

Abstract
Translational repression of maternal mRNAs is an essential regulatory mechanism during early embryonic development. Repression of the Drosophila nanos mRNA, required for the formation of the anterior–posterior body axis, depends on the protein Smaug binding to two Smaug recognition elements (SREs) in the nanos 3′ UTR. In a comprehensive mass spectrometric analysis of the SRE-dependent repressor complex, we identified Smaug, Cup, Me31B, Trailer hitch, eIF4E, and PABPC, in agreement with earlier data. As a novel component, the RNA-dependent ATPase Belle (DDX3) was found, and its involvement in deadenylation and repression of nanos was confirmed in vivo. Smaug, Cup, and Belle bound stoichiometrically to the SREs, independently of RNA length. Binding of Me31B and Tral was also SRE-dependent, but their amounts were proportional to the length of the RNA and equimolar to each other. We suggest that “coating” of the RNA by a Me31B•Tral complex may be at the core of repression.
Keywords: translational repression, maternal RNA, deadenylation
INTRODUCTION
Control of gene expression by translational regulation of mRNAs is found throughout biology, but is particularly important in oocyte development and early embryogenesis in animals. As the zygotic genome is not transcribed during very early development, mRNAs required at this time are produced during oocyte development (maternal mRNAs), and many are stockpiled in a repressed, “masked” state. During maturation of the oocyte to a fertilizable egg and the first phases of embryonic development, specific maternal mRNAs are translationally activated in a controlled manner. Many are also regulated by localization at specific sites and by degradation (Lasko 2011; Barckmann and Simonelig 2013; Laver et al. 2015).
In Drosophila, zygotic genome activation is a gradual process; full-scale zygotic transcription does not commence until nuclear division cycle 14, with the beginning of the cellular blastoderm stage (Ali-Murthy et al. 2013; Harrison and Eisen 2015; Laver et al. 2015). One maternal RNA governing early development is the nanos (nos) mRNA. Its regulation is essential for development: Formation of the anterior–posterior axis of the embryo depends on the Nos protein being produced exclusively at the posterior pole (Wang and Lehmann 1991). For this purpose, most of the nos mRNA, which is distributed throughout the embryo, is translationally repressed (Gavis and Lehmann 1994) and degraded over the first 2–3 h of development (Dahanukar and Wharton 1996; Bashirullah et al. 1999). At most, ∼4% of the nos mRNA is localized at the posterior pole (Bergsten and Gavis 1999; Trcek et al. 2015) and, due to stabilization and derepression by Oskar, serves as a localized source of Nos (Ephrussi and Lehmann 1992; Smith et al. 1992; Dahanukar et al. 1999). Both repression and degradation of nonlocalized nos mRNA depend on the protein Smaug (Smg) (Dahanukar and Wharton 1996; Smibert et al. 1996; Dahanukar et al. 1999; Smibert et al. 1999) and the Piwi-interacting RNA (piRNA) machinery (Rouget et al. 2010). Smg is essential for the maternal-to-zygotic transition (Benoit et al. 2009), causing repression and degradation of hundreds of maternal mRNAs (Tadros et al. 2007; Chen et al. 2014a). Smg regulates nos by binding two Smaug recognition elements (SREs) in the nos 3′ UTR and recruits the CCR4–NOT complex, which catalyzes mRNA deadenylation (Semotok et al. 2005; Jeske et al. 2006; Zaessinger et al. 2006). For translational repression, Smg binds the protein Cup (Nelson et al. 2004), and a miRNA-independent repressive role of Ago1 has also been reported (Pinder and Smibert 2013), but the mechanism of repression is not fully understood (Jeske et al. 2011).
Deadenylation and translational repression of nos can be observed in extracts from early Drosophila embryos (Jeske et al. 2006, 2011). Deadenylation and repression both depend on the SREs and, by inference, on Smg, but are independent of each other. Smg-associated Cup inhibits translation by binding the cap-binding translation initiation factor eIF4E and competitively displacing eIF4G (Nelson et al. 2004; Jeske et al. 2011). However, the 5′ cap as well as eIF4E and eIF4G are dispensable for SRE-dependent repression (Jeske et al. 2011); thus, an additional repression mechanism must exist. In support of this, the SRE-dependent repressor complex contains the proteins Me31B and Trailer hitch (Tral) in addition to Smg, Cup, and eIF4E (Jeske et al. 2011). Me31B and its orthologs are DEAD-box family RNA helicases/RNA-dependent ATPases and involved in translational repression in flies (Nakamura et al. 2001; Tritschler et al. 2009), yeast (Dhh1p) (Coller and Parker 2005), and vertebrates (DDX6/p54/RCK) (Minshall et al. 2001; Chen et al. 2014b; Mathys et al. 2014). Tral (S. cerevisiae Scd6p; C. elegans CAR1; vertebrate Rap55 or Lsm14) associates with Me31B and also represses translation (Audhya et al. 2005; Boag et al. 2005; Wilhelm et al. 2005; Tanaka et al. 2006; Weston and Sommerville 2006; Nissan et al. 2010; Hubstenberger et al. 2013; Ayache et al. 2015).
Formation of the SRE-dependent repressor complex in embryo extract is ATP-dependent and slow, requiring 20–30 min. Once formed, the complex is kinetically unusually stable, with an estimated t1/2 of ∼4 h. These observations suggest that the repressor complex is not governed by a simple association–dissociation equilibrium. Presumably due to the stability of the complex, repression is ∼20- to 50-fold, that is, ≥95% of the SRE-containing RNA is turned off. Importantly, SRE-dependent repression acts on translation initiation driven by the CRPV IRES. As this IRES can directly associate with ribosomes, independently of any initiation factors, the repressor complex likely affects either ribosome association or elongation (Jeske et al. 2011).
Here we report a systematic analysis of the composition of the SRE-dependent repressor complex. In addition to the previously known proteins, the DEAD-box protein Belle (Bel) was found in the complex. Genetic experiments confirmed that Bel participates in nos regulation in vivo. Me31B and Tral bind in multiple copies along the repressed RNA, presumably sequestering it in a form that is inaccessible for ribosomes.
RESULTS
Composition of the SRE-dependent repressor complex
For an analysis of the constituents of the SRE-dependent repressor complex, gradient centrifugation was used as a first purification step. In Figure 1, radiolabeled, m7G-capped luciferase RNAs were used carrying a nos 3′ UTR fragment with two SREs, either wild type (SRE+) or with an inactivating point mutation in each (SRE−). The RNAs had “internal” poly(A) tails, which stimulate translation like a 3′-terminal tail, but are protected from SRE-dependent deadenylation by flanking 3′ sequences (Fig. 1A; Jeske et al. 2011). As reported (Jeske et al. 2011), these RNAs develop the full extent of translational repression only upon a preincubation with embryo extract under conditions that do not permit translation (Fig. 1B,D; Supplemental Table S1). After a first preincubation, aliquots of the RNAs were sedimented through sucrose gradients (Fig. 1B,C), and the peak fractions were assayed for translation in embryo extract either directly or after a second preincubation. The results revealed strong translational repression independently of the second preincubation (Fig. 1B,D; Supplemental Table S1). Thus, the repressor complex formed during the first preincubation survived the long fractionation procedure, in agreement with its known kinetic stability (Jeske et al. 2011). The RNA sedimented faster than in earlier experiments showing SRE-dependent inhibition of 48S complex formation (Jeske et al. 2011), probably due to differences in experimental conditions. With precautions taken to suppress RNase activity in the extract (see Materials and Methods), RNA stability was not significantly different between the repressed SRE+ RNA and the SRE− control (Fig. 1E,F).
FIGURE 1.
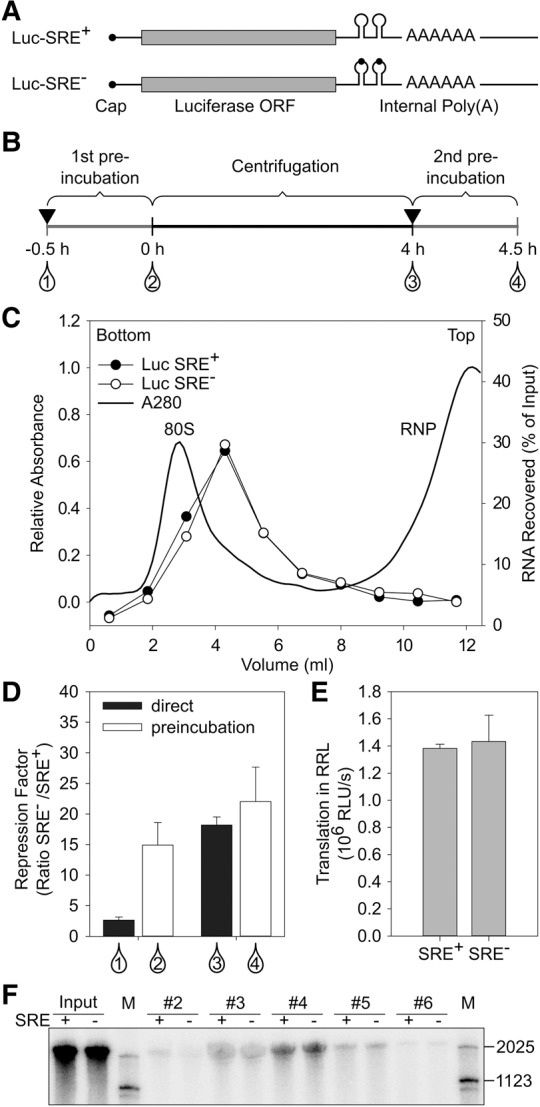
Reporter RNAs maintain their repressed state during gradient centrifugation. (A) Cartoon of luciferase reporter RNAs. (B) Scheme of the assay. Black triangles indicate addition of embryo extract, and drop symbols indicate samples withdrawn for translation and luciferase assays. Numbers refer to the data shown in D. (C) Repressor complexes formed on radiolabeled reporter RNAs were separated by sucrose-gradient sedimentation. Distributions of the two RNAs are overlaid. UV absorption indicates the positions of free RNPs and the 80S ribosome. (D) As shown in B, luciferase RNAs were tested for translational repression either directly or after preincubation in embryo extract (samples 1 and 2 in B). A second set of samples was preincubated in extract and then separated by gradient centrifugation. Aliquots from the peak fractions as in C were assayed for translation in embryo extract either with or without a second preincubation in fresh extract (samples 3 and 4 in B). Luciferase activities in these assays are listed in Supplemental Table S1. (E) RNAs were purified from equal volumes of the peak fractions of gradients as in C, and equal aliquots were assayed for translation in rabbit reticulocyte lysate, which does not exhibit SRE-dependent repression (Jeske et al. 2006). Thus, similar luciferase yields indicated similar RNA recoveries for both RNAs. Error bars represent the standard deviation of three independent experiments. (F) Radiolabeled luciferase RNA from the sucrose gradient shown in Figure 1C was purified and analyzed by denaturing gel electrophoresis and phosphorimaging. Numbers above the lanes indicate fraction numbers of the sucrose gradient. Note that the inclusion of “short RNA” (see Materials and Methods) strongly stabilized the RNA compared to earlier experiments (Jeske et al. 2011).
