

Abstract
Pre-harvest sprouting (PHS) is an important cause of quality loss in many cereal crops and is particularly prevalent and damaging in wheat. Resistance to PHS is therefore a valuable target trait in many breeding programs. The Phs-A1 locus on wheat chromosome arm 4AL has been consistently shown to account for a significant proportion of natural variation to PHS in diverse mapping populations. However, the deployment of sprouting resistance is confounded by the fact that different candidate genes, including the tandem duplicated Plasma Membrane 19 (PM19) genes and the mitogen-activated protein kinase kinase 3 (TaMKK3-A) gene, have been proposed to underlie Phs-A1. To further define the Phs-A1 locus, we constructed a physical map across this interval in hexaploid and tetraploid wheat. We established close proximity of the proposed candidate genes which are located within a 1.2 Mb interval. Genetic characterization of diverse germplasm used in previous genetic mapping studies suggests that TaMKK3-A, and not PM19, is the major gene underlying the Phs-A1 effect in European, North American, Australian and Asian germplasm. We identified the non-dormant TaMKK3-A allele at low frequencies within the A-genome diploid progenitor Triticum urartu genepool, and show an increase in the allele frequency in modern varieties. In United Kingdom varieties, the frequency of the dormant TaMKK3-A allele was significantly higher in bread-making quality varieties compared to feed and biscuit-making cultivars. Analysis of exome capture data from 58 diverse hexaploid wheat accessions identified fourteen haplotypes across the extended Phs-A1 locus and four haplotypes for TaMKK3-A. Analysis of these haplotypes in a collection of United Kingdom and Australian cultivars revealed distinct major dormant and non-dormant Phs-A1 haplotypes in each country, which were either rare or absent in the opposing germplasm set. The diagnostic markers and haplotype information reported in the study will help inform the choice of germplasm and breeding strategies for the deployment of Phs-A1 resistance into breeding germplasm.
Keywords: dormancy, seed, PM19, TaMKK3-A, pre-harvest sprouting, Triticum aestivum, haplotype
Introduction
Pre-harvest sprouting (PHS) refers to the too-early germination of physiologically matured grains while still on the ear, but before harvest. PHS is primarily caused by insufficient levels, or rapid loss, of seed dormancy and is an important cause of quality loss in many cereal crops (Li et al., 2004; Fang and Chu, 2008). This is particularly relevant in wheat due to its detrimental effects on bread-making potential which represents the most common use of wheat grains globally (Simsek et al., 2014). PHS is believed to be a modern phenomenon, as progenitor and wild wheat species generally display high levels of seed dormancy (Gatford et al., 2002; Lan et al., 2005). Selection for reduced seed dormancy during domestication and modern breeding programs allowed for more uniform seed germination and rapid crop establisment (Nave et al., 2016). However, this also resulted in higher level of susceptiblity to PHS in modern wheat varieties (Barrero et al., 2010). In addition to its detrimental effect on quality, PHS also reduces yield and affects seed viability, making resistance to PHS a high priority in many breeding programs.
Occurrence of PHS is heavily influenced by the environment. PHS is prevalent in wheat growing regions with high levels of rainfall during the period of grain maturation and after-ripening. Increased ambient temperature during this period can further increase the susceptibility of grains to sprouting (Barnard and Smith, 2009; Mares and Mrva, 2014). This enviromental dependency of PHS constitutes a constraint in selecting for PHS resistance in field conditions. In addition, resistance to PHS is highly quantitative and is controlled by numerous quantitative trait loci (QTL) located on all 21 chromosomes of bread wheat (Flintham et al., 2002; Kulwal et al., 2005; Mori et al., 2005; Kottearachchi et al., 2006; Ogbonnaya et al., 2007; Liu et al., 2008; Torada et al., 2008; Xiao-bo et al., 2008; Mohan et al., 2009; Munkvold et al., 2009; Knox et al., 2012; Kulwal et al., 2012; Gao et al., 2013; Lohwasser et al., 2013; Mares and Mrva, 2014; Kumar et al., 2015). This makes resistance to PHS one of the most multi-genic traits in wheat and further highlights the complexity in breeding for this trait.
Despite the multi-genic control of PHS resistance, a few major loci have been consistently shown to account for a significant proportion of natural variation to sprouting in diverse mapping populations. This include the homoeologous R (Red color) genes on the long arms of chromosome group 3 controlling seed coat color, Qphs.pseru-3AS (same as QPhs.ocs-3A.1) on chromosome 3AS and a locus on chromosome arm 4AL, designated as Phs-A1 (Flintham, 2000; Himi and Noda, 2005; Mori et al., 2005; Liu et al., 2008; reviewed by Mares and Mrva, 2014). Consistent with its strong effect, Phs-A1 has been identified in at least 11 bi-parental and multi-parent mapping populations derived from diverse germplasm from Australia, United Kingdom, Japan, China, Mexico, Canada and Europe (Torada et al., 2005; Ogbonnaya et al., 2007; Chen et al., 2008; Torada et al., 2008; Cabral et al., 2014; Albrecht et al., 2015; Barrero et al., 2015). Physiological evaluation of Phs-A1 shows that it delays the rate of dormancy loss during seed after-ripening when plants are grown across a wide range of temperatures (13°C–22°C; Shorinola et al., 2016).
Unlike the Qphs.pseru-3AS and R loci, which have been unequivocally cloned to be wheat Mother of Flowering Time (TaMFT; same as TaPHS1) and Myb10 transcription factor (TaMyb10), respectively (Himi and Noda, 2005; Nakamura et al., 2011; Liu et al., 2013), two different candidate genes have been proposed to underlie the effect of Phs-A1. Recently, two independent studies by Barrero et al. (2015) and Torada et al. (2016) identified the tandem duplicated Plasma Membrane 19 (PM19-A1 and PM19-A2) genes and a mitogen-activated protein kinase kinase 3 (TaMKK3-A) gene, respectively, as candidates for Phs-A1. The PM19 genes were identified through a combined genetic approach using multi-parent mapping populations and transcriptomic analysis of near-isogenic recombinant inbred lines. The TaMKK3-A gene was identified through a more traditional positional cloning strategy using bi-parental mapping populations. Each study confirmed the effect of the gene(s) on dormancy through either down-regulation of transcript levels through RNA interference (PM19) or transgenic complementation of the susceptible parent with the resistant allele (TaMKK3-A).
It is presently unclear whether the sprouting variation associated with Phs-A1 across diverse germplasm is due to allelic variation at PM19 or TaMKK3-A alone, or if it’s due to a combination of both genes (Torada et al., 2016). Fine-mapping studies (Shorinola et al., 2016) defined Phs-A1 to a genetic interval distal to PM19 for United Kingdom germplasm, consistent with the position of TaMKK3-A. However, a comprehensive understanding of Phs-A1 diversity taking into account both PM19 and TaMKK3-A genes across a wider set of germplasm is lacking.
In this study, we characterized the Phs-A1 physical interval in both hexaploid and tetraploid emmer wheat to establish the physical proximity of PM19 and TaMKK3-A. We developed markers for the candidate genes, and showed TaMKK3-A alleles to be diagnostic for sprouting resistance in a panel of parental lines from mapping populations in which Phs-A1 was identified. We used diploid, tetraploid and hexaploid accessions to further trace the origin of the sprouting susceptible TaMKK3-A allele and used exome capture data from the wheat HapMap panel (Jordan et al., 2015) to examine the haplotype variation across the Phs-A1 locus.
Materials and Methods
Physical Map Sequence Assembly and Annotation
A fingerprinted Bacterial Artificial Chromosome (BAC) library of flow-sorted 4A chromosome was used for constructing the Chinese Spring Phs-A1 physical map1. Using the high-throughput BAC screening approach described by Cvikova et al. (2015), a sequence database made from a three-dimensional pool of BAC clones comprising the Minimum Tilling Path (MTP) was searched for the sequences of PM19-A1 and TaMKK3-A. This identified two positive clones for PM19-A1 (TaaCsp4AL037H11 and TaaCsp4AL172K12) and three positive clones for TaMKK3-A (TaaCsp4AL032F12, TaaCsp4AL012P14 and TaaCsp4AL002F16; Supplementary Table S1). Using Linear Topology Contig (LTC; Frenkel et al., 2010) BAC clustering information for this library, we identified the BAC clusters (defined as a network of overlapping BACs forming a contiguous sequence) to which these BACs belong. The PM19-A1-containing BACs belong to BAC Cluster 16421 which has 20 BACs in its MTP while the TaMKK3-A-containing BACs belong to BAC Cluster 285 comprised of four MTP BACs (Supplementary Table S1).
