

Abstract
Ligand recognition has been extensively explored in G protein-coupled A1, A2A, and A2B adenosine receptors but not in the A3 receptor, which is cerebroprotective and cardioprotective. We mutated several residues of the human A3 adenosine receptor within transmembrane domains 3 and 6 and the second extracellular loop, which have been predicted by previous molecular modeling to be involved in the ligand recognition, including His95, Trp243, Leu244, Ser247, Asn250, and Lys152. The N250A mutant receptor lost the ability to bind both radiolabeled agonist and antagonist. The H95A mutation significantly reduced affinity of both agonists and antagonists. In contrast, the K152A (EL2), W243A (6.48), and W243F (6.48) mutations did not significantly affect the agonist binding but decreased antagonist affinity by ~3–38-fold, suggesting that these residues were critical for the high affinity of A3 adenosine receptor antagonists. Activation of phospholipase C by wild type (WT) and mutant receptors was measured. The A3 agonist 2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyladenosine stimulated phosphoinositide turnover in the WT but failed to evoke a response in cells expressing W243A and W243F mutant receptors, in which agonist binding was less sensitive to guanosine 5′-γ-thiotriphosphate than in WT. Thus, although not important for agonist binding, Trp243 was critical for receptor activation. The results were interpreted using a rhodopsin-based model of ligand-A3 receptor interactions.
The physiological effects of extracellular adenosine are mediated by four G protein-coupled receptors (GPCRs),1 i.e. A1, A2A, A2B, and A3 adenosine receptors. The A3 adenosine receptor, which is the most recently identified adenosine receptor subtype (1, 2), is implicated in a variety of important physiological processes (3–5). Activation of A3 adenosine receptors increases the release of inflammatory mediators, such as histamine, from rodent mast cells (6) and inhibits the production of tumor necrosis factor-α (2, 7). The activation of the A3 adenosine receptor is also suggested to be involved in immunosuppression (8) and in the response to ischemia of the brain (9, 10) and heart (11). It is becoming increasingly apparent that agonists or antagonists of A3 adenosine receptors have potential as therapeutic agents for the treatment of ischemic and inflammatory diseases (5, 8).
The development of agonists and antagonists for the A3 receptors has so far been directed by traditional medicinal chemistry. The availability of genetic information promises to facilitate understanding of the drug-receptor interaction leading to the rational design of a potentially therapeutically important class of drugs. Molecular modeling may further rationalize observed interactions between the receptor and a ligand. Previously, models derived for GPCRs based on structural homology with bacteriorhodopsin (12–14) had been helpful in understanding and predicting drug-receptor interactions for a variety of receptors. The high resolution crystal structure of bovine rhodopsin has been determined recently (15), providing a detailed atomic description of a GPCR in an inactive conformation and a solid basis for modeling the structure of other rhodopsin-like GPCRs. Such models can be used to help rationalize many observations made on the relationships between the conserved residues and the functional coupling properties. Conservation of functionally important sequences or residues within a certain receptor family is generally considered as a basis for the molecular mechanism leading to the receptor activation in the various subtypes of this receptor family. However, considering the diversity of ligands for different receptors or for different subtypes of a certain receptor family, it has also been accepted that different receptor subtypes may have quite specific structural and functional characteristics.
The ligand-binding sites on the A1, A2A, and A2B receptors have been characterized previously (16–20) using site-directed mutagenesis. However, the molecular basis for ligand recognition in the A3 adenosine receptor remains largely unknown. Only very recently, we created a “neoceptor” and several constitutively active mutant human A3 adenosine receptors by site-directed mutagenesis (21, 22), which provided new insight into the molecular recognition in the A3 receptor. In order to provide additional insights into ligand-A3 adenosine receptor interactions, site-directed mutagenesis was used to study the role of a number of residues in the transmembrane (TM) domains and the second extracellular loop (EL2, Fig. 1). The present study identified a number of residues essential for high affinity binding of agonist and/or antagonist, as well as the receptor activation process. The results were interpreted with the aid of a model of ligand-A3 receptor interactions based on the high resolution crystal structure of bovine rhodopsin.
Fig. 1. Heptahelical diagram of the human A3 adenosine receptor.

The putative transmembrane domains were modified according to the high resolution rhodopsin model (15). Amino acids mutated in the present study are circled. Residues 286–295 correspond to an extra helical domain in rhodopsin, which is discontinuous from TM7.