For the actual analysis of the repressor complex, similar RNAs as in Figure 1 were used (1-AUG nos and 1-AUG nos SRE−) that only differed by containing a shorter ORF and no 5′ cap; the cap is irrelevant for repression and stability of the repressor complex (Jeske et al. 2011). The RNAs were randomly biotinylated. After incubation in extract, these shorter RNAs showed an SRE-dependent difference in sedimentation, the SRE+ RNA sedimenting in the 80S region ahead of the control; this was more visible in an analytical gradient (Fig. 2A) than in the preparative experiment (Fig. 2B). Gradient fractions were selected as shown in Figure 2B, pooled from several runs and concentrated. Equal quantities of the SRE+ and SRE− RNPs, based on trace-labeling of the RNA, were affinity-purified on streptavidin beads, and proteins eluted by SDS were analyzed by gel electrophoresis (Fig. 2C) followed by liquid chromatography/tandem mass spectrometry (LC/MS/MS). Proteins detected were evaluated by label-free quantification (MaxQuant) (Cox and Mann 2008) based on the intensities of the MS signals and spectral counts, corrected for the molecular mass of each protein. In Figure 2E, the apparent abundance of each protein is plotted for the SRE+ RNA against the control. All proteins are listed in Supplemental Table S2.
FIGURE 2.

Analysis of the SRE-dependent repressor complex. (A) Radiolabeled, biotinylated RNAs (1-AUG nos and 1-AUG nos SRE−) were incubated for assembly of a repressor complex and separated on a sucrose gradient as in B, but the volume loaded was smaller (0.2 mL versus 1 mL). (B) Radiolabeled, biotinylated RNAs (1-AUG nos and 1-AUG nos SRE−) were separated on a preparative sucrose gradient (see Materials and Methods). Fractions pooled for the analysis of the repressor complex are indicated by the bracket. Error bars represent the standard deviation (n = 4). (C) Corresponding fractions from a total of 12 gradients from four independent experiments each for the SRE+ RNA and the SRE− control were pooled, and RNPs were purified on streptavidin beads. Equal amounts based on trace-labeling of the RNA were analyzed by SDS-PAGE and silver staining. Arrowheads indicate bands enriched in the SRE+ RNP that might correspond to Smaug (109 kDa), Trailer Hitch (69 kDa), and Me31B (52 kDa). (D) Specific association of proteins with the SRE+ RNA was confirmed by Western analysis in an independent pull-down assay. Smg, Cup, Me31B, and PABPC served as controls for Bel. (E) Proteins in the purified RNP fractions were analyzed by mass spectrometry and label-free quantification. Apparent protein abundance in SRE+ versus SRE− RNP was plotted on a log2 scale. Proteins enriched in the SRE+ RNP beyond P = 0.05 and NOT11 are labeled. The complete list of proteins represented in E is found in Supplemental Table S2. Different sets of proteins in the same data are highlighted in Supplemental Figure S1, and additional enriched proteins are listed in Supplemental Table S3.
As expected, Smg was among the most abundant proteins and most strongly enriched in the SRE+ RNP. Cup, Tral, and Me31B formed a tight cluster with an apparent abundance even higher than Smg and enriched in the SRE+ RNP. Three additional proteins were also abundant and enriched in the SRE+ RNP: First, enrichment of eIF4E-1 (Hernández et al. 2005) agrees with previous results (Jeske et al. 2011). As the RNA was not capped, the protein's presence was presumably due to protein–protein interactions, e.g., with Cup (Nelson et al. 2004; Chekulaeva et al. 2006). Second, the presence of PABPC was expected due to the internal poly(A) tail. An SRE-dependent enrichment of the protein agrees with the observation that a poly(A) tail facilitates repression (Jeske et al. 2006, 2011) and with the presence of PABPC in DDX6 complexes purified under stringent conditions (Ayache et al. 2015; Bish et al. 2015). In contrast, Western analyses of RNP complexes isolated by a simple pull-down procedure consistently showed the PABPC content to be independent of the SREs (Fig. 2D; Jeske et al. 2011). The procedure leading to the MS analysis took considerably longer than a simple pull-down and might thus reveal a more stable association of PABPC with the repressed RNA compared to the control. Third, a novel component, the RNA-dependent ATPase Belle (Bel) (Johnstone et al. 2005) was identified. Its specific association with the SRE+ RNA was confirmed by Western blot (Figs. 2D, 5).
FIGURE 5.

The SRE-dependent repressor complex sequesters the RNA through multiple copies of Me31B and Tral. (A) Three biotinylated RNAs of different lengths but each containing two SREs were used, together with matching SRE− controls, for repressor complex formation in embryo extract and streptavidin pull-down. Bound proteins were analyzed by Western blotting. Known amounts of recombinant proteins were used as standards. Analyses of Smg and Tral are shown as representative examples. (B) Stoichiometries of bound proteins were estimated from experiments as in A. Signals for SRE+ and mutant controls are shown. The horizontal lines mark a 1:1 molar ratio of protein to RNA. Error bars represent the standard deviation from three to five independent experiments ([*] P ≤ 0.05; [**] P ≤ 0.01; [***] P ≤ 0.001). Additional data are presented in Supplemental Figure S2. (C) An RNase I protection experiment was carried out as described in Materials and Methods. (D) Quantification of experiments as shown in C (average of n = 4 with three independent batches of embryo extract). Error bars represent the standard deviation. Data were fitted to a first-order decay with the last time point of both RNAs omitted. The half-life of the SRE+ RNA was 1.7-fold longer than that of the SRE− control. (E) The association of Caf1 and Not2 with the SRE+ RNA and SRE− control was examined as in A. Three streptavidin pull-down experiments were carried out with the 630 nt RNA and independent batches of embryo extract. Western blotting and comparison to standard curves was carried out for the proteins indicated. The average amount of Smg recovered was 200 ± 100 fmol. Tral was recovered at 1000 ± 150 fmol in the SRE+ sample and at 500 ± 120 fmol in the SRE− sample (data not shown). All three subunits of the CCR4–NOT complex were present below the smallest amount in the standard curves (50 fmol). In a separate Western blot, signals for Caf1, Not2 and Ccr4 were below 12.5 fmol (data not shown).
The CCR4–NOT complex is responsible for Smg-dependent deadenylation (Semotok et al. 2005; Zaessinger et al. 2006) and associates with SRE-containing RNAs (Jeske et al. 2011). Satisfyingly, all core components of the complex (Not1, Ccr4/Twin, Caf1/Pop2, Not2/Rga, Not3, and Caf40/Rcd-1) formed a cluster of similar SRE-specific enrichment and roughly similar abundance (Fig. 2E). However, reduced abundance of all subunits compared to the Smg/Cup cluster suggests that the CCR4–NOT complex is not part of the stable core of the repressor complex. Several other proteins were also enriched in the SRE+ RNP, but less so than either the Smg/Cup cluster or the CCR4–NOT complex (Fig. 2E; Supplemental Table S3). These proteins include the conserved CTLH (C terminal to LisH [Lissencephaly type-1-like homology motif]) complex (Francis et al. 2013) and the Cup paralog 4E-T (Kamenska et al. 2014, 2016). Dcp1, which has been found to be associated with a Me31B–Tral–Cup complex (Tritschler et al. 2008) was present, but the catalytic subunit of the decapping complex, Dcp2, was not detected at all. A low-level presence of Oskar may be related to its role in derepression of nos in the pole plasm.
Pat1 (HPat or Patr-1 in Drosophila) and EDC3, which compete with Tral for the same surface of Me31B (Tritschler et al. 2008, 2009; Haas et al. 2010; Jonas and Izaurralde 2013; Sharif et al. 2013), were present at much lower levels than Tral and weakly enriched on the SRE+ RNA (Supplemental Fig. S1C). Ypsilon schachtel (Yps) and Exuperantia (Exu) have been found in Me31B-containing RNPs (Nakamura et al. 2001; Wilhelm et al. 2005), but Yps was not enriched in the repressor complex (Supplemental Fig. S1C). Western blotting confirmed an equal association with both RNAs (data not shown). Exu was not detected, consistent with the absence of nos from immunoprecipitated Exu–Yps complexes (Wilhelm et al. 2000).
As expected (Nelson et al. 2004; Jeske et al. 2011), eIF4G was moderately depleted from the repressed RNP. All other initiation factors were less abundant than eIF4E and eIF4G and not enriched in either RNP (Supplemental Fig. S1A). Ribosomal proteins were depleted from the SRE+ RNP (Supplemental Fig. S1B,D). Ago 1 has been reported to participate in SRE-dependent repression (Pinder and Smibert 2013), but was not enriched in the SRE+ RNP. Other proteins involved in small RNA pathways were not enriched either (Supplemental Fig. S1C). This is not unexpected as the region of the nos 3′ UTR most strongly targeted by piRNAs (Rouget et al. 2010; Barckmann et al. 2015) was not present in our constructs.
In an independent experiment, Smg was immunoprecipitated from extract that had not been treated with RNase. Proteins were identified by LC/MS/MS and compared to a preimmune serum control. The results supported those of the streptavidin purification: Core components of the repressor complex were enriched with Smg; only the enrichment of Belle was weak. All core subunits of the CCR4–NOT complex and four subunits of the CTLH complex were also enriched (Fig. 3; Supplemental Table S4).
FIGURE 3.
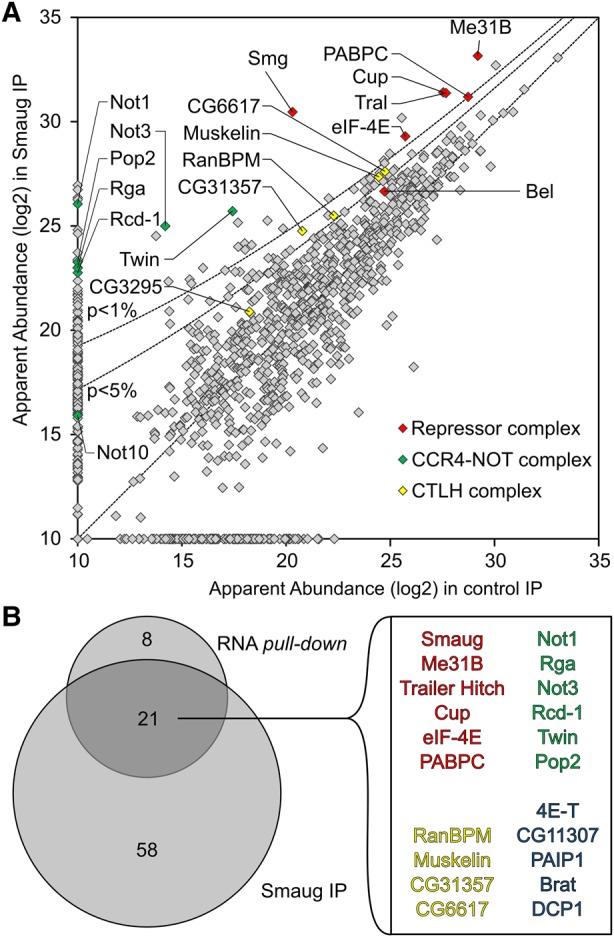
MS analysis of proteins coprecipitated with Smg. (A) Quantitative MS data were plotted for the Smg immunoprecipitation versus a preimmune control. Proteins that were also significantly enriched in the streptavidin pull-down of the SRE-dependent repressor complex are highlighted as in Figure 2E. P-value cutoffs are indicated as lines. (B) Venn diagram comparing proteins enriched beyond P = 0.05 in the Smg immunoprecipitation and in the streptavidin pull-down (Fig. 2E). The 21 proteins in the overlap are listed. Belle, NOT10, and the CTLH complex subunit CG3295 had P-values higher than 0.05. All proteins enriched in the Smg IP are listed in Supplemental Table S4.
Repressed nos mRNA exists as a monomeric RNP
The repressed RNPs sedimented rapidly, comparable to ribosomes (Fig. 2A). In the case of oskar mRNA, oligomerization of the repressed RNPs contributes to their rapid sedimentation (Chekulaeva et al. 2006; Besse et al. 2009). However, when a biotinylated SRE+ RNA was incubated in embryo extract together with a second SRE+ RNA, lacking biotin and distinguishable by size, streptavidin pull-down resulted in the purification of only the biotinylated RNA; no association with the second RNA was seen (Fig. 4A). We conclude that Smg-dependent repression does not involve RNA oligomerization.