DNA of the BACs was extracted using the Qiagen Plasmid Midi Kit (Qiagen, Cat. No. 12143). Eleven of the 20 MTP BACs containing and distal to PM19-A1 in the physical map of Cluster 16421 and the four MTP BACs of Cluster 285 were sequenced on the Illumina MiSeq with 250 bp paired-end reads. An average of 2,105,488 and 2,752,220 paired-end reads per BAC were produced for Cluster 16421 and 285 BACs, respectively. Illumina reads for each BAC were separately assembled using the CLC Bio genomic software 2. Before assembly, reads were filtered to remove contaminant sequences by mapping to the BAC vector (pIndigoBAC-5) sequence and the Escherichia coli genome. De novo assembly of reads after contaminant removal was done with the following assembly parameters: Word size: 64 bp; Bubble size: 250 bp; Mismatch cost: 2; Insertion cost: 3; Deletion cost 3; Length fraction: 90%; Similarity fraction: 95%.
The assembled contigs were repeat-masked by BLASTn analysis against the Triticeae Repeat Element Database (TREP 3; Wicker et al., 2000). Gene annotation was performed using the wheat gene models described by Krasileva et al. (2013) and by BLASTX analysis to NBCI nr.4 Gene models were also obtained by ab initio gene prediction with FGENESH (Solovyev et al., 2006). Only FGENESH gene models with protein sequence support from NCBI or Ensembl Plant protein databases5 were used. Gene models with greater than 90% protein or nucleotide sequence identity and more than 75% sequence coverage to already annotated genes on NCBI or Ensembl databases were considered as high confidence genes. Gene models that did not meet these criteria were considered as low confidence genes, and were not analyzed further.
TaMKK3-A Genotyping
A Kompetitive Allele Specific PCR (KASP; Smith and Maughan, 2015) assay was developed for genotyping the C to A (C > A) causal TaMKK3-A mutation reported by Torada et al. (2016). For this, two allele-specific reverse primers (TaMKK3-A-snp1-res: TTTTTGCTTCGCCCTTAAGG and TaMKK3-A-snpA1-sus: TTTTTGCTTCGCCCTTAAGT) each containing the allele-specific SNP at the 3′ end, were used in combination with a common A-genome specific forward primer (GCATAGAGATCTAAAGCCAGCA). To distinguish the amplification signal produced from each allele specific primer, FAM and HEX fluorescence dye probes (Ramirez-Gonzalez et al., 2015) were added to the 5′ end of TaMKK3-A-snpA1-res and TaMKK3-A-snpA1-sus, respectively. KASP assays were performed as previously described (Shorinola et al., 2016).
In addition to the KASP assay, a genome-specific Cleavage Amplified Polymorphism Sequence (CAPS) assay (Konieczny and Ausubel, 1993), designated as TaMKK3-A-caps, was developed. This CAPS marker is associated with the presence/absence of an Hpy166II restriction site which co-localizes with the C > A causal polymorphism in the fourth exon of TaMKK3-A. Genome-specific primer pairs (Forward: CACCAAAGAATAGAAATGCTCTCT and Reverse: AGGAGTAGTTCTCATTGCGG) were designed to amplify an 887-bp sequence including the fourth exon. PCR was performed with Phusion High Fidelity polymerase (NEB, United Kingdom; Cat No: M0530S) in a 50 μL volume containing 20% buffer, 0.2 mM of dNTP, 5 μM each of TaMKK3-A-cap forward and reverse primers, 3% of DMSO, 200 – 400 ng of genomic DNA and 0.5 unit of Phusion polymerase (NEB, United Kingdom; Cat No: M0530S). Thermal cycling was done with Eppendorf Mastercycler® Pro Thermal Cyclers with the following program: initial denaturation at 98°C for 2 min; 35 cycles of denaturation at 98°C for 30 s; Annealing at 62°C for 30 s and extension at 72°C for 60 s; final extension at 72°C for 10 min. Following PCR amplification, a 25 μL restriction digest reaction containing 21.5 μL of the final PCR reaction, 2.5 μL of CutSmart® Buffer (NEB, United Kingdom; Cat No: B7204S) and 10 units of Hpy166II was incubated at 37°C for 1 h. Digest products were separated on a 1.5% agarose gel.
PM19-A1 Genotyping
Detection of an 18 bp deletion on the promoter region of PM19-A1 was carried out using primers TaPM19-A1-5F (GAAACAGCTACCGTGTAAAGC) and TaPM19-A1-5R (TGGTGAAGTGGAGTGTAGTGG) reported by Barrero et al. (2015). PCR reaction mixture contained template DNA, 2.5 mM MgCl2, 1.5 mM dNTP, 1.5 μM of each primer, and 1 unit of Taq polymerase (NEB). The reaction mixture was made up to a total volume of 10 μl. The PCR conditions were as follows: 3 min at 94°C, followed by 30 cycles of 40 s at 94°C, 40 s at 60°C, and 1 min at 72°C. The last step was incubation for 7 min at 72°C. The PCR products were resolved on a 4% agarose gel and visualized with SYBR green I (Cambrex Bio Science, Rockland, ME, United States).
Germplasm for Genetic Characterization of TaMKK3 and PM19
We genotyped PM19 and TaMKK3-A across 23 wheat varieties previously reported to segregate for Phs-A1, including United Kingdom (Alchemy, Robigus, Option, Claire, Boxer, Soleil), Australian (Yitpi, Baxter, Chara, Westonia, Cranbrook, Aus1408, Janz, Cunningham, Halberd), Japanese (Kitamoe, Haruyokoi, OS21-5), Mexican/CIMMYT (W7984, Opata M85), Canadian (Leader), Chinese (SW95-50213) and Swiss varieties (Münstertaler). We also genotyped TaMKK3-A in accessions from progenitor species T. urartu (41 accessions; Au genome), T. turgidum ssp. dicoccoides (151 accessions; AABB genomes), 804 hexaploid accessions from the Watkins landrace collection (Wingen et al., 2014), and 457 modern European bread wheat varieties from the Gediflux collection released between 1945 and 2000 (Reeves et al., 2004).
Variant Calling and Haplotype Analysis
We examined the haplotype structure around the Phs-A1 locus in three different germplasm sets. These included 457 varieties in the Gediflux collection, a panel of 195 Australian varieties, and the wheat Haplotype Map (HapMap) panel consisting of 62 diverse global accessions (Jordan et al., 2015). For the HapMap panel, we selected polymorphic sites as follows. We extracted SNP information from published variant call files (VCF) produced from whole exome capture (WEC) resequencing dataset of the 62 HapMap lines6. For this, the corresponding IWGSC contig information for genes represented in the Phs-A1 physical map were first obtained and used to filter the HapMap VCF for SNP sites located within these contigs. We kept SNP sites with allele frequencies of >5% and accessions with >80% homozygous calls across SNPs. Allele information at the selected SNP loci was reconstructed for each line using the reference, alternate and genotype field information obtained from the VCF. Haplotype cluster analysis was done with Network 5.0.0.0 (Fluxus Technology Limited, United Kingdom) using the Median Joining Network Algorithm. Haplotypes in the Gediflux and Australian germplasm were defined using a subset of the HapMap SNPs which were most informative in distinguishing between the HapMap haplotypes and with >30% allele frequency.
Pedigree Visualization
Pedigree information was obtained from the Genetic Resources Information System for Wheat and Triticale (GRIS7) and the International Crop Information System (ICIS8). Pedigree visualization was performed with Helium (Shaw et al., 2014). The coefficient of parentage (COP) analysis (i.e., the probability that alleles of two individuals are identical by descent) was calculated for all pairwise comparisons of lines within the most prevalent haplotypes (Australian: H1/H2 and H5/H7; United Kingdom: H3 and H12). For accuracy, landraces or cultivars with unknown or ambiguous pedigrees were not included in the COP analysis. Diversity within haplotype groups was estimated by the mean calculation of all COPs within each matrix.
Results
TaMKK3-A and PM19 Are Located within a 1.2 Mb Physical Interval
We constructed an extended physical map across the Phs-A1 interval to investigate the physical proximity between the TaMKK3-A and PM19 candidate genes. Using PM19-A1 and TaMKK3-A sequences as queries, we screened in silico a BAC library of flow-sorted 4AL chromosome arm of the bread wheat cultivar Chinese Spring (CS). PM19-A1 and TaMKK3-A were found on two independent non-overlapping BAC clone clusters which were anchored on the high resolution radiation hybrid map of chromosome 4A (Balcárková et al., 2016). The MTP of Cluster 16421 (PM19) was comprised of 20 BAC clones whereas the MTP of Cluster 285 (TaMKK3-A) included four BAC clones (Figure 1A and Supplementary Table S1).
FIGURE 1.
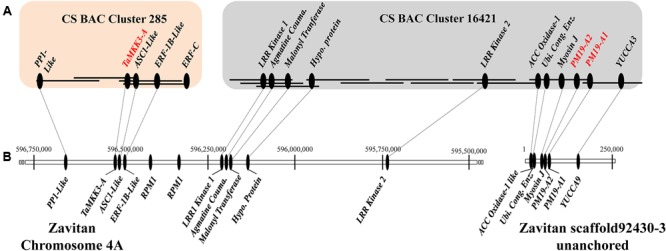
Physical map of the Phs-A1 interval in bread wheat Chinese Spring (CS) and wild emmer (Zavitan). (A) Phs-A1 interval in CS is covered by two non-overlapping BAC clusters: Cluster 285 (4 BACs) and Cluster 16421 (11 of the 20 MTP BACs). BACs are represented by solid lines while genes found on the BAC are represented by filled ovals. The proposed candidate genes for Phs-A1 are in red font. (B) Whole genome assembly of Zavitan wild emmer across the Phs-A1 interval. Genes present in both assemblies are joined by dotted lines.
