

Abstract
Persistent DNA damage induces profound alterations in gene expression that, in turn, influence tissue homeostasis, tumorigenesis, and cancer treatment outcome. However, the underlying mechanism for gene expression reprogramming induced by persistent DNA damage remains poorly understood. Here, using a highly effective bioluminescence-based reporter system and other tools, we report that persistent DNA damage inhibits nonsense-mediated RNA decay (NMD), an RNA surveillance and gene-regulatory pathway, in noncycling cells. NMD suppression by persistent DNA damage required the activity of the p38α MAPK. Activating transcription factor 3 (ATF3), an NMD target and a key stress-inducible transcription factor, was stabilized in a p38α- and NMD-dependent manner following persistent DNA damage. Our results reveal a novel p38α-dependent pathway that regulates NMD activity in response to persistent DNA damage, which, in turn, controls ATF3 expression in affected cells.
Keywords: DNA damage response, gene expression, mRNA decay, p38, senescence, ATF3, nonsense-mediated RNA decay, persistent DNA damage
Introduction
The cellular response to persistent DNA damage influences tissue homeostasis, tumorigenesis, and cancer progression (1–3). Persistent DNA damage occurs when new damage is constantly being introduced, for instance, by excessive mitogenic signals or by cancer treatments such as chemotherapy or radiation, or when DNA damage is difficult to repair (4–14). Proliferating cells are particularly susceptible to persistent DNA damage and usually undergo programmed cell death, as carrying genetic lesions through cell division causes severe genomic instability. However, nonproliferating cells can tolerate a certain dose of persistent DNA damage. In these non-cycling cells, persistent DNA damage reprograms gene expression to promote responses such as cellular senescence, characterized by exit from the cell cycle and profound changes in morphology, signaling, and gene expression, and the senescence-associated secretory phenotype (SASP),3 a collection of secreted factors such as cytokines, chemokines, growth factors, and proteases (1, 3, 15). Both senescence and SASP directly impact the tumorigenesis process and cancer treatment outcome by influencing tumor cell growth and modifying the tumor microenvironment (2, 15–22). These phenotypes and other responses to persistent DNA damage are a result of gene expression reprogramming brought about by signaling molecules such as p38, which regulate transcription, RNA stability, and translation (17, 23). The p38 MAPK, which contains four isoforms (p38α, p38β, p38γ, and p38δ), plays a key role in the cellular response to persistent DNA damage as well as other stresses (23). In certain contexts, where persistent DNA damage is present, p38 can promote cellular senescence and SASP expression (17, 24–31).
Originally identified as an RNA quality control pathway that detects and degrades aberrant mRNAs with premature translation termination codons (PTCs), nonsense-mediated RNA decay (NMD) has also emerged as a prominent gene-regulatory mechanism (32, 33). NMD controls the stability of many normal transcripts that contain features recognizable to the NMD machinery, such as upstream ORFs, 3′ UTR introns, alternative splice variants that introduce a PTC, or exceedingly long 3′ UTRs (33–35). By degrading transcripts bearing such NMD-inducing features, NMD contributes to gene expression profiles crucial for development and other cellular processes (36–46). In addition to “constitutively” degrading target mRNAs, NMD also contributes to gene expression changes in response to developmental and environmental cues that adjust NMD efficiency. For instance, NMD activity is repressed during neuronal differentiation as well as myogenesis to enhance the expression of NMD target transcripts that facilitate proper development (36, 41). These alterations of NMD activity appear to be essential in vertebrates, as many NMD-deficient mutants exhibit abnormal brain, eye, and cardiovascular development and are lethal (47). NMD also helps coordinate the cellular response to several forms of stress, including amino acid deprivation, hypoxia, and endoplasmic reticulum stress, by governing the expression levels of many stress response genes (39, 42, 44, 45, 48). Furthermore, when cells are exposed to severe stresses that cause apoptosis, NMD is attenuated as a result of proteolytic cleavage of the core NMD factor UPF1, leading to the up-regulation of a number of apoptosis genes (49).
In this work, we investigated the potential regulation of NMD by persistent DNA damage and its effects on gene expression. Using a highly effective reporter system, we found that NMD activity is attenuated by persistent DNA damage in nonproliferating cells, leading to stabilization and up-regulation of the transcripts of ATF3, a known NMD target and a key transcription factor that regulates gene expression, to promote the establishment of senescence and SASP expression after DNA damage (39, 44, 50–53). The repression of NMD by persistent DNA damage is mediated in part by p38α, although p38α activation alone is not sufficient to inhibit NMD. Our results reveal a novel p38-dependent pathway that regulates NMD activity in response to persistent DNA damage and contributes to gene expression alterations in non-cycling cells.
Results
Persistent DNA damage, but not transient DNA damage, inhibits NMD
To determine whether persistent DNA damage modulates NMD activity, we treated confluent nontransformed human RPE1 cells with the DNA-damaging agent bleomycin (63 μg/ml) for 24 h and then allowed the cells to recover in the absence of bleomycin for 4 days. Subsequently, NMD activity was assessed using a previously developed bioluminescent NMD reporter system (Fig. 1a) (54). Using this reporter, NMD is quantified by the ratio of red bioluminescence signal, produced by the CBR luciferase fused to a PTC-containing TCRβ minigene (CBR-TCR(PTC)), to green bioluminescence signal, produced by the CBG luciferase fused to a wild-type TCRβ minigene (CBG-TCR(WT)). As expected, bleomycin generated significant amounts of DNA damage and induced a robust DDR that persisted at the time of NMD analysis, as indicated by the phosphorylation of the histone variant H2AX (γH2AX), a widely used DDR marker (Fig. 1b). Strikingly, bleomycin-treated cells exhibited an ∼2-fold increase in the CBR:CBG ratio, indicative of NMD repression (Fig. 1c, left panel). This bioluminescence imaging result was corroborated by real-time qPCR analysis of the mRNA levels of the CBR-TCR(PTC) and CBG-TCR(WT) reporters (Fig. 1c, right panel). Bleomycin-mediated NMD suppression is not specific to RPE1 cells, as similar results were obtained in confluent human BJ fibroblasts (Fig. 1d). Together, these data suggest that persistent DNA damage represses NMD.
Figure 1.