FIGURE 4.
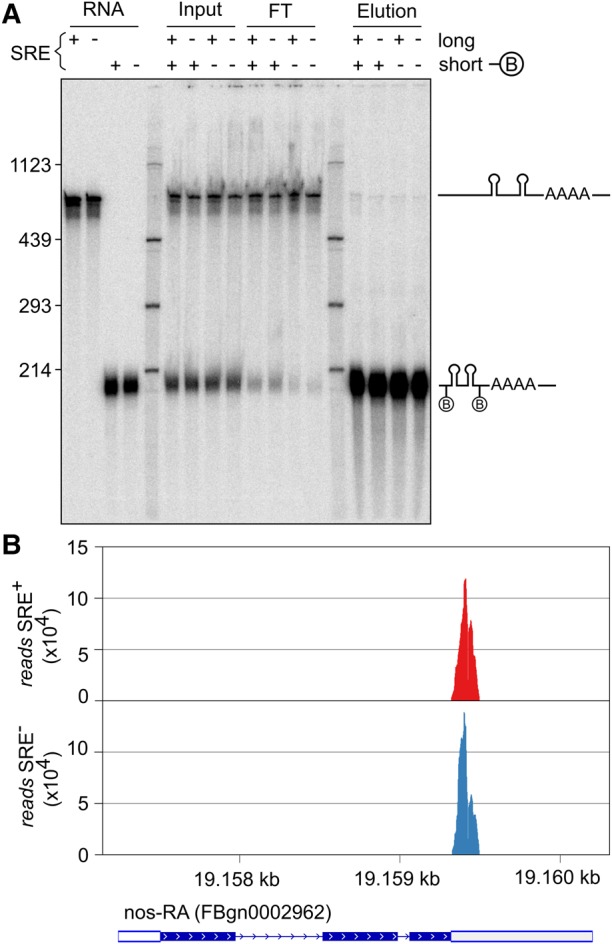
SRE-containing RNAs do not oligomerize. (A) A biotinylated RNA of 200 nt (SREonly; SRE+ or SRE−) and a nonbiotinylated RNA of 630 nt (AUGonly; SRE+ or SRE−) were incubated together in embryo extract under conditions permitting assembly of the repressor complex. Streptavidin pull-downs were performed to enrich the biotinylated RNA together with potentially associated RNAs. RNA was eluted in formamide loading buffer at 95°C. The lanes labeled “RNA” show the purified RNAs used, “input” shows the RNAs after incubation in extract, “FT” is the flow-through of the pull-down, and “elution” shows the bound fraction. The figure shows one experiment of two. (B) RNA was purified from affinity-purified SRE+ and SRE− RNPs and deep-sequenced. Reads mapping to the nos gene are displayed. For the experiment, bait RNAs were used at 10 nM. The abundance of nos has been estimated as 2 nM (Trcek et al. 2015). With an approximately twofold dilution upon extract preparation and an additional twofold dilution in the assay, endogenous nos sequences should have been detectable if an association with the bait RNA had taken place.
As an unbiased test for a potential association of the SRE+ RNA with other RNAs, total RNA was isolated from purified repressor complexes and from SRE− controls and analyzed by deep sequencing. Although sequencing was targeted to small RNAs, nos sequences were also recovered; these were limited to the part contained in the bait RNA, and no difference between SRE+ RNA and SRE− control was observed (Fig. 4B). Calculations (see figure legend) indicated that endogenous nos RNA present in the extract would have been detectable if it had been associated with the bait RNA. Thus, the repressed nos RNP does not oligomerize with other repressed RNPs. The lack of nos oligomerization is consistent with in vivo data (Little et al. 2015). No SRE-dependent enrichment of other RNAs was observed, making it unlikely that trans-acting RNAs are involved in SRE-dependent repression in vitro.
Multiple copies of Me31B and Tral associate with the repressed RNA
Me31B orthologs can oligomerize on their own or when bound to RNA, and the ability of protein variants to oligomerize correlates with their ability to repress translation. The proteins appear to bind in multiple copies along RNA in vivo (Minshall and Standart 2004; Ernoult-Lange et al. 2012).
In order to determine whether oligomerization of repressor proteins on the reporter RNAs might play a role in SRE-dependent translational repression, we estimated the stoichiometries of proteins in the repressor complex: Three different biotinylated radiolabeled RNAs were used, each containing two copies of the SREs: SREonly (200 nt), the 1-AUG nos RNA used for the MS analysis (630 nt) and the luciferase reporter RNA (1956 nt); corresponding SRE− RNAs served as controls. All RNAs were allowed to assemble repressor complexes before affinity purifications were carried out. The quantities of immobilized RNAs were determined from their specific radioactivities, and amounts of associated proteins were estimated by Western blotting and comparison to standard curves of purified recombinant material. Representative data are shown in Figure 5A and Supplemental Figure S2, and a summary of the average stoichiometries is presented in Figure 5B. RNA association of Smg was SRE-dependent, but independent of RNA length: The stoichiometry was between 1 and 2 for all three RNAs. Within the accuracy of the experiment, this was equimolar with the SREs (see legend to Supplemental Fig. S2). Binding of Cup was also approximately stoichiometric with the SREs. Bel bound independently of RNA length, but tended to be less abundant; with the longest RNA, specific binding was no longer distinguishable from background. In contrast to Smg, Cup, and Bel, both Tral and Me31B clearly bound in a length-dependent manner, in excess of Smg and the SREs and approximately equimolar to each other. As these data indicate binding of multiple copies of Me31B and Tral along the RNA, it is unclear whether the amounts associated with the SRE− RNAs should be subtracted as background or not. Without background subtraction, the stoichiometry of Me31B/Tral binding to RNA was near one copy of Me31B and Tral per 100 nucleotides.
“Coating” of RNA by Me31B and Tral may form an inert, “masked” RNP that is at the core of translational repression, sterically preventing ribosome access to the RNA. For want of a reagent more comparable in size to a ribosome (3 × 106 Da), accessibility of the repressed RNA was probed with the endonuclease RNase I (as an MPB fusion protein; 72,000 Da). The repressed SRE+ RNA proved to be considerably more resistant to the nuclease than the SRE− control (Fig. 5C,D). This agrees with an earlier observation that an SRE+ RNA, when simply incubated in embryo extract under conditions of unchecked endogenous nuclease activity, was moderately more stable than an SRE− control (Jeske et al. 2011). These data strongly argue in favor of sequestration of the RNA by a protein complex.
The stoichiometries indicate that Me31B and Tral cooperate as a defined subcomplex within the repressor complex. Indeed, treatment of a Smg immunoprecipitate with the cross-linker BuUrBu (Müller et al. 2010) identified a cross-link between Tral and Me31B consistent with the interaction dependent on the FDF motif of Tral (Fig. 6A; Supplemental Fig. S3; Tritschler et al. 2008, 2009). Thus, this interaction is likely to be relevant within the context of the repressor complex. When Tral and GST-Me31B were coexpressed in insect cells by means of baculovirus vectors, Tral was copurified with GST-Me31B on glutathione beads, suggesting the existence of a stable complex (Fig. 6B,C). Copurification was not affected by elevated salt concentration or RNase A.
FIGURE 6.

Me31B and Tral form a complex. (A) Structure of a complex between the C-terminal domain of DDX6 and an EDC3 peptide containing the FDF motif (PDB 2WAX) (Tritschler et al. 2009). Tral uses the same motif to bind Me31B. The black line represents the cross-link identified (Supplemental Fig. S3), with the Cα − Cα distance indicated. (B) Sf21 cells were infected with baculoviruses expressing GST-Me31B, Tral, or both as indicated. “Total” refers to an SDS lysate. Purifications on glutathione beads were carried out from native lysates. Proteins were analyzed by Western blotting for Me31B (top) or Tral (bottom). Drosophila embryo extract (DEE) and noninfected SF21 cells served as controls. (C) Glutathione bead eluates were analyzed by SDS polyacrylamide gel electrophoresis and Coomassie staining.
The components of the repressor complex were abundant among the soluble proteins of the embryo extract, as estimated by quantitative Western blotting (with an error of approximately two; see Materials and Methods): Smg was present at 0.08 µM; Cup, 2 µM; Bel, 0.8 µM; Me31B, 3.5 µM, Tral, 7.6 µM. We estimate that extracts were approximately twofold diluted compared to egg content. In comparison, an mRNA concentration of roughly 0.4 µM in a Drosophila egg can be estimated on the basis of an egg volume of 0.01 µL (Azevedo et al. 1996), a total RNA content of 0.19 µg per egg (Hough-Evans et al. 1980), and the assumption that 2% of this is mRNA with an average length of 3000 nt. The ratio of protein to RNA concentration is consistent with Smg acting on a sizeable fraction of maternal mRNAs (Tadros et al. 2007; Chen et al. 2014a) and with Cup participating in translational repression exerted by other RNA binding proteins, e.g., Bruno (Nakamura et al. 2004; Wilhelm et al. 2005; Chekulaeva et al. 2006). The abundance of both Me31B and Tral is consistent with the two proteins binding in multiple copies and as a complex to repressed mRNAs. The high concentration of Me31B, exceeding that of mRNA, is consistent with data in other organisms (Ernoult-Lange et al. 2012 and references cited therein).
The MS data suggest that the CCR4–NOT complex is not part of the core repressor complex. Association of the CCR4–NOT complex with the 630 nt RNA was examined by quantitative Western blotting. In agreement with the MS analysis, CCR4, Caf1, and Not2 bound the RNA in an SRE-dependent manner, but were clearly substoichiometric (Fig. 5E and data not shown).
Bel is required for nos mRNA translational repression in vivo
Belle is a DDX3-type RNA helicase. These proteins have been reported to be involved in translation, but both activating and repressive roles have been described. To examine a direct role of Bel in nos mRNA control in the embryo, we used two strong or null alleles of bel, bel6 and belL4740, which cause larval lethality. In addition, the hypomorphic allele belneo30 was used, which leads to female sterility when combined with stronger alleles (Johnstone et al. 2005; Ihry et al. 2012). Consistent with this, transheterozygous belneo30/6 and belneo30/L4740 females were sterile: When crossed with wild-type males, they produced embryos (referred to as belneo30/6 and belneo30/L4740 embryos) that failed to eclose (Fig. 7A). belneo30/6 embryos showed a stronger phenotype than belneo30/L4740, most of them being fragile and having short or no dorsal appendages.
FIGURE 7.

