

Abstract
The z-disc is a structural component at the lateral borders of the sarcomere and is important for mechanical stability and contractility of both cardiac and skeletal muscles. Of note, the sarcomeric z-disc also represents a nodal point in cardiomyocyte function and signaling. Mutations of numerous z-disc proteins are associated with cardiomyopathies and muscle diseases. To identify additional z-disc proteins that might contribute to cardiac disease, we employed an in silico screen for cardiac-enriched cDNAs. This screen yielded a previously uncharacterized protein named cardiac-enriched FHL2-interacting protein (CEFIP), which exhibited a heart- and skeletal muscle-specific expression profile. Importantly, CEFIP was located at the z-disc and was up-regulated in several models of cardiomyopathy. We also found that CEFIP overexpression induced the fetal gene program and cardiomyocyte hypertrophy. Yeast two-hybrid screens revealed that CEFIP interacts with the calcineurin-binding protein four and a half LIM domains 2 (FHL2). Because FHL2 binds calcineurin, a phosphatase controlling hypertrophic signaling, we examined the effects of CEFIP on the calcineurin/nuclear factor of activated T-cell (NFAT) pathway. These experiments revealed that CEFIP overexpression further enhances calcineurin-dependent hypertrophic signal transduction, and its knockdown repressed hypertrophy and calcineurin/NFAT activity. In summary, we report on a previously uncharacterized protein CEFIP that modulates calcineurin/NFAT signaling in cardiomyocytes, a finding with possible implications for the pathogenesis of cardiomyopathy.
Keywords: calcineurin, cardiac hypertrophy, cardiomyocyte, cardiomyopathy, cardiovascular disease, signaling, C10orf71, CEFIP, z-disc
Introduction
The sarcomeric z-disc has primarily been defined as a structural component at the lateral borders of the sarcomere that is important for mechanical stability and contractility of cardiac and skeletal muscle. In the last decade, the z-disc has emerged as nodal point of cardiomyocyte signal transduction and in the pathogenesis of cardiomyopathy (1). It has become apparent that components of the z-disc like muscle lim protein (MLP)3 (2) or filamin C (3) and the costameric protein melusin (4) take part in signaling pathways or processes of mechanical stress sensing and its response. Many novel heart- and skeletal muscle-specific proteins have been discovered and suggested to play an important role in various signaling pathways, e.g. the previously described protein family of calsarcins (5) that directly interacts with the serine/threonine phosphatase calcineurin. Calsarcin-1 knock-out mice are sensitized to pathological stimuli such as pressure overload, resulting in an excessive hypertrophic response and severe cardiomyopathy (6). Overexpression of calsarcin-1 is sufficient to inhibit Gq agonist-induced hypertrophy and suppress calcineurin signaling in the heart (7). The calcium/calmodulin-dependent phosphatase calcineurin and its downstream targets, transcription factors of the nuclear factor of activated T-cells (NFAT) family, play essential roles in cardiomyocyte signaling (8, 9). Dephosphorylation of these transcription factors by calcineurin promotes their translocation into the nucleus and induces the pro-hypertrophic gene program. Overexpression of constitutively active calcineurin in mouse heart leads to substantial ventricular hypertrophy, massive cardiac enlargement, and heart failure (8). In contrast, mice lacking the most prevalent calcineurin isoform (CnAβ) are dramatically impaired in their ability to establish cardiac hypertrophy induced by pressure overload and angiotensin II or isoproterenol infusions (10). Similarly, the immunosuppressive agents cyclosporine A or tacrolimus (FK506) can block hypertrophic growth in cardiomyocytes by inhibiting calcineurin through the formation of complexes with immunophilins (11).
To identify previously unknown sarcomeric proteins, we screened expressed sequence tag (EST) databases for uncharacterized proteins predominantly found in cardiac and skeletal muscle cDNA libraries. Using a bioinformatics approach, we found an open reading frame coding for a 1435-aa protein, which we termed CEFIP (cardiac-enriched FHL2-interacting protein). This cardiac- and muscle-specific protein is dysregulated in human ischemic and dilated cardiomyopathy as well as in murine models of heart failure and hypertrophy. By performing yeast two-hybrid (Y2H) screens, we identified a well-known protein FHL2 (four and a half-LIM domains 2), which is primarily expressed in the heart, as one of the binding partners of CEFIP. FHL2 has recently been described to bind calcineurin and repress pathological cardiac growth. In contrast, we found that the overexpression of CEFIP led to dramatic induction of the hypertrophic marker genes and NFAT-dependent signaling. Taken together, we identified CEFIP as a novel z-disc protein and established its role as an activator of calcineurin-dependent hypertrophy in cardiomyocytes.
RESULTS
CEFIP is a novel cardiac- and skeletal muscle-specific protein
Numerous z-disc proteins have recently been identified as a cause for cardiomyopathies and cardiac diseases. Based on the notion that almost all of these are expressed in a tissue-restricted pattern, we performed an in silico screen for uncharacterized heart-specific expressed genes (12). Data mining and extraction mainly relied on T-STAG (Tissue-S) and Unigene EST databases. Utilizing this approach, we recently also discovered the cardiac z-disc protein Fbxl-22, which controls turnover of specific sarcomeric proteins (13). Several of the newly identified expressed sequence tags correspond to the Homo sapiens Unigene cluster Hs.585480 and the Mus musculus Unigene cluster Mm.318319 encoding C10orf71. Here, we describe a so far uncharacterized gene that we termed CEFIP (Cardiac enriched FHL2-interacting protein; human NM_001135196.1 and mouse NM_001195097.1). These NCBI reference sequences were used for designing probes containing the open reading frame. Northern blot analyses of multiple human and mouse tissues indeed showed a heart- and skeletal muscle-specific expression pattern (Fig. 1A). These results could be confirmed in quantitative real-time PCR experiments of multiple mouse tissue samples (Fig. 1B). To also analyze the expression of CEFIP at the protein level, Western blottings of several mouse tissue samples were performed. Because there was no antibody for CEFIP available, we generated a novel rabbit antiserum using a 354-aa fragment of mouse N-terminal CEFIP as immunogen. To verify the specificity of the antiserum, a multiple tissue Western blotting was probed with the novel CEFIP antibody, and another blot with antibody preincubated with the CEFIP fragment used for immunization served as control. Again, these blots revealed strong expression of CEFIP in mouse heart and skeletal muscle tissue, whereas the peptide displacement confirms the antibody's specificity at ∼250 kDa (Fig. 1C). The full open reading frame encodes a 1435-aa (human NP_001128668.1) and a 1412-aa (mouse NP_001182026.1) protein, respectively. The alignment of the CEFIP protein sequence reveals that it is highly evolutionarily conserved among different species (Fig. 1D and supplemental Fig. 1). Surprisingly, it only contains a domain of unknown function (DUF4585, pfam15232) that is shared with proline-rich basic protein 1 (PROB1), a protein with unknown function that is also mainly expressed in striated muscles, but no other known protein domains.
Figure 1.

