In addition, tsetse harbor a simple gut microbiota. A predominant gut microbiota member, the Gammaproteobacterium Wigglesworthia spp., has coevolved with tsetse for a significant portion of Glossina radiation proving critical to tsetse fitness. Although multiple roles have been described for Wigglesworthia within colony flies, little research has been dedicated towards functional characterization within wild tsetse. Here, dual RNA-Seq was performed to characterize the tsetse-Wigglesworthia symbiosis within flies captured in Nguruman, Kenya. A significant correlation in Gene Ontology (GO) distribution between tsetse and Wigglesworthia was observed, with homogeneous enrichment in metabolic and transport categories, likely supporting a hallmark of the symbiosis-bidirectional metabolic exchange. Within field flies, highly transcribed Wigglesworthia loci included those involved in B vitamin synthesis and in substrate translocation, including amino acid transporters and multidrug efflux pumps, providing a molecular means for interaction. The universal expression of several Wigglesworthia and G. pallidipes orthologs, putatively involved in nutrient provisioning and resource allocation, was confirmed in sister tsetse species. These transcriptional profiles varied through host age and mating status likely addressing varying symbiont demands and also confirming their global importance within Glossina. This study, not only supports symbiont nutrient provisioning roles, but also serves as a foundation for insight into novel roles and molecular mechanisms associated with vector–microbiota interactions. The role of symbiont B vitamin provisioning towards impacting host epigenetics is discussed. Knowledge of vector–microbiota interactions may lead to the discovery of novel targets in pest control.
Keywords: tsetse, Wigglesworthia, symbiosis, microbiota
Introduction
Bacteria are the most prevalent animal symbionts (Moya etal. 2008). Among the best examples of reciprocal selection occurring between animals and bacteria are the intimate alliances of insects and their ancient obligate endosymbionts (Douglas 1998; Moran and Sloan 2015). These associations drive evolutionary modifications between partners that serve to enhance the functional roles that form the pinnacle of the species–species integration. Only a small fraction of insects harbor these long-established and essential associations (Douglas 1998; Moran etal. 2008), primarily those that require nutritional supplementation due to limited and unbalanced diets deficient in vitamins or amino acids, such as exclusive feeding on blood, phloem, or xylem. These symbionts are often localized to specific host tissues or organs (often called the bacteriome) that maximize functional efficiency by centralizing activity. Despite the functional characterization of the critical roles provided by these symbioses, little is known concerning the molecular dialog between host and symbiont that may facilitate integration in the wild.
Tsetse flies (Diptera: Glossinidae) are medically significant as the obligate vector of African trypanosomes (Trypanosoma spp.), the etiological agents of Human African trypanosomiasis (HAT) and Animal African trypanosomiasis (AAT). In addition to potential infections with trypanosomes, tsetse harbor a naturally simple digestive tract microbiota consisting of only a few bacterial species (Weiss etal. 2011; Aksoy etal. 2014). These bacteria have distinct coevolutionary histories with tsetse and, likewise, contributory roles towards their insect host biology (reviewed in Rio etal. [2016]). Two Gammaproteobacteria are associated with the digestive track of the tsetse fly; Wigglesworthia spp. and Sodalis glossinidius (Aksoy 1995a; Cheng and Aksoy 1999). Infections with Wolbachia are also common in tsetse reproductive tissue (Cheng etal. 2000). These parasitic Wolbachia infections may cause the arrest of embryonic development following incongruous mating in a condition known as cytoplasmic incompatibility (Alam etal. 2011). The simplicity in the microbiota composition is likely generated through two unique facets of tsetse fly biology; the obligate vertebrate blood feeding lifestyle of adults and the live birth of progeny following intrauterine larval development (known as adenotrophic viviparity [Benoit etal. 2015]), both of which severely curtail environmental microbial exposure.
The most predominant member of the tsetse alimentary tract microbiota (Rio etal. 2006; Aksoy etal. 2014) is the obligate mutualist Wigglesworthia spp. that has codiversified with tsetse for a significant portion of Glossina radiation, dating back 50–80 million years (Chen etal. 1999). The small Wigglesworthia genome (∼0.7 Mb) retains only those genes necessary for habitation within the tsetse (Akman etal. 2002; Rio etal. 2012). Both the tsetse and Wigglesworthia partners rely on each other for evolutionary persistence, exemplified by their interwoven reproductive fitness. The Wigglesworthia symbiont is tightly harnessed to the tsetse host due to extreme reductive genome evolution (Akman etal. 2002; Rio etal. 2012) that makes a free-living lifestyle highly unlikely. Likewise, removal of the Wigglesworthia symbionts from the tsetse fly through selective antibiotic cocktails severely impedes fly reproduction (Hill etal. 1973; Nogge 1978; Nogge 1981) that can be partially restored through B vitamin cocktails or yeast supplementation (Nogge 1981; Pais etal. 2008) indicating essential metabolic interdependency between these partners.
Within tsetse, intracellular Wigglesworthia are localized to a specialized organ (bacteriome) at the anterior gut (Aksoy 1995a). A second Wigglesworthia population is harbored exclusively within milk glands that empty into the uterus (Attardo etal. 2008). These modified accessory glands found only within females, provide nourishment to the larva while also serving as a vessel for vertically transmitting gut microbiota (Ma and Denlinger 1974; Attardo etal. 2008; Balmand etal. 2013). To date, most of our understanding of the tsetse-Wigglesworthia association has been generated primarily from experimental assays conducted with colony flies reared under controlled rearing conditions. In this setting, key Wigglesworthia roles towards tsetse reproduction, digestion, development and immunological priming have been identified (reviewed in Rio etal. [2016]).
Studies that investigate the relationship between animals and their microbiota are largely performed using model systems that have been maintained in laboratory settings for extensive periods. Little research has been dedicated towards validating whether these model systems accurately reflect wild counterparts within their native habitats. Field-based studies may be used to validate or disprove conclusions obtained through the microbiota studies of captive or artificially bred animals. Equally as important, these field-based studies can also identify novel functional significance of the microbiota towards host biology that may be masked, or simply unattainable, whereas rearing in captivity.
In the efforts to deepen our understanding of wild animals and their microbiota interactions within their natural ecology, a parallel Illumina-based RNA-Seq approach was used to examine the molecular integration of the tsetse fly-Wigglesworthia mutualism in field collected flies. Here, we characterize the genome activity of the tsetse-Wigglesworthia association within the bacteriomes of wild caught adult tsetse (Glossina pallidipes) captured in Nguruman, Kenya. To further assess the universality of RNA-Seq results across tsetse species, we characterize the expression profiles of genes associated with novel putative functional roles for Wigglesworthia and Glossina in sister tsetse species. These high-throughput transcriptome analyses provide a global picture of host and symbiont interconnectedness by providing insights into important functional roles including concerted transcriptional profiles involved in nutrient provisioning and resource allocation within their natural habitat. These studies aim to identify molecular candidate targets that disrupt the crucial symbiosis within the medically important tsetse fly providing novel vector control strategies.
Materials and Methods
Insect Collection
Adult tsetse flies, Glossina pallidipes (Diptera: Glossinidae) were isolated from Nguruman escarpment, Kenya in March 2015 using Ngu traps. Bacteriomes were microscopically removed and placed in RNAlater (Invitrogen, CA). Flies included in this study were all trypanosome free as verified through microscopic analyses of their midguts.
Molecular Phylogenetic Analyses
The bacteriomes of G. pallidipes field collected flies were dissected and DNA was extracted following the Holmes–Bonner protocol (Holmes and Bonner 1973). DNA samples were subject to PCR amplification with general eubacterial 16S rRNA primers, 27F′ and 1492R′ (Lane 1990; Weisburg etal. 1991) (Ta= 50 °C; 28 cycles). Amplicons were ligated into pGEM-T vector (Promega, WI) and Escherichia coli JM109 cells (Promega, WI) were transformed. Colonies were verified for a 16S rRNA insertion and sequenced at the West Virginia University’s Department of Biology Genomics Center on an ABI 3130xl analyzer Applied BiosystemsCA) using a 3.1 BigDye protocol (Applied Biosystems). All sequences were quality trimmed and assembled into contigs using CLUSTALW (available at www.ebi.ac.uk/Tools/msa/muscle/; last accessed January 3, 2017). A consensus 16S rRNA nucleotide sequence for the G. pallidipes Wigglesworthia isolate was deposited in the NCBI Genbank database under accession number MF148851.