Individual BACs were sequenced, assembled, repeat-masked and annotated for coding sequences. In eleven of the 20 MTP BACs of Cluster 16421 sequenced, nine high-confidence genes were found in addition to the PM19-A1 and PM19-A2. These included YUCCA3-like, Myosin-J Heavy Chain protein, Ubiquitin Conjugating Enzyme, Amino-Cyclopropane Carboxylate Oxidase 1 like (ACC Oxidase-1), two Leucine-Rich Repeat Kinases (LRR kinase 1 and LRR kinase 2), Agmatine Coumaroyl Transferase, Malonyl Coenzyme A:anthocyanin 3-O-glucoside-6″-O-malonyltransferase and a gene encoding for a hypothetical protein. In addition to TaMKK3-A, Cluster 285 contained four additional genes including Protein Phosphatase1-Like (PP1-Like), Activating Signal Co-integrator 1- Like (ASC1-Like), Ethylene Responsive Factor-1B-Like (ERF-1B-Like) and a gene fragment showing high sequence similarity to ERF-1B-Like and as such designated as ERF-C. Together, this highlights the presence of at least 16 protein-coding genes across the Phs-A1 interval in hexaploid bread wheat (Figure 1).
We also characterized the Phs-A1 interval in the recently constructed assembly of a wild emmer wheat, Zavitan (Figure 1B; Hen-Avivi et al., 2016; Avni et al., 2017). This allowed comparative analysis of the Phs-A1 interval in tetraploid and hexaploid wheat species. Fifteen of the 16 genes found in the CS physical map were located on two Zavitan scaffolds. Nine of these 15 genes were positioned across a 0.93 Mb interval on the Zavitan 4A pseudomolecule. These included 4 genes from BAC Cluster 285 and five genes from BAC Cluster 16421 (Figure 1). The remaining six genes spanned a 0.13 Mb interval on an unanchored scaffold. On average, the coding sequence identity between CS and Zavitan was 99.7% across the genes shared by both assemblies. We could not find sequence for ERF-C in the Zavitan assembly at similar identity. We annotated two genes encoding for disease resistance protein RPM1 in the Zavitan sequence corresponding to the gap between the two CS BAC clusters. Combining the CS and Zavitan physical maps, the physical region between TaMKK3-A and the PM19 genes was covered and estimated to be approximately 1.2 Mb (Figure 1).
TaMKK3-A Is Most Closely Associated with Phs-A1
Torada et al. (2016) reported a C > A mutation in position 660 of the TaMKK3-A coding sequence (C660A) as being causative of the Phs-A1 effect. Using alignments of the three wheat genomes, we developed a genome-specific and co-dominant KASP assay for this SNP designated as TaMKK3-A-snp1. The TaMKK3-A-snp1 assay is co-dominant as it distinguished between heterozygotes and homozygotes F2 progenies in the Alchemy × Robigus population previously reported to segregate for Phs-A1 (Shorinola et al., 2016; Figure 2A). We also developed a CAPS marker (Konieczny and Ausubel, 1993) for TaMKK3-A to enable genotyping of Phs-A1 using a gel-based assay. This marker, designated as TaMKK3-A-cap, amplifies a genome-specific 887 bp region and is designed to discriminate for the presence of an Hpy166II site (GTNNAC) which is lost by the C660A mutation. Dormant lines with the C allele maintain the Hpy166II site which leads to digestion of the 887 bp amplicon into fragments of 605 and 282 bp (Figure 2B). Conversely, non-dormant lines with the A allele lose the Hpy166II site and hence remain intact (887 bp) after digestion. As with the KASP assay, the CAPS marker was co-dominant when used to genotype F2 progenies (Figure 2B).
FIGURE 2.
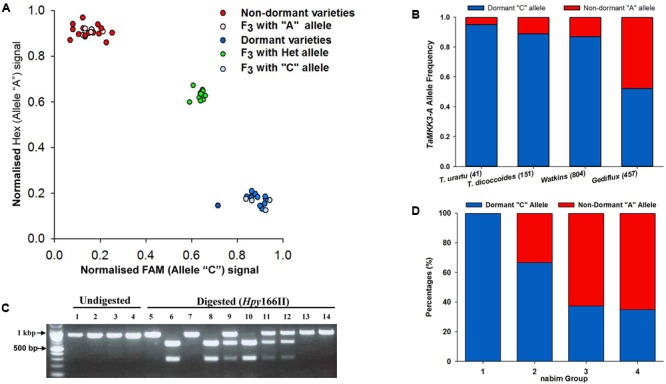
Marker development and allele distribution of MKK3-A in ancestral and historic germplasm. (A) Genotype plot of varieties and a F3 population segregating for Phs-A1 using the TaMKK3-A KASP assay. (B) Development of co-dominant CAPS marker based on Hpy166II restriction digest of the C660A SNP. Lanes 1 - 4 contain undigested fragments of Robigus, Alchemy, Claire and Option, respectively, while Lanes 5 – 14 contain digested fragments of Robigus, Alchemy, Claire and Option (lanes 5–8) and six segregating F3 plants from an Alchemy × Robigus cross (lanes 9–16). (C) Allele frequency of the causal C660A SNP in T. urartu and T. turgidum ssp. dicoccoides accessions and the Watkins and Gediflux collections. The number of lines genotyped in each germplasm set is in parenthesis. (D) TaMKK3-A allele distribution in the four wheat end-use groups (nabim 1–4) in the United Kingdom. The red and blue bars in (C,D) represents the non-dormant “A” allele” and dormant “C” allele, respectively.
Using the KASP assay, we genotyped a panel comprised of the parents of 11 bi-parental mapping populations and a MAGIC population in which Phs-A1 had previously been reported (Table 1). The TaMKK3-A-snp1 was polymorphic and diagnostic for Phs-A1 in all parental lines. Consistent with Torada et al. (2016), non-dormant sprouting-susceptible parents carry the TaMKK3-A “A” allele while all the dormant sprouting-resistant parents carry the TaMKK3-A-snp1 “C” allele (Table 1). We genotyped the same panel for the promoter deletion in PM19-A1 previously proposed to be causal of PHS susceptibility (Barrero et al., 2015). We found the PM19-A1 deletion to be linked with the non-dormant TaMKK3-A A allele in most, but not all, of these populations. The putative linkage was broken in the dormant Kitamoe, OS21-5 and SW95-50213 parents, whose dormancy phenotypes are not consistent with their PM19-A1 promoter deletion status, but can be explained by their TaMKK3-A genotype (Table 1). This association was confirmed genetically in the SW95-50213 × AUS1408 cross. This population, which did not segregate for the dormancy phenotype in the original work by Mares et al. (2005), is monomorphic for the dormant C allele at TaMKK3-A, but segregates for the PM19-A1 deletion. Similarly, parents of the two populations OS21-5 × Haruyokoi and Kitamoe × Münstertaler segregating for the dormancy phenotype in the work by Torada et al. (2005) are monophormic for the PM19-A1 deletions, but segregate accordingly for the TaMKK3-A causal mutation. These results strongly support TaMKK3-A as the most likely causal gene for Phs-A1 across this highly informative panel.
Table 1.