Persistent DNA damage inhibits NMD. a, schematic of a dual-color bioluminescence-based reporter for measuring NMD activity in human cells. b, left panel, representative images of immunofluorescence staining for DAPI or γH2AX in RPE1 cells after 24-h treatment with either H2O or bleomycin (Bleo), followed by 96-h recovery. Right panel, Western blot showing γH2AX levels in lysates collected after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. c, left panel, ratios of CBR:CBG bioluminescence signals in RPE1 cells expressing the NMD reporter (hereafter referred to as RPE1 reporter cells for simplicity) after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of four independent experiments. ***, p ≤ 0.001 (paired t test). Right panel, ratios of CBR-TCR(PTC):CBG-TCR(WT) mRNA expression in RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of four independent experiments. **, p ≤ 0.01 (paired t test). d, ratios of CBR:CBG bioluminescence signals (top panel) and γH2AX levels (bottom panel) in BJ fibroblasts expressing the NMD reporter after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of three independent experiments. **, p ≤ 0.01 (paired t test). e, ratios of CBR:CBG bioluminescence signals (top panel) and γH2AX levels (bottom panel) in RPE1 reporter cells after 120-h treatment with either DMSO or 60 nm CPT. Data represent the mean ± S.D. of four independent experiments. **, p ≤ 0.01 (paired t test). f, ratios of CBR:CBG bioluminescence signals (top panel) and γH2AX levels (bottom panel) in RPE1 reporter cells 144 h after exposure to 0 Gy, 0.5 Gy, or 10 Gy of ionizing radiation (IR). Data represent the mean ± S.D. of three independent experiments. *, p ≤ 0.05 (paired t test).
To further demonstrate that persistent DNA damage attenuates NMD activity, we used other methods to induce persistent DNA damage and examined NMD efficiency using our bioluminescent reporter. Continuous treatment of RPE1 cells with a low concentration (60 nm) of the topoisomerase I inhibitor camptothecin (CPT) for 5 days also attenuated NMD activity (Fig. 1e). Similarly, exposing cells to high doses (10 Gy) of ionizing radiation 6 days prior to NMD analysis resulted in NMD repression (Fig. 1f). In contrast, a 0.5-Gy dose of ionizing radiation, which initially induced the DDR (data not shown) but did not cause enough damage to persist for 6 days, did not affect NMD activity (Fig. 1f). Taken together, these data strongly suggest that NMD is inhibited in response to persistent DNA damage in non-cycling human cells.
To distinguish between the effects of persistent DDRs and immediate DDRs on NMD, we again treated RPE1 cells with bleomycin for 24 h but assessed NMD activity immediately following the treatment. High levels of γH2AX signal detected by immunofluorescence staining and Western blotting indicated that these conditions generate a strong DDR (Fig. 2a). However, a similar level of NMD activity was detected in bleomycin-treated and H2O-treated (control) cells, indicating that NMD activity is not altered immediately after DNA damage (Fig. 2b). This suggests that DNA damage signals do not immediately influence NMD activity.
Figure 2.

Transient DNA damage does not affect NMD. a, left panel, representative images of immunofluorescence staining for DNA or γH2AX in RPE1 cells after 24-h treatment with either H2O or bleomycin (Bleo). Right panel, Western blot showing γH2AX levels in lysates collected after 24-h treatment with either H2O or bleomycin. b, ratios of CBR:CBG bioluminescence signals in RPE1 reporter cells after 24-h treatment with either H2O or bleomycin. Data represent the mean ± S.D. of three independent experiments. NS, p > 0.05 (paired t test). c, left panel, ratios of CBR:CBG bioluminescence signals (top panel) and γH2AX levels (bottom panel) in RPE1 reporter cells after 1-h treatment with either H2O or bleomycin, followed by 3-h recovery. Data represent the mean ± S.D. of three independent experiments. NS, p > 0.05 (paired t test). Right panel, ratios of CBR:CBG bioluminescence (top panel) and γH2AX levels (bottom panel) signals in RPE1 reporter cells after 1-h treatment with either H2O or bleomycin followed by 120-h recovery. Data represent the mean ± S.D. of three independent experiments. NS, p > 0.05 (paired t test).
We next determined whether a low level of transient DNA damage, which can be readily repaired, exerts a delayed effect on NMD activity or whether DNA damage must persist to induce NMD repression. To this end, RPE1 cells were treated for 1 h with the same dose of bleomycin as above and allowed to recover for 3 h (to detect an immediate response) or 5 days (to detect a delayed response). These conditions generated a robust DNA damage response initially at the 4-h time point, but little or no DNA damage persisted to day 5 (see the γH2AX signal in Fig. 2c). In this setting, no difference in NMD efficiency was observed at either time point (Fig. 2c). Taken together, these data indicate that persistent DNA damage, but not transient DNA damage, induces NMD inhibition.
NMD repression is not a common feature of cellular senescence
Persistent DNA damage induces cellular senescence, which is accompanied by substantial changes in cellular morphology, metabolism, and gene expression (15). It is possible that the NMD attenuation caused by persistent DNA damage stems from the establishment of a senescent state rather than directly from DNA damage signaling. Results of senescence-associated (SA) β-gal staining indicates that our treatment conditions used to generate persistent DNA damage efficiently induced cellular senescence (Fig. 3a). To determine whether NMD inhibition is a facet of the senescence phenotype, even in the absence of DNA damage, we induced senescence by infecting RPE1 cells with lentiviruses expressing the cell cycle inhibitor p16 and then measured NMD activity using our bioluminescent reporter. Like bleomycin treatment, p16 overexpression efficiently induced senescence (Fig. 3b). However, no DDR was induced by p16 overexpression, as indicated by the lack of γH2AX signal in the cells (Fig. 3c). In contrast to cells with persistent DNA damage, no significant changes in NMD activity were detected in p16-expressing senescent cells compared with nonsenescent control cells expressing the empty vector (Fig. 3c). These results suggest that NMD repression is not an obligate consequence of cellular senescence; rather, NMD activity is likely regulated directly by signals emanating from persistent DNA lesions.
Figure 3.