The evolutionary model used for Bayesian analyses (General time-reversible plus invariant sites plus gamma; GTR + I+ G) of 16S rRNA sequences was determined using the Akaike Information Criterion in MRMODELTEST 2.3 (Nylander 2004). Bayesian analyses were performed in MRBAYES 3.2.6 (Ronquist and Huelsenbeck 2003) with the number of categories used to approximate the gamma distribution set at four. Additionally, six Markov chains (Larget and Simon 1999) were run for 3,000,000 generations. Posterior probability (PP) values were calculated, with stabilization of model parameters (i.e., burn-in) occurring at 2,800,000 generations (standard deviation of split frequencies < 0.01). Every 100th tree following stabilization (burn-in) was sampled to calculate a 50% majority-rule consensus tree. All trees were constructed using the program FIGTREE v1.4.3 (http://tree.bio.ed.ac.uk/software/figtree/; last accessed January 3, 2017).
RNA Extraction, Illumina Library Preparation, Sequencing, and Genome Aligning
The bacteriomes of 12 mature tsetse flies were pooled for one biological sample, resulting in the construction of six biological samples analyzed from male and female adult samples. Bacteriome samples were homogenized and total RNA was extracted using a MasterPure RNA purification kit (Epicentre, Madison, WI) according to the manufacturer’s protocol for tissue samples. DNA was removed from the RNA samples using a Turbo DNA-free kit (Ambion, Austin, TX) following the rigorous DNase treatment option. RNA of sufficient quality for cDNA synthesis was confirmed using the Agilent 2000 Bioanalyzer RNA Nano chip. RNA samples were subsequently processed with a Ribo-Zero magnetic kit for Gram-negative bacteria (Epicentre, Madison, WI) according to the manufacturer’s protocol. The resulting mRNA-enriched RNA was then purified using an RNeasy MinEluteCleanup kit (Qiagen, Valencia, CA) and eluted in RNAse-free H2O. The eluted RNA (∼1 µg) was then processed using a Kapa stranded mRNA-seq kit (Kapa Biosystems, Wilmington, MA), with the omission of the poly(A) pulldown, by the WVU Genomics Core Facility. The resulting cDNA libraries were sequenced using the Illumina HiSeq 1000 platform (2 by 51 bp) at Marshall University. Following sequencing, raw reads were postprocessed in order to remove Illumina adapters/primer sequences.
FASTQC analysis was performed on the RNA-Seq data sets to validate read quality. In order to capture both Wigglesworthia and tsetse fly reads, the Kallisto–Sleuth pipeline (Bray etal. 2016) was first used for the identification and quantification of G. pallidipes-specific read counts based on the genome available at Vectorbase (https://www.vectorbase.org/organisms/glossina-pallidipes, last accessed February 16, 2015, Gpal1.2 version). Following the parsing out of tsetse-specific reads from the total pool of bacteriome reads, the remaining sequences were mapped to the Wigglesworthia morsitans genome (NC_016893.1; Rio etal. 2012) using the STAR alignment tool (Dobin and Gingeras 2015). TPM (Transcripts per Million) was used as a measure of gene expression (Li etal. 2010). Relative fold differences in gene expression between samples were determined as a ratio of each TPM.
Differential Expression and Gene Ontology Analyses
Differential expression profiles of specific loci between male and female bacteriomes were identified using DESeq (Anders and Huber 2010), Kallisto–Sleuth, and ANOVA with an internal multiple tests correction in R using custom scripts (only performed for the Wigglesworthia data set with scripts available upon request). Transcripts were considered differentially expressed if showing an adjusted P value ≤ 0.05. A web-scraping Python script (available at https://github.com/nathantspencer/auto_vectorbase; last accessed October 26, 2016.) merged biologically relevant information obtained from VectorBase to all tsetse genes with significant differences in expression levels between the bacteriomes of different sexes. For those loci that lacked any associated VectorBase annotation, NCBI blastx analyses to the nonredundant protein sequences (nr) database were performed with results filtered to retain only hits with an E-value of < 1e−10 and a BitScore of >50. TRANSPORTDB 2.0 (Elbourne etal. 2017) (http://www.membranetransport.org/transportDB2/index.html; last accessed June 15 2017) was used to describe the predicted cytoplasmic transport protein complement within the Wigglesworthia (WGM) genome with those identified then used to assess transporter expression within field flies. Further, sequences were assigned to Gene Ontology (GO) terms falling within biological process, molecular function and cellular component according to GO hierarchy using BLAST2GO (Conesa etal. 2005; https://www.blast2go.com/home; last accessed March 14 2017). Differentially expressed GO terms were identified using Fisher’s exact test followed by a False Discovery Rate (FDR) corrections. Raw RNA-Seq data were uploaded to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (Bio-project PRJNA387614).
Real-Time qRT-PCR Gene Expression Analyses of Select Wigglesworthia Genes
Tsetse flies (G. morsitans) were reared in standard conditions and mated as described previously (Snyder etal. 2015). Tsetse flies, of known ages and female fecundity status, were sacrificed and bacteriomes dissected. Total RNA was isolated following the TRIzol protocol (Invitrogen, CA) and treated with DNAseI (Ambion, TX). First strand cDNA synthesis was performed with 140 ng total RNA, a 2 μM primer cocktail of gene specific 3′ end primers (supplementary table S6, Supplementary Material online) and Superscript Reverse Transcriptase II (Invitrogen) following manufacturer instructions. qRT-PCR was performed with SsoFast EvaGreen supermix (Bio-Rad), 0.4 mM gene-specific primers designed by the Primer3 server (supplementary table S6, Supplementary Material online), and 2 μl of cDNA template in a Bio-Rad CFX96 real-time PCR detection system (Bio-Rad, CA) with 35 amplification cycles. Primers were checked with the BLAST tool at NCBI to exclude potential unspecific amplification. Additionally, primer specificity was confirmed by a melting curve analysis where the dwell temperatures increased from 65 to 95 °C in 0.5 °C increments every 5 s. Primer efficiency was evaluated using the standard curve method and ranged from 90 to 110%. The threshold cycle (2−ΔΔCT) method was used to calculate relative expression. The Wigglesworthia rpsC (30S ribosomal subunit) was used as the reference gene. At least five individual bacteriomes were processed per group, with each sample being analyzed in triplicate, and the average quantification cycle (CT) obtained. The fold change in gene expression, as compared with the same sex teneral stage (i.e., newly emerged, nonfed adult), was determined for each sample. Negative controls were included in all amplification reactions. Values are represented as the mean ± the standard error of the mean (SEM) with statistical significance determined with ANOVA followed by Tukey’s multiple comparisons post hoc analyses.
Investigation of Tsetse Transporter Expression within Tsetse Tissues
To examine the expression of the Glossina species orthologs of reduced folate carrier and thiamine transporter (i.e., GPAI043750 and GPAI022398, respectively), semiquantitative reverse transcription PCR (RT-PCR) analyses were performed for GMOY005445, GMOY009200, GFUI022020, and GFUI042523. Tsetse flies were sacrificed and tissues (bacteriomes and heads) dissected. Total RNA was isolated following the TRIzol protocol (Invitrogen, Carlsbad, CA), treated with DNase I (Ambion), and verified to be free of DNA contamination through PCR using RNA only as the template. First-strand cDNA synthesis was performed with 140 ng RNA, a 2 μM primer cocktail of gene-specific 3′ end primers and SuperScript II reverse transcriptase (Invitrogen). Second-strand synthesis was performed with primers specific for the complementary 5′ end of the gene and 2 μl of cDNA template for 35 amplification cycles. Primers for GMOY005445 included: forward: 5′-CTCAAAGCCACCACCTTGTT-3′, reverse: 5′-CAACGATGACAAGACGGCTA-3′; for GMOY009200: forward: 5′-GAAGGTGATTGGTGGACTGG-3′, reverse: 5′-TGAGGATACAGAGGCAGGAA-3′; for GFUI022020: forward: 5′-TAGCGGCTACACAAATGCCA-3′, reverse: 5′-AGCCACACGTCTTCAACGAT-3′; and for GFUI042523: forward: 5′-TCGGGTAACATCGCACACAA-3′, reverse: 5′-CTTCCTCCAAATGACGGGCT-3′. The constitutively expressed β-Tubulin gene (forward: 5′-GATAACGAGGCCCTGTACGA-3′, reverse: 5′-GATAACGAGGCCCTGTACGA-3′) was used as a loading control and to verify RNA integrity. At least six bacteriomes were processed per group. Negative controls were included in all amplification reactions.
Statistical Analyses
All statistical analyses were performed with GraphPad Prism software (version 6.0) with the identification of specific analyses performed indicated throughout text.