TaMKK3-A and PM19 alleles in Phs-A1 association panel.
| Population | Variety | Origin | Status∗ | TaMKK3-A Allele | PM19-A1 promoter InDel | Reference |
|---|---|---|---|---|---|---|
| Alchemy × Robigus | Alchemy | UK | Dormant | C | Insertion | Shorinola et al., 2016 |
| Robigus | UK | Non-dormant | A | Deletion | ||
| Option × Claire | Option | UK | Dormant | C | Insertion | Shorinola et al., 2016 |
| Claire | UK | Non-dormant | A | Deletion | ||
| MAGIC Population | Yitpi | AUS | Dormant | C | Insertion | Barrero et al., 2015 |
| Baxter | AUS | Non-dormant | A | Deletion | ||
| Chara | AUS | Non-dormant | A | Deletion | ||
| Westonia | AUS | Non-dormant | A | Deletion | ||
| Opata × W7984 | W7984 | MEX | Dormant | C | Insertion | Lohwasser et al., 2013 |
| Opata | MEX | Non-dormant | A | Deletion | ||
| OS21-5 × Haruyokoi | OS21-5 | JPN | Dormant | C | Deletion | Torada et al., 2008 |
| Haruyokoi | JPN | Non-dormant | A | Deletion | ||
| Leader × Haruyokoi | Leader | CAN | Dormant | C | Insertion | Torada et al., 2008 |
| Haruyokoi | JPN | Non-dormant | A | Deletion | ||
| Kitamoe × Münstertaler | Kitamoe | JPN | Dormant | C | Deletion | Torada et al., 2005 |
| Münstertaler | SUI | Non-dormant | A | Deletion | ||
| Cranbrook × Halberd | Halberd | AUS | Dormant | C | Insertion |
Mares et al., 2005; Zhang et al., 2008 |
| Cranbrook | AUS | Non-dormant | A | Deletion | ||
| Janz × AUS1408 | Aus1408 | AUS, SA | Dormant | C | Insertion | Mares et al., 2005; Ogbonnaya et al., 2007 |
| Janz | AUS | Non-dormant | A | Deletion | ||
| SW95-50213 × Cunningham | SW95-50213 | CHN | Dormant | C | Deletion | Mares et al., 2005 |
| Cunningham | AUS | Non-dormant | A | Deletion | ||
| SW95-50213 × AUS1408# | SW95-50213 | CHN | Dormant | C | Deletion | Mares et al., 2005 |
| Aus1408 | AUS, SA | Dormant | C | Insertion | ||
| Boxer × Soleil | Soleil | UK | Dormant | C | Insertion | Flintham, 2000 |
| Boxer | UK | Non-dormant | A | Deletion |
∗Phenotypic classifications are based on comparisons between the parental varieties of each population through genetic studies. #Phs-A1 was not detected in this population as the DH lines were generally dormant. However, limited number of lines showed transgressive segregation relative to the dormant phenotypes of the two parents.
Origin and Distribution of the TaMKK3-A Alleles in Ancestral and Modern Germplasm
To examine the origin, distribution and allele frequencies of the causative TaMKK3-A C660A SNP, we genotyped a set of 41 T. urartu (diploid: AA genome) and 151 T. turgidum ssp. dicoccoides (tetraploid: AABB genome) accessions. These represent the diploid and tetraploid progenitors of the modern bread wheat A genome on which Phs-A1 is located. Torada et al. (2016) previously suggested that the non-dormant A allele was the mutant form since the dormant C SNP was conserved across different species. Across T. urartu accessions, the C allele was predominant (39 accessions) while the non-dormant A allele was present in only two accessions (5% allele frequency; Figure 2C). Similarly, across T. dicoccoides accessions, the dormant C allele was found in 134 accessions while the non-dormant allele was found in 17 accessions (11% allele frequency; Figure 2C). Our results are consistent with Torada et al. (2016) in that the non-dormant A allele is derived from the wild type C allele. In addition, the presence of the A allele across both progenitor species suggests that the mutation predates the hybridization and domestication events that gave rise to modern bread wheat.
We also genotyped the Watkins Collection representing a set of global bread wheat landraces collected in the 1920s and 1930s (Wingen et al., 2014), as well as the Gediflux collection comprised of modern European bread wheat varieties released between 1945 and 2000 (Reeves et al., 2004). The allele frequency of the non-dormant A allele was 13% in the Watkins landrace collection (Figure 2C and Supplementary Table S2), comparable to that in T. dicoccoides (11%). However, the non-dormant A allele frequency in the Gediflux collection was 48% across 457 varieties (Figure 2C). This represents a marked increase of the non-dormant allele in the more modern European collection when compared to the 15% A allele frequency of the European sub-population within the Watkins collection (Supplementary Table S2).
To determine if the TaMKK3-A dormant allele was associated with improved end-use quality, we genotyped 41 United Kingdom varieties representing the four United Kingdom market classes (Figure 2D, nabim groups 1–4; nabim, 2014). Of the 13 bread-making quality varieties (groups 1 and 2), 11 (85%) had the dormant TaMKK3-A allele. This frequency was significantly higher (Contingency table χ2 = 8.497; P < 0.01) than in the 28 biscuit and animal feed varieties (groups 3 and 4) in which the TaMKK3-A dormant allele was only present in 10 varieties (36%).
Phs-A1 Haplotypes in Global Germplasm
We next examined the allelic diversity across the extended Phs-A1 interval (including TaMKK3-A and PM19) with the aim of elucidating the haplotype structure across this region. For this, we used the SNP Haplotype Map (HapMap) dataset obtained from WEC resequencing of 62 diverse germplasm (Jordan et al., 2015). From this SNP dataset, we obtained data for eight of the sixteen genes found in the Phs-A1 interval (PP1-like, TaMKK3-A, ASC1-like, ERF-C, LRR Kinase 1, LRR Kinase 2, PM19-A2 and PM19-A1) corresponding to 51 SNPs. To improve the accuracy of the haplotype analysis, we selected accessions with >80% homozygous calls across the selected genes and SNPs with >5% allele frequency. This filtering resulted in 39 SNPs across the eight genes in 58 accessions.
Across the Phs-A1 interval (PP1-like to PM19-A1) we identified 14 distinct haplotypes (H1–14; Figure 3A). Haplotypes were comprised of a mix of cultivars, landrace, breeding lines and synthetic population in varying proportion (Figure 3B and Supplementary Table S3). H1 represented the major haplotype present in 33% of all accessions examined, whereas five haplotypes were relatively infrequent (<5%; H2, H5, H6, H9, H13). Six of the selected SNPs were found in TaMKK3-A including the causal C660A SNP in the fourth exon and five additional intron SNPs. These six SNPs defined four distinct TaMKK3-A haplotypes (Figure 3C and Supplementary Table S3; TaMKK3-A_HapA – D) in the HapMap collection with only one having the non-dormant A allele (TaMKK3-A_HapA). The non-dormant A allele was present in 50% of the HapMap population, consistent with the Gediflux collection (48%).
FIGURE 3.
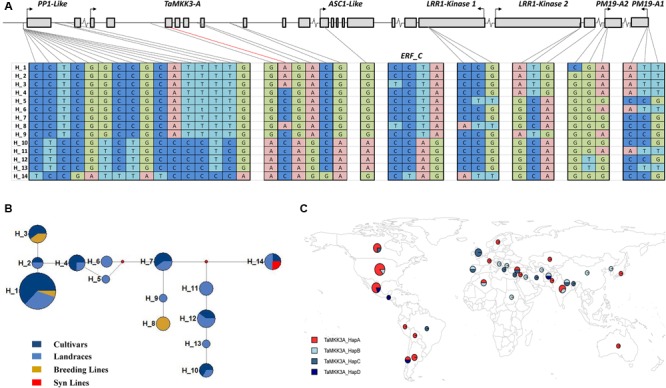
Phs-A1 haplotype analysis. (A) Structure of 14 haplotypes identified in the HapMap population across 39 SNP loci in the Phs-A1 interval. SNP loci are ordered based on their physical position on the Zavitan assembly. Exons, intron and intergenic regions are represented by filled boxes, solid lines and breaks, respectively. Note that ERF-C is not on the Zavitan assembly. (B) Haplotype network of the 14 haplotypes. The size of each circle corresponds to the number of lines in each haplotype. Blue, light blue, amber and red represent cultivars, landraces, breeding and synthetic line in each haplotype, respectively. (C) Geographical distribution of the four TaMKK3-A haplotypes (Supplementary Table S3). The size of the circle represents the sample size obtained within each country while each section represents the proportion of the country sample size with the specified haplotype.
Haplotype Structure at the Phs-A1 Interval in United Kingdom and Australian Germplasm
To characterize a larger set of European (Gediflux) and Australian germplasm, we selected seven informative polymorphisms across seven genes from the HapMap dataset and developed KASP assays for these (Supplementary Table S4). Using these seven assays, we defined 16 haplotypes in the European Gediflux collection (Supplementary Table S5). This included eleven haplotypes previously identified in the global HapMap dataset and five haplotypes unique to this European germplasm set, although these were relatively infrequent (Figure 4). The United Kingdom subpopulation within the Gediflux collection comprised 176 varieties and contained 11 of the 15 haplotypes identified. Six haplotypes include the dormant TaMKK3-A C allele (63% of United Kingdom varieties), with the majority of these varieties sharing haplotype H12 (89 of 110 varieties), consistent with the wider Gediflux population (Figure 4). This suggests one main source of PHS resistance in United Kingdom and European germplasm.
FIGURE 4.
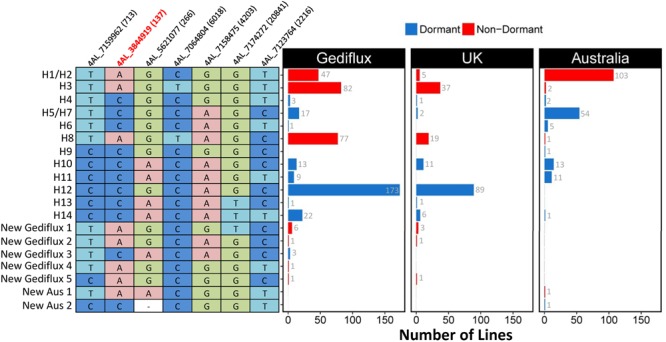
Relationship between the European, United Kingdom, Australian and HapMap Phs-A1 Haplotypes. The distribution (bar charts) of the HapMap and unique haplotypes found in the Gediflux (European), United Kingdom and Australian germplasm using genotype information of seven of the 39 HapMap SNPs within the Phs-A1 interval. Red bars correspond to the non-dormant “A” allele, whereas blue bars correspond to the dormant “C” allele.