CEFIP is highly enriched in heart and skeletal muscle. A, confirmation of the specific cardiac- and skeletal muscle-enriched expression pattern of human and mouse CEFIP by Northern blotting of multiple human and mouse tissues. B, qPCR shows a 163-fold induction in heart and a 384-fold induction of CEFIP in mouse tissue normalized to spleen. C, Western blot analysis of CEFIP in mouse tissue extracts confirms the heart- and skeletal muscle-enriched expression pattern. The specificity of the antibody is confirmed by preincubation of the antiserum with the peptide that had been used as immunogen blocking specific reactivity. D, percentage identity of protein sequence between H. sapiens, M. musculus, Rattus norvegicus, Macaca mulatta, Canis lupus, and Bos taurus shows high conservation among these species.
CEFIP is located at the cardiac z-disc and dysregulated in in vivo models of cardiomyopathy
To examine the subcellular localization of CEFIP, we used the newly generated antibody for immunostaining in cardiomyocytes (supplemental Fig. 2). Staining of cultivated neonatal rat cardiomyocytes with the anti-CEFIP antiserum showed a signal at the z-disc co-labeled with the well-characterized α-actinin2 antibody (Fig. 2A) (7, 14). Similarly, co-immunostaining in isolated adult rat ventricular cardiomyocytes with anti-α-actinin2 and anti-CEFIP revealed an overlapping fluorescence pattern (Fig. 2B). Subcellular immunolocalization in cryosections of intact mouse heart again confirmed z-disc localization for CEFIP (Fig. 2C).
Figure 2.
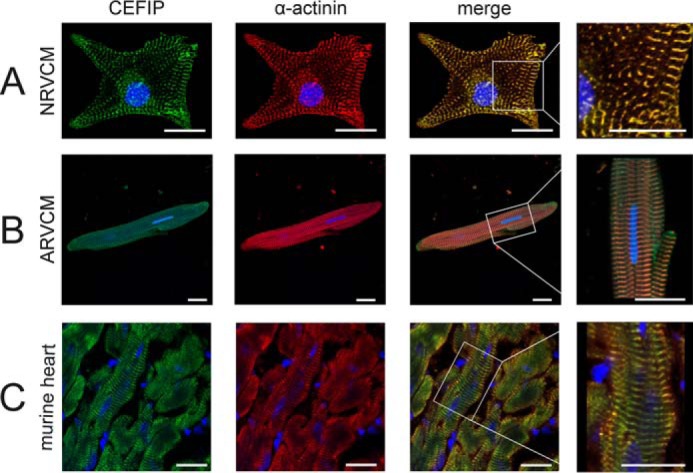
CEFIP localizes to the sarcomeric z-disc. Staining of NRVCM (A), ARVCM (B), and intact mouse heart tissue (C) with a CEFIP antibody reveals a strong signal at the sarcomeric z-disc, validated by co-localization with α-actinin (scale bar, 20 μm).
To identify a potential patho-physiological relevance of CEFIP in vivo, we determined its expression in established models of cardiac diseases. CEFIP was significantly up-regulated in mouse models of hypertrophy like biomechanical stress due to transverse aortic constriction (TAC), calcineurin-TG mice (CnA-TG), and models of dilated cardiomyopathy, including calsarcin-1 knock-out (CS1-KO (15)) and muscle LIM protein knock-out (MLP-KO) mice (Fig. 3, A–D). Consistently, the expression level of CEFIP mRNA was significantly higher in samples of myocardial tissue from human patients suffering from ischemic (ICM) or dilated (DCM) cardiomyopathy compared with non-failing controls (Fig. 3E).
Figure 3.

CEFIP is up-regulated in models of cardiomyopathies. Expression of CEFIP was determined in various established mouse models of cardiomyopathy and heart failure via qPCR. A, CEFIP mRNA levels are increased in mice that received TAC compared with sham-operated mice (1.86 ± 0.1-fold, p < 0.001, n = 6 per group). B–D, similarly, CEFIP expression is up-regulated in transgenic mice overexpressing constitutively active calcineurin (CnA) in the heart (1.63 ± 0.1-fold, p < 0.001, n = 12 per group), in calsarcin-1 (CS1) knock-out mice (1.93 ± 0.4-fold, p < 0.01, n = 11 per group), and in muscle LIM protein (MLP) knock-out mice (2.94 ± 0.5-fold, p < 0.05, n = 6 per group). E, CEFIP mRNA levels are also elevated in human heart tissue samples of patients suffering from ischemic or dilative cardiomyopathy (ICM: 2.3 ± 0.3-fold, p = 0.01, n = 6 NF/10 ICM; DCM: 2.25 ± 0.2-fold, p < 0.01, n = 6 NF/10 DCM). NF indicates non-failing hearts. Error bars show means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
CEFIP modulates cardiomyocyte hypertrophy
Previously, it was shown that sarcomeric z-disc proteins play an important role in cardiomyocyte function and signaling (1). To elucidate the role of CEFIP in cardiomyocytes, we generated adenoviral constructs for overexpression or knockdown of CEFIP allowing gain and loss of function experiments. NRVCMs were infected by AdCEFIP or by a control virus overexpressing β-galactosidase (AdLacZ) and treated with the hypertrophy-inducing agent phenylephrine (PE, 100 μm). The adenoviral infection with AdCEFIP led to an overexpression of CEFIP up to 8-fold on the mRNA level and 9.7-fold on the protein level (Fig. 4, A and B), which was accompanied by an induction of members of the hypertrophic gene program such as NppA/ANF (2.36-fold), NppB/BNP (1.63-fold), Acta1 (1.9-fold), and RCAN1.4 (2-fold) (Fig. 4C). Similarly, overexpression of CEFIP significantly increased the cell-surface area (CSA), equivalent to the increased CSA due to stimulation with PE (100 μm, 24 h). Moreover, the presence of PE with AdCEFIP treatment further enhanced cardiomyocyte hypertrophy (Fig. 4, C and D). Conversely, down-regulation of CEFIP by 72% at the mRNA level and 64% at the protein level (Fig. 4, E and F), due to synthetic miRNA overexpression, revealed no differences in the expression pattern of the hypertrophic gene markers under baseline conditions, but it blunted increased expression following stimulation with PE (Fig. 4G). Interestingly, CSA is decreased in AdmiR-CEFIP-treated cardiomyocytes compared with cells treated with AdmiR-Control at baseline (Fig. 4H). Of note, AdmiR-CEFIP treatment abrogated cardiomyocyte hypertrophy induced by phenylephrine (Fig. 4H).
Figure 4.