Results
General Transcriptome Features
Six cDNA libraries were generated from the total homogenates of sex-specific bacteriomes isolated from Nguruman, Kenya field-captured adult tsetse flies. These libraries consisted of the homogenized bacteriomes of same sex adults of different ages, mating, and feeding states all obtained within a 24 h sampling period in March 2015. The libraries were sequenced using Illumina HiSeq technology, resulting in ∼23 million high-quality paired-end reads (2 x 51 bp in length) for each biological sample, with a combined total for all libraries of 140,008,786 cleaned reads. Barcodes and adapters were trimmed during the demultiplexing process. A total of 139,635,056 reads (∼99.7% of cleaned reads) were identified as having tsetse origin whereas 373,730 reads (∼0.3% of cleaned reads) mapped to Wigglesworthia genes (fig. 1A). There were no significant differences in the mean abundance of either Wigglesworthia or tsetse reads between male and female specific libraries (Student’s t-test; P = 0.7952 and P = 0.9302, respectively).
Fig. 1.

—A summary of RNA-Seq libraries. (A) Number (Log10 base) of total RNA-Seq reads mapping to either tsetse (G. pallidipes) or Wigglesworthia reference genomes. 1 Standard Error of the Mean (S.E.M.) bars are depicted. (B) Percentage of the Wigglesworthia genome found to be expressed within each of the bacteriome libraries, shared and uniquely (relative to other same sex libraries) expressed proportions are indicated. (C) A total of 273 genes lacked reads in any library, the distribution of their Clusters of Orthologous Genes (COGs) designations are conveyed in the pie chart.
Wigglesworthia-Based Analyses
To investigate the phylogenetic relationship of the G. pallidipes Wigglesworthia isolate to other Gammaproteobacteria, we first performed Bayesian analyses based on 16S rRNA sequences generated through this study and additional sequences obtained from the GenBank database. Tsetse bacteriomes, obtained from G. pallidipes collected from Nguruman, Kenya, were dissected and DNA extracted for the PCR amplification and sequencing of eubacteria 16S rRNA. Single amplicons with highest nucleotide identities to the Wigglesworthia 16S rRNA isolated from other tsetse species were consistently obtained when amplifying from the bacteriome-generated DNA. Phylogenetic analyses of 16S rRNA sequences, generated through Bayesian analyses, indicates a close relation with a high posterior probability value (0.9483) between the Wigglesworthia G. pallidipes and G. morsitans (WGM) isolates (supplementary fig. S1, Supplementary Material online). Depicting a substantially more distant relation to the Wigglesworthia isolate from G. pallidipes is the G. brevipalpis Wigglesworthia isolate (WGB, the other available reference genome). WGB is located within an adjacent sister clade reflective of the ancestral state of G. brevipalpis within the tsetse lineage (Petersen etal. 2007; Dyer etal. 2008) and codiversification of this obligate symbiont through tsetse radiation (Chen etal. 1999). The higher sequence identity between the Wigglesworthia G. pallidipes and WGM 16S rRNA sequences (98% sequence identity vs. 95% sequence identity between G. pallidipes and WGB) validated the use of the WGM isolate as a reference genome for the mapping of Illumina reads.
All libraries had equivalent levels (44–48%) of the Wigglesworthia genome expressed (fig. 1B) with no significant differences between male and female bacteriomes in the percentage of the genome being expressed (Chi-square; P = 0.6390). Small portions of each library (2–6%) were exclusive in expression relative to other libraries within the same sex (fig. 1B). A total of 418 genes were expressed within at least 1 bacteriome library, corresponding to ∼60% of the Wigglesworthia genome being transcribed during tsetse adulthood. Moreover, a total of 273 Wigglesworthia loci, distributed among 18 Cluster of Orthologous Genes (COGs), lacked Illumina reads within all the bacteriome libraries (fig. 1C and supplementary table S1, Supplementary Material online). The most widely represented COG lacking expression was translation, ribosomal structure and biogenesis, representing ∼26% of COGs.
Gene Ontology (GO) Annotation
BLAST2GO (Conesa etal. 2005) was used to assign 370 protein-coding Wigglesworthia genes to associated Gene Ontology (GO) classifications for cellular components, molecular functions and biological processes. The resulting 1,106 GO terms ranged from levels 2 to 14 (supplementary fig. S2, Supplementary Material online), including parent and child terms, with 403 accessions within biological process, 538 in molecular function, and 165 in cellular components. A total of 23 GO terms were obtained when examining root level 4, with 3 to cellular components, 14 to biological processes and 6 to molecular functions (fig. 2A). Among cellular components, cytoplasm was the most enriched category (∼58% of all genes within cellular components). Within biological processes, cellular nitrogen compound metabolism (∼15% of all genes within biological processes) was the top process. Lastly within molecular functions, nucleotide binding and anion binding (∼21% of all genes within molecular functions for both categories) was the top process identified. An enrichment analysis using Fisher’s exact test with a false-discovery rate (FDR) correction found no significant differences between male and female bacteriomes (Fisher’s exact test, P > 0.05) in the number of Wigglesworthia genes expressed and their respective GO distribution. Within all adult bacteriomes, there was a universal enrichment of expressed Wigglesworthia genes with GO terms associated with metabolism and transport (i.e., 57% of total GO categories at root level 4). Cellular nitrogen compound metabolism was the most highly represented GO category (∼10% of all genes with GO terms at root level 4), a group that includes loci involved in the synthesis of amino acids, vitamins and cofactors.
Fig. 2.

—Gene ontology (GO) distribution of Wigglesworthia transcriptome. (A) GO classifications at root level 4 for each Wigglesworthia protein-coding gene (n = 370) expressed. GO terms were determined using BLAST2GO followed by enrichment analyses using Fisher’s exact test with a false rate discovery correction. Red boxes indicate GO classifications associated with metabolism and the blue circle highlights transport. (B) GO classifications normalized to average TPM values. There were no statistically significant differences in mean TPM values either within a given category between male and female bacteriomes or between different categories within the same sex.
To examine whether GO classifications differed in their mean expression levels, categories were averaged in their respective TPM values and comparatively analyzed (fig. 2B). Individual Student’s t-test analyses found no significant differences (P > 0.05) in the mean TPM values of individual GO categories between male and female bacteriomes (fig. 2B). Additionally, no significant statistical differences in the mean expression between GO categories within a sex (for males; P = 0.9968, for females; P = 0.9908) were observed.
Tsetse Sex-Specific Differences in Wigglesworthia Expression
Principal Component Analysis (PCA) revealed that only 24% (PC2) of the Wigglesworthia count variation between male and female bacteriome libraries (fig. 3A) could be accounted for by sex. The variation in symbiont expressed genes between tsetse male and female bacteriomes is likely driven by genes that either lacked expression or were overexpressed in the bacteriome of one sex relative to the other. A total of nine genes was expressed by Wigglesworthia exclusively within female or male bacteriomes (table 1), albeit with relatively low TPM values (i.e., 8.1–34.6, whereas the average TPM of libraries was 2,392). Within only female bacteriomes, Wigglesworthia reads pertaining to; lolD, an ATP binding component/lipoprotein transporter, pyrG, a CTP synthetase and WIGMOR_RS03465, a putative membrane protein transporter, were identified. Within only male bacteriomes, reads associated with; trkA, a potassium uptake protein, pyrI, a component of pyrimidine biosynthesis, dnaB, a DNA helicase, rseP, a zinc metalloprotease, ahpC, an alkyl hydroperoxide reductase, and mrdB, involved in cell elongation/peptidoglycan synthesis were found. Additionally, a small number of Wigglesworthia genes significantly differed in expression between female and male bacteriomes (fig. 3B and supplementary table S2, Supplementary Material online). Upon comparison of the results generated by the different analyses (DESeq, Kallisto–Sleuth and R analyses), a consensus of four Wigglesworthia loci were found to differ in expression between tsetse sexes; flhD, flagellar transcriptional activator, motA, the proton conductor of flagella motor, WIGMOR_RS02980, a cysteine ABC transporter binding protein and infB, a translation initiation factor IF2, which were all more highly expressed within female bacteriomes.
Fig. 3.