By combining haplotype and pedigree information for these lines we could trace, to a reasonable degree of accuracy, the founder lines for the most common resistant haplotypes in United Kingdom germplasm (Supplementary Figure S1). We identified the origin of the major resistant haplotype in the United Kingdom germplasm (H12) as ViImorin-27, a French winter wheat variety released in the late 1920s (Figure 5 and Supplementary Figure S2). Vilmorin-27 was a direct parent and the donor of haplotype H12 for Cappelle–Desprez, a major founder variety for wheat breeding programs in Northern France and the United Kingdom released in 1948. Haplotype H12 has since remained an important part of United Kingdom breeding programs through varieties such as Rendezvous and Riband (released between 1985 and 1987).
FIGURE 5.
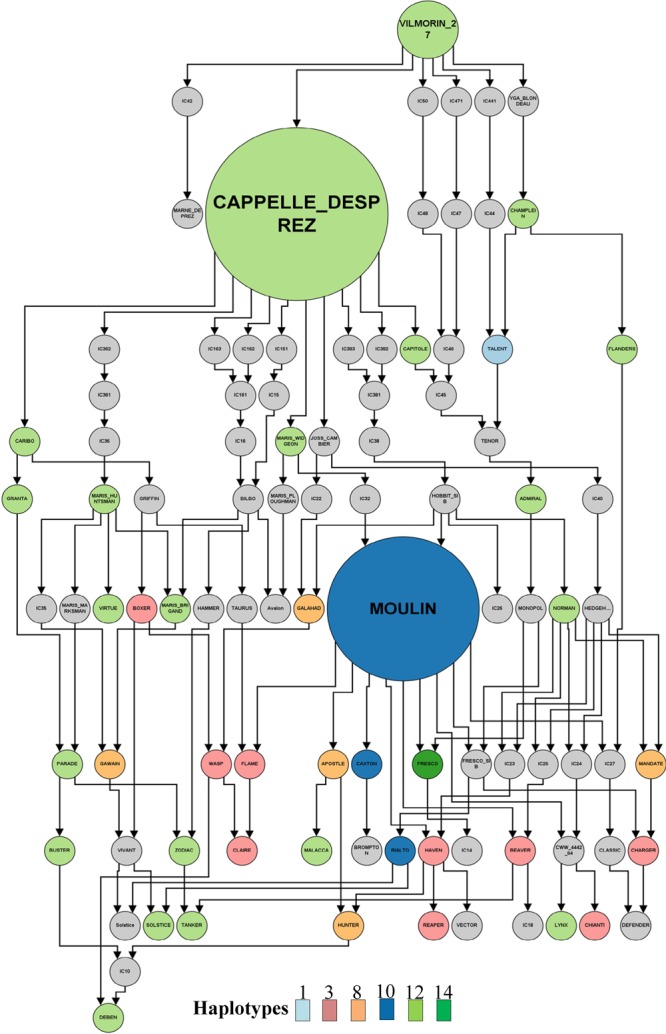
Pedigree of selected United Kingdom and European varieties highlighting the origin of the major resistant haplotype (H12). Each circle represents a variety and colors represent the different haplotypes. Nodes are size based on the number of varieties derived from the node.
Within the 195 Australian varieties we identified 12 haplotypes including ten previously identified HapMap haplotypes, and two Australian-specific haplotypes at low frequency (<1%, Supplementary Table S6 and Figure S3). Eight haplotypes present in 88 varieties (45%) have the dormant TaMKK3-A C allele while the other four haplotypes present in 107 varieties (55%) have the non-dormant TaMKK3-A A allele (Supplementary Figure S4). This represents a near balanced distribution of both alleles in Australian germplasm. In this set, 71% of lines with the dormant TaMKK3-A C alleles were traced to Federation (or Purple Straw) ancestry. Across the entire set, the alternative, non-dormant allele was more associated with the presence of cv. Gabo or CIMMYT-derived material in the pedigree. These lines had a more recent average release date of 1976 compared to the lines with the dormant allele (average release date 1941).
The mean COP for the Australian and United Kingdom Gediflux set of lines was 0.10 and 0.11 respectively (Table 2). Within each germplasm set, the lines with the most prevalent haplotypes had higher COP values, indicating a higher degree of relatedness amongst these lines relative to the entire collection (Table 2).
Table 2.
Mean Coefficient of Parentage (COP) within Australian and United Kingdom germplasm, and between groups of the most prevalent haplotypes containing dormant (C) and non-dormant (A) SNPs at TaMKK3-A.
| Germplasm | Haplotype | TaMKK3-A | Mean | Comparisons (n) |
|---|---|---|---|---|
| SNP | COP | |||
| Australia | All | A/C | 0.10 | 13530 |
| Australia | H5/H7 | C | 0.17 | 350 |
| Australia | H1/H2 | A | 0.15 | 2700 |
| United Kingdom | All | A/C | 0.11 | 1596 |
| United Kingdom | H12 | C | 0.20 | 496 |
| United Kingdom | H3 | A | 0.21 | 55 |
Discussion
Physical Map
We characterized the Phs-A1 interval by constructing a 1.5 Mb physical map spanning the PM19 and TaMKK3-A candidate genes (Barrero et al., 2015; Torada et al., 2016) and including 16 protein-coding genes. We observed near perfect sequence and gene content conservation in the interval between hexaploid and tetraploid physical maps. A similar overall collinearity between bread wheat, barley and Brachypodium was also observed except for the interval between ACC Oxidase-1 and ERF-C where the gene content in each species diverged (Supplementary Figure S5). The PM19 candidates where conserved across these species, whereas TaMKK3-A was only present in barley and wheat.
Sequence information from the BAC-based CS assembly and the whole genome shotgun Zavitan assemblies was used in a complementary manner. Neither assembly was fully contiguous across the Phs-A1 interval, but the gaps were different in the two assemblies allowing the spanning of the complete interval. This lack of contiguity was also present in the IWGSC WGA v0.49, TGAC (Clavijo et al., 2017) and Refeqv1.0 assemblies, where intervals covering TaMKK3-A and ASC1-Like were unanchored. While the new whole genome assemblies offer major improvements in contiguity, the available BAC physical maps will be of value to assign unanchored scaffolds or solve inconsistencies in regions were contiguity is broken.
TaMKK3-A Determines Phs-A1 Effect across Diverse Germplasm
The 1.5 Mb physical interval which defines Phs-A1 includes the proposed candidates PM19 and TaMKK3-A, as well as other genes with potential roles in dormancy/germination regulation. For example, ACC Oxidase-1 catalyzes the last steps in the biosynthesis of ethylene – a germination promoting hormone (Matilla and Matilla-Vázquez, 2008; Linkies and Leubner-Metzger, 2012; Corbineau et al., 2014). However, using two bi-parental mapping populations we showed linkage of Phs-A1 to the interval between PP1-Like – LRR Kinase 2 in United Kingdom populations, thereby excluding the PM19 and ACC Oxidase-1 loci as candidate genes (Shorinola et al., 2016). This was consistent with Torada et al. (2016) who identified TaMKK3-A as the causal gene in their mapping population and work in barley which identified the barley homolog (MKK3) as the causal gene for the seed dormancy QTL SD2 (Nakamura et al., 2016).
In support of this, the causal TaMKK3-A C660A SNP is perfectly associated with the phenotypes of 19 diverse parents of 11 mapping population in which Phs-A1 had previously been identified. This was also the case for the parents of the MAGIC population (Yipti, Chara, Westonia, Baxter) previously used to propose the PM19 loci as the causal gene (Barrero et al., 2015). Barrero et al. (2015) proposed a promoter deletion in PM19-A1 affecting motifs important for ABA responsiveness as the cause of non-dormancy in sprouting susceptible genotypes. The PM19-A1 deletion and the non-dormant TaMKK3-A A allele are in complete linkage in all the non-dormant parents from the multiple mapping populations. However, the PM19-A1 promoter deletion did not account for the dormant phenotype of Kitamoe, OS21-5 and SW95-50213 (Table 1). These dormant varieties have the PM19-A1 promoter deletion associated with low dormancy, but carries the dormant TaMKK3-A allele. These natural recombinants suggest that TaMKK3-A is the causal Phs-A1 gene. SW95-50213 is a Chinese landrace which is an important source of Phs-A1-mediated dormancy in Australian breeding programs. When SW95-50213 was crossed to a line carrying both TaMKK3-A and PM19 dormant alleles (AUS1408), no grain dormancy QTL could be identified (Mares et al., 2005). Despite the segregation of the PM19-A1 promoter polymorphism in this population, all lines displayed dormant to intermediate dormancy phenotype consistent with the TaMKK3-A genotype of their parents. Taken together, this evidence confirms the tight linkage between TaMKK3-A, PM19, and the Phs-A1 phenotype, and suggest that TaMKK3-A, but not PM19, is the causal gene underlying sprouting variation associated with Phs-A1 in diverse European, North American, Australian, and Asian germplasm.