Bel is required for nos mRNA translational repression in vivo. (A) Phenotypic quantification of embryos coming from belneo30/6 or belneo30/L4740 mutant females crossed with wild-type males. Numbers refer to the embryos examined. (B) nos mRNA quantification using RT-qPCR in wild-type and bel mutant embryos spanning 1 h intervals up to 4 h of development. RpL32 was used as a control mRNA for normalization. Means are from three to four biological replicates. The error bars represent SEM. (*) P < 0.05; (**) P < 0.01 using the bilateral Student's t-test. (C) PAT assays measuring nos mRNA poly(A) tail lengths in wild-type and bel mutant embryos spanning 1 h intervals up to 4 h of development. PAT assay profiles using ImageJ are shown in Supplemental Figure S4A. sop encodes a ribosomal protein and was used as a control mRNA. (D) In situ hybridization of nos mRNA (top panels) and immunostaining with anti-Nos (bottom panels) of wild-type and bel mutant 0–2 h embryos. Quantification of immunostaining is indicated below the images. (E) Western blots of wild-type and bel mutant 0–2 h embryos probed with antibodies against six components of the nos repressor complex, including the CCR4–NOT complex. Anti-α-tubulin (Tub) was used as a loading control. (F) Confocal images of syncytial embryos co-stained with rabbit anti-Bel and guinea pig anti-Smg. Bottom panels show a higher magnification. Quantification using the Pearson correlation coefficient (PCC) indicated significant partial colocalization (PCC = 0.52). Anterior is to the left. The scale bars represent 30 and 10 µm in top and bottom panels, respectively. (G) Quantification of nos and Hsp83 mRNAs using RT-qPCR in anti-GFP immunoprecipitations from belCC00869 embryos that express GFP-Bel and control (wild type) embryos that do not express GFP. RpL32 mRNA was used for normalization. mRNA levels in control embryos were set to one. Means are from two biological replicates quantified in triplicates. The error bars represent SEM. (***) P < 0.001 using the bilateral Student's t-test.
To address a role of Bel in nos mRNA deadenylation and decay, we quantified nos mRNA by RT-qPCR in wild-type and bel mutant embryos spanning 1 h intervals during the first 4 h of embryogenesis. nos mRNA decay was prominent after 2 h in wild-type embryos, but was strongly impaired in bel mutant embryos (Fig. 7B). Accordingly, poly(A) test assays, used to measure nos mRNA poly(A) tail lengths in embryos up to 4 h of development, showed that deadenylation was inhibited in bel mutant embryos (Fig. 7C; Supplemental Fig. S4A). In situ hybridization of 0–2 h embryos suggested that nos mRNA stabilization might even start before 2 h of embryogenesis, since the staining was darker in bel mutants than in wild-type embryos (Fig. 7D). Translational repression of nos was also impaired in bel mutant embryos: Immunostaining with anti-Nos antibody revealed ectopic, increased Nos levels in belneo30/L4740 embryos (Fig. 7D). belneo30/6 embryos showed heterogeneous staining: A large proportion (80%) were irregularly or not stained, but the remaining 20% showed again high levels of ectopic Nos protein throughout the embryo (Fig. 7D). The heterogeneity in belneo30/6 embryos could be due to earlier defects during oogenesis (Johnstone et al. 2005) or to a potential gain-of-function nature of the bel6 allele: bel6 has a stop codon after the first third of the coding sequence, which encodes a 4E-BP domain (Yarunin et al. 2011; Ihry et al. 2012). Thus, a truncated protein in bel6 might dominantly affect translation through binding to eIF4E. Analyses of Nos protein levels by Western blots were in agreement with the staining pattern of the majority of embryos, showing increased levels in 0–2 h belneo30/L4740 embryos and reduced levels in 0–2 h belneo30/6 mutant embryos (Supplemental Fig. S4B). Defects in nos regulation in bel mutant embryos did not result from reduced levels of other components of the repressor complex or of the CCR4–NOT complex (Fig. 7E).
These results show that Bel participates in the repression of nos mRNA in the somatic part of the embryo and thus imply that Bel is present there. A GFP-tagged Bel protein has been reported to be distributed throughout the syncytial embryo (Johnstone et al. 2005). Immunostaining of embryos with anti-Bel and anti-Smg antibodies validated the cytoplasmic distribution of Bel throughout the embryo and its partial colocalization with Smg (Fig. 7F).
In an independent experiment to ask whether nos mRNA is bound to Bel in embryos, we used the GFP protein-trap bel allele belCC00869, in which GFP is inserted in frame in the N-terminal part of Bel (Buszczak et al. 2007). RNA immunoprecipitation with anti-GFP antibody showed an enrichment of nos mRNA over Rpl32 mRNA in 0–2 h embryos expressing GFP-Bel (belCC00869) compared to control embryos. Another Smg target, Hsp83 (Semotok et al. 2005), was also enriched (Fig. 7G).
Taken together, these results support a functional role of Bel in the nos repressor complex in vivo, acting on both translational repression and deadenylation.
DISCUSSION
We have identified seven stoichiometric components of the SRE-dependent repressor complex that are likely to explain its ATP-dependent formation, high stability and repressive potency: Smg, which directly recognizes the SREs; Cup, which associates with Smg; the DEAD-box ATPase Me31B and its partner Tral; a second DEAD-box ATPase, Bel; and finally the cap-binding initiation factor eIF4E and, with less certainty, the cytoplasmic poly(A) binding protein, PABPC. The repressor complex analyzed is functional since translation of the RNA on which it has assembled is fully repressed (Fig. 1D). Thus, assembly of the seven proteins identified constitutes the slow step of translation repression. The same complex likely facilitates deadenylation, since Smg and the SREs are also important for deadenylation of nos by CCR4–NOT. Accordingly, all core components of the CCR4–NOT complex were associated with the repressor complex.
The CCR4–NOT complex can also contribute to translational repression, independently of its deadenylase activity (Cooke et al. 2010; Braun et al. 2011; Chekulaeva et al. 2011; Kuzuoğlu-Öztürk et al. 2016). However, CCR4–NOT was clearly substoichiometric and thus may not be essential for translational repression. The assay of the repressor complex only tests for constituents incorporated in the slow step, though; as the translation assay requires incubation of the gradient-purified repressed RNP with embryo extract, we cannot exclude that other components of the extract may associate with the stable complex and participate in its repressive activity, i.e., a protein that is not among the stable core components of the repressed RNP may still play a role in repression.
Five subunits of the conserved CTLH complex were enriched in the purified repressor complex, but substoichiometric with respect to the core components. The yeast edition of the complex is a ubiquitin ligase (Santt et al. 2008; Chen et al. 2017). Smg is degraded during cell cycle 14 (Dahanukar et al. 1999; Benoit et al. 2009), and most other core constituents of the repressor complex also strongly decrease during the maternal-to-zygotic transition (Gouw et al. 2009). The CTLH complex might be involved in the degradation of these proteins.
The DEAD-box RNA helicase Bel was the only newly discovered constituent of the repressor complex. Enrichment of the protein was less pronounced compared to the other core components, but genetic data confirmed that Bel is required for both translational repression and deadenylation of nos mRNA in vivo. Bel orthologs Ded1p and DDX3 are known to be involved in translation, but their precise role is unclear, since both depletion and overexpression inhibit translation (for reviews, see Soto-Rifo and Ohlmann 2013; Sharma and Jankowsky 2014). Bel and its orthologs can be localized in RNP granules containing repressed mRNAs and promote granule formation (Soto-Rifo and Ohlmann 2013; Sharma and Jankowsky 2014). Bel and C. elegans LAF-1 have been suggested to play a role in the translational repression of specific mRNAs, bruno and tra-2, respectively, but evidence for a direct role has been lacking so far (Goodwin et al. 1997; Yarunin et al. 2011). A cooperation of Ded1p with Dhh1p (Me31B) in translational repression is suggested by genetic and physical interactions (Tseng-Rogenski et al. 2003; Beckham et al. 2008; Drummond et al. 2011).
The idea that maternal mRNA in unfertilized eggs is not translated because it is masked by a “protective protein coat” was proposed more than 50 years ago (Spirin 1966). Whereas mechanisms have been analyzed that repress maternal mRNAs by targeting, directly or via the poly(A) tail, the 5′ cap function (Wilhelm and Smibert 2005; Lasko 2011; Barckmann and Simonelig 2013), proteins coating and sequestering the RNA have not been identified with certainty. Circumstantial biochemical evidence has supported the concept, though: Repressed RNPs formed in vitro sediment rapidly, suggesting association of the RNA with many proteins and tight packaging (Chekulaeva et al. 2006; this paper). Repressed RNAs are also moderately resistant to nucleases (Chekulaeva et al. 2006; Jeske et al. 2011; this paper). Repression of CRPV IRES-dependent translation, which is independent of all initiation factors, is consistent with exclusion of ribosomes (Jeske et al. 2011). Here we present evidence suggesting that the protective protein coat is formed by a complex of Me31B and Tral. The SREs nucleate the assembly of multiple copies of a Me31B•Tral complex on the RNA. The ortholog of Me31B is a component of stored Xenopus oocyte mRNPs (Weston and Sommerville 2006). The presence of Me31B and its partner Tral in the repressor complex is also consistent with previous reports of these two proteins interacting and causing translational repression (see Introduction). DDX6-type proteins bind RNA even in the absence of ATP (Dutta et al. 2011; Ernoult-Lange et al. 2012; Sharif et al. 2013). Tral presumably contributes directly to RNA coating, as it contains two types of potential RNA binding domains, an N-terminal Lsm domain and two RGG domains. Evidence for RNA binding by Tral orthologs has been published (Audhya et al. 2005; Tanaka et al. 2006). As the complex affords protection even against a relatively small endonuclease, we propose that it prevents translation by sterically excluding ribosomes, in agreement with the original idea of masking (Spirin 1966, 1994). Specificity of repression for the nos RNA depends on sequence-specific binding of Smg, but on the basis of biochemical similarities we suspect that other repressors may use similar mechanisms (Chekulaeva et al. 2006; Minshall et al. 2007).
A conceptual assembly of the repressor complex (Fig. 8) starts with Smg binding to the SREs. Smg binds Cup, which, in turn, associates with the Lsm domain of Tral (Tritschler et al. 2008; Igreja and Izaurralde 2011). Tral uses its FDF motif to bind Me31B (Tritschler et al. 2008, 2009; Igreja and Izaurralde 2011; this paper), but Me31B can also directly interact with Cup (Nishimura et al. 2015; Ozgur et al. 2015; Kamenska et al. 2016). The mechanism of Me31B•Tral polymerization remains to be analyzed. Cup also brings in eIF4E (Wilhelm et al. 2003; Nakamura et al. 2004; Nelson et al. 2004; Zappavigna et al. 2004; Igreja and Izaurralde 2011; Kinkelin et al. 2012). Bel may join the complex via interactions with eIF4E (Sharma and Jankowsky 2014) or Me31B (Drummond et al. 2011). Candidates for recruiting the CCR4–NOT complex include Me31B (Chen et al. 2014b; Mathys et al. 2014; Rouya et al. 2014; Ozgur et al. 2015; Waghray et al. 2015) and Smg (Semotok et al. 2005; Zaessinger et al. 2006). Me31B and Tral are both present in the embryo at very high concentrations, and micromolar concentrations of Me31B or Tral (Scd6p) can inhibit translation nonspecifically in vitro (Coller and Parker 2005; Nissan et al. 2010). It will be interesting to find out how assembly of the stable Me31B•Tral oligomer is restricted to SRE-containing mRNAs.
FIGURE 8.
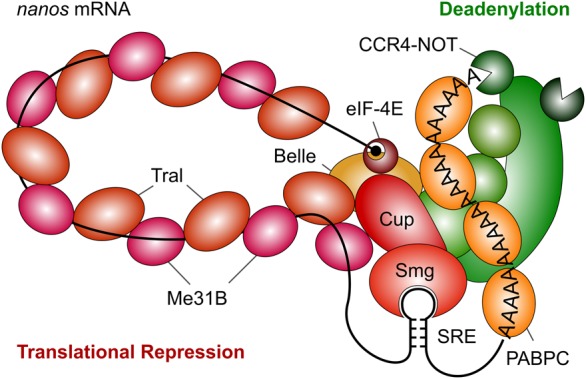
Model of the SRE-dependent repressor complex. The cartoon is based on the results of this paper and the references cited in the Discussion. Note that the accuracy of our Western blots is limited, thus the stoichiometry of protein binding depicted in the figure should not be interpreted narrowly.