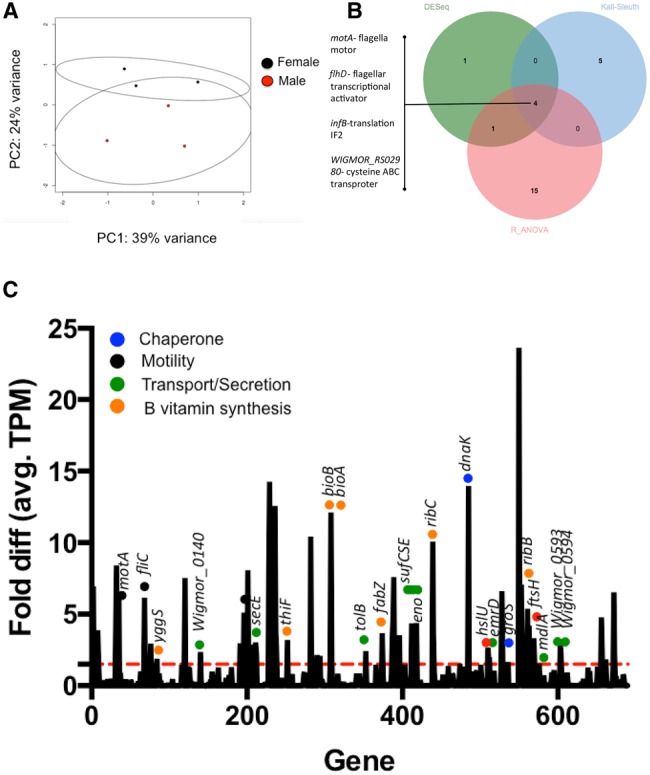
—Tsetse sex-specific differences in Wigglesworthia expression. (A) Principal components analysis (PCA) of Wigglesworthia expression within male and female bacteriomes. Each point represents the Wigglesworthia transcriptomic signature of one RNA-Seq library. The directionality of separation was observed in the Principal Component (PC) 2, with 24% variance. 95% confidence circles are indicated. (B) A small number of differentially expressed Wigglesworthia genes were found between female and male bacteriomes. A consensus of 4 Wigglesworthia loci was found to be significantly higher expressed in female bacteriome libraries utilizing different analytical methods; DESeq (Differential expression of RNA-Seq data), Kall–Sleuth (Kallisto–Sleuth), and R_ANOVA (R statistical package, Analysis of variance). (C) Histogram showing the distribution of fold diff (avg. TPM) across the Wigglesworthia genome for compiled RNA-Seq libraries. The Wigglesworthia genome is depicted linearized, colored circles are associated with specific functional groups, the red dashed line indicates 1.5 fold diff. (avg. TPM).
Table 1.
Wigglesworthia Genes Exclusively Expressed within the Bacteriomes of Either Female or Male G. pallidipesa
| COG Category | Gene | Sequence Identifier | Mean TPM | Description |
|---|---|---|---|---|
| Female | ||||
| Cell wall/membrane/ envelope biogenesis | lolD | WIGMOR_RS00540 | 21.5 | ATP binding component lipoprotein transporter |
| Nucleotide transport and metabolism | pyrG | WIGMOR_RS02070 | 13.9 | CTP synthetase |
| Hypothetical | WIGMOR_RS03465 | 25.9 | Putative membrane protein transporter | |
| Male | ||||
| Nucleotide transport and metabolism | trkA | WIGMOR_RS00660 | 8.1 | Potassium uptake protein |
| pyrI | WIGMOR_RS00770 | 34.6 | Pyrimidine biosynthesis | |
| Replication, recomb. and repair | dnaB | WIGMOR_RS01295 | 11.6 | DNA helicase |
| rseP | WIGMOR_RS01905 | 9.7 | Zinc metalloprotease | |
| Defense mechanisms | ahpC | WIGMOR_RS01355 | 26.4 | Alkyl hydroperoxide reductase |
| Cell cycle, control and division | mrdB | WIGMOR_RS3065 | 14.3 | Cell elongation/ peptidoglycan synthesis |
Expression observed in ≥2 libraries.
Due to the little variation in expression between tsetse sexes, we decided to then pool all the libraries to characterize global Wigglesworthia expression patterns of adult bacteriomes within field tsetse. The genes, groEL and cspE, had the highest abundance of mapped reads with TPM values of 70- and 75-fold higher than the average TPM, respectively. The groEL gene encodes a member of the chaperonin-60 family and has previously been associated with constitutive overproduction by Wigglesworthia throughout tsetse development (Aksoy 1995b) and also by other insect-associated obligate endosymbionts (Ishikawa and Yamaji 1985; Charles etal. 1997). Chaperonins assist in the correct assembly of proteins, including the refolding of denatured polypeptides, and their translocation (Goloubinoff etal. 1989; Goloubinoff etal. 1989; Nakamoto etal. 2014), which may be of particular importance given the high mutational biases within the genomes of many of these ancient obligate symbionts (Fares etal. 2002, 2005; Tokuriki and Tawfik 2009; McCutcheon and Moran 2011). Previous reports of groEL as one of the more highly transcribed Wigglesworthia genes serve to validate our RNA-Seq results. Interestingly, cspE, annotated as a cold shock protein and DNA-binding transcriptional repressor, is the gene under the highest purifying selection (dN/dS = 0) within the Wigglesworthia genomes (Akman et al 2002; Rio etal. 2012). Upon excluding the saturating signals of groEL and cspE which introduce bias in the determination of the average TPM values, 65 loci were found to have ≥ 1.5 fold higher TPM values in comparison to the average TPM (fig. 3C and supplementary table S3, Supplementary Material online). Many of these genes are associated with roles as chaperones (dnaK, groES), proteases involved in the synthesis of the cell envelope (hslU and ftsH), supporting active division by Wigglesworthia within tsetse bacteriomes, and in cellular motility (motA and fliC). Additional genes, exhibiting ≥ 1.5 fold higher TPM values in comparison to the average TPM, included those putatively involved in protein translocation (secE), amino acid transport (Wigmor_0593, Wigmor_0594), and multidrug transport pumps (emrD, a member of the major facilitator superfamily [MFS] and mdlA, a member of the ATP-binding cassette [ABC] superfamily), which have likely been repurposed for the transfer of molecules other than antibiotic drugs. In other Gram-negative bacteria, including Pseudomonas aeruginosa, Neisseria gonorrhoeae, and Salmonella typhimurium, these efflux pumps have been reused to excrete host-produced toxins, enabling the adaptation and survival within these niches (reviewed in Sun etal. [2014]). Loci involved in the recognition of environmental stimuli (the sensor histidine kinase cpxA) and towards B vitamin synthesis (i.e., thiamine [B1], riboflavin [B2], pyridoxine [B6], biotin [B7], and folate [B9]) also exhibited >1.5 fold TPM values. Further, loci involved in the assembly of [Fe–S] clusters (sufCES) had >1.5 fold TPM values. In Wigglesworthia, the sulfur utilization factor (suf) unit is the only intact Fe–S cluster biosynthetic machinery available within the genome, with the exception of the cysteine desulfurase, iscS, traditionally associated with the iron sulfur cluster (Isc) formation system (Johnson etal. 2005). Fe–S clusters are present in proteins with a great variety of functions including electron transfer, posttranscriptional gene regulation, providers of free iron, DNA damage repair, and substrate binding during catalysis (reviewed in Brzoska etal. [2006]). Moreover, Fe–S containing proteins are direct players in many key aspects of metabolism, participating in the biosynthesis of essential cofactors and enzymes. For example, B vitamin metabolism involves Fe–S cluster containing enzymes. Specifically, the pyrimidine moiety for the thiamine cofactor is generated through the conversion of AIR into HMP-P, which is catalyzed by ThiC, an enzyme that holds a 4Fe–4S cluster (Raschke etal. 2007; Chatterjee etal. 2008). Thiamine biosynthesis also requires IscS as a S donor via ThiI (Palenchar etal. 2000), as well as the participation of another Fe–S protein called ThiH (Leonardi etal. 2003; Park etal. 2003; Leonardi and Roach 2004). Similarly, the last step in biotin synthesis is catalyzed by biotin synthase (BioB), another iron–sulfur cluster containing enzyme. At this step, the sulfur atom comes from one of two distinct iron–sulfur clusters present in the enzyme, a 2Fe–2S and a 4Fe–4S (Jarrett 2015), for the conversion of dethiobiotin to biotin (Ugulava etal. 2001; Jameson etal. 2004).
Lastly, 38 of the 55 putative transporters (∼69%) identified within the Wigglesworthia genome using TRANSPORTERDB (Elbourne etal. 2017) had Illumina reads mapping back to these loci (supplementary fig. S3, Supplementary Material online). A total of 10 of these 38 (∼26%) transporters were identified as having ≥ 1.5 TPM in comparison to the average TPM of bacteriome libraries, including the aforementioned efflux pumps (mdlA and emrD), a GlnQ family glutamine transport ATP-binding protein (Wigmor_0594), a cystine ABC transporter substrate binding protein (Wigmor_0593) and several components of energy generating ATP synthase.
qPCR Validation of Wigglesworthia Gene Expression within Heterospecific Tsetse
To further verify the transcriptional activity of genes, we describe the expression of select genes within the bacteriomes of a heterospecific tsetse fly, G. morsitans. A total of 8 Wigglesworthia genes (ribB (3,4-dihydroxy-2-butanone-4-phosphate synthase), ribC (riboflavin synthase, alpha subunit), bioB (biotin synthase), bioA (adenosylmethionine-8-amino-7-oxononanoate aminotransferase), mdlA (putative ATP-binding component of multidrug ABC transporter), cspE (DNA binding transcriptional repressor), fliC (flagellin), and Wigmor_0594 (GlnQ family glutamine transport ATP binding protein) identified by RNA-Seq analyses to have ≥1.5 fold greater than average TPM were chosen for real time quantitative gene expression assays. Because we were interested in identifying whether Wigglesworthia symbionts within different tsetse species also actively transcribed these genes and, further, whether specific adult ages and/or fecundity status were likely driving an increase in library TPM values (information that could not be captured with our field collections), these analyses were performed with RNA isolated from tsetse colony flies of known ages and mating status.