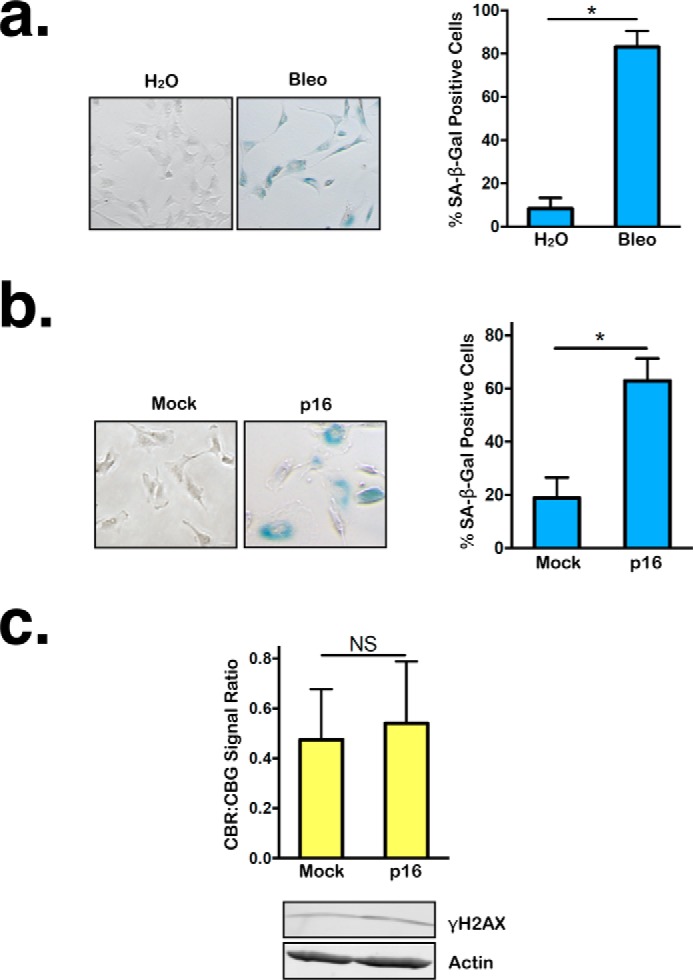
Although p16 overexpression, like persistent DNA damage, can induce cellular senescence, it does not alter NMD activity. a, left panel, representative SA β-gal staining images of RPE1 reporter cells after 24-h treatment with either H2O or bleomycin (Bleo), followed by 96-h recovery. Right panel, percentage of SA β-gal–positive RPE1 cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of three independent experiments. *, p ≤ 0.05 (paired t test). b, left panel, representative images of RPE1 reporter cells expressing either empty vector or p16 after SA β-gal staining. Right panel, percentage of SA β-gal–positive RPE1 cells expressing either empty vector or p16. Data represent the mean ± S.D. of three independent experiments. *, p ≤ 0.05 (paired t test). c, top panel, ratios of CBR:CBG bioluminescence signals in RPE1 reporter cells expressing either empty vector or p16. Data represent the mean ± S.D. of three independent experiments. NS, p > 0.05 (paired t test). Bottom panel, Western blot showing γH2AX levels in cells overexpressing either empty vector or p16.
Persistent DNA damage inhibits NMD in a p38α-dependent manner
The MAPK p38 is a key player in the cellular response to persistent DNA damage (23). Consistent with previous findings, bleomycin-treated RPE1 cells exhibited robust p38 activation and signaling, as evidenced by the increase in p38 phosphorylation at Thr-180/Tyr-182 and phosphorylation of HSP27 at Ser-82, a downstream target of p38 (Fig. 4a) (17, 55). To test the possibility that p38 mediates NMD suppression in cells with persistent DNA damage, we depleted p38α, a ubiquitously expressed and major isoform of p38, in RPE1 cells using a previously validated shRNA (Fig. 4b) (17, 56). p38α knockdown significantly restored NMD activity in the presence of persistent DNA damage, albeit partially (Fig. 4c). In further support of the role of p38α as a mediator of NMD suppression, CDD111 (also known as SD0006), a p38α-specific kinase inhibitor that prevents downstream p38 signaling but not phosphorylation of p38 itself, partially restored NMD activity in the presence of persistent DNA damage (Fig. 4d) (57). Importantly, disruption of p38α activity in the absence of DNA damage did not significantly alter NMD activity (Fig. 4, c and d). These results indicate that p38α is a novel regulator of NMD in response to persistent DNA damage. In addition, the requirement of p38α for NMD suppression further demonstrates that the effects of persistent DNA damage on NMD are not simply caused by an increase in the production of mutant mRNAs in the damaged cells that may overwhelm the NMD machinery.
Figure 4.

NMD inhibition by persistent DNA damage is mediated in part by p38α. a, levels of total p38α and phosphorylated p38 and HSP27 in RPE1 cells treated with either H2O or bleomycin (Bleo) for 24 h, followed by 96-h recovery. b, shRNA-mediated knockdown of p38α in RPE1 cells. c, ratios of CBR:CBG bioluminescence signals in control and p38α knockdown RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of four independent experiments. **, p ≤ 0.01 (paired t test). d, ratios of CBR:CBG bioluminescence signals in DMSO- or CDD111-treated RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Data represent the mean ± S.D. of three independent experiments. *, p < 0.5; **, p ≤ 0.01 (paired t test).
p38 activation is not sufficient to inhibit NMD
It has been shown that p38 activation is sufficient to induce certain aspects of the persistent DNA damage response, such as expression and maintenance of several SASP factors (17, 31). To determine whether p38 activation is also sufficient to attenuate NMD, we expressed a constitutively active version of MKK6 (MKK6-CA), an upstream kinase that directly phosphorylates and activates p38 (including p38α), in RPE1 cells and then assessed NMD activity via reporter imaging. Cells were infected with adenoviruses expressing either LacZ (control) or MKK6-CA and incubated for 7 days to induce an extended period of p38 activation that mimics the prolonged p38 activation in cells harboring persistent DNA damage. MKK6-CA expression induced a level of p38 activation comparable with that induced by bleomycin treatment; however, it failed to alter NMD activity (Fig. 5, a–c). These data suggest that p38 activation alone is not sufficient to inhibit NMD and that a second signal induced by persistent DNA damage is needed to work in concert with p38 to suppress NMD.
Figure 5.

p38 activation is not sufficient to inhibit NMD. a, levels of MKK6, both total p38α and phosphorylated p38 and HSP27 in RPE1 cells expressing LacZ or constitutively active MKK6-CA for 7 days. b, comparison of the levels of total p38α and phosphorylated p38 in RPE1 cells overexpressing either LacZ or MKK6-CA for 7 days with that in RPE cells treated with either H2O or bleomycin (Bleo) for 24 h, followed by 96-h recovery. c, ratios of CBR:CBG bioluminescence signals in RPE1 reporter cells expressing either LacZ or MKK6-CA for 7 days. Data represent the mean ± S.D. of three independent experiments. NS, p > 0.05 (paired t test).