CEFIP overexpression results in cardiomyocyte hypertrophy, whereas knockdown of CEFIP abolishes phenylephrine-induced hypertrophy. A and B, adenoviral gene transfer results in an 8.0 ± 0.2-fold overexpression of CEFIP by quantitative real time PCR (p < 0.001, n = 9) and a 9.8 ± 0.4-fold overexpression on protein level (p < 0.005, n = 9). C, qPCR reveals an increased expression of hypertrophic marker genes such as NppA, NppB, Acta1, and RCAN1.4 under the influence of overexpressed CEFIP (NppA: 2.36 ± 0.3-fold, p < 0.01; NppB: 1.63 ± 0.2-fold, p = 0.05; Acta1: 1.9 ± 0.2-fold, p < 0.05; RCAN1.4: 1.95 ± 0.3-fold, p < 0.05; n = 12). Stimulation with phenylephrine exaggerates the effect of CEFIP overexpression. D, consistently, excess CEFIP increases the cell surface of NRVCMs significantly (1.39 ± 0.02-fold, p < 0.001, n >1000). E and F, knockdown via synthetic miRNA against CEFIP leads to a 72 ± 5%-fold (p < 0.001, n = 9) down-regulation observed via qPCR and a 64.2 ± 1.6% on protein level (p < 0.001, n = 9). G, expression analyses via qPCR reveal that mRNA expression levels of hypertrophic marker genes like NppA, NppB, Acta1, and RCAN1.4 remain unaffected upon adenoviral down-regulation of CEFIP after stimulation with phenylephrine. H, relative CSA is decreased in the absence of CEFIP (−11.5 ± 1.2%, p < 0.001, n >1000). Phenylephrine causes a significant increase of the CSA in control NRVCMs. The knockdown of CEFIP inhibits cardiomyocyte hypertrophy. Statistical significance was calculated by two-way ANOVA. Error bars show means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
CEFIP is a highly dynamic protein
To establish either a potential structural or a more dynamic role for CEFIP, we studied the dynamic behavior of CEFIP with the help of FRAP. Cultured myotubes derived from immortalized mouse myoblasts were transduced with CEFIP-EGFP. Importantly, also in these skeletal muscle cells, EGFP-tagged CEFIP targeted to z-discs. Single z-discs in these transduced cells were bleached with a laser using a spinning disc confocal microscope (Fig. 5A and supplemental Movie S1). Analysis of CEFIP recovery showed a biphasic curve. The biexponential fit accounts for a fast and a slow phase (Fig. 5B). Because the initial fast recovery is to some extent caused by lateral diffusion of unbound EGFP-tagged protein (3), we focused on the slow halftime (t½ slow) of CEFIP that indicates the exchange process of bound proteins with the soluble fraction. The exchange of CEFIP was extremely fast with a t½ slow of 32 ± 5 s, while 91 ± 3% of the protein was in the mobile fraction (Fig. 5, C and D). This half-time is comparable with that previously found for FLNc and aciculin/PGM5 in mammalian cultured muscle cells, whereas that of α-actinin2 and especially titin is considerably slower (Fig. 5E). Therefore, CEFIP seems to belong to the group of highly dynamic z-disc proteins involved in mechanosensing, signaling, and myofibrillar remodeling (3, 16).
Figure 5.

CEFIP is a highly dynamic protein that interacts with FHL2. A–E, FRAP analysis of CEFIP dynamics in z-discs of cultured myotubes expressing EGFP-tagged CEFIP. A, CEFIP is targeted to z-discs. ROI for bleaching was limited to a single z-disc, and fluorescence recovery was followed over time. Time series of fluorescence micrographs are shown before bleaching (pre-bleaching), immediately after bleaching (post-bleaching), and 5 and 20 s after bleaching and after maximum fluorescence recovery. B, typical recovery profile of CEFIP in z-discs. C, slow halftime of CEFIP in z-disc of 32.2 s (median) is shown as box plot of all experiments (n = 8). D, estimation of CEFIP mobility in z-discs indicates that 91% of the protein is in the mobile fraction (n = 8). E, overview of exchange half-lives (t1/2) and mobile fractions (Mf) of known heart related proteins. F, because of auto-activation of the full-length CEFIP, three yeast two-hybrid screens were performed using different protein fragments of CEFIP as bait. In two screens, several clones encoding FHL2 were found. G, FHL2 was confirmed as a partner of full-length CEFIP via co-immunoprecipitation (IP) from HEK293 cells expressing full-length Myc-tagged CEFIP and HA-tagged FHL2. H, co-immunostaining of adult rat ventricular cardiomyocytes confirms co-localization of CEFIP and FHL2 in z-disc. Scale bars, 5 μm (A), 10 μm (H). WB, Western blotting.
CEFIP directly interacts with FHL2 and induces calcineurin-dependent hypertrophy
To unravel a potential function of CEFIP in cardiomyocyte signaling, we performed Y2H experiments to identify interacting proteins. Because of auto-activation of the full-length construct, we used three different protein fragments (289–966, 966–2160, and 2160–3168 nt) as bait (Fig. 5F and Table 1). Interestingly, in screens of two fragments (966–2160 and 2160–3168 nt), we found several prey clones coding for the well-known titin and filamin-binding protein FHL2 (four and a half-LIM domains 2). Of note, this heart-specific protein also localizes to the z-disc (17). We confirmed the interaction between HA-tagged FHL2 and Myc-tagged CEFIP via co-immunoprecipitation (Fig. 5G). Furthermore, immunostaining of adult rat ventricular cardiomyocytes confirmed co-localization of CEFIP and FHL2 at the sarcomeric z-disc (Fig. 5H). FHL2 has recently been described to directly bind to calcineurin and to repress pathological cardiac growth (17). Given this information and the pro-hypertrophic effects CEFIP exerted on cardiomyocytes, we next examined whether calcineurin signaling is affected by CEFIP. NRVCMs co-infected with a reporter adenovirus expressing luciferase under control of an NFAT-responsive promoter showed a 2.0-fold up-regulation of NFAT-luciferase activity on adenoviral overexpression of CEFIP. This effect was further enhanced in the presence of constitutively active calcineurin (Fig. 6A). Similar to treatment with phenylephrine, stimulation with calcineurin and simultaneously overexpressed CEFIP led to “super”-induction of the hypertrophy marker genes NppA/ANF (1.7-fold), NppB/BNP (1.8-fold), and RCAN1.4 (3-fold) (Fig. 6B). Consistently, cardiomyocyte hypertrophy, as assessed by CSA measurements, was exaggerated in the presence of both calcineurin and CEFIP (Fig. 6C). Conversely, we observed a dose-dependent reduction of NFAT-luc activity upon miRNA-mediated CEFIP knockdown, even in presence of calcineurin overexpression (Fig. 6D). Likewise, the induction of hypertrophic marker genes and the increase in cell-surface area caused by stimulation with calcineurin was attenuated in the absence of CEFIP (Fig. 6, E and F).
Table 1.
List of potential CEFIP-binding partners
Three independent yeast two-hybrid screens were performed using different fragments of human CEFIP (289–966, 966–2160, and 2160–3168 nt) as bait against human cardiac cDNA library.