Breeding Implications
Given the identification of a number of T. urartu accessions with the non-dormant A allele, it is likely that the C660A mutation originates from this diploid ancestor and predates the domestication and hybridization events that gave rise to modern bread wheat. This is similar to the causal mutation in TaPHS1, the major gene controlling PHS resistance QTL on 3AS which was also found in diploid (T. monococcum) and hexaploid wheat species. However, unlike TaMKK3-A, it is believed that the diploid and hexaploid mutations in TaPHS1 arose independently as these mutations could not be found in the diploid (T. urartu) and tetraploid (T. dicoccoides) A-genome progenitor of the modern bread wheat (Liu et al., 2015).
The non-dormant allele frequency was below 15% in accessions and landraces collected previous to 1920, but rose sharply to close to 50% in more modern germplasm. It is tempting to speculate that this could be due to selective pressure by breeders over the past 70 years for the non-dormant A allele in European and Australian environments. This pressure could be driven by selection for genotypes with more rapid and uniform germination that would be associated with the non-dormant allele. However, allele frequencies for both alleles have remained overall balanced given the improved end-use quality associated with the dormant allele. This hypothesis is supported by the fact that 85% of United Kingdom bread-making varieties carry the dormant allele, compared to only 35% of feed and biscuit-making varieties.
To facilitate breeding for PHS resistance, we developed co-dominant KASP and CAPS markers for the causal TaMKK3-A mutation, as well as KASP markers for the wider region. We identified 14 Phs-A1 haplotypes in a global germplasm panel with four haplotypes for the TaMKK3 gene itself, of which only one included the C660A non-dormant SNP. Comparison of Australian and United Kingdom haplotypes highlighted distinct frequencies in both sets with the most prevalent haplotypes containing the dormant TaMKK3-A allele differing in both countries. Haplotype H5/H7 is most frequent in Australian varieties, whereas haplotype H12 dominates in the United Kingdom. Interestingly, these haplotypes are either rare (<5% H5/H7 in United Kingdom) or absent (H12 not present in Australia) in the other country, suggesting distinct sources of PHS resistance in Australian and United Kingdom breeding programs. Understanding haplotypes structure across genes of agronomic interest is increasingly possible with the latest advances in wheat genomics (Clavijo et al., 2017; Uauy, 2017). It is also increasingly relevant given potential negative linkage drag associated with major phenology traits (Voss-Fels et al., 2017). The markers and knowledge generated in this study should facilitate the choice of parental genotypes for the deployment of TaMKK3-A in commercial cultivars.
The earliest line in the Australian set (Golden Drop, released 1840) carries the favorable TaMKK3-A ‘C’ SNP and also the most prevalent haplotype (H5/H7) at this locus. Golden Drop was derived from a Purple Straw/Yandilla cross and its sister line, Federation (released in 1901) became the foundation of many successful Australian cultivars due to earlier maturity and thus ability to avoid drought stress late in the growing season. Not only was Federation wheat better adapted to the Australian climate, it also had improved grain quality for milling, and so become widely adopted by breeders (Eagles et al., 2009).
The next major introduction of germplasm into Australia occurred in the 1970’s, as CIMMYT material was deployed widely by breeders seeking traits affecting height, quality and disease resistance (Brennan and Fox, 1998). Important CIMMYT parents in Australian breeding include Sonora-64, Pitic, Pavon-76, WW15 and WW80. Pedigree analysis suggests that such material could be the source of the most prevalent haplotype in Australia (H1/H2) containing the non-favorable TaMKK3-A allele. A high proportion of modern Australian cultivars with the non-dormant haplotype suggests opportunities may exist for the incorporation of favorable alleles at the locus. In this context, current breeding programs in Australia are using SW95-50213 and also Aus1408 as a source of dormancy (Hickey et al., 2009), lines that carry the favourable allele at the TaMKK3-A locus.
Future Outlook
The dormant TaMKK3-A C allele is predominant in all the progenitor and historic germplasm evaluated in this study, suggesting that it represents the ancestral allele as proposed by Torada et al. (2016). The N220K amino acid substitution (C660A mutation) in the kinase domain results in a gain-of-function allele which reduces dormancy in wheat. This is in contrast with barley where the non-dormant MKK3 allele is ancestral and the N260T substitution in the kinase domain results in a loss-of-function allele leading to increased seed dormancy (Nakamura et al., 2016). This provides an additional example of how for the same biological process, gain-of-function (dominant) mutations have been more readily selected in polyploid wheat compared to recessive variation in diploid barley (Borrill et al., 2015). The fact that the same gene has been selected in both species also suggests that the kinase activity of TaMKK3-A can be modulated to fine-tune the level of seed dormancy in temperate cereals. A better understanding of the activity and regulation of TaMKK3-A and its homoeologs could allow the identification of mutants (Krasileva et al., 2017) or the creating of gene edited alleles (Zong et al., 2017) with different levels of activity or the design of novel alleles with different degrees of dormancy.
In addition, the cloning of three of the major genes controlling PHS resistance (TaMyb10, TaPHS1 and TaMKK3-A) in wheat now offers a unique opportunity to examine how allelic combination of these genes can be used to modulate the levels of seed dormancy. Using a RIL population segregating for Phs-A1 and Qphs.pseru-3AS, Mori et al. (2005) demonstrated the additive interaction between these loci. This suggests that the markers generated in this study and others (Himi et al., 2011; Liu et al., 2015) will be valuable in deploying different levels of seed dormancy across different agro-ecological zones.
Author Contributions
OS led the genotype and pedigree analysis of the United Kingdom varieties, annotation of BAC sequences, developed the KASP and CAPS marker, and analyzed the HapMap data; JH performed pedigree analysis of Australian; JT and MH performed genotyping of Australian varieties; MV, BB, and KH constructed the 4AL physical map of CS; AD constructed the physical map of tetraploid wheat Zavitan; AT performed genotyping of Japanese varieties; JB led the work on Australian varieties; OS, CU, JH, and JB contributed to the writing of the manuscript; OS and CU designed the experiments.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer MG and handling editor declared their shared affiliation.
Acknowledgments
We thank Chloe Riviere for help with plant husbandry and Dr. Sergio Galvez-Rojas for advice on data visualization. We also thank Katerina Viduka for the assistance in the genotyping, Dr. Ben Trevaskis and Dr. Howard Eagles for assistance in the pedigree analysis of the Australian collection, and Dr. Frank Gubler for the helpful discussions and support. We thank the International Wheat Genome Sequencing Consortium (IWGSC) for pre-publication access to the wheat genome RefSeq v0.4 and v1.0.