ATF3 mRNA is stabilized by persistent DNA damage in a p38α-dependent manner
The stress-induced transcription factor ATF3 is an NMD target and is up-regulated in cells in response to persistent DNA damage (39, 44, 58). The observed inhibitory effects of persistent DNA damage on NMD activity lead us to predict that ATF3 (and likely many other NMD targets) will be stabilized under this condition. To test whether this is the case for ATF3 mRNAs, we generated persistent DNA damage in RPE1 cells with bleomycin and used real-time qPCR to determine what percentage of mRNAs remain undegraded at different time points after treatment with actinomycin D, which prevents new RNA synthesis. Consistent with ATF3 mRNAs being targets of NMD, ATF3 transcripts exhibited a dramatic increase in stability and steady-state expression levels in bleomycin-treated cells, which have low levels of NMD activity, compared with H2O-treated cells, which have normal NMD activity (Fig. 6a). No statistically significant stabilization was observed for ORCL mRNA, which is not a NMD target, after bleomycin treatment (Fig. 6a). Furthermore, we found that the elevated expression and stabilization of ATF3 mRNAs in response to persistent DNA damage are partially dependent on p38α, which mediates NMD inhibition. Knockdown of p38α using an shRNA or inhibition of p38α kinase activity using CDD111 partially reversed the bleomycin-dependent stabilization (6 h after actinomycin D treatment) and up-regulation of ATF3 transcripts (Fig. 6, b–d). In contrast, ORCL mRNA remained unaffected by these treatments (Fig. 6, b and d). These data suggest that p38α inhibits ATF3 mRNA degradation, at least in part, through NMD attenuation in response to persistent DNA damage, thereby augmenting its expression.
Figure 6.

NMD suppression by p38α in response to persistent DNA damage contributes to ATF3 mRNA stabilization. a, left panel, percentage of ATF3 and ORCL mRNAs remaining in RPE1 reporter cells after 24-h treatment with either H2O or bleomycin (Bleo), followed by 96-h recovery. Following the recovery period, actinomycin D was added, and total RNA was collected at the indicated times, followed by quantitative RT-PCR analysis. The difference in ATF3 stability between H2O- or bleomycin-treated cells is statistically significant (p ≤ 0.01, paired t test) for each time point. No significant stabilization of ORCL mRNA was observed between H2O- or bleomycin-treated cells. Data represent the mean ± S.D. of three independent experiments. Right panel, steady-state levels of ATF3 transcripts in RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Expression in the H2O-treated cells was normalized to 1. Data represent the mean ± S.D. of five independent experiments. ***, p ≤ 0.001 (paired t test). b, left panel, percentage of ATF3 and ORCL mRNAs remaining in DMSO- and CDD111-treated RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery and then actinomycin D treatment for 6 h. Samples were collected immediately before or after 6-h treatment with actinomycin D. Right panel, steady-state levels of ATF3 transcripts in DMSO- and CDD111-treated RPE1 cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Expression in the H2O-treated cells was normalized to 1. Data represent the mean ± S.D. of five independent experiments. **, p ≤ 0.01; ***, p ≤ 0.001 (paired t test). NS, p > 0.05. c, shRNA-mediated knockdown of p38α in RPE1 cells. d, left panel, percentage of ATF3 and ORCL mRNAs remaining in control and p38α knockdown RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Samples were collected as in b. Right panel, steady-state levels of ATF3 transcripts in control and p38α knockdown-treated RPE1 reporter cells after 24-h treatment with either H2O or bleomycin, followed by 96-h recovery. Expression in the H2O-treated cells was normalized to 1. Data represent the mean ± S.D. of four independent experiments. *, p ≤ 0.05; **, p ≤ 0.01 (paired t test). NS, p > 0.05. e, shRNA-mediated knockdown of SMG1 in RPE1 cells. f, percentage of ATF3 and ORCL mRNAs remaining in control- and SMG1 knockdown RPE1 cells that were treated with either H2O or bleomycin for 24 h, followed by 96-h recovery. Actinomycin D was added to cells to suppress transcription, and total RNA was collected immediately before or 6 h after addition of actinomycin D, followed by quantitative RT-PCR analysis. Data represent the mean ± S.D. of four independent experiments. *, p ≤ 0.05; NS, p > 0.05 (paired t test). g, model for the regulation of NMD activity by p38α in response to persistent DNA damage that leads to the stabilization of ATF3 mRNAs and subsequent changes in transcription and gene expression.
NMD attenuation by persistent DNA damage contributes to the stabilization of ATF3 transcripts
To further demonstrate that NMD inhibition contributes to ATF3 mRNA stabilization in response to persistent DNA damage, we knocked down the essential NMD factor SMG1 in RPE1 cells and assessed the stability of ATF3 transcripts after bleomycin treatment. Consistent with ATF3 being an NMD target, SMG1 knockdown stabilized ATF3 transcripts in the absence of DNA damage (Fig. 6, e and f). If NMD inhibition contributes to ATF3 stabilization after persistent DNA damage, then one would expect that ATF3 transcripts will exhibit a similar level of stability in control and SMG1 knockdown cells after bleomycin treatment because NMD is also inhibited in control knockdown cells with persistent DNA damage. However, if persistent DNA damage stabilizes ATF3 mRNA independently of NMD inhibition, then SMG1 knockdown cells will show a higher level of stability of ATF3 transcripts after persistent DNA damage, as the transcripts will be stabilized both by attenuated NMD activity (because of SMG1 knockdown) and the NMD-independent mechanism. The result shown in Fig. 6f indicates that SMG1 knockdown did not cause a further increase in ATF3 mRNA stability after bleomycin treatment compared with control knockdown cells, reinforcing the idea that NMD inhibition by persistent DNA damage contributes to the stabilization of ATF3 transcripts. However, compared with the effects of SMG1 knockdown, bleomycin treatment induced a higher level of stabilization of ATF3 mRNAs (Fig. 6f), suggesting that additional mechanisms exist to further stabilize ATF3 transcripts after persistent DNA damage (see “Discussion”). Taken together, the data described above strongly suggest that NMD attenuation contributes to ATF3 up-regulation, via p38 activation, in response to persistent DNA damage (Fig. 6g).
Discussion
In this study, we found that persistent DNA damage, but not transient DNA damage, induces NMD repression and that this repression contributes to the stabilization of the mRNA of the transcription factor ATF3. In addition, we found that the inhibition of NMD by persistent DNA damage requires p38α MAPK but is independent of cellular senescence.