| Prey | Gene description | Gene ID | Protein | Accession no. |
|---|---|---|---|---|
| Bait CEFIP (nt 289–966) | ||||
| ACTN2 | Actinin α2 | 88 | α-actinin-2 isoform 1 | NP_001094 |
| DES | Desmin | 1674 | Desmin | NP_1918 |
| DYNLL1 | Dynein light chain LC8-type 1 | 8655 | Dynein light chain LC8-type 1 | AAI04245 |
| EDC4 | Enhancer of mRNA decapping 4 | 23644 | Enhancer of mRNA decapping 4 | AAH64567 |
| EFEMP2 | EGF-containing fibulin-like extracellular matrix protein 2 | 30008 | Fibulin-4 | CAA10791 |
| HSPB3 | Heat shock protein family B (small) member 3 | 8988 | Heat shock protein β-3 | NP_006299.1 |
| HSPB2 | Heat shock protein family B (small) member 2 | 3316 | Heat shock 27-kDa protein 2 | AAI09394 |
| IMMT | Inner membrane mitochondrial protein | 10989 | Proliferation-inducing gene 4 | AAP69987 |
| IMMT | Inner membrane mitochondrial protein | 10989 | Inner membrane protein, mitochondrial (mitofilin) | AAH02412 |
| LZTS2 | Leucine zipper tumor suppressor 2 | 84445 | LAPSER1 | AAK31577 |
| MPRIP | Myosin phosphatase Rho-interacting protein | 23164 | Myosin phosphatase Rho-interacting protein isoform | NP_958431 |
| PDE4DIP | Phosphodiesterase 4D-interacting protein | 9659 | Phosphodiesterase 4D-interacting protein | AAH78660 |
| VIM | Vimentin | 7431 | Vimentin | CAI39485 |
| VPS51 | VPS51, GARP complex subunit | 738 | Chromosome 11 open reading frame 2 | AAH07198 |
| Bait CEFIP (nt 966–2160) | ||||
| ACTN2 | Actinin α2 | 88 | α-Actinin-2 isoform 1 | NP_001094 |
| FHL2 | Four and a half LIM domains 2 | 2274 | FHL2 isoform 5 | ABB73038 |
| GYPC | Glycophorin C | 2995 | Glycophorin-C isoform 1 | NP_002092 |
| HSPB3 | Heat shock protein family B (small) member 3 | 8988 | Heat shock protein β-3 | NP_006299.1 |
| IFITM1 | Interferon-induced transmembrane protein 1 | 8519 | Interferon-induced transmembrane protein 1 | AAH00897 |
| MYL2 | Myosin light chain 2 | 4633 | cardiac myosin light chain 2 | AAA91832.1 |
| Bait CEFIP (nt 2160–3168) | ||||
| ACTN2 | Actinin α2 | 88 | α-Actinin-2 isoform 1 | NP_001094 |
| FHL2 | Four and a half LIM domains 2 | 2274 | FHL2 isoform 5 | ABB73038 |
| HSPB3 | Heat shock protein family B (small) member 3 | 8988 | Heat shock protein β-3 | NP_006299.1 |
| MPC1 | Mitochondrial pyruvate carrier1 (brain protein 44-like protein) | 51660 | Brain protein 44-like | AAH00810 |
| TAOK3 | TAO kinase 3 | 51347 | STE20-like kinase | AAF14559 |
Figure 6.

CEFIP interacting partner FHL2 shows no additive effect on the NFAT-luciferase activity. A, for luciferase reporter assays NRVCMs were co-infected with adenoviruses expressing NFAT reporter-based firefly luciferase, Renilla luciferase, constitutively active calcineurin, and CEFIP or β-galactosidase (LacZ as control). Renilla luciferase activity was used for normalization. Overexpression of CEFIP increases the NFAT-luciferase activity 2.02 ± 0.1-fold (n = 9, p < 0.01). B, overexpression of constitutively active calcineurin produces induction of hypertrophic marker genes (NppA, NppB, and RCAN1.4). Overexpression of CEFIP on top of calcineurin exaggerates this effect (NppA: 1.66 ± 0.1-fold, n = 9, p < 0.01; NppB: 1.77 ± 0.2-fold, n = 9, p < 0.05; RCAN1.4: 3.1 ± 0.6-fold, n = 9, p < 0.05). C, overexpression of calcineurin results in increased cell-surface area. CEFIP enhances hypertrophic growth induced by calcineurin in NRVCMs (1.2 ± 0.02, n >2800, p < 0.001). D, infection of NRVCMs with different quantities of synthetic miRNA down-regulating CEFIP (12.5, 25, 50 m.o.i.) reduces dose-dependently the NFAT-luciferase activity down to 30.96 ± 4.5% (n = 9, p < 0.001). E, adenoviral knockdown of CEFIP blunts the induction of NppA, NppB, and RCAN1.4 caused by constitutively active calcineurin as shown by qPCR. F, consistently, down-regulation of CEFIP inhibits hypertrophic growth induced by calcineurin in neonatal rat ventricular cardiomyocytes. G, luciferase expression driven by constitutively active calcineurin is amplified in NRVCMs infected with an adenovirus coding for CEFIP (9.14 ± 0.9-fold, n = 14, p < 0.001). This induction is not further increased by the addition of siRNA knockdown of FHL2. H, induction of the calcineurin driven luciferase activity by absence of FHL2 could not be blunt by simultaneous knockdown of CEFIP. Statistical significance was calculated by two-way ANOVA. Error bars show means ± S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To further analyze the interaction between FHL2 and CEFIP, we performed luciferase assays in the absence of FHL2 when CEFIP or CnA is either overexpressed or knocked down. FHL2 was 80% down-regulated by silencer siRNA. As reported earlier, knockdown of FHL2 increased NFAT-luciferase activity (Fig. 6G). Interestingly, simultaneous overexpression of CEFIP and down-regulation of FHL2 revealed a slight additive increase of luciferase activity (Fig. 6G). Conversely, knockdown of CEFIP abrogated the hypertrophic effect by the absence of FHL2 (Fig. 6H). However, overexpression of both CnA and CEFIP when FHL2 is knocked down had no significant effect compared with overexpression of CnA alone in the absence of FHL2 (Fig. 6G). Moreover, knockdown of CEFIP did not completely attenuate the activation of NFAT-luc due to the knockdown of FHL2 in the presence of CnA (Fig. 6H). Thus, it is unlikely that FHL2 and CEFIP function in a linear pathway. Instead, we hypothesize that CEFIP plays an independent role to fine-tune calcineurin/NFAT signaling in cardiomyocytes (Fig. 7).
Figure 7.

Model of the calcineurin–NFAT signaling pathway and the potential role of CEFIP. A model diagram depicts the cardiac localization and function of CEFIP. CEFIP is localized to the z-disc and plays an important role in calcineurin–NFAT signaling. CEFIP directly activates calcineurin, which in turn induces the expression of hypertrophic gene markers in cardiomyocytes via dephosphorylation of NFAT. Notably, CEFIP interacts and co-localizes with FHL2 that negatively regulates calcineurin signaling. However, FHL2 and CEFIP modulate calcineurin signaling in opposing and independent ways.
Discussion
Here, we introduce CEFIP as a novel striated muscle-enriched sarcomeric z-disc protein. This previously uncharacterized protein appears to play an important role in cardiomyocyte hypertrophy via modulation of calcineurin-dependent signaling.
CEFIP is a novel z-disc protein
Recently, the perception of the z-disc has experienced a transformation from the traditionally, merely mechanical role into a nodal point in cardiomyocyte signaling (1) and a “hotspot” of cardiomyopathies. Several z-disc components have been discovered to participate in the processes of mechanical stress sensing and signal transduction (3, 18, 19). Because of its relatively large size, one might assume that CEFIP plays a role as a structural component of the z-disc. However, FRAP experiments revealed unforeseen dynamics and mobility of CEFIP compared with other sarcomeric proteins in mammalian (3, 18, 19) and avian (20, 21) muscle cells implying a role in signaling rather than a mere structural function within the myofibril.