Funding. This work was supported by the United Kingdom Biotechnology and Biological Sciences Research Council (BBSRC), AHDB-HGCA, KWS, Lantmännen, Limagrain and RAGT (BB/I01800X/1, BB/J004588/1, BB/J004596/1, BB/P013511/1, BB/P016855/1). BB and MV were supported by grant LO1204 from the National Program of Sustainability I, by the Czech Science Foundation (14-07164S). OS was supported by the John Innes Foundation. The work performed in Australia was supported by the Grains and Research Development Corporation.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01555/full#supplementary-material
References
- Albrecht T., Oberforster M., Kempf H., Ramgraber L., Schacht J., Kazman E., et al. (2015). Genome-wide association mapping of preharvest sprouting resistance in a diversity panel of European winter wheats. J. Appl. Genet. 56 277–285. 10.1007/s13353-015-0286-5 [DOI] [PubMed] [Google Scholar]
- Avni R., Nave M., Barad O., Baruch K., Twardziok S. O., Gundlach H., et al. (2017). Wild emmer genome architecture and diversity elucidate wheat evolution and domestication. Science 357 93–97. 10.1126/science.aan0032 [DOI] [PubMed] [Google Scholar]
- Balcárková B., Frenkel Z., Škopová M., Abrouk M., Kumar A., Chao S., et al. (2016). A high resolution radiation hybrid map of wheat chromosome 4A. Front. Plant Sci. 7:2063 10.3389/fpls.2016.02063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard A., Smith M. F. (2009). The effect of rainfall and temperature on the preharvest sprouting tolerance of winter wheat in the dryland production areas of the Free State Province. Field Crops Res. 112 158–164. 10.1016/j.fcr.2009.02.011 [DOI] [Google Scholar]
- Barrero J. M., Cavanagh C., Verbyla K. L., Tibbits J. F., Verbyla A. P., Huang B. E., et al. (2015). Transcriptomic analysis of wheat near-isogenic lines identifies PM19-A1 and A2 as candidates for a major dormancy QTL. Genome Biol. 16 93 10.1186/s13059-015-0665-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrero J. M., Jacobsen J., Gubler F. (2010). “Seed dormancy: approaches for finding new genes in cereals,” in Plant Developmental Biology - Biotechnological Perspectives Vol. 1 eds Pua E. C., Davey M. R. (Berlin: Springer; ) 361–381. 10.1007/978-3-642-02301-9_18 [DOI] [Google Scholar]
- Borrill P., Adamski N., Uauy C. (2015). Genomics as the key to unlocking the polyploid potential of wheat. New Phytol. 208 1008–1022. 10.1111/nph.13533 [DOI] [PubMed] [Google Scholar]
- Brennan J. P., Fox P. N. (1998). Impact of CIMMYT varieties on the genetic diversity of wheat in Australia, 1973-1993. Aust. J. Agric. Res. 49 175–178. 10.1071/A97065 [DOI] [Google Scholar]
- Cabral A. L., Jordan M. C., Mccartney C. A., You F. M., Humphreys D. G., Maclachlan R., et al. (2014). Identification of candidate genes, regions and markers for pre-harvest sprouting resistance in wheat (Triticum aestivum L.). BMC Plant Biol. 14:340 10.1186/s12870-014-0340-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. X., Cai S. B., Bai G. H. (2008). A major QTL controlling seed dormancy and pre-harvest sprouting resistance on chromosome 4A in a Chinese wheat landrace. Mol. Breed. 21 351–358. 10.1007/s11032-007-9135-5 [DOI] [Google Scholar]
- Clavijo B. J., Venturini L., Schudoma C., Accinelli G. G., Kaithakottil G., Wright J., et al. (2017). An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 27 885–896. 10.1101/gr.217117.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbineau F., Xia Q., Bailly C., El-Maarouf-Bouteau H. (2014). Ethylene, a key factor in the regulation of seed dormancy. Front. Plant Sci. 5:539 10.3389/fpls.2014.00539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvikova K., Cattonaro F., Alaux M., Stein N., Mayer K. F., Dolezel J., et al. (2015). High-throughput physical map anchoring via BAC-pool sequencing. BMC Plant Biol. 15:99 10.1186/s12870-015-0429-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagles H. A., Cane K., Vallance N. (2009). The flow of alleles of important photoperiod and vernalisation genes through Australian wheat. Crop Pasture Sci. 60 646–657. 10.1071/CP09014 [DOI] [Google Scholar]
- Fang J., Chu C. (2008). Abscisic acid and the pre-harvest sprouting in cereals. Plant Signal. Behav. 3 1046–1048. 10.4161/psb.3.12.6606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flintham J. (2000). Different genetic components control coat-imposed and embryo-imposeddormancy in wheat. Seed Sci. Res. 10 43–50. 10.1017/S0960258500000052 [DOI] [Google Scholar]
- Flintham J., Adlam R., Bassoi M., Holdsworth M., Gale M. D. (2002). Mapping genes for resistance to sprouting damage in wheat. Euphytica 126 39–45. 10.1023/A:1019632008244 [DOI] [Google Scholar]
- Frenkel Z., Paux E., Mester D., Feuillet C., Korol A. (2010). LTC: a novel algorithm to improve the efficiency of contig assembly for physical mapping in complex genomes. BMC Bioinformatics 11:584 10.1186/1471-2105-11-584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X., Hu C. H., Li H. Z., Yao Y. J., Meng M., Dong L., et al. (2013). Factors affecting pre-harvest sprouting resistance in wheat (Triticum aestivum l.): a review. J. Anim. Plant Sci. 23 556–565. [Google Scholar]
- Gatford K. T., Eastwood R. F., Halloran G. M. (2002). Germination inhibitors in bracts surrounding the grain of Triticum tauschii. Funct. Plant Biol. 29 881–890. 10.1071/PP01011 [DOI] [PubMed] [Google Scholar]
- Hen-Avivi S., Savin O., Racovita R. C., Lee W. S., Adamski N. M., Malitsky S., et al. (2016). A metabolic gene cluster in the wheat W1 and the barley cer-cqu loci determines beta-diketone biosynthesis and glaucousness. Plant Cell 28 1440–1460. 10.1105/tpc.16.00197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey L. T., Dieters M. J., Delacy I. H., Kravchuk O. Y., Mares D. J., Banks P. M. (2009). Grain dormancy in fixed lines of white-grained wheat (Triticum aestivum L.) grown under controlled environmental conditions. Euphytica 168 303–310. 10.1007/s10681-009-9929-0 [DOI] [Google Scholar]
- Himi E., Maekawa M., Miura H., Noda K. (2011). Development of PCR markers for Tamyb10 related to R-1, red grain color gene in wheat. Theor. Appl. Genet. 122 1561–1576. 10.1007/s00122-011-1555-2 [DOI] [PubMed] [Google Scholar]
- Himi E., Noda K. (2005). Red grain colour gene (R) of wheat is a Myb-type transcription factor. Euphytica 143 239–242. 10.1007/s10681-005-7854-4 [DOI] [Google Scholar]
- Jordan K. W., Wang S., Lun Y., Gardiner L. J., Maclachlan R., Hucl P., et al. (2015). A haplotype map of allohexaploid wheat reveals distinct patterns of selection on homoeologous genomes. Genome Biol. 16 48 10.1186/s13059-015-0606-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox R. E., Clarke F. R., Clarke J. M., Fox S. L., Depauw R. M., Singh A. K. (2012). Enhancing the identification of genetic loci and transgressive segregants for preharvest sprouting resistance in a durum wheat population. Euphytica 186 193–206. 10.1007/s10681-011-0557-0 [DOI] [Google Scholar]
- Konieczny A., Ausubel F. M. (1993). A procedure for mapping Arabidopsis mutations using co-dominant ecotype-specific PCR-based markers. Plant J. 4 403–410. 10.1046/j.1365-313X.1993.04020403.x [DOI] [PubMed] [Google Scholar]
- Kottearachchi N. S., Uchino N., Kato K., Miura H. (2006). Increased grain dormancy in white-grained wheat by introgression of preharvest sprouting tolerance QTLs. Euphytica 152 421–428. 10.1007/s10681-006-9231-3 [DOI] [Google Scholar]
- Krasileva K., Buffalo V., Bailey P., Pearce S., Ayling S., Tabbita F., et al. (2013). Separating homeologs by phasing in the tetraploid wheat transcriptome. Genome Biol. 14:R66 10.1186/gb-2013-14-6-r66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasileva K. V., Vasquez-Gross H. A., Howell T., Bailey P., Paraiso F., Clissold L., et al. (2017). Uncovering hidden variation in polyploid wheat. Proc. Natl. Acad. Sci. U.S.A. 114 E913–E921. 10.1073/pnas.1619268114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulwal P., Ishikawa G., Benscher D., Feng Z., Yu L.-X., Jadhav A., et al. (2012). Association mapping for pre-harvest sprouting resistance in white winter wheat. Theor. Appl. Genet. 125 793–805. 10.1007/s00122-012-1872-0 [DOI] [PubMed] [Google Scholar]
- Kulwal P. L., Kumar N., Gaur A., Khurana P., Khurana J. P., Tyagi A. K., et al. (2005). Mapping of a major QTL for pre-harvest sprouting tolerance on chromosome 3A in bread wheat. Theor. Appl. Genet. 111 1052–1059. 10.1007/s00122-005-0021-4 [DOI] [PubMed] [Google Scholar]