Similar to Wigglesworthia transcript abundance within G. pallidipes adult bacteriomes, the G. morsitans colony flies exhibited expression of all eight loci throughout adulthood (fig. 4). Only 1 gene, bioB, a biotin synthase, demonstrated no significant differences in gene expression upon the comparison of flies of different adult ages and female fecundity status. Although in both sexes, there was an increase in bioB expression as tsetse aged, a trend even more significantly apparent with bioA (involved in the conversion of 8-amino-7-oxononanoate to 7,8-diamino nonanoate within the biotin [B7] metabolic pathway), indicating a higher demand for symbiont-mediated production of biotin through the progression of tsetse adulthood. Further, there was a significantly higher expression of Wigglesworthia bioA within the bacteriomes of 2-week-old mated relative to same age virgin females supporting an additional role for biotin during pregnancy. The cspE locus, encoding a cold shock protein/DNA binding transcriptional repressor, demonstrated significant differences in expression only between different aged males with 4-week-old males exhibiting the highest fold change in gene expression relative to younger males. Four other Wigglesworthia loci examined; Wigmor_0594, ribB, ribC, and mdlA, exhibited significant expression differences within the bacteriomes of female adults of different ages and mating status, but no significant differences upon the comparison of distinctly aged males, suggesting more prominent roles in female biology. The two Wigglesworthia loci involved in riboflavin (B2) synthesis, ribB, involved in the conversion of ribulose 5-phosphate to 3,4-dihydroxy-2 butanone-4-phosphate, and ribA, mediating the final step in riboflavin synthesis from 6,7-dimethyl-8-ribityl lumazine, had the highest level of expression within the bacteriomes of 2 week pregnant females. The higher expression of these genes suggests a greater need for symbiont-produced riboflavin during pregnancy potentially increasing maternal resources and aiding in embryogenesis. The two loci involved in transport functions, mdlA and Wigmor_0594, had significantly higher expression levels within the bacteriomes of 4-week-old virgin females and teneral and 2-week-old mated females, respectively. Interestingly, fliC had significantly higher expression within the bacteriomes of younger relative to older flies within both sexes (P ≤ 0.05). Cumulatively, these results characterize the gene expression dynamics within a heterospecific tsetse and show that Wigglesworthia orthologs remain transcribed and may additionally vary in transcript abundance to adapt to the needs of different aged adults and fecundity status. These biological differences may have also driven the increase in read abundance within our RNA-Seq libraries from the field. Importantly, these analyses provide insight into specific symbiont biochemical processes that vary in activity depending on specific host traits that could not be obtained through the homogenization of bacteriomes of adult flies, which were pooled regardless of age or mating status, within our field studies. Like many insect-symbionts, the small and compact genome of Wigglesworthia is a result of a long and complex evolutionary history associated with tsetse (reviewed in McCutcheon and Moran 2011). It is hypothesized that the majority of genes retained in Wigglesworthia’s small genome simply preserve the relationship with the host. Yet, the demands on the symbiosis, and consequently effects towards impacting symbiont gene expression, may vary in response to host age, mating and nutritional status.
Fig. 4.

—Expression kinetics of Wigglesworthia genes within a sister tsetse species, G. morsitans morsitans. qRT-PCR based fold change gene expression was analyzed through the threshold cycle (2−ΔΔCT) method. The results for fliC, Wigmor_0594, ribB, ribC, mdlA, cspE, bioB, and bioA were normalized to rpsC within the bacteriomes of various aged adult males, females and mating status and compared with same sex tenerals. Each point represents the expression (Average of triplicate Cts) of an individual fly with mean (represented by the horizontal bar) and ± 1 Standard Error of the Mean (SEM) indicated. N > 5 bacteriomes per group, asterisks indicate statistically significant differences between the bacteriomes. ***P < 0.001, **P < 0.01 and *P < 0.05; Gm: Glossina morsitans, wk: week.
G. pallidipes-Based Analyses
A total of 14,119 G. pallidipes gene transcripts had Illumina reads mapping back, representing ∼71% of the total potential tsetse transcripts (16 Feb 2015, VectorBase, https://www.vectorbase.org/organisms/glossina-pallidipes/iaea/gpali12) being expressed within the bacteriome organ. Female flies had reads to 594 more gene transcripts than males (13,098 vs. 12,504), with 1,600 and 1,000 transcripts unique to females and male bacteriomes, respectively.
GO Annotation
BLAST2GO (Conesa etal. 2005) was used to assign 13,786 protein-coding tsetse genes to associated GO classifications for cellular components, molecular functions and biological processes. The resulting 23,771 GO terms ranged from levels 2 to 15 (supplementary fig. S4, Supplementary Material online), including parent and child terms, with 8,405 accessions within biological process, 10,840 in molecular function, and 4,509 in cellular components. A total of 18 GO terms for tsetse transcripts were obtained when examining root level 4, with four to cellular components, eight to biological processes, and six to molecular functions (fig. 5A). Among cellular components, integral component of membrane was the most enriched category (∼32% of all genes within cellular components). Within biological processes, macromolecule metabolic process (∼22% of all genes within biological processes) was the most enriched process. Lastly within molecular functions, nucleic acid binding (∼19% of all genes within molecular functions) was the top process identified. Within all adult bacteriomes, and highly mirroring the Wigglesworthia symbiont profile (fig. 5A), there was a universal enrichment of expressed tsetse genes with GO terms associated with metabolism and transport (i.e., 44% of total GO categories at root level 4) likely facilitating the coordination and shuffling of metabolite production between these two organisms during the symbiosis. More specifically, GO category proportions between tsetse and Wigglesworthia have a positive correlation to each other when analyzed using simple linear regression (SLR) (r = 0.4149, P = 0.0438 for a test of 0 SLR slope). Lastly, multiple expressed tsetse loci were associated with roles in conveying substrates into and out of tsetse bacteriocytes with a number relevant to vitamin transport including thiamine (thiamine pyrophosphate carrier proteins [GPAI038258 and GPAI008049], and thiamine transporter 1 [GPAI022398], riboflavin [solute carrier family 52, riboflavin transporter member 3-A {GPAI045057}], proton-coupled folate transporter [GPAI036223], reduced folate carrier [GPAI043750], solute carrier family 52, riboflavin transporter, member 3-A [GPAI045057] and multivitamin transporters [GPAI012112, GPAI014644, and GPAI028931]. Using Web Apollo [Lee etal. 2013] to identify orthologs within other tsetse species, we also confirmed that reduced folate carrier [GPAI043750]) and thiamine transporter-1 (GPAI022398) were both expressed within the bacteriomes of adults of two other tsetse species, G. morsitans and G. fuscipes (fig. 6) supporting the significance of these genes throughout Glossina. Further, the expression of both these loci within the head samples of G. fuscipes indicates a systemic expression that may address the ubiquitous nature of vitamin requirements throughout the body.
Fig. 5.

—Gene ontology (GO) distribution of G. pallidipes transcriptome. (A) GO classifications at root level 4 for each G. pallidipes and Wigglesworthia gene expressed. The red box indicates GO classifications associated with metabolism and the circle highlights transport. (B) GO classifications of expressed tsetse genes normalized to average TPM values. There were no statistically significant differences in mean TPM values within a given category between male and female bacteriomes (Mann–Whitney; P > 0.05). A Kruskal–Wallis one-way analysis of variance (ANOVA) and Dunn’s multiple comparisons test were performed to determine whether mean TPM values differed between categories within a sex. F-tests were applied to assess the homogeneity of variances. The normality of TPM distributions was determined with a goodness-of-fit test. Mean TPM values were logtransformed to satisfy normality. SEMs are depicted. (C) A small number of differentially expressed G. pallidipes genes were found between female and male bacteriomes. A consensus of 17 G. pallidipes loci was found to significantly differ between male and female bacteriomes upon comparisons utilizing different analytical methods.
Fig. 6.

—Semiquantitative RT-PCR analysis of tsetse transporters within tissues of different Glossina species. Columns show orthologs of reduced folate carrier (GPAI043750) and thiamine transporter 1 (GPAI022398). Rows correspond to the Glossina species indicated with orthologs identified using VectorBase and Web Apollo. The results shown are representative samples (n > 6) of each group. (A and B) TPM of the transporter loci in the six bacteriome libraries from Nguruman, Kenya field flies. (C–F) panels show the corresponding expression in two species of 2 week old tsetse flies from colony lines of G. morsitans (WVU) and G. fuscipes (Yale). Top row corresponds to the indicated transporter while the bottom row is β-tubulin expression (serving as a RNA integrity control). All samples are bacteriomes, except for those labeled with an asterisk (*), which represents RNA isolated from heads.