Our finding of NMD regulation by persistent DNA damage expands our understanding of the persistent DNA damage response and the physiological role of the NMD pathway. In addition to degrading mutant nonsense mRNAs, NMD controls the expression of many normal transcripts. By demonstrating the suppression of NMD activity in the presence of persistent DNA damage, we illuminate another mechanism for the reprogramming of gene expression in the persistent DNA damage response. Our results strongly suggest that persistent DNA damage reduces NMD activity toward ATF3 transcripts, thereby increasing their stability. As a transcription factor, ATF3 regulates the cellular responses to multiple forms of stress, including DNA damage, by controlling the expression of many genes (59). Thus, the up-regulation of ATF3 caused by NMD inhibition is expected to contribute to expression changes for many other genes in response to persistent DNA damage, which may directly promote the survival and adaptation of damaged cells. This regulation of NMD and ATF3 by persistent DNA damage may also have important relevance to tumorigenesis and cancer treatment. The mainstays of cancer treatment are radiation and chemotherapy, both of which generate DNA damage. However, the efficacy of DNA-damaging therapies is hampered by frequent cancer relapse, which is in part caused by the remodeling of the tumor microenvironment following treatment (16, 18, 20, 60). The gene expression changes in non-cycling stromal cells caused by therapy-induced persistent DNA damage promote cellular senescence and the SASP, both of which play a crucial role in shaping the tumor microenvironment (2, 60). Stabilization of ATF3 transcripts because of NMD attenuation under this condition may promote expression of SASP factors that are target genes of ATF3 (50). A number of key SASP factors, such as IL-6 and CXCL2, have been suggested to be direct targets of NMD (2, 44, 45). Given that many of these SASP factors are pro-tumorigenic, restoring NMD activity in stromal cells in the tumor microenvironment could be beneficial during cancer treatment (15).
Our study has also identified a novel role for p38α in regulating NMD activity. As a key player in the persistent DNA damage response, p38 controls senescence induction and SASP expression in part by regulating gene expression (23, 61, 62). p38 induces gene expression changes by altering the activities of transcription factors and RNA binding proteins to regulate transcription, mRNA stability, and translation (23). The suppression of NMD by p38α identified in this study is a new p38-mediated mechanism to regulate mRNA stability (Figs. 4 and 6). In addition to NMD, p38α also controls the stability of many mRNAs after persistent DNA damage by reducing the occupancy of the RNA-binding protein AUF1 on their 3′ UTR, which normally promotes their degradation (17). Interestingly, it has been shown that AUF1 also binds to the 3′ UTR of ATF3 mRNA and that this AUF1 binding is reduced in response to amino acid deprivation, leading to stabilization of ATF3 transcripts (63). It will be interesting to determine whether the p38–AUF1 axis also plays a role in ATF3 mRNA stabilization in the persistent DNA damage response. The combined effects on transcriptional activation and mRNA stabilization of ATF3 mRNA lead to up-regulation of ATF3 and downstream gene expression alterations after persistent DNA damage. Although p38α is required for NMD attenuation after persistent DNA damage, p38α activation alone is not sufficient to suppress NMD (Fig. 5). Presumably, p38α functions in concert with another signal generated by persistent DNA damage to control NMD activity. Further dissection of the mechanism of NMD regulation by p38α and other factors will provide crucial insights into the reprogramming of gene expression in the persistent DNA damage response, which is directly relevant to the understanding and treatment of cancer.
Experimental procedures
Cell culture, adenovirus and lentivirus production, and infection
RPE1 cells were maintained in DMEM nutrient mixture F-12 Ham (Sigma, D6421) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, 2.5 mm l-glutamine, and 7.5% FBS and grown in a 5% CO2 incubator at 37 °C. Human foreskin BJ fibroblasts were maintained in 70% DMEM (Sigma, D5796), 15% medium 199 (Sigma, M7528), and 15% FBS supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin and grown in a 5% CO2 incubator at 37 °C. Human HEK293T and HEK293 cells were cultured in DMEM (Sigma, D5796) with 10% FBS at 37 °C with 5% CO2.
An adenoviral construct encoding the bioluminescent NMD reporter was generated by inserting the reporter into the pAdenoX-PRLS-ZsGreen1 vector using the In-Fusion HD cloning kit (Clontech) according to the protocol of the manufacturer. A linearized adenoviral vector with exposed inverted terminal repeats was transfected into HEK293 cells for viral production, followed by viral amplification in the same cell line. Target cells were infected with adenoviruses for 24 h before bioluminescence imaging.
To knock down p38α or SMG1 in RPE1 cells, lentiviruses expressing a non-targeting control shRNA (5′-CAACAAGAUGAAGAGCACCAA-3′), an shRNA targeting p38α (5′-GUUACGUGUGGCAGUGAAGAA-3′), or an shRNA targeting SMG1 (5′-GCCGAGAUGUUG AUCCGAAUA-3′) were generated in HEK293T cells as described previously (54). Briefly, HEK293T cells were co-transfected with an shRNA-encoding lentiviral vector and packaging plasmids (pCMV-dR8.2 and pCMV-VSVG) using TransIT-LT1 transfection reagent (Mirus). Virus-containing supernatant was collected 48 and 72 h after transfection. Filtered viruses were used to infect RPE1 cells, followed by puromycin selection.
To generate recombinant retroviruses expressing the CDK inhibitor p16, HEK293T cells were co-transfected with either pBabe-puro or pBabe-puro-p16 and packaging plasmids (pCMV-UMVC and pCMV-VSVG) using TransIT-LT1 transfection reagent. Virus-containing supernatant was collected 48 and 72 h after transfection. RPE1 cells were then infected with the viruses, followed by puromycin selection.
For the MKK6 experiments, RPE1 cells were plated at a density of 150,000 cells/well in a 6-well plate and allowed to grow for 48 h before addition of either LacZ or MKK6-CA adenovirus (gifts from Dr. Tatiana Efimova) at 1:1000 dilution. The virus was removed after 24 h, and cells were allowed to recover for 6 days before NMD analysis.
Cell plating conditions and induction of persistent DNA damage
Bleomycin treatment
For all 24-h bleomycin treatments, RPE1 cells or BJ fibroblast cells were plated at a density of 150,000 cells/well in 6-well plates and allowed to grow to confluency for 72 h before addition of bleomycin. For 1-h bleomycin treatments, cells were plated at a density of 100,000 cells/well in 6-well plates and allowed to grow for 48 h before beginning treatment. Bleomycin (B8416, Sigma) was dissolved in water at 15 units/ml (9.4 mg/ml) and diluted in medium 1:149.25 to a final concentration of ∼63 μg/ml for treatment. CDD111 (10 mm stock solutions in DMSO) was diluted 1:1000 in medium to a final concentration of 10 μm for treatment. Medium containing DMSO or CDD111 was refreshed daily.