- Kumar S., Knox R., Clarke F., Pozniak C., Depauw R., Cuthbert R., et al. (2015). Maximizing the identification of QTL for pre-harvest sprouting resistance using seed dormancy measures in a white-grained hexaploid wheat population. Euphytica 205 287–309. 10.1007/s10681-015-1460-x [DOI] [Google Scholar]
- Lan X. J., Wei Y. M., Liu D. C., Yan Z. H., Zheng Y. L. (2005). Inheritance of seed dormancy in Tibetan semi-wild wheat accession Q1028. J. Appl. Genet. 46 133–138. [PubMed] [Google Scholar]
- Li C., Ni P., Francki M., Hunter A., Zhang Y., Schibeci D., et al. (2004). Genes controlling seed dormancy and pre-harvest sprouting in a rice-wheat-barley comparison. Funct. Integr. Genomics 4 84–93. 10.1007/s10142-004-0104-3 [DOI] [PubMed] [Google Scholar]
- Linkies A., Leubner-Metzger G. (2012). Beyond gibberellins and abscisic acid: how ethylene and jasmonates control seed germination. Plant Cell Rep. 31 253–270. 10.1007/s00299-011-1180-1 [DOI] [PubMed] [Google Scholar]
- Liu S., Cai S., Graybosch R., Chen C., Bai G. (2008). Quantitative trait loci for resistance to pre-harvest sprouting in US hard white winter wheat Rio Blanco. Theor. Appl. Genet. 117 691–699. 10.1007/s00122-008-0810-7 [DOI] [PubMed] [Google Scholar]
- Liu S., Sehgal S. K., Li J., Lin M., Trick H. N., Yu J., et al. (2013). Cloning and characterization of a critical regulator for preharvest sprouting in wheat. Genetics 195 263–273. 10.1534/genetics.113.152330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Sehgal S. K., Lin M., Li J., Trick H. N., Gill B. S., et al. (2015). Independent mis-splicing mutations in TaPHS1 causing loss of preharvest sprouting (PHS) resistance during wheat domestication. New Phytol. 208 928–935. 10.1111/nph.13489 [DOI] [PubMed] [Google Scholar]
- Lohwasser U., Rehman Arif M. A., Börner A. (2013). Discovery of loci determining pre-harvest sprouting and dormancy in wheat and barley applying segregation and association mapping. Biol. Plant. 57 663–674. 10.1007/s10535-013-0332-2 [DOI] [Google Scholar]
- Mares D., Mrva K. (2014). Wheat grain preharvest sprouting and late maturity alpha-amylase. Planta 240 1167–1178. 10.1007/s00425-014-2172-5 [DOI] [PubMed] [Google Scholar]
- Mares D., Mrva K., Cheong J., Williams K., Watson B., Storlie E., et al. (2005). A QTL located on chromosome 4A associated with dormancy in white- and red-grained wheats of diverse origin. Theor. Appl. Genet. 111 1357–1364. 10.1007/s00122-005-0065-5 [DOI] [PubMed] [Google Scholar]
- Matilla A. J., Matilla-Vázquez M. A. (2008). Involvement of ethylene in seed physiology. Plant Sci. 175 87–97. 10.1016/j.plantsci.2008.01.014 [DOI] [Google Scholar]
- Mohan A., Kulwal P., Singh R., Kumar V., Mir R., Kumar J., et al. (2009). Genome-wide QTL analysis for pre-harvest sprouting tolerance in bread wheat. Euphytica 168 319–329. 10.1007/s10681-009-9935-2 [DOI] [Google Scholar]
- Mori M., Uchino N., Chono M., Kato K., Miura H. (2005). Mapping QTLs for grain dormancy on wheat chromosome 3A and the group 4 chromosomes, and their combined effect. Theor. Appl. Genet. 110 1315–1323. 10.1007/s00122-005-1972-1 [DOI] [PubMed] [Google Scholar]
- Munkvold J., Tanaka J., Benscher D., Sorrells M. (2009). Mapping quantitative trait loci for preharvest sprouting resistance in white wheat. Theor. Appl. Genet. 119 1223–1235. 10.1007/s00122-009-1123-1 [DOI] [PubMed] [Google Scholar]
- nabim (2014). Wheat Varieties. Available at: http://www.nabim.org.uk/wheat/wheat-varieties [accessed April 19 2017]. [Google Scholar]
- Nakamura S., Abe F., Kawahigashi H., Nakazono K., Tagiri A., Matsumoto T., et al. (2011). A wheat homolog of MOTHER OF FT AND TFL1 acts in the regulation of germination. Plant Cell 23 3215–3229. 10.1105/tpc.111.088492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura S., Pourkheirandish M., Morishige H., Kubo Y., Nakamura M., Ichimura K., et al. (2016). Mitogen-activated protein kinase kinase 3 regulates seed dormancy in barley. Curr. Biol. 26 775–781. 10.1016/j.cub.2016.01.024 [DOI] [PubMed] [Google Scholar]
- Nave M., Avni R., Ben-Zvi B., Hale I., Distelfeld A. (2016). QTLs for uniform grain dimensions and germination selected during wheat domestication are co-located on chromosome 4B. Theor. Appl. Genet. 129 1303–1315. 10.1007/s00122-016-2704-4 [DOI] [PubMed] [Google Scholar]
- Ogbonnaya F. C., Imtiaz M., Depauw R. M. (2007). Haplotype diversity of preharvest sprouting QTLs in wheat. Genome 50 107–118. 10.1139/g06-142 [DOI] [PubMed] [Google Scholar]
- Ramirez-Gonzalez R. H., Segovia V., Bird N., Fenwick P., Holdgate S., Berry S., et al. (2015). RNA-Seq bulked segregant analysis enables the identification of high-resolution genetic markers for breeding in hexaploid wheat. Plant Biotechnol. J. 13 613–624. 10.1111/pbi.12281 [DOI] [PubMed] [Google Scholar]
- Reeves J., Chiapparino E., Donini P., Ganal M., Guiard J., Hamrit S., et al. (2004). “Changes over time in the genetic diversity of four major European crops: a report from the GEDIFLUX Framework 5 Project,” in Proceedings of the 17th EUCARPIA General Congress, Tulln, Austria, 8–11 September 2004 eds Vollmann J., Grausgruber H., Ruckenbauer P. (Vienna: University of Natural Resources and Applied Life Sciences; ) 3–7. [Google Scholar]
- Shaw P. D., Graham M., Kennedy J., Milne I., Marshall D. F. (2014). Helium: visualization of large scale plant pedigrees. BMC Bioinformatics 15:259 10.1186/1471-2105-15-259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorinola O., Bird N., Simmonds J., Berry S., Henriksson T., Jack P., et al. (2016). The wheat Phs-A1 pre-harvest sprouting resistance locus delays the rate of seed dormancy loss and maps 0.3 cM distal to the PM19 genes in UK germplasm. J. Exp. Bot. 67 4169–4178. 10.1093/jxb/erw194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek S., Ohm J.-B., Lu H., Rugg M., Berzonsky W., Alamri M. S., et al. (2014). Effect of pre-harvest sprouting on physicochemical properties of starch in wheat. Foods 3 194–207. 10.3390/foods3020194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S. M., Maughan P. J. (2015). SNP genotyping using KASPar assays. Methods Mol. Biol. 1245 243–256. 10.1007/978-1-4939-1966-6_18 [DOI] [PubMed] [Google Scholar]
- Solovyev V., Kosarev P., Seledsov I., Vorobyev D. (2006). Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 7(Suppl. 1):S10 10.1186/gb-2006-7-s1-s10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torada A., Ikeguchi S., Koike M. (2005). Mapping and validation of PCR-based markers associated with a major QTL for seed dormancy in wheat. Euphytica 143 251–255. 10.1007/s10681-005-7872-2 [DOI] [Google Scholar]
- Torada A., Koike M., Ikeguchi S., Tsutsui I. (2008). Mapping of a major locus controlling seed dormancy using backcrossed progenies in wheat (Triticum aestivum L.). Genome 51 426–432. 10.1139/G08-007 [DOI] [PubMed] [Google Scholar]
- Torada A., Koike M., Ogawa T., Takenouchi Y., Tadamura K., Wu J., et al. (2016). A causal gene for seed dormancy on wheat chromosome 4A encodes a MAP kinase kinase. Curr. Biol. 26 782–787. 10.1016/j.cub.2016.01.063 [DOI] [PubMed] [Google Scholar]
- Uauy C. (2017). Wheat genomics comes of age. Curr. Opin. Plant Biol. 36 142–148. 10.1016/j.pbi.2017.01.007 [DOI] [PubMed] [Google Scholar]
- Voss-Fels K. P., Qian L., Parra-Londono S., Uptmoor R., Frisch M., Keeble-Gagnère G., et al. (2017). Linkage drag constrains the roots of modern wheat. Plant Cell Environ. 40 717–725. 10.1111/pce.12888 [DOI] [PubMed] [Google Scholar]
- Wicker T., Matthews D. E., Keller B. (2000). TREP: a database for Triticeae repetitive elements. Trends Plant Sci. 7 561–562. 10.1016/S1360-1385(02)02372-5 [DOI] [Google Scholar]
- Wingen L. U., Orford S., Goram R., Leverington-Waite M., Bilham L., Patsiou T. S., et al. (2014). Establishing the A. E. Watkins landrace cultivar collection as a resource for systematic gene discovery in bread wheat. Theor. Appl. Genet. 127 1831–1842. 10.1007/s00122-014-2344-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao-bo R., Xiu-Jin L., Deng-Cai L., Jia-Li W., You-Liang Z. (2008). Mapping QTLs for pre-harvest sprouting tolerance on chromosome 2D in a synthetic hexaploid wheat × common wheat cross. J. Appl. Genet. 49 333–341. 10.1007/BF03195631 [DOI] [PubMed] [Google Scholar]
- Zhang X. Q., Li C., Tay A., Lance R., Mares D., Cheong J., et al. (2008). A new PCR-based marker on chromosome 4AL for resistance to pre-harvest sprouting in wheat (Triticum aestivum L.). Mol. Breed. 22 227–236. 10.1007/s11032-008-9169-3 [DOI] [Google Scholar]
- Zong Y., Wang Y., Li C., Zhang R., Chen K., Ran Y., et al. (2017). Precise base editing in rice, wheat and maize with a Cas9- cytidine deaminase fusion. Nat. Biotechnol. 35 438–440. 10.1038/nbt.3811 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.