To examine whether categories differed in their mean expression levels between males and females, GO classifications were averaged in their respective TPM values and comparatively analyzed (fig. 5B). Individual Mann–Whitney analyses found no significant differences (P > 0.05) in the mean TPM values of distinct GO categories expressed between tsetse male and female bacteriomes (fig. 5B). Interestingly, female flies had genes binning within the nucleus GO category (n = 713) where as males lacked any associated loci within this category. Within a sex, multiple GO categories significantly differed in their mean TPM values (P < 0.0001 for both males and female flies, supplementary table S4, Supplementary Material online contains detailed P values).
Tsetse Sex-Specific Differences in Gene Expression within Bacteriomes
To identify tsetse genes that were found to differ in expression within the bacteriomes of the different sexes, DESeq and Kallisto–Sleuth analyses were used for the comparison of transcriptome libraries. Kallisto–Sleuth identified 82 loci, whereas DESeq identified 32 genes that were differentially expressed with statistical significance, with 17 in consensus between the two approaches (fig. 5C and supplementary table S5, Supplementary Material online). Of the loci found to be in consensus between the two analyses, the majority (13/17, 76%) exhibited significantly higher expression within male bacteriomes, including orthologs to suppressor of zeste 2, Su(z)2 (GPAI038413), multiple trypsins (GPAI012727 and GPAI012728), loci involved in lipid metabolism; low density lipoprotein receptor adaptor protein 1 (GPAI030993) and lipin (GPAI004503), and orthologs to the previously described male significant (Fontaine etal. 2003; Blagden etal. 2009); outer dense fiber protein 3-like protein 2 (GPAI039186) and a La related protein (GPAI010220). Within female bacteriomes, tsetse loci putatively involved in immunity related salivary c-type lectin (GPAI033252) and the transcriptional regulator protein FAM76A (GPAI025219) demonstrated significantly higher expression upon comparison to males.
Discussion
Animal alliances with bacteria are significant sources of biological diversification, having shaped evolutionary history (McFall-Ngai etal. 2013) as well as facilitating or constraining future adaptation to environmental change (Kiers etal. 2010; Wernegreen 2012; Kikuchi etal. 2016). Much of what we know about host–microbiota interactions is generated from the lab environment with the use of artificially bred or captive animals. Here, we characterize the transcriptomes obtained through parallel Illumina deep sequencing of two obligate symbiotic partners, the tsetse fly, G. pallidipes, and its ancient bacterial symbiont, Wigglesworthia, from Kenyan field populations. Although much has been discovered on the functional contributions of this important member of the tsetse microbiota through empirical investigations of established tsetse fly lines, little research has been dedicated towards understanding the coordinated activities between partners in the wild.
Through empirical studies involving colony flies, Wigglesworthia symbionts have been identified as indispensable players in tsetse energy processing by nutritionally supplementing the strict diet of its host with B vitamins including thiamine (B1), pyridoxine (B6) and folate (B9) (Snyder etal. 2010, 2012, 2015; Michalkova etal. 2014) which are lacking in vertebrate blood. As Wigglesworthia is essential for the development and fecundity of its tsetse host, we hypothesize that genes expressed within the bacteriome of field tsetse are integral components of the host–symbiont interface mediating nutrient synthesis and transfer, communication and regulatory control.
The interchange of metabolites within a microbial symbiosis is often the motivating factor for its formation and subsequent persistence. In the tsetse-Wigglesworthia association, the bacteria lie free within the cytoplasm of bacteriocytes (Aksoy 1995a), very different from many other endosymbionts that are surrounded by a secondary membrane which is host derived (Hinde 1971; von Dohlen etal. 2001; Baumann 2005). An additional distinction from other endosymbionts is that Wigglesworthia, despite drastic genome reductions, still encodes products that are integral to Gram-negative cell wall structure including lipopolysaccharide and peptidoglycan (Akman etal. 2002; Rio etal. 2012). Thus for Wigglesworthia-generated metabolites to gain access into the host cytoplasm of bacteriocytes, these substances would only have to pass through the bacterial dual membrane. Our field results indicate transcriptional activity for the majority of Wigglesworthia transporters and even more robust expression for transporters involved in multidrug efflux, glutamine and cystine ABC-binding and ATP synthases. These transporters are likely key mediators in the biochemical communication and metabolite translocation between partners. Following export from the Wigglesworthia cells, systemic distribution of provisioned nutrients is still necessary to address the nutritional demands of other body regions. We demonstrate, that in both field and colony flies of different species, tsetse transcribe various transporters within the bacteriome whose orthologs have been characterized to be involved in the specific transfer of vitamins/cofactors and likely mediating their dispersal to other tissues. Further, these transporters are also expressed in other fly tissue, indicating a role in the widespread distribution of symbiont-produced nutrients. Future studies should examine how tsetse and Wigglesworthia coordinate these different metabolic and physiological steps, particularly in regards to time, space and stoichiometry. In an analogous insect system, involving aphids and their ancient obligate Buchnera bacterial symbionts, the insect hosts have been shown to control the biosynthesis of essential amino acids through substrate feedback inhibition, specifically by regulating the supply of the precursor glutamine into bacteriocytes (Price etal. 2014). Here, we also show, using flies of known age and mating status, Wigglesworthia transcriptional activity does fluctuate likely addressing differences in nutrient requirements through host age and reproduction.
The majority of genes found to be expressed by both Wigglesworthia and tsetse within bacteriomes are involved in metabolism and transport, indicating that, similar to what has been demonstrated with tsetse colony lines, the production and transfer of nutrients is also pinnacle to this association within the field. An obvious symbiotic exchange between tsetse and Wigglesworthia is in amino acids (Wigglesworthia is an auxotroph for the majority of amino acids) and B vitamins (blood meals are deficient) as indicated through genome analyses, several lab based empirical studies and with the field studies described here. For instance, thiamine synthesis may either involve the integration of pyridoxal phosphate (B6), or purine metabolism, with cysteine that is likely provided by either the tsetse host or from the blood meal and imported through the Wigglesworthia cystine ABC transporter. These pathways converge for condensing the phosphorylated derivatives of 4-amino-5-hydroxymethylypyrimidine (HMP(PP) and 4-methyl-5-(β-hydroxyethyl) (THZ(P)) moieties for the formation of thiamine monophosphate (TMP). TMP can then be supplied to tsetse and the neighboring Sodalis symbionts (Snyder etal. 2010, 2012), which are both thiamine auxotrophs. Similarly, the majority of enzymatic activity necessary for biotin and riboflavin biosynthesis was characterized within the bacteriome tissues of field flies. It is important to note that there were steps that were lacking within Wigglesworthia transcriptome libraries in B vitamin biosynthesis pathways that could be due to a multitude of factors including; contributions by the tsetse host or Sodalis, inadequacy in the depth of coverage of transcriptome libraries, the spatial/temporal stability of the mRNA and/or protein (i.e., shorter half-lives), or the possibility of a not yet described enzyme performing the necessary interconversion.
Symbionts may have major implications towards host epigenetics (Kim etal. 2016). For example, the parasitic Wolbachia manipulates a host microRNA to interfere with a DNA methlytransferase thereby affecting Dengue virus manipulation in Aedes aegypti (Zhang etal. 2013). It remains to be seen whether Wigglesworthia metabolite production may also affect tsetse epigenetics, especially when this symbiont is the major or sole provider of B vitamins. For example, biotin may be added to histones to induce posttranslational modification that affects gene expression by changing the accessibility of genes towards transcription factors (Smith etal. 2007; Filenko etal. 2011). Additionally, folate provides methyl groups used for DNA methylation (Crider etal. 2012). Within insects, epigenetic modifications have been associated with significant biological processes including aging (Lin etal. 2005; Lockett etal. 2016) and caste determination (Lyko etal. 2010). Our field studies not only increase our basic knowledge of tsetse biology, but additionally, strongly support results generated with colony lines in regards to the nutrient exchange involved in the symbiosis. The tsetse microbiota may ultimately be used for innovative symbiont-based control mechanisms beyond paratransgenesis (Aksoy etal. 2008). Research efforts focused on eliminating essential microbiota members or inhibiting their important functional roles for vector fitness, particularly by altering life span, fertility or vector competency, may provide new paradigms towards tsetse-specific suppression methods.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank KALRO-BRI and Drs. Brian Weiss and Geoffrey Attardo for assistance in field collections, Niel Infante for help in bioinformatics, James Michael for sequencing efforts, Dr. Brittany Ott for input on Bayesian analyses and Dr. Kenneth Ryan for his expertise on statistics. We gratefully acknowledge the supply of G. morsitans and G. fuscipes pupae for tsetse fly colony lines from the Institute of Zoology, Slovak Academy of Sciences. Nathan Spencer created the publically available web scraper for Vector Base. Research in the Rio lab is supported through National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award number R01AI118789 (to R.V.M.R.).