The presence of two ATP-dependent RNA helicases, Bel and Me31B, in the repressor complex probably accounts for its ATP-dependence. (Note that the apparent ATP-dependence of Smg- and miRNA-dependent deadenylation has recently been shown to be a misinterpretation of the data [Niinuma and Tomari 2017]. ATP-dependence of repressor complex formation thus needs to be reexamined, but is unlikely to suffer from the same misinterpretation.) Me31B and/or Bel might also be responsible for the kinetic stability of the repressor complex. An attractive model is provided by the exon junction complex (EJC), which is frozen on the RNA because its central component, the DEAD-box ATPase eIF4AIII, is locked in a post-hydrolysis state by other EJC constituents (Ballut et al. 2005; Nielsen et al. 2009). Due to the cooperativity of ATP and RNA binding, this fixes the EJC on the RNA. Ded1 and two other DEAD-box helicases tested were able to form very long-lived complexes with RNA in the presence of ATP analogs (Liu et al. 2014). Thus, this “clamping” function may be a general feature of DEAD-box helicases. We speculate that a component of the repressor complex may inhibit the dissociation of ATP or its hydrolysis products from Me31B and/or Bel to prevent the disintegration of the repressor complex.
Polymerization of Me31B and Tral along the RNA, nucleated by Smaug binding in the 3′ UTR, conceptually solves a problem that, to our knowledge, has barely been discussed in the literature, although it is faced by all 3′ UTR-bound protein complexes repressing translation initiation: Cartoons depicting the mechanism of action of such complexes invariably show an interaction of the 3′ end with the 5′ end, accompanied by the formation of an RNA loop. Any such interaction has to be intramolecular, i.e., the 3′ UTR-bound repressor complex has to find the 5′ end of its “own” mRNA in the face of competition from “foreign” 5′ ends. (For an interesting alternative, see Macdonald et al. 2016.) One possibility for such an intramolecular interaction to occur would be “through space”: The two opposite ends of the flexible mRNA molecule diffuse randomly through the cytoplasm. As they are tethered to each other via the RNA body, an intramolecular interaction would be favored by a high local concentration of the cis 5′ end with respect to the regulatory 3′ UTR site. However, the efficiency with which this leads to an intramolecular interaction depends on variables like the length of the RNA and the concentration of competing 5′ ends. One would suspect that a more reliable mechanism should have evolved, in particular with a repressor complex as stable as the one described here: Any trans interaction established by mistake would not only result in a wrong RNA being repressed, but presumably also in a nos mRNA active in the wrong place. Polymerization of the Me31B•Tral complex along the RNA would constitute a fool-proof mechanism guaranteeing that the repressive action of the SREs is strictly intramolecular.
MATERIALS AND METHODS
RNA
All RNA constructs (SRE only; 1-AUG nos; luciferase reporter; all with two wild-type SREs or with a point mutation in each SRE) have been previously described (Jeske et al. 2006, 2011). RNAs were synthesized with T3 RNA polymerase. Biotin-16-UTP (Jena Bioscience) and [α-32P]-UTP were incorporated during transcription at a reduced concentration of UTP. For incorporation of a similar number of biotin molecules per RNA, UTP was adjusted according to the number of uridines in the RNA (Luc: 1 mM, 1-AUG: 0.25 mM, SREonly: 0.1 mM) at a constant concentration of biotin-16-UTP (20 µM). When desired, an m7G cap was also incorporated cotranscriptionally. RNAs were gel-purified.
“Short RNA” was produced by partial hydrolysis of yeast RNA: 75 mg of yeast total RNA was dissolved in 5 mL 20 mM Tris–HCl, pH 8.0. 200 µL of 2.5 M NaOH was added, and the mixture incubated for 50 min at 40°C. Two hundred microliters of 5 M HCl was added and incubation continued for 10 min at 40°C. After addition of 600 µL 3 M sodium acetate, the RNA was purified by phenol/chloroform extraction and isopropanol precipitation.
Embryo extract and in vitro translation
Extracts were prepared as previously described (Jeske and Wahle 2008) except that embryos (Canton S) were 15 to 135 min old, and the lysate was centrifuged twice (20,000g, 30 min, 4°C). Aliquots were frozen in liquid N2 and stored at −80°C.
Luciferase reporter RNAs were incubated at 25°C in 40% embryo extract, 16 mM Hepes pH 7.4, 50 mM potassium acetate, 1 mM magnesium acetate, 0.8 mM ATP, 0.25 mg/mL yeast tRNA, 0.2 mg/mL “short RNA,” 0.08 g/L creatine kinase, 1 mM DTT, 80 U/mL RNase inhibitor. Reactions were started with or without preincubation by the addition of 20 mM phosphocreatine and amino acids (20 µM each), incubated for 30 min at 25°C and stopped on ice. Luciferase activity was assayed with the Promega kit.
Purification of the repressor complex
Radiolabeled, biotinylated RNAs (10 nM) were incubated under conditions in which no translation takes place (“preincubation conditions”; 60% embryo extract, 26 mM Hepes-KOH pH 7.4, 81 mM potassium acetate, 1.6 mM magnesium acetate, 1.3 mM ATP, 1 mM DTT, 80 U/mL RNase inhibitor, 0.2 mg/mL “short RNA”) for 25 min at 25°C. The inclusion of “short RNA” improved RNA stability and complex recovery. Aliquots (1 mL) were loaded on 5%–45% sucrose gradients (12 mL per tube in TL buffer: 16 mM Hepes-KOH pH 7.4, 50 mM potassium acetate, 1 mM magnesium acetate, 0.8 mM ATP) and centrifuged for 3 h at 40,000 rpm, 4°C (Beckman SW40Ti). Gradients were harvested from the bottom in 20 fractions. Fractions 5–10 were pooled, frozen in liquid N2 and stored at −80°C. Pools from three gradients were combined and concentrated in Amicon centrifugal filters. Streptavidin beads (GE Healthcare; 30 µL packed volume) were blocked with TL buffer containing 0.1 mg/mL yeast RNA and 0.1 mg/mL methylated BSA and washed with TL buffer. Beads were incubated with equal amounts, based on trace-labeling of the RNA, of the concentrated pools and 0.1 mg/mL yeast RNA for 15 min at room temperature, pelleted and resuspended in 200 µL of TL buffer with yeast RNA as described above. Beads were pelleted, resuspended in the same buffer and centrifuged through a 30% sucrose cushion in TL buffer (200 µL). They were washed once more with the same buffer in a fresh tube, once with wash buffer (50 mM Hepes–KOH pH 7.4, 150 mM KCl, 54 mM potassium acetate, 1 mM magnesium acetate, 30 µg/mL heparin, 0.1 mg/mL yeast RNA), and once with wash buffer without RNA. Proteins were eluted in 10 mM Tris–HCl pH 8.0, 0.5% SDS at 80°C for 10 min.
For “simple” pull-down assays, the same procedure was used, but gradient centrifugation was omitted.
Mass spectrometry
Streptavidin-purified repressor proteins from four preparations (12 gradients) for each RNA were pooled and separated in an SDS–polyacrylamide gel. Each gel lane was cut into 12 pieces, and the proteins were in-gel digested with trypsin (Shevchenko et al. 2006). Disulfides were reduced with DTT and cysteines alkylated with iodoacetamide. Peptides were analyzed by LC/MS/MS on an U3000 RSLC Nano-HPLC system coupled to an Orbitrap Fusion Tribrid mass spectrometer equipped with a nano-electrospray ionization source (Thermo Fisher Scientific). The samples were loaded onto a trapping column (Acclaim PepMap C8, 300 µm × 5 mm, 5 µm, 100 Å) and washed for 15 min with 0.1% trifluoroacetic acid (TFA) at a flow rate of 30 µL/min. Trapped peptides were eluted on the separation column (Acclaim PepMap C18, 75 µm × 250 mm, 2 µm, 100 Å), which had been equilibrated with 99% A (0.1% formic acid). Peptides were separated with a linear gradient: 0%–35% B (100% acetonitrile, 0.08% formic acid) for 90 min at 40°C and a flow rate of 300 nL/min. Full MS data were acquired in the orbitrap (R = 60,000), MS/MS spectra (HCD, 30% normalized collision energy) were recorded in the linear trap for 5 sec (most intense signals).
MS data were analyzed with MaxQuant 1.5.2.8 (Cox and Mann 2008) (RRID: SCR:014485). For protein identification, data were searched against the Uniprot proteome (www.uniprot.org) of D. melanogaster (20,042 protein entries; accessed January 19, 2015). The inverted sequences of all proteins were used for decoy analysis. Mass accuracy was set to 20 ppm and 0.5 Da for precursor and fragment ions, respectively. Carbamidomethylation was set as fixed modification, and methionine oxidation and N-terminal acetylation were set as variable modifications. The search included common contaminating proteins, but these were omitted from the plots shown. Raw files from the analysis of 12 gel pieces of one lane each for WT and MUT were combined into one experiment for MaxQuant analysis. For calculation of the apparent protein abundance, the resulting peptide intensities for each protein were multiplied by the number of peptide spectral matches (PSMs) and normalized to the molecular weight of the protein. The calculated apparent abundance values were plotted, on a log2 scale, for proteins bound to SRE-containing RNA versus mutant RNA. For estimation of the P-value for SRE-dependent enrichment, the proteins were separated in 33 equally sized bins along the diagonal axis. The distance of each protein from the diagonal in each bin follows a normal distribution with a mean of 0 for all nonspecifically bound proteins. The distances were fitted against the normal distribution to obtain σ². To estimate P-values for each protein enrichment, the σ²-values were fitted with the equation y = a × b−c·(bin-45). The squared differences to the model were multiplied with the number of proteins per bin to weight the data in the nonlinear least squares fit. Data are available via ProteomeXchange with identifier PXD006596.
Analysis of RNA associated with the repressor complex
Repressor complex was isolated as described above from 400 µL of reaction mixture without gradient centrifugation, and no yeast RNA was used during the pull-down and washing procedures. RNA was eluted with TRIzol for 10 min at 80°C. Five hundred nanograms of total RNA was used in the small RNA protocol with the TruSeq Small RNA Sample Prepkit v2 (Illumina) according to the manufacturer's instructions. The barcoded libraries were size restricted between 140 and 165 bp, purified and quantified using the Library Quantification Kit (Illumina/Universal, KAPA Biosystems). Library pooling, cluster generation, high-throughput sequencing of 2 × 100 bp and demultiplexing of raw reads was done according to Stokowy et al. (2014).
Reads were stripped of the 3′ linker (TGGAATTCTCGGGTGCCAAGGAACTCCAGTCAC) using Cutadapt, and the resulting RNA sequences were mapped to the Drosophila melanogaster genome using Bowtie (100% match; release 5). Reads were first annotated to tRNA, rRNA, snoRNA, snRNA, and miRNAs. piRNAs were the remaining reads that were 23–29 nt in length. piRNAs were mapped to TE using Bowtie with up to three mismatches. Uniquely mapped piRNAs were mapped to piRNA clusters using cluster coordinates from Brennecke et al. (2007). mRNA-derived small RNAs were uniquely mapped reads that mapped in sense orientation to genes. Small RNA counts were normalized to 1 million mapped reads.
RNase protection assay
Two nanomolars of radiolabeled 1-AUG-RNA (SRE+ or SRE−) were incubated under preincubation conditions (without DTT and RNase inhibitor) for 25 min at 25°C. Seventy microliters of the reaction were mixed with 35 µL of RNase If (NEB) at a final concentration of 0.66 U/µL. Fifteen-microliter aliquots of the reaction were stopped at different time points in SDS-containing 2× proteinase K buffer with 20 µg Proteinase K, 20 µg glycogen and an unrelated radiolabeled RNA as extraction control. After incubation at 37°C for 30 min, the sample was ethanol precipitated and analyzed on a denaturing 5% polyacrylamide gel.