CEFIP interacts with FHL2 and regulates hypertrophic signaling
Because the function of CEFIP was largely unknown, we first applied a yeast two-hybrid screen using three independent protein fragments of CEFIP as baits in order to identify possible protein interaction partners. We identified FHL2 as a binding partner of CEFIP and could verify this interaction by co-immunoprecipitation and co-localization. Interestingly, at least two independent binding sites for FHL2 must exist on CEFIP, as two non-overlapping baits revealed this new interaction partner. FHL2, like CEFIP, is localized at the z-disc and highly enriched in the heart (22). FHL2 interacts with a multitude of proteins that are localized to the cell membrane, cytoplasm, and sarcomere as well as to the nucleus (23). Calcineurin, one of the key players in cardiac hypertrophy (8), ranks among these proteins (17). Yet the role of FHL2 in cardiac hypertrophy is still controversial. The absence of FHL2 in vivo does not produce a pathological phenotype at baseline. Furthermore, FHL2-deficient mice develop hypertrophy to the same extent as wild-type mice following TAC (24). In contrast, the hypertrophic response is exaggerated in FHL2 knock-out mice when stimulated with the β-adrenergic receptor agonist isoproterenol (25). While under hypertrophic conditions, the absence of FHL2 seems to produce none or maladaptive effects, and the induction of FHL2 appears to be beneficial. Overexpression of FHL2 blunts hypertrophy upon stimulation with phenylephrine or MEK1 (26). Consistently, the inhibition of hypertrophy in ROCK2-deficient mice upon stimulation with angiotensin II is mediated by enhanced expression of FHL2 (27). Interestingly, an increase or reduction of CEFIP expression is associated with opposite effects in terms of cardiomyocyte hypertrophy compared with FHL2.
CEFIP affects calcineurin signaling
Our results indicate that overexpression of CEFIP is sufficient to induce cardiomyocyte hypertrophy. Moreover, the hypertrophic effects of phenylephrine are exaggerated by CEFIP overexpression. Conversely, knockdown of CEFIP blunts hypertrophy induced by phenylephrine.
Gq/G11 agonists like angiotensin II or phenylephrine mediate hypertrophy through several molecular pathways (28), including the calcineurin–NFAT cascade (29). In line with this notion, knockdown of CEFIP is sufficient to inhibit hypertrophy induced by calcineurin, which is also an interaction partner of FHL2 (17). Activation of calcineurin leads to dephosphorylation of NFAT transcription factors and their translocation to the nucleus, where they initiate the induction of hypertrophy-associated genes (30). Overexpression of CEFIP increases NFAT–luciferase–reporter activity and even further enhances the effect of constitutively active calcineurin. Similar results have been obtained by knockdown of FHL2 before (17) and could be reproduced by us. However, combination of CEFIP overexpression with FHL2 knockdown during overexpression of constitutively active calcineurin was not able to further augment promoter activity, which might be due to a ceiling effect. Of note, the increase of calcineurin-dependent NFAT promotor activity caused by the absence FHL2 is abolished by knockdown of CEFIP. Taken together, the interaction of FHL2 with calcineurin as well as with CEFIP establishes a complex of these three proteins at the z-disc, where FHL2 and CEFIP generate opposing effects on calcineurin/NFAT signaling (Fig. 7).
CEFIP in cardiac disease
We also provide data that CEFIP is induced in human failing hearts due to ischemic or dilated cardiomyopathy. We found a similar expression pattern in different animal models with heart failure. Again, FHL2 expression seems to be changed in the opposite direction: FHL2 is down-regulated in human non-ischemic heart failure (31). Moreover, a missense mutation of FHL2 was identified in a patient with familial dilated cardiomyopathy (32). FHL2 is suppressed in hypertrophic cardiomyopathy as well (33).
Integrating the heart- and skeletal muscle-specific expression of CEFIP and its induction in models of cardiac disease, we hypothesize that CEFIP acts as a mediator in cardiac disease. It may be part of the maladaptive hypertrophic response of cardiomyocytes to biomechanical stress that accompanies valvular or ischemic heart disease. Moreover, one may speculate that gain of function mutations of CEFIP trigger cardiac hypertrophy. To clarify the role of CEFIP in heart disease mutation analyses of CEFIP in patients with dilated or hypertrophic cardiomyopathy might be helpful.
Because we could clearly show that overexpression of CEFIP is sufficient to induce hypertrophy by modulating the calcineurin pathway, CEFIP represents a potential target for a therapeutic intervention such as siRNA attempts. Because of its heart and skeletal muscle specificity, negative or unwanted side effects might be minimized.
In summary, we describe here a novel cardiac- and skeletal muscle-specific protein termed CEFIP, which functions as a regulator of calcineurin-dependent hypertrophy. On the one hand, increased CEFIP expression led to an induction of cardiomyocyte hypertrophy; on the other hand, the absence of CEFIP repressed pharmacological agonist- and calcineurin-induced hypertrophy. Additional in vivo experiments will help to clarify the role of CEFIP in cardiac-signaling pathways and in the pathogenesis of cardiomyopathy.
Experimental procedures
Northern blot analysis
Multiple tissue Northern blots (BioChain) containing human or mouse poly(A) RNA were hybridized with [32P]dCTP-labeled (Rediprime II Random Prime Labeling System, Amersham Biosciences) cDNA probes corresponding to the ORF of human or mouse CEFIP at 65 °C overnight. Washing was repeated with 2× SSC, 0.1% SDS and 0.2× SSC, 0.1% SDS at 65 °C. Autoradiography was conducted at −80 °C for 24–168 h with an intensifying screen.
Isolation and culture of NRVCMs
NRVCMs were isolated according to a previously published protocol (19). Hearts from 1- to 2-day-old Wistar rats (Charles River Laboratories) were excised and minced in ADS buffer (120 mm NaCl, 20 mm HEPES, 8 mm NaH2PO4, 6 mm glucose, 5 mm KCl, 0.8 mm MgSO4, pH 7.4). An enzymatic solution containing collagenase type II (0.5 mg/ml, Worthington) and pancreatin (0.6 mg/ml, Sigma) in sterile ADS buffer was used to digest the tissue. A Percoll (GE Healthcare) gradient centrifugation step was applied to remove contaminating fibroblasts from cardiomyocytes. NRVCMs were resuspended and cultured in Dulbecco's modified Eagle's medium (DMEM) containing 10% FCS, 1% penicillin/streptomycin, and 1% l-glutamine (PAA) for 24 h. After 24 h, cells were infected with adenoviruses in serum-free medium for a further 24 h and afterward stimulated with phenylephrine for 48 h.
Isolation and culture of adult rat ventricular cardiomyocytes (ARVCMs)
ARVCMs were isolated using collagenase and plated on laminin-coated dishes as described (34). ARVCMs were cultured in a HEPES-modified medium 199 (M199, Sigma S7528, supplemented with 5 mm taurine, 5 mm carnitine, 5 mm creatine, 5 mm N-mercaptopropionyl glycine, 0.1 μm insulin, 10,000 units/ml penicillin, and 10 mg/ml streptomycin, pH 7.25).