Literature Cited
- Akman L, et al. 2002. Genome sequence of the endocellular obligate symbiont of tsetse, Wigglesworthia glossinidia. Nat Genet. 32(2): 402–407. [DOI] [PubMed] [Google Scholar]
- Aksoy E, et al. 2014. Analysis of multiple tsetse fly populations in Uganda reveals limited diversity and species–specific gut microbiota. Appl Environ Microbiol. 80(14): 4301–4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksoy S. 1995a. Wigglesworthia gen nov. and Wigglesworthia glossinidia sp. nov., taxa consisting of the mycetocyte-associated, primary endosymbiont of tsetse flies. Int J Syst Bacteriol. 45(4): 848–851. [DOI] [PubMed] [Google Scholar]
- Aksoy S. 1995b. Molecular analysis of the endosymbionts of tsetse flies: 16S rDNA locus and over-expression of a chaperonin. Insect Mol Biol. 4(1): 23–29. [DOI] [PubMed] [Google Scholar]
- Aksoy S, Weiss B, Attardo G.. 2008. Paratransgenesis applied for control of tsetse transmitted sleeping sickness. Adv Exp Med Biol. 627: 35–48. [DOI] [PubMed] [Google Scholar]
- Alam U, et al. 2011. Wolbachia symbiont infections induce strong cytoplasmic incompatibility in the tsetse fly Glossina morsitans. PLoS Pathog. 7(12): e1002415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W.. 2010. Differential expression analysis for sequence count data. Genome Biol. 11(10): R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attardo GM, et al. 2008. Analysis of milk gland structure and function in Glossina morsitans: milk protein production, symbiont populations and fecundity. J Insect Physiol. 54(8): 1236–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmand S, Lohs C, Aksoy S, Heddi A.. 2013. Tissue distribution and transmission routes for the tsetse fly endosymbionts. J Invertebr Pathol. 112(Suppl): S116–S122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann P. 2005. Biology bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 59: 155–189. [DOI] [PubMed] [Google Scholar]
- Benoit JB, Attardo GM, Baumann AA, Michalkova V, Aksoy S.. 2015. Adenotrophic viviparity in tsetse flies: potential for population control and as an insect model for lactation. Annu Rev Entomol. 60: 351–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagden SP, et al. 2009. Drosophila Larp associates with poly(A)-binding protein and is required for male fertility and syncytial embryo development. Dev Biol. 334(1): 186–197. [DOI] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, Pachter L.. 2016. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 34(5): 525–527. [DOI] [PubMed] [Google Scholar]
- Brzoska K, Meczynska S, Kruszewski M.. 2006. Iron-sulfur cluster proteins: electron transfer and beyond. Acta Biochim Pol. 53(4): 685–691. [PubMed] [Google Scholar]
- Charles H, Heddi A, Guillaud J, Nardon C, Nardon P.. 1997. A molecular aspect of symbiotic interactions between the weevil Sitophilus oryzae and its endosymbiotic bacteria: over-expression of a chaperonin. Biochem Biophys Res Commun. 239(3): 769–774. [DOI] [PubMed] [Google Scholar]
- Chatterjee A, et al. 2008. Reconstitution of ThiC in thiamine pyrimidine biosynthesis expands the radical SAM superfamily. Nat Chem Biol. 4(12): 758–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XA, Song L, Aksoy S.. 1999. Concordant evolution of a symbiont with its host insect species: molecular phylogeny of genus Glossina and its bacteriome-asssociated endosymbiont Wigglesworthia glossinidia. J Mol Evol. 48(1): 49–58. [DOI] [PubMed] [Google Scholar]
- Cheng Q, et al. 2000. Tissue distribution and prevalence of Wolbachia infections in tsetse flies, Glossina spp. Med Vet Entomol. 14(1): 44–50. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Aksoy S.. 1999. Tissue tropism, transmission, and expression of foreign genes invivo in midgut symbionts of tsetse flies. Insect Mol Biol. 8(1): 125–132. [DOI] [PubMed] [Google Scholar]
- Conesa A, et al. 2005. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21(18): 3674–3676. [DOI] [PubMed] [Google Scholar]
- Crider KS, Yang TP, Berry RJ, Bailey LB.. 2012. Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role. Adv Nutr. 3(1): 21–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Gingeras TR.. 2015. Mapping RNA-seq Reads with STAR. Curr Protoc Bioinformatics 51: 11-14–11-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE. 1998. Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic bacteria Buchnera. Annu Rev Entomol. 43(1): 17–37. [DOI] [PubMed] [Google Scholar]
- Dyer NA, et al. 2008. Molecular phylogenetics of tsetse flies (Diptera: Glossinidae) based on mitochondrial (COI, 16S, ND2) and nuclear ribosomal DNA sequences, with an emphasis on the palpalis group. Mol Phylogenet Evol. 49(1): 227–239. [DOI] [PubMed] [Google Scholar]
- Elbourne LD, Tetu SG, Hassan KA, Paulsen IT.. 2017. TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 45(D1): D320–D324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fares MA, Moya A, Barrio E.. 2005. Adaptive evolution in GroEL from distantly related endosymbiotic bacteria of insects. J Evol Biol. 18(3): 651–660. [DOI] [PubMed] [Google Scholar]
- Fares MA, Ruiz-González MX, Moya A, Elena SF, Barrio E.. 2002. Endosymbiotic bacteria: groEL buffers against deleterious mutations. Nature 417(6887): 398. [DOI] [PubMed] [Google Scholar]
- Filenko NA, et al. 2011. The role of histone H4 biotinylation in the structure of nucleosomes. PLoS One 6(1): e16299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine JM, Rest JS, Welsh MJ, Benndorf R.. 2003. The sperm outer dense fiber protein is the 10th member of the superfamily of mammalian small stress proteins. Cell Stress Chaperones 8(1): 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, Christeller JT, Gatenby AA, Lorimer GH.. 1989. Reconstitution of active dimeric ribulose bisphosphate carboxylase from an unfoleded state depends on two chaperonin proteins and Mg-ATP. Nature 342(6252): 884–889. [DOI] [PubMed] [Google Scholar]
- Goloubinoff P, Gatenby AA, Lorimer GH.. 1989. Groe heat-shock proteins promote assembly of foreign prokaryotic ribulose bisphosphate carboxylase oligomers in Escherichia coli. Nature 337(6202): 44–47. [DOI] [PubMed] [Google Scholar]
- Hill P, Saunders DS, Campbell JA.. 1973. The production of “symbiont-free” Glossina morsitans and an associated loss of female fertility. Trans Roy Soc Trop Med Hygiene 67(5): 727–728. [DOI] [PubMed] [Google Scholar]
- Hinde R. 1971. The fine structure of the mycetome symbiotes of the aphids Brevicoryne brassicae, Myzus persicae, and Macrosiphum rosae. J Insect Physiol. 17(10): 2035–2050. [DOI] [PubMed] [Google Scholar]
- Holmes DS, Bonner J.. 1973. Preparation, molecular weight, base composition, and secondary structure of giant ribonucleic acid. Biochemistry 12(12): 2330–2338. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Yamaji M.. 1985. Symbionin, an aphid endosymbiont-specific protein. 1. Production of insects deficient in symbiont. Insect Biochem. 15(2): 155. [Google Scholar]
- Jameson GNL, Cosper MM, Hernández HL, Johnson MK, Huynh BH.. 2004. Role of the [2Fe-2S] cluster in recombinant Escherichia coli biotin synthase. Biochemistry 43(7): 2022–2031. [DOI] [PubMed] [Google Scholar]
- Jarrett JT. 2015. The biosynthesis of thiol- and thioether-containing cofactors and secondary metabolites catalyzed by radical S-adenosylmethionine enzymes. J Biol Chem. 290(7): 3972–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DC, Dean DR, Smith AD, Johnson MK.. 2005. Structure, function, and formation of biological iron-sulfur clusters. Annu Rev Biochem. 74: 247–281. [DOI] [PubMed] [Google Scholar]
- Kiers ET, Palmer TM, Ives AR, Bruno JF, Bronstein JL.. 2010. Mutualisms in a changing world: an evolutionary perspective. Ecol Lett. 13(12): 1459–1474. [DOI] [PubMed] [Google Scholar]
- Kikuchi Y, et al. 2016. Collapse of insect gut symbiosis under simulated climate change. MBio 7(5): e01578-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Thairu MW, Hansen AK.. 2016. Novel insights into insect–microbe interactions: role of epigenomics and small RNAs. Front Plant Sci. 7: 1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ. (1990). 16S/23S rRNA sequencing In: Goodfellow ESM, editor. Nucleic acid techniques in bacterial systematics. Chichester (UK: ): John Wiley and Sons. [Google Scholar]
- Larget B, Simon DL.. 1999. Markov chain Monte Carlo algorithms for the bayesian analysis of phylogenetic trees. Mol Biol Evol. 16(6): 750–759. [Google Scholar]