Western blots and immunostaining
The Western blots in Figure 7 and Supplemental Figure S4 and immunostaining of embryos were performed as previously described (Benoit et al. 2005). In other experiments, SDS–polyacrylamide gels were blotted overnight in 25 mM Tris, 192 mM glycine onto PVDF membranes and blocked in 5% milk in TBST. After primary antibody incubation, blots were washed with TBST and incubated with fluorescently labeled secondary antibodies (IR-Dye; LI-COR), washed and scanned on a LICOR scanner. The following proteins were used as standards for quantitative Western blots: Me31B, Tral, and Bel were the E. coli-produced proteins used for immunization (see below). His-tagged Ccr4, Not2, and Caf 1 were produced in E. coli from pET19 and purified under denaturing (Ccr4, Not2) or native conditions (Caf1). Flag-Smg and Flag-Cup were coexpressed in the baculovirus system and affinity-purified as a mixture. All standard proteins were quantitated by Coomassie staining of SDS–polyacrylamide gels and comparison to an unrelated standard protein; dye binding to different proteins under acidic conditions has been estimated to vary by about a factor of 2 (Chial and Splittgerber 1993). The Me31B standard was used to estimate the Me31B concentration in a batch of embryo extract which was then used as a standard in the analysis of pull-down assays. All other standard proteins were used directly.
Antibodies against Cup were obtained from Akira Nakamura (Nakamura et al. 2004) or Robin Wharton (Verrotti and Wharton 2000); against Nos from Akira Nakamura; against PABPC from Nahum Sonenberg (Imataka et al. 1998) or Matthias Hentze (Duncan et al. 2009); against Yps from James Wilhelm (Wilhelm et al. 2000). Antibodies against Me31B (Nakamura et al. 2001; Harnisch et al. 2016), Smg (Chartier et al. 2015), Ccr4, Caf1 (affinity-purified), Not2 (Temme et al. 2004), and Not3 (Jeske et al. 2006) have been described. Additional guinea pig antibody against Smg was from Craig Smibert (Tadros et al. 2007). Antibodies against Bel were initially obtained from Paul Lasko (Johnstone et al. 2005), and antibodies against Tral were initially from Elisa Izaurralde (Tritschler et al. 2008). Additional antibodies against Bel and Tral were generated as follows: N-terminally His-tagged variants of the proteins were expressed in E. coli. Cells from a 400 mL culture were resuspended in 100 mM Tris–HCl pH 7.0, 1 mM EDTA, incubated with 1.5 mg lysozyme, and lysed by ultrasonification. After DNase I treatment of the lysate, 20 mM EDTA, 2% Triton, and 500 mM NaCl were added, insoluble proteins were pelleted for 10 min at 31,000g and washed with 100 mM Tris–HCl pH 7.0, 20 mM EDTA. The pellets were dissolved in urea buffer (8 M urea, 100 mM Tris–HCl pH 8.0, 100 mM Na2HPO4) and proteins bound to Ni–NTA–Agarose. The columns were washed with urea buffer and urea buffer with 20 mM imidazole and proteins eluted with urea buffer plus 250 mM imidazole. After concentration with AMICON centrifugal filters, the proteins were diluted with PBS to <4 M urea, and 800 µg was used by Eurogentec S.A. (Belgium) to immunize two rats (Tral) or two rabbits (Bel).
For MS analysis of Smg-associated proteins, 2 × 60 µL of Protein A-Sepharose beads (GE Healthcare) were washed three times with TL-buffer (see Purification of the Repressor Complex section above). Beads (60 µL) were incubated with 20 µL of anti-Smaug serum or the corresponding preimmune serum in 500 µL TL buffer for 2 h at ∼8°C, washed twice with wash buffer (see Purification of the Repressor Complex section) and twice with TL buffer. The beads were incubated with 500 µL Drosophila embryo extract, diluted 1:4 in TL buffer for 2 h at ∼8°C, transferred to Protein-LoBind tubes (Eppendorf) and washed with TL-buffer (two times), wash buffer (three times), and TL-buffer (two times). In one sample, bound proteins were cross-linked for 2 h at 8°C with 0.5 µM BuUrBu cross-linker (Müller et al. 2010) in 500 µL of TL Buffer. Bound proteins were denatured in 100 µL 8 M urea, 0.4 M ammonium bicarbonate, disulfides were reduced with 10 mM DTT and alkylated with 55 mM iodoacetamide. The sample was diluted to 0.8 M urea and digested with trypsin overnight at 37°C. LC/MS/MS analysis was performed as described above except that MS/MS spectra (HCD, 30% normalized collision energy) were acquired in the Orbitrap analyzer (R = 60,000) for 5 sec (most intense signals). Data were evaluated as described above. Cross-links were analyzed by means of the MeroX software (Götze et al. 2015) with the following parameters: Trypsin was set as the protease, the BuUrBu cross-linker was selected, methionine oxidation was considered as variable modification, cysteine alkylation was set as static modification, the signal-to-noise ratio was set to 1.5, precursor precision was set to 3 ppm, and fragment ion precision was set to 20 ppm. The analysis was performed with activated RISE-mode, and data were searched against a sequence database containing the set of enriched proteins from the RNA-pull-down experiment as well as sequences of 200 standard protein contaminants (cRAP-database http://www.thegpm.org/crap/index.html), which resulted in one highly confident cross-linked peptide pair between Me31B and Tral.
For RNA immunoprecipitations, 50 µl Protein A Mag Sepharose beads (GE Healthcare) were prewashed twice in RIP buffer (25 mM Hepes pH 6.8, 250 mM sucrose, 1 mM MgCl2, 1 mM DTT, 150 mM NaCl, 0.1% Triton-X 100, containing freshly added complete protease inhibitor cocktail EDTA free [Roche] and RNasin [Promega]). Ten microliters of mouse anti-GFP (monoclonal antibody 3E6; Invitrogen) were added, the mixture was incubated in 500 µL RIP buffer for 2 h at 4°C on a wheel, and beads were washed twice in RIP buffer. Embryos (0–2 h old) were homogenized on ice in four volumes of RIP buffer and incubated for 20 min on ice. The homogenate was centrifuged twice at 10,000g for 5 min and precleared on 50 µL of RIP buffer-washed Protein A Mag Sepharose beads for 45 min at 4°C on a wheel. Beads were removed, and the extract was mixed with the anti-GFP antibody beads and incubated for 1.5 to 2.5 h at 4°C with rotation. The beads were washed eight times with RIP buffer, extracted with TRIzol, and the RNA was isopropanol-precipitated in the presence of glycogen and resuspended in 12 µL H2O.
Expression of GST-Me31B and Tral
The coding sequence of Me31B with an N-terminal GST tag and PreScission site was cloned into pFastBac1 and transferred into the Bac-to-Bac baculovirus expression system (Thermo Fisher Scientific). Expression clones for untagged Tral were generated by the same procedure. For protein expression, Sf21 cells were infected at MOI = 1 and harvested 3 d later. For Western blots, equal numbers of cells were lysed in SDS gel loading buffer. For GST pulldown, cells were sonicated in GST buffer (50 mM HEPES pH 7.5, 150 mM KCl, 1.5 mM MgCl2, 10% saccharose). The cleared lysate was incubated with Glutathione Sepharose (GE Healthcare) for 2 h at 4°C. After incubation, beads were washed three times briefly and two times for 10 min in GST buffer. Bound proteins were eluted in SDS gel loading buffer and analyzed by Western blot or SDS-PAGE and Coomassie staining. Copurification of Tral with GST-Me31B was not affected by the inclusion of RNase A or elevated salt concentration (400 mM KCl).
Fly stocks
The w1118 stock was used as control. bel mutant alleles were bel6, belL4740 and belneo30 (Bloomington Stock Center). The GFP protein-trap allele belCC00869 corresponds to a GFP insertion after the first coding exon at amino acid 15; it contains the complete Bel coding sequence (Buszczak et al. 2007).
In situ hybridization, PAT assays, and RT-qPCR
Whole-mount in situ hybridization was performed by standard methods. The probe was an antisense RNA made from the pN5 nos cDNA clone (Wang and Lehmann 1991). Poly(A) test (PAT) assays and RT–qPCR were performed as previously described (Rouget et al. 2010) on two to four independent RNA preparations. Reverse transcription for qPCR was done using random hexamers (Invitrogen) and Superscript-III (Invitrogen). Real-time PCR (qPCR) was performed with the LightCycler System (Roche Molecular Biochemical) using RpL32 as a control mRNA. Primers were as follows: Hsp83-fw-qPCR: CAACAAGCAGCGTCTGAAAAG; Hsp83-rev-qPCR: AGCCTGGAATGCAAAGGTC; nos1128F: CGGAGCTTCCAATTCCAGTAAC; nos1281R: AGTTATCTCGCACTGAGTGGCT; RpL32F: CTTCATCCGCCACCAGTC; RpL32R: CGACGCACTCTGTTGTCG; nosPostPAT: TTTTGTTTACCATTGATCAATTTTTC; sopPAT: GGATTGCTACACCTCGGCCCGT.
Microscopy and image processing
Fluorescent images were acquired using a Carl Zeiss LSM 780 LASER scanning confocal microscope (Montpellier RIO Imaging facility) and a 40× PLAN Apochromatic 1.3 oil immersion objective. The acquisition software was Zen. Contrast and relative intensities were processed with ImageJ software. Light microscope images were acquired using Leica Leitz DMRB Fluorescence Microscope with Nomarsky lens. Colocalization was quantified by FIJI as follows: Background was subtracted with a rolling ball radius of 30 µm, and the Pearson correlation coefficient (PCC) was calculated using the colloc2 plugin with auto-thresholding. Mean PCC was calculated from three images.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank F. Kluge and D. Taenzler for excellent assistance with experiments; P. Enke for generating baculovirus clones; K. Krohn, IZKF, University of Leipzig, for performing Illumina sequencing; M. Hentze, E. Izaurralde, P. Lasko, A. Nakamura, C. Smibert, N. Sonenberg, R. Wharton, and J. Wilhelm for reagents; H. Ashe for fly stocks; F. Falvo for the BuUrBu cross-linker; and M. Jeske and C. Eckmann for comments on earlier versions of the manuscript. This work was supported by grants from the Deutsche Forschungsgemeinschaft (WA 548/13-2 within FOR 855; WA 548/16-1; GRK 1591) to E.W. and by CNRS-University of Montpellier UMR9002, Association Nationale de la Recherche et de la Technologie (ANR-15-CE12-0019-01), Fondation pour la Recherche Médicale (Equipe FRM 2013 DEQ20130326534), and Fondation ARC pour la Recherche sur le Cancer (ARC Libre 2009, no. 3192) to M.S. J.D. was supported by Fondation ARC.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.062208.117.