Immunofluorescence microscopy
NRVCMs, ARVCMs, and mouse tissue cryosections were fixed with 4% paraformaldehyde in PBS for 10 min at room temperature, permeabilized, and blocked with 2.5% BSA and 0.1% Triton X-100 in PBS for 1 h at room temperature. The slides were incubated with a monoclonal mouse antibody for α-actinin 2 (1:400, Sigma) together with a polyclonal CEFIP (rabbit 1:100) antibody at 4 °C overnight. The CEFIP antibody was generated by immunizing rabbits with a purified, bacterially expressed recombinant protein representing the first 354 amino acids of mouse CEFIP (custom immunization by Biogenes, Berlin, Germany). Its specificity was confirmed by Western blotting. Secondary antibodies against mouse immunoglobulins conjugated with Cy3 (Dianova) and rabbit immunoglobulins conjugated with FITC (Dianova) were used at a dilution of 1:500 for 1 h at room temperature. Simultaneously, nuclei were labeled with DAPI (4′,6′-diamidino-2-phenylindole, Vector Laboratories), and after serial washing with PBS, the slides were mounted with FluorPreserve reagent (Merck). Confocal images were taken with a Zeiss LSM800 laser-scanning microscope with a Plan-Apochromat ×40/1.4 oil differential interference contrast (UV) visible-IR objective at room temperature. Cropped regions from the overviews are newly acquired images. Cell-surface areas were captured and analyzed on BZ-9000-E HS all-in-one fluorescence microscope (Keyence) at ×60 magnification (Plan-Apo oil immersion ×60, NA 1.4; Nikon) or ×20 magnification (CFI Plan-Apo ×20, NA 0.75; Nikon).
RNA isolation and real-time PCR
Total RNA from NRVCMs was isolated using QIAzol lysis reagent (Qiagen), according to the manufacturer's protocol, and was DNase I-digested. After purification, 1 μg of RNA was reverse-transcribed into cDNA using the Superscript III first strand cDNA synthesis kit (Life Technologies, Inc.). For qRT-PCR, the EXPRESS SYBR GreenER Reagent (Life Technologies, Inc.) or iQ Multiplex Powermix (Bio-Rad) was used in the CFX96 real time cycler (Bio-Rad).
Cloning of human and mouse CEFIP
The complete open reading frame of CEFIP was amplified from heart cDNA using the following gene-specific primers: mouse CEFIP_F, 5′-GCT GGC ACC ATG CAG GGA AAC AAA AAA TGC AC-3′, and mouse CEFIP ms_R, 5′-GCT GGG TCG CCT CAA GAA ACA CCC TCT GTG GCA AAG-3′; human CEFIP_F, 5′-GCT GGC ACC ATG ATG CAA GGA AAT AAG AAG TGC ACA GAC G-3′, and human CEFIP ms_R, 5′-GCT GGG TCG CCT CAA GAA ATG CCT TCT GTG GCA AAG-3′. The PCR product was recombined into a pDONR221 gateway vector using the Gateway Technology (Life Technologies, Inc.). For protein expression, CEFIP was shuttled into an expression plasmid or entry vector to design constructs coding for N-terminal fusion tags (HA, MYC, and V5). An adenovirus encoding the full-length CEFIP was generated with the pAd/CMV/V5-DEST Gateway vector kit following the manufacturer's instructions. As a control, a β-galactosidase-V5 encoding adenovirus was used for overexpression experiments.
Cloning of synthetic CEFIP knockdown miRNA
Oligonucleotides against CEFIP were designed using Invitrogen's BLOCK-iT RNAi Designer (miR-CEFIP_TOP, TGC TGA GCA GAA GTT GAC ACC ACT TTG TTT TGG CCA CTG ACT GAC AAA GTG GTC AAC TTC TGC T and GGC CAA AAC AAA GTG GTG TCA ACT TCT GCT C) and cloned into a pcDNA 6.2-GWmiR vector according to the manufacturer's protocol. This construct was recombined into pDONR221 entry vector and shuttled into pAd/CMV/V5 adenoviral expression vector. For knockdown experiments, the pcDNA6.2 GWmiR plasmid, which is predicted not to target any known mammalian gene, served as a control.
Fluorescence recovery after photobleaching (FRAP) and data analysis
C2C12 myotubes were transduced with CEFIP-EGFP and kept at 37 °C and 5% CO2. FRAP experiments were performed essentially as described (3, 16) using a Cell Observer Spinning Disk Confocal Microscope (Carl Zeiss, Jena, Germany) equipped with an external 473-nm laser coupled via a scanner (UGA-40, Rapp OptoElectronic, Hamburg, Germany) and evaluated using Zen 2012 software (Carl Zeiss). For FRAP analysis, eight independent experiments were performed. The region of interest (ROI) for bleaching was limited to a single z-disc. For each cell, 1–3 ROIs were chosen and bleached using a 473-nm laser (100 milliwatts) with 100% intensity (Rapp OptoElectronic) for a pulse time of 1 ms with eight iterations. Images were taken before and immediately after bleaching, and fluorescence recovery was monitored for 230 s with an interval time of 0.1–1 s until the signal was fully recovered. Raw data were transformed into normalized FRAP curves as described previously (3, 16).
Yeast two-hybrid library assays
Automated Y2H screens were performed as described (35). Human cDNA libraries from human heart and skeletal muscle (Clontech) as well as a library of individual cloned full-length open reading frames from cDNAs of 5000 different genes (DKFZ Heidelberg) were screened to a minimal coverage of 5 million clones per library. As full-length human CEFIP showed auto-activation, the screen was performed using three nucleotide (nt) fragments of CEFIP (289–966, 966–2160, and 2160–3168 nt). To mate yeast strains harboring the bait protein and the prey library, exponentially growing cultures of an absorbance (600 nm) of 1 were combined, pelleted by centrifugation, and resuspended in an equal volume of YPDA medium containing 20% PEG 6000. Mating mixes were incubated at 30 °C with gentle agitation for exactly 3 h before washing and resuspending cells in selective medium. Briefly, clones displaying differential growth on selective plates lacking histidine, leucine, and tryptophan were picked and grown in selective medium lacking leucine and tryptophan, and plasmid DNA was isolated and subsequently electroporated into DH10B Escherichia coli (Gibco via Invitrogen, Karlsruhe, Germany). The obtained clones were sequenced and retransformed with the CEFIP construct to confirm the interaction. Interaction pairs were chosen to be real pairs if out of two independent bacterial preparations from the same yeast clone the sequenced clone is identical, or where the sequence of two preys that interacted with the same bait is highly related.
Luciferase reporter gene assays
NRVCMs were infected with adenoviruses coding for either CEFIP (25 m.o.i.) for overexpression experiments or synthetic miRNA against CEFIP (100 m.o.i.) for knockdown experiments. An adenovirus encoding LacZ as well as an adenovirus encoding unspecific miRNA was used as controls. AdNFATluc carrying firefly luciferase and AdRen-luc carrying Renilla luciferase were co-infected. 72 h after infection, cells were harvested in passive lysis buffer (Promega). The luciferase assay was performed using the dual-luciferase reporter assay (Promega) according to the manufacturer's protocol on the infinite m200 PRO system (Tecan). The experiments were performed in quadruplicate and repeated at least three times.