- Lee E, et al. 2013. Web Apollo: a web-based genomic annotation editing platform. Genome Biol. 14(8): R93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi R, Fairhurst SA, Kriek M, Lowe DJ, Roach PL.. 2003. Thiamine biosynthesis in Escherichia coli: isolation and initial characterisation of the ThiGH complex. FEBS Lett. 539(1–3): 95–99. [DOI] [PubMed] [Google Scholar]
- Leonardi R, Roach PL.. 2004. Thiamine biosynthesis in Escherichia coli: invitro reconstitution of the thiazole synthase activity. J Biol Chem. 279(17): 17054–17062. [DOI] [PubMed] [Google Scholar]
- Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN.. 2010. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26(4): 493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MJ, Tang LY, Reddy MN, Shen CK.. 2005. DNA methyltransferase gene dDnmt2 and longevity of Drosophila. J Biol Chem. 280(2): 861–864. [DOI] [PubMed] [Google Scholar]
- Lockett GA, Almond EJ, Huggins TJ, Parker JD, Bourke AF.. 2016. Gene expression differences in relation to age and social environment in queen and worker bumble bees. Exp Gerontol. 77: 52–61. [DOI] [PubMed] [Google Scholar]
- Lyko F, Foret S, Kucharski R, Wolf S, Falckenhayn C, Maleszka R.. 2010. The honey bee epigenomes: differential methylation of brain DNA in queens and workers. PLoS Biol. 8(11): e1000506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma WC, Denlinger DL.. 1974. Secretory discharge and microflora of milk gland in tsetse flies. Nature 247(5439): 301–303. [Google Scholar]
- McCutcheon JP, Moran NA.. 2011. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 10(1): 13–26. [DOI] [PubMed] [Google Scholar]
- McFall-Ngai M, et al. 2013. Animals in a bacterial world, a new imperative for the life sciences. Proc Natl Acad Sci U S A. 110(9): 3229–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalkova V, Benoit JB, Weiss BL, Attardo GM, Aksoy S.. 2014. Obligate symbiont-generated vitamin B6 is critical to maintain proline homeostasis and fecundity in tsetse flies. Appl Environ Microbiol. 80(18): 5844–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran NA, McCutcheon JP, Nakabachi A.. 2008. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 42: 165–190. [DOI] [PubMed] [Google Scholar]
- Moran NA, Sloan DB.. 2015. The hologenome concept: helpful or hollow? PLoS Biol. 13(12): e1002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya A, Pereto J, Gil R, Latorre A.. 2008. Learning how to live together: genomic insights into prokaryote-animal symbioses. Nat Rev Genet. 9(3): 218–229. [DOI] [PubMed] [Google Scholar]
- Nakamoto H, et al. 2014. Physical interaction between bacterial heat shock protein (Hsp) 90 and Hsp70 chaperones mediates their cooperative action to refold denatured proteins. J Biol Chem. 289(9): 6110–6119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogge G. 1978. Aposymbiotic tsetse flies, Glossina morsitans morsitans obtained by feeding on rabbits immunized specifically with symbionts. J Insect Physiol. 24(4): 299–304. [DOI] [PubMed] [Google Scholar]
- Nogge G. 1981. Significance of symbionts for the maintenance of the optimal nutritional state for successful reproduction in hematophagous arthropods. Parasitology 82: 101–104. [Google Scholar]
- Nylander JAA. 2004. MrModeltest v2. Uppsala, Sweden: Evolutionary biology centre, Uppsala University. [Google Scholar]
- Pais R, Lohs C, Wu Y, Wang J, Aksoy S.. 2008. The obligate mutualist Wigglesworthia glossinidia influences reproduction, digestion, and immunity processes of its host, the tsetse fly. Appl Environ Microbiol. 74(19): 5965–5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palenchar PM, Buck CJ, Cheng H, Larson TJ, Mueller EG.. 2000. Evidence that ThiI, an enzyme shared between thiamin and 4-thiouridine biosynthesis, may be a sulfurtransferase that proceeds through a persulfide intermediate. J Biol Chem. 275(12): 8283–8286. [DOI] [PubMed] [Google Scholar]
- Park JH, Dorrestein PC, Zhai H, Kinsland C, McLafferty FW, Begley TP.. 2003. Biosynthesis of the thiazole moiety of thiamin pyrophosphate (vitamin B1). Biochemistry 42(42): 12430–12438. [DOI] [PubMed] [Google Scholar]
- Petersen FT, Meier R, Kutty SN, Wiegmann BM.. 2007. The phylogeny and evolution of host choice in the Hippoboscoidea (Diptera) as reconstructed using four molecular markers. Mol Phylogenet Evol. 45(1): 111–122. [DOI] [PubMed] [Google Scholar]
- Price DR, Feng H, Baker JD, Bavan S, Luetje CW, Wilson AC.. 2014. Aphid amino acid transporter regulates glutamine supply to intracellular bacterial symbionts. Proc Natl Acad Sci U S A. 111(1): 320–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschke M, Burkle L, Muller N, Nunes-Nesi A, Fernie AR, Arigoni D, Amrhein N, Fitzpatrick TB.. 2007. Vitamin B1 biosynthesis in plants requires the essential iron sulfur cluster protein, THIC. Proc Natl Acad Sci U S A. 104(49): 19637–19642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio RV, Attardo GM, Weiss BL.. 2016. Grandeur alliances: symbiont metabolic integration and obligate arthropod hematophagy. Trends Parasitol. 32(9): 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio RV, et al. 2012. Insight into the transmission biology and species-specific functional capabilities of tsetse (Diptera: glossinidae) obligate symbiont Wigglesworthia. MBio 3(1): e00240-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio RV, Wu YN, Filardo G, Aksoy S.. 2006. Dynamics of multiple symbiont density regulation during host development: tsetse fly and its microbial flora. Proc Biol Sci. 273(1588): 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist F, Huelsenbeck JP.. 2003. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19(12): 1572–1574. [DOI] [PubMed] [Google Scholar]
- Smith EM, Hoi JT, Eissenberg JC, Shoemaker JD, Neckameyer WS, Ilvarsonn AM, Harshman LG, Schlegel VL, Zempleni J.. 2007. Feeding Drosophila a biotin-deficient diet for multiple generations increases stress resistance and lifespan and alters gene expression and histone biotinylation patterns. J Nutr. 137(9): 2006–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AK, Deberry JW, Runyen-Janecky L, Rio RVM.. 2010. Nutrient provisioning facilitates homeostasis between tsetse fly (Diptera: Glossinidae) symbionts. Proc Biol Sci. 277(1692): 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AK, McLain C, Rio RV.. 2012. The tsetse fly obligate mutualist Wigglesworthia morsitans alters gene expression and population density via exogenous nutrient provisioning. Appl Environ Microbiol. 78(21): 7792–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder AK, Rio RVM, Goodrich-Blair H.. 2015. “Wigglesworthia morsitans” folate (vitamin B9) biosynthesis contributes to tsetse host fitness. Appl Environ Microbiol. 81(16): 5375–5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Deng Z, Yan A.. 2014. Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem Biophys Res Commun. 453(2): 254–267. [DOI] [PubMed] [Google Scholar]
- Tokuriki N, Tawfik DS.. 2009. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 459(7247): 668–673. [DOI] [PubMed] [Google Scholar]
- Ugulava NB, Gibney BR, Jarrett JT.. 2001. Biotin synthase contains two distinct iron-sulfur cluster binding sites: chemical and spectroelectrochemical analysis of iron-sulfur cluster interconversions. Biochemistry 40(28): 8343–8351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Dohlen CD, Kohler S, Alsop ST, McManus WR.. 2001. Mealybug beta-proteobacterial endosymbionts contain gamma-proteobacterial symbionts. Nature 412(6845): 433–436. [DOI] [PubMed] [Google Scholar]
- Weisburg WG, Barns SM, Pelletier DA, Lane DJ.. 1991. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 173(2): 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss BL, Wang J, Aksoy S.. 2011. Tsetse immune system maturation requires the presence of obligate symbionts in larvae. PLoS Biol. 9(5): e1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernegreen JJ. 2012. Mutualism meltdown in insects: bacteria constrain thermal adaptation. Curr Opin Microbiol. 15(3): 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Hussain M, O'Neill SL, Asgari S.. 2013. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proc Natl Acad Sci U S A. 110(25): 10276–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.