REFERENCES
- Ali-Murthy Z, Lott SE, Eisen MB, Kornberg TB. 2013. An essential role for zygotic expression in the pre-cellular Drosophila embryo. Plos Genet 9: e1003428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audhya A, Hyndman F, McLeod IX, Maddox AS, Yates JR III, Desai A, Oegema K. 2005. A complex containing the Sm protein CAR-1 and the RNA helicase CGH-1 is required for embryonic cytokinesis in Caenorhabditis elegans. J Cell Biol 171: 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayache J, Bénard M, Ernoult-Lange M, Minshall N, Standart N, Kress M, Weil D. 2015. P-body assembly requires DDX6 repression complexes rather than decay or Ataxin2/2L complexes. Mol Biol Cell 26: 2579–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azevedo RBR, French V, Partridge L. 1996. Thermal evolution of egg size in Drosophila melanogaster. Evolution 50: 2338–2345. [DOI] [PubMed] [Google Scholar]
- Ballut L, Marchadier B, Baguet A, Tomasetto C, Séraphin B, Le Hir H. 2005. The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat Struct Mol Biol 12: 861–869. [DOI] [PubMed] [Google Scholar]
- Barckmann B, Simonelig M. 2013. Control of maternal mRNA stability in germ cells and early embryos. Biochim Biophys Acta 1829: 714–724. [DOI] [PubMed] [Google Scholar]
- Barckmann B, Pierson S, Dufourt J, Papin C, Armenise C, Port F, Grentzinger T, Chambeyron S, Baronian G, Desvignes JP, et al. 2015. Aubergine iCLIP reveals piRNA-dependent decay of mRNAs involved in germ cell development in the early embryo. Cell Rep 12: 1205–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashirullah A, Halsell SR, Cooperstock RL, Kloc M, Karaiskakis A, Fisher WW, Fu W, Hamilton JK, Etkin LD, Lipshitz HD. 1999. Joint action of two RNA degradation pathways controls the timing of maternal transcript elimination at the midblastula transition in Drosophila melanogaster. EMBO J 18: 2610–2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckham C, Hilliker A, Cziko AM, Noueiry A, Ramaswami M, Parker R. 2008. The DEAD-box RNA helicase Ded1p affects and accumulates in Saccharomyces cerevisiae P-bodies. Mol Biol Cell 19: 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit B, Mitou G, Chartier A, Temme C, Zaessinger S, Wahle E, Busseau I, Simonelig M. 2005. An essential cytoplasmic function for the nuclear poly(A) binding protein, PABP2, in poly(A) tail length control and early development in Drosophila. Dev Cell 9: 511–522. [DOI] [PubMed] [Google Scholar]
- Benoit B, He CH, Zhang F, Votruba SM, Tadros W, Westwood JT, Smibert CA, Lipshitz HD, Theurkauf WE. 2009. An essential role for the RNA-binding protein Smaug during the Drosophila maternal-to-zygotic transition. Development 136: 923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten SE, Gavis ER. 1999. Role for mRNA localization in translational activation but not spatial restriction of nanos RNA. Development 126: 659–669. [DOI] [PubMed] [Google Scholar]
- Besse F, López de Quinto S, Marchand V, Trucco A, Ephrussi A. 2009. Drosophila PTB promotes formation of high-order RNP particles and represses oskar translation. Genes Dev 23: 195–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bish R, Cuevas-Polo N, Cheng Z, Hambardzumyan D, Munschauer M, Landthaler M, Vogel C. 2015. Comprehensive protein interactome analysis of a key RNA helicase: detection of novel stress granule proteins. Biomolecules 5: 1441–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boag PR, Nakamura A, Blackwell TK. 2005. A conserved RNA-protein complex component involved in physiological germline apoptosis regulation in C. elegans. Development 132: 4975–4986. [DOI] [PubMed] [Google Scholar]
- Braun JE, Huntzinger E, Fauser M, Izaurralde E. 2011. GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol Cell 44: 120–133. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. 2007. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128: 1089–1103. [DOI] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J, Planck J, Owen S, Skora AD, Nystul TG, Ohlstein B, Allen A, et al. 2007. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics 175: 1505–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartier A, Klein P, Pierson S, Barbezier N, Gidaro T, Casas F, Carberry S, Dowling P, Maynadier L, Bellec M, et al. 2015. Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis. PLoS Genet 11: e1005092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chekulaeva M, Hentze MW, Ephrussi A. 2006. Bruno acts as a dual repressor of oskar translation, promoting mRNA oligomerization and formation of silencing particles. Cell 124: 521–533. [DOI] [PubMed] [Google Scholar]
- Chekulaeva M, Mathys H, Zipprich JT, Attig J, Colic M, Parker R, Filipowicz W. 2011. miRNA repression involves GW182-mediated recruitment of CCR4–NOT through conserved W-containing motifs. Nat Struct Mol Biol 18: 1218–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Dumelie JG, Li X, Cheng MH, Yang Z, Laver JD, Siddiqui NU, Westwood JT, Morris Q, Lipshitz HD, et al. 2014a. Global regulation of mRNA translation and stability in the early Drosophila embryo by the Smaug RNA-binding protein. Genome Biol 15: R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Boland A, Kuzuoğlu-Öztürk D, Bawankar P, Loh B, Chang C-T, Weichenrieder O, Izaurralde E. 2014b. A DDX6–CNOT1 complex and W-binding pockets in CNOT9 reveal direct links between miRNA target recognition and silencing. Mol Cell 54: 737–750. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Wu X, Wadas B, Oh JH, Varshavsky A. 2017. An N-end rule pathway that recognizes proline and destroys gluconeogenic enzymes. Science 355: eaal3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chial HJ, Splittgerber AG. 1993. A comparison of the binding of Coomassie brilliant blue to proteins at low and neutral pH. Anal Biochem 213: 362–369. [DOI] [PubMed] [Google Scholar]
- Coller J, Parker R. 2005. General translational repression by activators of mRNA decapping. Cell 122: 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke A, Prigge A, Wickens M. 2010. Translational repression by deadenylases. J Biol Chem 285: 28506–28513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Dahanukar A, Wharton RP. 1996. The Nanos gradient in Drosophila embryos is generated by translational regulation. Genes Dev 10: 2610–2620. [DOI] [PubMed] [Google Scholar]
- Dahanukar A, Walker JA, Wharton RP. 1999. Smaug, a novel RNA-binding protein that operates a translational switch in Drosophila. Mol Cell 4: 209–218. [DOI] [PubMed] [Google Scholar]
- Drummond SP, Hildyard J, Firczuk H, Reamtong O, Li N, Kannambath S, Claydon AJ, Beynon RJ, Eyers CE, McCarthy JE. 2011. Diauxic shift-dependent relocalization of decapping activators Dhh1 and Pat1 to polysomal complexes. Nucleic Acids Res 39: 7764–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan KE, Strein C, Hentze MW. 2009. The SXL-UNR corepressor complex uses a PABP-mediated mechanism to inhibit ribosome recruitment to msl-2 mRNA. Mol Cell 36: 571–582. [DOI] [PubMed] [Google Scholar]
- Dutta A, Zheng S, Jain D, Cameron CE, Reese JC. 2011. Intermolecular interactions within the abundant DEAD-box protein Dhh1 regulate its activity in vivo. J Biol Chem 286: 27454–27470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ephrussi A, Lehmann R. 1992. Induction of germ cell formation by oskar. Nature 358: 387–392. [DOI] [PubMed] [Google Scholar]
- Ernoult-Lange M, Baconnais S, Harper M, Minshall N, Souquere S, Boudier T, Bénard M, Andrey P, Pierron G, Kress M, et al. 2012. Multiple binding of repressed mRNAs by the P-body protein Rck/p54. RNA 18: 1702–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis O, Han F, Adams JC. 2013. Molecular phylogeny of a RING E3 ubiquitin ligase, conserved in eukaryotic cells and dominated by homologous components, the muskelin/RanBPM/CTLH complex. PLoS One 8: e75217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavis ER, Lehmann R. 1994. Translational regulation of nanos by RNA localization. Nature 369: 315–318. [DOI] [PubMed] [Google Scholar]
- Goodwin EB, Hofstra K, Hurney CA, Mango S, Kimble J. 1997. A genetic pathway for regulation of tra-2 translation. Development 124: 749–758. [DOI] [PubMed] [Google Scholar]
- Götze M, Pettelkau J, Fritzsche R, Ihling CH, Schäfer M, Sinz A. 2015. Automated assignment of MS/MS cleavable cross-links in protein 3D-structure analysis. J Am Soc Mass Spectr 26: 83–97. [DOI] [PubMed] [Google Scholar]
- Gouw JW, Pinkse MW, Vos HR, Moshkin Y, Verrijzer CP, Heck AJ, Krijgsveld J. 2009. In vivo stable isotope labeling of fruit flies reveals post-transcriptional regulation in the maternal-to-zygotic transition. Mol Cell Proteomics 8: 1566–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas G, Braun JE, Igreja C, Tritschler F, Nishihara T, Izaurralde E. 2010. HPat provides a link between deadenylation and decapping in metazoa. J Cell Biol 189: 289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harnisch C, Cuzic-Feltens S, Dohm JC, Götze M, Himmelbauer H, Wahle E. 2016. Oligoadenylation of 3′ decay intermediates promotes cytoplasmic mRNA degradation in Drosophila cells. RNA 22: 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison MM, Eisen MB. 2015. Transcriptional activation of the zygotic genome in Drosophila. Curr Top Dev Biol 113: 85–112. [DOI] [PubMed] [Google Scholar]
- Hernández G, Altmann M, Sierra JM, Urlaub H, Diez del Corral R, Schwartz P, Rivera-Pomar R. 2005. Functional analysis of seven genes encoding eight translation initiation factor 4E (eIF4E) isoforms in Drosophila. Mech Dev 122: 529–543. [DOI] [PubMed] [Google Scholar]
- Hough-Evans BR, Jacobs-Lorena M, Cummings MR, Britten RJ, Davidson EH. 1980. Complexity of RNA in eggs of Drosophila melanogaster and Musca domestica. Genetics 95: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubstenberger A, Noble SL, Cameron C, Evans TC. 2013. Translation repressors, an RNA helicase, and developmental cues control RNP phase transitions during early development. Dev Cell 27: 161–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igreja C, Izaurralde E. 2011. CUP promotes deadenylation and inhibits decapping of mRNA targets. Genes Dev 25: 1955–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihry RJ, Sapiro AL, Bashirullah A. 2012. Translational control by the DEAD Box RNA helicase belle regulates ecdysone-triggered transcriptional cascades. PLoS Genet 8: e1003085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imataka H, Gradi A, Sonenberg N. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J 17: 7480–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeske M, Wahle E. 2008. Cell-free deadenylation assays with Drosophila embryo extracts. Methods Enzymol 448: 107–118. [DOI] [PubMed] [Google Scholar]
- Jeske M, Meyer S, Temme C, Freudenreich D, Wahle E. 2006. Rapid ATP-dependent deadenylation of nanos mRNA in a cell-free system from Drosophila embryos. J Biol Chem 281: 25124–25133. [DOI] [PubMed] [Google Scholar]
- Jeske M, Moritz B, Anders A, Wahle E. 2011. Smaug assembles an ATP-dependent stable complex repressing nanos mRNA translation at multiple levels. EMBO J 30: 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone O, Deuring R, Bock R, Linder P, Fuller MT, Lasko P. 2005. Belle is a Drosophila DEAD-box protein required for viability and in the germ line. Dev Biol 277: 92–101. [DOI] [PubMed] [Google Scholar]