CPT treatment
RPE1 cells were plated at a density of 50,000 cells/well in 6-well plates and cultured for 72 h before beginning treatment. CPT (60 μm in DMSO) was diluted 1:1000 in medium to a final concentration of 60 nm for treatment. Medium containing DMSO or CPT was refreshed daily.
Irradiation
RPE1 cells were plated at a density of 150,000 cells/well in 6-well plates and allowed to grow to confluency for 72 h before irradiation. Cells were irradiated with doses of either 0.5 Gy or 10 Gy at 0.625 Gy/min using a Gammacell 40 irradiator.
Reporter adenoviral transduction
For NMD analysis, the NMD reporter was introduced into cells via adenoviral infection. Adenoviruses expressing the NMD reporter were added to cell growth medium 24 h prior to bioluminescence imaging.
Bioluminescence imaging and spectral deconvolution for signal unmixing
Cells were incubated with 150 μg/ml D-luciferin for 10 min at 37 °C, and bioluminescence signals were measured using a charge-coupled device camera-based bioluminescence imaging system (IVIS 50, Caliper) with appropriate open, red, or green filters and exposure settings (exposure time: 30 s, 60 s, 60 s; binning: 8; field of view: 15; f/stop: 1). Regions of interest were drawn over images of wells, and bioluminescence signals were quantified using the Living Image (Caliper) and Igor (Wavemetrics) analysis software packages as described previously (54, 64). Spectral unmixing was performed using a previously developed ImageJ plugin as described previously (54, 64). Detailed original results are available upon request.
Reverse transcription, qPCR, and Western blotting
Total RNA was isolated using the NucleoSpin RNA kit from Clontech (740955), and cDNA was synthesized using the PrimeScript RT reagent kit from Clontech (RR037A) according to the instructions of the manufacturer. qPCR reactions were performed in triplicate using a two-step PCR protocol (melting temperature, 95 °C; annealing and extension temperature, 60 °C; cycle number, 40) on an ABI VII7 real-time PCR system with PowerUp SYBR Green Master Mix (Thermo Scientific). The mRNA levels of the housekeeping gene GAPDH were used for normalization. Primers for ATF3 (forward, 5′-GCCATTGGAGAGCTGTCTTC-3′; reverse, 5′-GGG CCATCTGGAACATAAGA-3′), ORCL (forward, 5′-GGCAGCAGATGAAATCTGAA-3′; reverse, 5′-TCCAGAATGTGATTTTTGCAG-3′), and GAPDH (forward, 5′-AACAGCCTCAAGATCATCAGC-3′; reverse, 5′-GATGATGTTCTGGAGAGCC-3′) were purchased from Integrated DNA Technologies. Detailed original results are available upon request.
Western blotting was performed using the Li-Cor Odyssey system. Briefly, cells were lysed with SDS sample buffer (90 mm Tris, 20% glycerol, 2% SDS, and 5% β-mercaptoethanol). Samples were run on SDS-PAGE gel and transferred to a PDVF membrane. Membranes were blocked in casein buffer and subsequently probed with primary antibody diluted in casein buffer. Antibodies against γH2AX (9718, 1:1000), p38α (9218, 1:1000), phospho-p38 (4511, 1:1000), HSP27 (2402, 1:1500), phospho-HSP27 (9709, 1:1000), MKK6 (9264, 1:1000), and SMG1 (9149, 1:1000) were purchased from Cell Signaling Technology. Ku70 (MMS-263R) and β-Actin (MA5-15739, 1:5000) antibodies were purchased from Thermo Scientific and Covance, respectively.
Author contributions
Z. Y. designed and supervised the study. A. N., A. C., and Z. Y. performed the experiments and analyzed the data. K. C. F., S. A. S., and D. P. W. provided critical reagents and technical assistance. A. N., A. C., and Z. Y. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Lynne Maquat (University of Rochester), Dr. Tatiana Efimova (George Washington University), and Dr. Robert Weiss (Cornell University) for providing reagents and critical discussions and Shankar Parajuli for technical assistance.
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- SASP
- senescence-associated secretory phenotype
- PTC
- premature translation termination codon
- NMD
- nonsense-mediated RNA decay
- DDR
- DNA damage response
- CBG
- click beetle green
- CBR
- click beetle red
- TCR
- T-cell receptor
- qPCR
- quantitative PCR
- CPT
- camptothecin
- SA
- senescence-associated
- CA
- constitutively active.