Co-immunoprecipitation
HEK293 cells were transfected with plasmids encoding the potential CEFIP interacting proteins (N-terminal fusions) by using JetPEI (PolyPlus Transfection) according to the manufacturer's instructions. Cells were harvested 48 h post-transfection with ice-cold ELB lysis buffer (50 mm HEPES, pH 7.0, 250 mm NaCl, 1% Nonidet P-40, 5 mm EDTA, Protease Inhibitor Mixture (Roche Applied Science, catalog no. 1836170)) containing Phosphatase Inhibitor Mixture 2 and 3 (Sigma, catalog no. P5726; P0044). After incubation on ice for 30 min, lysates were centrifuged for 5 min at 16,200 × g at 4 °C to remove cellular debris. A total of 3000 μg of cleared protein extract was incubated with anti-HA-agarose (Sigma, catalog no. A2095) for 2 h at 4 °C with moderate agitation. After four cycles of washing with lysis buffer, protein complexes were eluted in 1× bead volume SDS sample buffer. For Western blot experiments, the beads were separated by 2 min of centrifugation at 16,200 × g at room temperature, and the supernatant containing the protein fraction was analyzed by immunoblotting.
Western blotting
To determine specificity of CEFIP expression, protein extracts from NRVCMs or tissue samples were isolated and proceeded in Lysis-Buffer (20 mm Tris, pH 7.5, 12.5% (v/v) glycerol, 10 mm dithiothreitol, 500 mm NaCl, 1% (v/v) Nonidet P-40 (Sigma), 10 mm DTT, as well as protease inhibitor mixture (Roche Applied Science) and phosphatase inhibitor mixture 2 and 3 (Sigma)). Protein concentration was measured by Bradford protein assay kit according to the manufacturer's guidelines (Bio-Rad). Samples were resolved by SDS-PAGE, transferred onto a PVDF membrane (GE Healthcare), and incubated with respective target-specific primary antibody. Subsequently, the membrane was incubated in secondary HRP-coupled antibody followed by visualization using a chemiluminescence kit (ECL, GE Healthcare) and detection with a FluorChemQ imaging system (Alpha Innotech).
Statistical analyses
All results are the means ± S.E. Statistical analyses were performed using two-tailed Student's t test, one-way or two-way ANOVA followed by Student-Newman-Keuls post hoc tests when appropriate. p values < 0.05 were considered statistically significant.
Author contributions
F. D. conducted experiments, analyzed and interpreted the data, and wrote the manuscript. C. K. designed the study and interpreted data. C. R. performed the experiments and analyzed the data. S. H. performed experiments. S. S. provided the human samples. D. O. F., P. F. M. v. d. V., J. B., and S. M. performed and interpreted the FRAP experiments and generated the CEFIP-antibody. N. F. devised the strategy and designed the study. All authors critically revised the manuscript.
Supplementary Material
Acknowledgments
We thank Gabriele Brunke, Christin Tannert, and Vanessa Mangels for excellent technical assistance.
This work was supported in part by Deutsche Forschungsgemeinschaft (German Research Foundation) Forschergruppe 1352: “Structure, Function and Regulation of the Myofibrillar z-disc Interactome.” The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains supplemental Movie S1 and Figs. S1 and S2.
- MLP
- muscle lim protein
- CEFIP
- cardiac-enriched FHL2-interacting protein
- ANOVA
- analysis of variance
- NFAT
- nuclear factor of activated T-cell
- aa
- amino acid
- TAC
- transverse aortic constriction
- qPCR
- quantitative PCR
- ICM
- ischemic cardiomyopathy
- DCM
- dilated cardiomyopathy
- NRVCM
- neonatal rat ventricular cardiomyocyte
- ARVCM
- adult rat ventricular cardiomyocyte
- PE
- phenylephrine
- nt
- nucleotide
- m.o.i.
- multiplicity of infection
- CSA
- cell-surface area
- EGFP
- enhanced GFP
- FRAP
- fluorescence recovery after photobleaching
- ROI
- region of interest
- EST
- expressed sequence tag
- Y2H
- yeast two-hybrid
- F
- forward
- R
- reverse.
References
- 1. Frank D., Kuhn C., Katus H. A., and Frey N. (2006) The sarcomeric Z-disc: a nodal point in signalling and disease. J. Mol. Med. 84, 446–468 [DOI] [PubMed] [Google Scholar]
- 2. Knöll R., Hoshijima M., and Chien K. R. (2002) Muscle LIM protein in heart failure. Exp. Clin. Cardiol. 7, 104–105 [PMC free article] [PubMed] [Google Scholar]
- 3. Leber Y., Ruparelia A. A., Kirfel G., van der Ven P. F., Hoffmann B., Merkel R., Bryson-Richardson R. J., and Fürst D. O. (2016) Filamin C is a highly dynamic protein associated with fast repair of myofibrillar microdamage. Hum. Mol. Genet. 25, 2776–2788 [DOI] [PubMed] [Google Scholar]
- 4. Brancaccio M., Fratta L., Notte A., Hirsch E., Poulet R., Guazzone S., De Acetis M., Vecchione C., Marino G., Altruda F., Silengo L., Tarone G., and Lembo G. (2003) Melusin, a muscle-specific integrin β1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat. Med. 9, 68–75 [DOI] [PubMed] [Google Scholar]
- 5. Frey N., Richardson J. A., and Olson E. N. (2000) Calsarcins, a novel family of sarcomeric calcineurin-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 14632–14637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Frey N., Barrientos T., Shelton J. M., Frank D., Rütten H., Gehring D., Kuhn C., Lutz M., Rothermel B., Bassel-Duby R., Richardson J. A., Katus H. A., Hill J. A., and Olson E. N. (2004) Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat. Med. 10, 1336–1343 [DOI] [PubMed] [Google Scholar]
- 7. Frank D., Kuhn C., van Eickels M., Gehring D., Hanselmann C., Lippl S., Will R., Katus H. A., and Frey N. (2007) Calsarcin-1 protects against angiotensin-II induced cardiac hypertrophy. Circulation 116, 2587–2596 [DOI] [PubMed] [Google Scholar]
- 8. Molkentin J. D., Lu J. R., Antos C. L., Markham B., Richardson J., Robbins J., Grant S. R., and Olson E. N. (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93, 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ritter O., Hack S., Schuh K., Röthlein N., Perrot A., Osterziel K. J., Schulte H. D., and Neyses L. (2002) Calcineurin in human heart hypertrophy. Circulation 105, 2265–2269 [DOI] [PubMed] [Google Scholar]
- 10. Bueno O. F., Wilkins B. J., Tymitz K. M., Glascock B. J., Kimball T. F., Lorenz J. N., and Molkentin J. D. (2002) Impaired cardiac hypertrophic response in calcineurin Aβ-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 99, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu J., Farmer J. D. Jr, Lane W. S., Friedman J., Weissman I., and Schreiber S. L. (1991) Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66, 807–815 [DOI] [PubMed] [Google Scholar]