- Jonas S, Izaurralde E. 2013. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev 27: 2628–2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenska A, Lu WT, Kubacka D, Broomhead H, Minshall N, Bushell M, Standart N. 2014. Human 4E-T represses translation of bound mRNAs and enhances microRNA-mediated silencing. Nucleic Acids Res 42: 3298–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenska A, Simpson C, Vindry C, Broomhead H, Bénard M, Ernoult-Lange M, Lee BP, Harries LW, Weil D, Standart N. 2016. The DDX6–4E-T interaction mediates translational repression and P-body assembly. Nucleic Acids Res 44: 6318–6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkelin K, Veith K, Grünwald M, Bono F. 2012. Crystal structure of a minimal eIF4E-Cup complex reveals a general mechanism of eIF4E regulation in translational repression. RNA 18: 1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzuoğlu-Öztürk D, Bhandari D, Huntzinger E, Fauser M, Helms S, Izaurralde E. 2016. miRISC and the CCR4–NOT complex silence mRNA targets independently of 43S ribosomal scanning. EMBO J 35: 1186–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasko P. 2011. Posttranscriptional regulation in Drosophila oocytes and early embryos. Wiley Interdiscip Rev RNA 2: 408–416. [DOI] [PubMed] [Google Scholar]
- Laver JD, Marsolais AJ, Smibert CA, Lipshitz HD. 2015. Regulation and function of maternal gene products during the maternal-to-zygotic transition in Drosophila. Curr Top Dev Biol 113: 43–84. [DOI] [PubMed] [Google Scholar]
- Little SC, Sinsimer KS, Lee JJ, Wieschaus EF, Gavis ER. 2015. Independent and coordinate trafficking of single Drosophila germ plasm mRNAs. Nat Cell Biol 17: 558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Putnam AA, Jankowsky E. 2014. DEAD-box helicases form nucleotide-dependent, long-lived complexes with RNA. Biochemistry 53: 423–433. [DOI] [PubMed] [Google Scholar]
- Macdonald PM, Kanke M, Kenny A. 2016. Community effects in regulation of translation. eLife 5: e10965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys H, Basquin J, Ozgur S, Czarnocki-Cieciura M, Bonneau F, Aartse A, Dziembowski A, Nowotny M, Conti E, Filipowicz W. 2014. Structural and biochemical insights to the role of the CCR4–NOT complex and DDX6 ATPase in microRNA repression. Mol Cell 54: 751–765. [DOI] [PubMed] [Google Scholar]
- Minshall N, Standart N. 2004. The active form of Xp54 RNA helicase in translational repression is an RNA-mediated oligomer. Nucleic Acids Res 32: 1325–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Thom G, Standart N. 2001. A conserved role of a DEAD box helicase in mRNA masking. RNA 7: 1728–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshall N, Reiter MH, Weil D, Standart N. 2007. CPEB interacts with an ovary-specific eIF4E and 4E-T in early Xenopus oocytes. J Biol Chem 282: 37389–37401. [DOI] [PubMed] [Google Scholar]
- Müller MQ, Dreiocker F, Ihling CH, Schäfer M, Sinz A. 2010. Cleavable cross-linker for protein structure analysis: reliable identification of cross-linking products by tandem MS. Anal Chem 82: 6958–6968. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Amikura R, Hanyu K, Kobayashi S. 2001. Me31B silences translation of oocyte-localizing RNAs through the formation of cytoplasmic RNP complex during Drosophila oogenesis. Development 128: 3233–3242. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Sato K, Hanyu-Nakamura K. 2004. Drosophila Cup is an eIF4E binding protein that associates with Bruno and regulates oskar mRNA translation in oogenesis. Dev Cell 6: 69–78. [DOI] [PubMed] [Google Scholar]
- Nelson MR, Leidal AM, Smibert CA. 2004. Drosophila Cup is an eIF4E-binding protein that functions in Smaug-mediated translational repression. EMBO J 23: 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen KH, Chamieh H, Andersen CB, Fredslund F, Hamborg K, Le Hir H, Andersen GR. 2009. Mechanism of ATP turnover inhibition in the EJC. RNA 15: 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niinuma S, Tomari Y. 2017. ATP is dispensable for both miRNA- and Smaug-mediated deadenylation reactions. RNA 23: 866–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Padamsi Z, Fakim H, Milette S, Dunham WH, Gingras AC, Fabian MR. 2015. The eIF4E-binding protein 4E-T is a component of the mRNA decay machinery that bridges the 5′ and 3′ termini of target mRNAs. Cell Rep 11: 1425–1436. [DOI] [PubMed] [Google Scholar]
- Nissan T, Rajyaguru P, She M, Song H, Parker R. 2010. Decapping activators in Sacchromyces cerevisiae act by multiple mechanisms. Mol Cell 39: 773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozgur S, Basquin J, Kamenska A, Filipowicz W, Standart N, Conti E. 2015. Structure of a human 4E-T/DDX6/CNOT1 complex reveals the different interplay of DDX6-binding proteins with the CCR4-NOT complex. Cell Rep 13: 703–711. [DOI] [PubMed] [Google Scholar]
- Pinder BD, Smibert CA. 2013. microRNA-independent recruitment of Argonaute 1 to nanos mRNA through the Smaug RNA-binding protein. EMBO Rep 14: 80–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouget C, Papin C, Boureux A, Meunier AC, Franco B, Robine N, Lai EC, Pelisson A, Simonelig M. 2010. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 467: 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouya C, Siddiqui N, Morita M, Duchaine TF, Fabian MR, Sonenberg N. 2014. Human DDX6 effects miRNA-mediated gene silencing via direct binding to CNOT1. RNA 20: 1398–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santt O, Pfirrmann T, Braun B, Juretschke J, Kimmig P, Scheel H, Hofmann K, Thumm M, Wolf DH. 2008. The yeast GID complex, a novel ubiquitin ligase (E3) involved in the regulation of carbohydrate metabolism. Mol Biol Cell 19: 3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semotok JL, Cooperstock RL, Pinder BD, Vari HK, Lipshitz HD, Smibert CA. 2005. Smaug recruits the CCR4/POP2/NOT deadenylase complex to trigger maternal transcript localization in the early Drosophila embryo. Curr Biol 15: 284–294. [DOI] [PubMed] [Google Scholar]
- Sharif H, Ozgur S, Sharma K, Basquin C, Urlaub H, Conti E. 2013. Structural analysis of the yeast Dhh1-Pat1 complex reveals how Dhh1 engages Pat1, Edc3 and RNA in mutually exclusive interactions. Nucleic Acids Res 41: 8377–8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma D, Jankowsky E. 2014. The Ded1/DDX3 subfamily of DEAD-box RNA helicases. Crit Rev Biochem Mol Biol 49: 343–360. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. 2006. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 1: 2856–2860. [DOI] [PubMed] [Google Scholar]
- Smibert CA, Wilson JE, Kerr K, Macdonald PM. 1996. Smaug protein represses translation of unlocalized nanos mRNA in the Drosophila embryo. Genes Dev 10: 2600–2609. [DOI] [PubMed] [Google Scholar]
- Smibert CA, Lie YS, Shillinglaw W, Henzel WJ, Macdonald PM. 1999. Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro. RNA 5: 1535–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JL, Wilson JE, Macdonald PM. 1992. Overexpression of oskar directs ectopic activation of nanos and presumptive pole cell formation in Drosophila embryos. Cell 70: 849–859. [DOI] [PubMed] [Google Scholar]
- Soto-Rifo R, Ohlmann T. 2013. The role of the DEAD-box RNA helicase DDX3 in mRNA metabolism. Wiley Interdiscip Rev RNA 4: 369–385. [DOI] [PubMed] [Google Scholar]
- Spirin AS. 1966. On “masked” forms of messenger RNA in early embryogenesis and in other differentiating systems. Curr Top Dev Biol 1: 1–38. [PubMed] [Google Scholar]
- Spirin AS. 1994. Storage of messenger RNA in eukaryotes: envelopment with protein, translational barrier at 5′ side, or conformational masking by 3′ side? Mol Rep Dev 38: 107–117. [DOI] [PubMed] [Google Scholar]
- Stokowy T, Eszlinger M, Świerniak M, Fujarewicz K, Jarzab B, Paschke R, Krohn K. 2014. Analysis options for high-throughput sequencing in miRNA expression profiling. BMC Res Notes 7: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr-Weaver T, Hughes TR, Westwood JT, Smibert CA, Lipshitz HD. 2007. SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev Cell 12: 143–155. [DOI] [PubMed] [Google Scholar]
- Tanaka KJ, Ogawa K, Takagi M, Imamoto N, Matsumoto K, Tsujimoto M. 2006. RAP55, a cytoplasmic mRNP component, represses translation in Xenopus oocytes. J Biol Chem 281: 40096–40106. [DOI] [PubMed] [Google Scholar]
- Temme C, Zaessinger S, Simonelig M, Wahle E. 2004. A complex containing the CCR4 and CAF1 proteins is involved in mRNA deadenylation in Drosophila. EMBO J 23: 2862–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trcek T, Grosch M, York A, Shroff H, Lionnet T, Lehmann R. 2015. Drosophila germ granules are structured and contain homotypic mRNA clusters. Nat Commun 6: 7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Eulalio A, Helms S, Schmidt S, Coles M, Weichenrieder O, Izaurralde E, Truffault V. 2008. Similar modes of interaction enable Trailer Hitch and EDC3 to associate with DCP1 and Me31B in distinct protein complexes. Mol Cell Biol 28: 6695–6708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritschler F, Braun JE, Eulalio A, Truffault V, Izaurralde E, Weichenrieder O. 2009. Structural basis for the mutually exclusive anchoring of P body components EDC3 and Tral to the DEAD box protein DDX6/Me31B. Mol Cell 33: 661–668. [DOI] [PubMed] [Google Scholar]
- Tseng-Rogenski SS, Chong JL, Thomas CB, Enomoto S, Berman J, Chang TH. 2003. Functional conservation of Dhh1p, a cytoplasmic DExD/H-box protein present in large complexes. Nucleic Acids Res 31: 4995–5002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrotti AC, Wharton RP. 2000. Nanos interacts with cup in the female germline of Drosophila. Development 127: 5225–5232. [DOI] [PubMed] [Google Scholar]
- Waghray S, Williams C, Coon JJ, Wickens M. 2015. Xenopus CAF1 requires NOT1-mediated interaction with 4E-T to repress translation in vivo. RNA 21: 1335–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Lehmann R. 1991. Nanos is the localized posterior determinant in Drosophila. Cell 66: 637–647. [DOI] [PubMed] [Google Scholar]
- Weston A, Sommerville J. 2006. Xp54 and related (DDX6-like) RNA helicases: roles in messenger RNP assembly, translation regulation and RNA degradation. Nucleic Acids Research 34: 3082–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JE, Smibert CA. 2005. Mechanisms of translational regulation in Drosophila. Biol Cell 97: 235–252. [DOI] [PubMed] [Google Scholar]
- Wilhelm JE, Mansfield J, Hom-Booher N, Wang S, Turck CW, Hazelrigg T, Vale RD. 2000. Isolation of a ribonucleoprotein complex involved in mRNA localization in Drosophila oocytes. J Cell Biol 148: 427–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JE, Hilton M, Amos Q, Henzel WJ. 2003. Cup is an elF4E binding protein required for both the translational repression of oskar and the recruitment of Barentsz. J Cell Biol 163: 1197–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm JE, Buszczak M, Sayles S. 2005. Efficient protein trafficking requires trailer hitch, a component of a ribonucleoprotein complex localized to the ER in Drosophila. Dev Cell 9: 675–685. [DOI] [PubMed] [Google Scholar]
- Yarunin A, Harris RE, Ashe MP, Ashe HL. 2011. Patterning of the Drosophila oocyte by a sequential translation repression program involving the d4EHP and Belle translational repressors. RNA Biol 8: 904–912. [DOI] [PubMed] [Google Scholar]
- Zaessinger S, Busseau I, Simonelig M. 2006. Oskar allows nanos mRNA translation in Drosophila embryos by preventing its deadenylation by Smaug/CCR4. Development 133: 4573–4583. [DOI] [PubMed] [Google Scholar]
- Zappavigna V, Piccioni F, Villaescusa JC, Verrotti AC. 2004. Cup is a nucleocytoplasmic shuttling protein that interacts with the eukaryotic translation initiation factor 4E to modulate Drosophila ovary development. Proc Natl Acad Sci 101: 14800–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.