References
- 1. Rossiello F., Herbig U., Longhese M. P., Fumagalli M., and d'Adda di Fagagna F. (2014) Irreparable telomeric DNA damage and persistent DDR signalling as a shared causative mechanism of cellular senescence and ageing. Curr. Opin. Genet. Dev. 26, 89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Davalos A. R., Coppe J. P., Campisi J., and Desprez P. Y. (2010) Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 29, 273–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rodier F., Coppé J. P., Patil C. K., Hoeijmakers W. A., Muñoz D. P., Raza S. R., Freund A., Campeau E., Davalos A. R., and Campisi J. (2009) Persistent DNA damage signaling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fumagalli M., Rossiello F., Clerici M., Barozzi S., Cittaro D., Kaplunov J. M., Bucci G., Dobreva M., Matti V., Beausejour C. M., Herbig U., Longhese M. P., and d'Adda di Fagagna F. (2012) Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation. Nat. Cell Biol. 14, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Herbig U., Jobling W. A., Chen B. P., Chen D. J., and Sedivy J. M. (2004) Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol. Cell 14, 501–513 [DOI] [PubMed] [Google Scholar]
- 6. Hewitt G., Jurk D., Marques F. D., Correia-Melo C., Hardy T., Gackowska A., Anderson R., Taschuk M., Mann J., and Passos J. F. (2012) Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 3, 708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bartkova J., Rezaei N., Liontos M., Karakaidos P., Kletsas D., Issaeva N., Vassiliou L. V., Kolettas E., Niforou K., Zoumpourlis V. C., Takaoka M., Nakagawa H., Tort F., Fugger K., Johansson F., et al. (2006) Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 [DOI] [PubMed] [Google Scholar]
- 8. Chen Y., Chen P. L., Chen C. F., Jiang X., and Riley D. J. (2008) Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 7, 3194–3201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Di Micco R., Fumagalli M., Cicalese A., Piccinin S., Gasparini P., Luise C., Schurra C., Garre' M., Nuciforo P. G., Bensimon A., Maestro R., Pelicci P. G., and d'Adda di Fagagna F. (2006) Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642 [DOI] [PubMed] [Google Scholar]
- 10. Martín M., Terradas M., Iliakis G., Tusell L., and Genescà A. (2009) Breaks invisible to the DNA damage response machinery accumulate in ATM-deficient cells. Genes Chromosomes Cancer 48, 745–759 [DOI] [PubMed] [Google Scholar]
- 11. Martin N. T., Nakamura K., Paila U., Woo J., Brown C., Wright J. A., Teraoka S. N., Haghayegh S., McCurdy D., Schneider M., Hu H., Quinlan A. R., Gatti R. A., and Concannon P. (2014) Homozygous mutation of MTPAP causes cellular radiosensitivity and persistent DNA double-strand breaks. Cell Death Dis. 5, e1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu M., Hales B. F., and Robaire B. (2014) Effects of four chemotherapeutic agents, bleomycin, etoposide, cisplatin, and cyclophosphamide, on DNA damage and telomeres in a mouse spermatogonial cell line. Biol. Reprod. 90, 72. [DOI] [PubMed] [Google Scholar]
- 13. Minieri V., Saviozzi S., Gambarotta G., Lo Iacono M., Accomasso L., Cibrario Rocchietti E., Gallina C., Turinetto V., and Giachino C. (2015) Persistent DNA damage-induced premature senescence alters the functional features of human bone marrow mesenchymal stem cells. J. Cell Mol. Med. 19, 734–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Soubeyrand S., Pope L., and Haché R. J. (2010) Topoisomerase IIα-dependent induction of a persistent DNA damage response in response to transient etoposide exposure. Mol. Oncol. 4, 38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Muñoz-Espín D., and Serrano M. (2014) Cellular senescence: from physiology to pathology. Nat. Rev. Mol. Cell Biol. 15, 482–496 [DOI] [PubMed] [Google Scholar]
- 16. Canino C., Mori F., Cambria A., Diamantini A., Germoni S., Alessandrini G., Borsellino G., Galati R., Battistini L., Blandino R., Facciolo F, Citro G., Strano S., Muti P., Blandino G., and Cioce M. (2012) SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 31, 3148–3163 [DOI] [PubMed] [Google Scholar]
- 17. Alspach E., Flanagan K. C., Luo X., Ruhland M. K., Huang H., Pazolli E., Donlin M. J., Marsh T., Piwnica-Worms D., Monahan J., Novack D. V., McAllister S. S., and Stewart S. A. (2014) p38MAPK plays a crucial role in stromal-mediated tumorigenesis. Cancer Discov. 4, 716–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bent E. H., Gilbert L. A., and Hemann M. T. (2016) A senescence secretory switch mediated by PI3K/AKT/mTOR activation controls chemoprotective endothelial secretory responses. Genes Dev. 30, 1811–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Coppé J. P., Patil C. K., Rodier F., Sun Y., Muñoz D. P., Goldstein J., Nelson P. S., Desprez P. Y., and Campisi J. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gilbert L. A., and Hemann M. T. (2010) DNA damage-mediated induction of a chemoresistant niche. Cell 143, 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krtolica A., Parrinello S., Lockett S., Desprez P. Y., and Campisi J. (2001) Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. U.S.A. 98, 12072–12077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu D., and Hornsby P. J. (2007) Senescent human fibroblasts increase the early growth of xenograft tumors via matrix metalloproteinase. Cancer Res. 67, 3117–3126 [DOI] [PubMed] [Google Scholar]
- 23. Cuadrado A., and Nebreda A. R. (2010) Mechanisms and functions of p38 signalling. Biochem. J. 429, 403–417 [DOI] [PubMed] [Google Scholar]
- 24. Deng Q., Liao R., Wu B. L., and Sun P. (2004) High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J. Biol. Chem. 279, 1050–1059 [DOI] [PubMed] [Google Scholar]
- 25. Harada G., Neng Q., Fujiki T., and Katakura Y. (2014) Molecular mechanisms for the p38-induced cellular senescence in normal human fibroblast. J. Biochem. 156, 283–290 [DOI] [PubMed] [Google Scholar]
- 26. Iwasa H., Han J., and Ishikawa F. (2003) Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 8, 131–144 [DOI] [PubMed] [Google Scholar]
- 27. Kwong J., Hong L., Liao R., Deng Q., Han J., and Sun P. (2009) π38α and p38γ mediate oncogenic ras-induced senescence through differential mechanisms. J. Biol. Chem. 284, 11237–11246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lanna A., Henson S. M., Escors D., and Akbar A. N. (2014) AMPK-TAB1 activated p38 drives human T cell senescence. Nat. Immunol. 15, 965–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spallarossa P., Altieri P., Barisione C., Passalacqua M., Aloi C., Fugazza G., Frassoni F., Podestà M., Canepa M., Ghigliotti G., and Brunelli C. (2010) p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PLoS ONE 5, e15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Alimbetov D., Davis T., Brook A. J., Cox L. S., Faragher R. G., Nurgozhin T., Zhumadilov Z., and Kipling D. (2016) Suppression of the senescence-associated secretory phenotype (SASP) in human fibroblasts using small molecule inhibitors of p38 MAP kinase and MK2. Biogerontology 17, 305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Freund A., Patil C. K., and Campisi J. (2011) p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 30, 1536–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kervestin S., and Jacobson A. (2012) NMD: a multifaceted response to premature translational termination. Nat. Rev. Mol. Cell Biol. 13, 700–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schweingruber C., Rufener S. C., Zünd D., Yamashita A., and Mühlemann O. (2013) Nonsense-mediated mRNA decay: mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim. Biophys. Acta 1829, 612–623 [DOI] [PubMed] [Google Scholar]