- 12. Will R. D., Eden M., Just S., Hansen A., Eder A., Frank D., Kuhn C., Seeger T. S., Oehl U., Wiemann S., Korn B., Koegl M., Rottbauer W., Eschenhagen T., Katus H. A., and Frey N. (2010) Myomasp/LRRC39, a heart- and muscle-specific protein, is a novel component of the sarcomeric M-band and is involved in stretch sensing. Circ. Res. 107, 1253–1264 [DOI] [PubMed] [Google Scholar]
- 13. Spaich S., Will R. D., Just S., Spaich S., Kuhn C., Frank D., Berger I. M., Wiemann S., Korn B., Koegl M., Backs J., Katus H. A., Rottbauer W., and Frey N. (2012) F-box and leucine-rich repeat protein 22 is a cardiac-enriched F-box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo. Circ. Res. 111, 1504–1516 [DOI] [PubMed] [Google Scholar]
- 14. Mohapatra B., Jimenez S., Lin J. H., Bowles K. R., Coveler K. J., Marx J. G., Chrisco M. A., Murphy R. T., Lurie P. R., Schwartz R. J., Elliott P. M., Vatta M., McKenna W., Towbin J. A., and Bowles N. E. (2003) Mutations in the muscle LIM protein and α-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 80, 207–215 [DOI] [PubMed] [Google Scholar]
- 15. Schoensiegel F., Bekeredjian R., Schrewe A., Weichenhan D., Frey N., Katus H. A., and Ivandic B. T. (2007) Atrial natriuretic peptide and osteopontin are useful markers of cardiac disorders in mice. Comp. Med. 57, 546–553 [PubMed] [Google Scholar]
- 16. Molt S., Bührdel J. B., Yakovlev S., Schein P., Orfanos Z., Kirfel G., Winter L., Wiche G., van der Ven P. F., Rottbauer W., Just S., Belkin A. M., and Fürst D. O. (2014) Aciculin interacts with filamin C and Xin and is essential for myofibril assembly, remodeling and maintenance. J. Cell Sci. 127, 3578–3592 [DOI] [PubMed] [Google Scholar]
- 17. Hojayev B., Rothermel B. A., Gillette T. G., and Hill J. A. (2012) FHL2 binds calcineurin and represses pathological cardiac growth. Mol. Cell. Biol. 32, 4025–4034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kuhn C., Frank D., Dierck F., Oehl U., Krebs J., Will R., Lehmann L. H., Backs J., Katus H. A., and Frey N. (2012) Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res. Cardiol. 107, 262. [DOI] [PubMed] [Google Scholar]
- 19. Kuhn C., Frank D., Will R., Jaschinski C., Frauen R., Katus H. A., and Frey N. (2009) DYRK1A is a novel negative regulator of cardiomyocyte hypertrophy. J. Biol. Chem. 284, 17320–17327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang J., Dube D. K., Mittal B., Sanger J. M., and Sanger J. W. (2011) Myotilin dynamics in cardiac and skeletal muscle cells. Cytoskeleton 68, 661–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. da Silva Lopes K., Pietas A., Radke M. H., and Gotthardt M. (2011) Titin visualization in real time reveals an unexpected level of mobility within and between sarcomeres. J. Cell Biol. 193, 785–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chan K. K., Tsui S. K., Lee S. M., Luk S. C., Liew C. C., Fung K. P., Waye M. M., and Lee C. Y. (1998) Molecular cloning and characterization of FHL2, a novel LIM domain protein preferentially expressed in human heart. Gene 210, 345–350 [DOI] [PubMed] [Google Scholar]
- 23. Tran M. K., Kurakula K., Koenis D. S., and de Vries C. J. (2016) Protein-protein interactions of the LIM-only protein FHL2 and functional implication of the interactions relevant in cardiovascular disease. Biochim. Biophys. Acta 1863, 219–228 [DOI] [PubMed] [Google Scholar]
- 24. Chu P. H., Bardwell W. M., Gu Y., Ross J. Jr., and Chen J. (2000) FHL2 (SLIM3) is not essential for cardiac development and function. Mol. Cell. Biol. 20, 7460–7462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kong Y., Shelton J. M., Rothermel B., Li X., Richardson J. A., Bassel-Duby R., and Williams R. S. (2001) Cardiac-specific LIM protein FHL2 modifies the hypertrophic response to beta-adrenergic stimulation. Circulation 103, 2731–2738 [DOI] [PubMed] [Google Scholar]
- 26. Purcell N. H., Darwis D., Bueno O. F., Müller J. M., Schüle R., and Molkentin J. D. (2004) Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the LIM-only protein FHL2 in cardiomyocytes. Mol. Cell. Biol. 24, 1081–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Okamoto R., Li Y., Noma K., Hiroi Y., Liu P. Y., Taniguchi M., Ito M., and Liao J. K. (2013) FHL2 prevents cardiac hypertrophy in mice with cardiac-specific deletion of ROCK2. FASEB J. 27, 1439–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Frey N., and Olson E. N. (2003) Cardiac hypertrophy: the good, the bad, and the ugly. Annu. Rev. Physiol. 65, 45–79 [DOI] [PubMed] [Google Scholar]
- 29. De Windt L. J., Lim H. W., Bueno O. F., Liang Q., Delling U., Braz J. C., Glascock B. J., Kimball T. F., del Monte F., Hajjar R. J., and Molkentin J. D. (2001) Targeted inhibition of calcineurin attenuates cardiac hypertrophy in vivo. Proc. Natl. Acad. Sci. U.S.A. 98, 3322–3327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Heineke J., and Ritter O. (2012) Cardiomyocyte calcineurin signaling in subcellular domains: from the sarcolemma to the nucleus and beyond. J. Mol. Cell. Cardiol 52, 62–73 [DOI] [PubMed] [Google Scholar]
- 31. Bovill E., Westaby S., Crisp A., Jacobs S., and Shaw T. (2009) Reduction of four-and-a-half-LIM-protein 2 expression occurs in human left ventricular failure and leads to altered localization and reduced activity of metabolic enzymes. J. Thorac. Cardiovasc. Surg. 137, 853–861 [DOI] [PubMed] [Google Scholar]
- 32. Arimura T., Hayashi T., Matsumoto Y., Shibata H., Hiroi S., Nakamura T., Inagaki N., Hinohara K., Takahashi M., Manatsu S. I., Sasaoka T., Izumi T., Bonne G., Schwartz K., and Kimura A. (2007) Structural analysis of four and half LIM protein-2 in dilated cardiomyopathy. Biochem. Biophys. Res. Commun. 357, 162–167 [DOI] [PubMed] [Google Scholar]
- 33. Friedrich F. W., Reischmann S., Schwalm A., Unger A., Ramanujam D., Münch J., Müller O. J., Hengstenberg C., Galve E., Charron P., Linke W. A., Engelhardt S., Patten M., Richard P., van der Velden J., et al. (2014) FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 109, 451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luedde M., Flögel U., Knorr M., Grundt C., Hippe H. J., Brors B., Frank D., Haselmann U., Antony C., Voelkers M., Schrader J., Most P., Lemmer B., Katus H. A., and Frey N. (2009) Decreased contractility due to energy deprivation in a transgenic rat model of hypertrophic cardiomyopathy. J. Mol. Med. 87, 411–422 [DOI] [PubMed] [Google Scholar]
- 35. Albers M., Kranz H., Kober I., Kaiser C., Klink M., Suckow J., Kern R., and Koegl M. (2005) Automated yeast two-hybrid screening for nuclear receptor-interacting proteins. Mol. Cell. Proteomics 4, 205–213 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.