- 34. Hug N., Longman D., and Cáceres J. F. (2016) Mechanism and regulation of the nonsense-mediated decay pathway. Nucleic Acids Res. 44, 1483–1495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lykke-Andersen S., and Jensen T. H. (2015) Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat. Rev. Mol. Cell Biol. 16, 665–677 [DOI] [PubMed] [Google Scholar]
- 36. Bruno I. G., Karam R., Huang L., Bhardwaj A., Lou C. H., Shum E. Y., Song H. W., Corbett M. A., Gifford W. D., Gecz J., Pfaff S. L., and Wilkinson M. F. (2011) Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol. Cell 42, 500–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Colak D., Ji S. J., Porse B. T., and Jaffrey S. R. (2013) Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 153, 1252–1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eom T., Zhang C., Wang H., Lay K., Fak J., Noebels J. L., and Darnell R. B. (2013) NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. eLife 2, e00178, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gardner L. B. (2008) Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol. Cell. Biol. 28, 3729–3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Giorgi C., Yeo G. W., Stone M. E., Katz D. B., Burge C., Turrigiano G., and Moore M. J. (2007) The EJC factor eIF4AIII modulates synaptic strength and neuronal protein expression. Cell 130, 179–191 [DOI] [PubMed] [Google Scholar]
- 41. Gong C., Kim Y. K., Woeller C. F., Tang Y., and Maquat L. E. (2009) SMD and NMD are competitive pathways that contribute to myogenesis: effects on PAX3 and myogenin mRNAs. Genes Dev. 23, 54–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karam R., Lou C. H., Kroeger H., Huang L., Lin J. H., and Wilkinson M. F. (2015) The unfolded protein response is shaped by the NMD pathway. EMBO Rep. 16, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Long A. A., Mahapatra C. T., Woodruff E. A. 3rd, Rohrbough J., Leung H. T., Shino S., An L., Doerge R. W., Metzstein M. M., Pak W. L., and Broadie K. (2010) The nonsense-mediated decay pathway maintains synapse architecture and synaptic vesicle cycle efficacy. J. Cell Sci. 123, 3303–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mendell J. T., Sharifi N. A., Meyers J. L., Martinez-Murillo F., and Dietz H. C. (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat. Genet. 36, 1073–1078 [DOI] [PubMed] [Google Scholar]
- 45. Wang D., Zavadil J., Martin L., Parisi F., Friedman E., Levy D., Harding H., Ron D., and Gardner L. B. (2011) Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol. Cell. Biol. 31, 3670–3680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wong J. J., Ritchie W., Ebner O. A., Selbach M., Wong J. W., Huang Y., Gao D., Pinello N., Gonzalez M., Baidya K., Thoeng A., Khoo T. L., Bailey C. G., Holst J., and Rasko J. E. (2013) Orchestrated intron retention regulates normal granulocyte differentiation. Cell 154, 583–595 [DOI] [PubMed] [Google Scholar]
- 47. Hwang J., and Maquat L. E. (2011) Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr. Opin. Genet. Dev. 21, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Oren Y. S., McClure M. L., Rowe S. M., Sorscher E. J., Bester A. C., Manor M., Kerem E., Rivlin J., Zahdeh F., Mann M., Geiger T., and Kerem B. (2014) The unfolded protein response affects readthrough of premature termination codons. EMBO Mol. Med. 6, 685–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Popp M. W., and Maquat L. E. (2015) Attenuation of nonsense-mediated mRNA decay facilitates the response to chemotherapeutics. Nat. Commun. 6, 6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Buganim Y., Madar S., Rais Y., Pomeraniec L., Harel E., Solomon H., Kalo E., Goldstein I., Brosh R., Haimov O., Avivi C, Polak-Charcon S., Goldfinger N., Barshack I., and Rotter V. (2011) Transcriptional activity of ATF3 in the stromal compartment of tumors promotes cancer progression. Carcinogenesis 32, 1749–1757 [DOI] [PubMed] [Google Scholar]
- 51. Kim K. H., Park B., Rhee D. K., and Pyo S. (2015) Acrylamide induces senescence in macrophages through a process involving ATF3, ROS, p38/JNK, and a telomerase-independent pathway. Chem. Res. Toxicol. 28, 71–86 [DOI] [PubMed] [Google Scholar]
- 52. Lu D., Wolfgang C. D., and Hai T. (2006) Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J. Biol. Chem. 281, 10473–10481 [DOI] [PubMed] [Google Scholar]
- 53. Yan C., Lu D., Hai T., and Boyd D. D. (2005) Activating transcription factor 3, a stress sensor, activates p53 by blocking its ubiquitination. EMBO J. 24, 2425–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Nickless A., Jackson E., Marasa J., Nugent P., Mercer R. W., Piwnica-Worms D., and You Z. (2014) Intracellular calcium regulates nonsense-mediated mRNA decay. Nat. Med. 20, 961–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Guay J., Lambert H., Gingras-Breton G., Lavoie J. N., Huot J., and Landry J. (1997) Regulation of actin filament dynamics by p38 map kinase-mediated phosphorylation of heat shock protein 27. J. Cell Sci. 110, 357–368 [DOI] [PubMed] [Google Scholar]
- 56. Yu Y., Rajapakse A. G., Montani J. P., Yang Z., and Ming X. F. (2014) p38 mitogen-activated protein kinase is involved in arginase-II-mediated eNOS-uncoupling in obesity. Cardiovasc. Diabetol. 13, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Burnette B. L., Selness S., Devraj R., Jungbluth G., Kurumbail R., Stillwell L., Anderson G., Mnich S., Hirsch J., Compton R., De Ciechi P., Hope H., Hepperle M., Keith R. H., Naing W., et al. (2009) SD0006: a potent, selective and orally available inhibitor of p38 kinase. Pharmacology 84, 42–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chang B. D., Swift M. E., Shen M., Fang J., Broude E. V., and Roninson I. B. (2002) Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc. Natl. Acad. Sci. U.S.A. 99, 389–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hai T., Wolfgang C. D., Marsee D. K., Allen A. E., and Sivaprasad U. (1999) ATF3 and stress responses. Gene Expr. 7, 321–335 [PMC free article] [PubMed] [Google Scholar]
- 60. Chen F., Zhuang X., Lin L., Yu P., Wang Y., Shi Y., Hu G., and Sun Y. (2015) New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Reinhardt H. C., Aslanian A. S., Lees J. A., and Yaffe M. B. (2007) p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11, 175–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Morandell S., Reinhardt H. C., Cannell I. G., Kim J. S., Ruf D. M., Mitra T., Couvillon A. D., Jacks T., and Yaffe M. B. (2013) A cre-versible approach identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 5, 868–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Pan Y. X., Chen H., and Kilberg M. S. (2005) Interaction of RNA-binding proteins HuR and AUF1 with the human ATF3 mRNA 3′-untranslated region regulates its amino acid limitation-induced stabilization. J. Biol. Chem. 280, 34609–34616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gammon S. T., Leevy W. M., Gross S., Gokel G. W., and Piwnica-Worms D. (2006) Spectral unmixing of multicolored bioluminescence emitted from heterogeneous biological sources. Anal. Chem. 78, 1520–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]