EXPERIMENTAL PROCEDURES
Materials
The vector pcDNA3 was obtained from Invitrogen. Human A3 adenosine receptor cDNA was provided by M. Atkinson, A. Townsend-Nicholson, and P. R. Schofield (Garvan Medical Institute, Sydney, Australia) and was subcloned in pcDNA3 as pcDNA3/hA3R. All oligonucleotides used were synthesized by Bioserve Biotechnologies (Laurel, MD). [125I]N6-(4-Amino-3-iodobenzyl)adenosine-5′-N-methyluronamide ([125I]I-AB-MECA; 2000 Ci/mmol) and [3H]8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2.1-i]purin-5-one ([3H]PSB-11) (23) were from Amersham Biosciences; myo-[3H]inositol (20 Ci/mmol) was obtained from American Radiolabeled Chemicals (St. Louis, MO). 2-Chloro-N6-(3-iodobenzyl)adenosine-5′-N-methyluronamide (Cl-IB-MECA) and GTPγS were from Sigma. MRS1898 was synthesized as described previously (24). All the enzymes used in this study were obtained from New England Biolabs (Beverly, MA). QuikChange™ site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). A monoclonal antibody (12CA5) against a hemagglutinin epitope and adenosine deaminase (ADA) were obtained from Roche Diagnostics (Indianapolis, IN), and goat anti-mouse IgG antibody conjugated with horseradish peroxidase was from Sigma.
Numbering Scheme of GPCRs
For sequence alignments of selected regions of A3 adenosine receptors and other GPCRs, the standardized numbering system of van Rhee and Jacobson (25) was used to identify residues in the transmembrane domains (TMs) of various receptors. Each residue is identified by two numbers as follows: the first corresponds to the TM in which it is located; the second indicates its position relative to a particular highly conserved residue in that helix, arbitrarily assigned to 50. For example, His3.37 is the histidine in TM3, located 13 residues before the conserved arginine R3.50; Trp(6.48) corresponds to Trp243.
Molecular Modeling
Briefly, a model of the human A3 receptor was built in homology to the recently published x-ray structure of bovine rhodopsin (15) as described (21) using the Sybyl 6.6 modeling package. The model included the seven TMs (built and minimized individually and then grouped to form a helical bundle by adding one at a time) and the second extracellular loop, EL2 (conformation was initially modeled according to the corresponding domain of rhodopsin including the Cys83–Cys166 disulfide bond). Models of the ligands were constructed using the “Sketch Molecule” module of Sybyl. The ligands were minimized in Sybyl (using MOPAC calculated partial atomic charges) and were rigidly docked into the helical bundle using graphic manipulation coupled to continuous energy monitoring. Manual adjustments of ligand conformation were followed by additional minimization runs of up to 1500 steps, using the Tripos force field with Amber all-atom force parameters until the root mean square value of the conjugate gradient (CG) was <0.1 kcal/mol/Å. A fixed dielectric constant = 4.0 was used throughout these calculations.
Transient Expression of Wild Type (WT) and Mutant Receptors in COS-7 Cells
COS-7 cells (10−6) were grown in 100-mm cell culture dishes containing Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 μmol/ml glutamine. After 24 h, cells were washed with phosphate-buffered saline (with calcium) and then transfected with plasmid DNA (10 μg/dish) using the DEAE-dextran method (26) for 1 h. The cells were then treated with 100 μM chloroquine for 3 h in culture medium and cultured for an additional 48 h at 37 ° C and 5% CO2.
Membrane Preparation
After 48 h of transfection COS-7 cells were washed two times with phosphate-buffered saline (without calcium) and harvested by trypsinization. Harvested cells were homogenized using a Polytron homogenizer and then centrifuged at 16,000 × g for 20 min. The resulting pellet was resuspended in the 50 mM Tris·HCl buffer (pH 8.0) in the presence of 3 units/ml ADA and stored at −80 °C in aliquots. The protein concentration was determined by using the method of Bradford (27).
[125I]I-AB-MECA Binding Assay
For the agonist binding assay (21), each tube contained 50 μl of membrane suspension (8–12 μg of protein), 25μl of [125I]I-AB-MECA (for competition studies, final concentration 1.0 nM), and 25 μl of increasing concentrations of the test ligands in Tris·HCl buffer (50 mM, pH 8.0) containing 10 mM MgCl2, 1 mM EDTA. For saturation analysis of [125I]I-AB-MECA binding, concentrations ranging from 0.1 to 20 nM were used. Kd values of the radioligand were determined for all mutant receptors. Nonspecific binding was determined using 10 μM Cl-IB-MECA in buffer. The mixtures were incubated at 37 °C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburg, MD). Filters were washed three times with 9 ml of ice-cold buffer. Radioactivity was determined in a Beckman 5500B γ-counter.
Binding of the Radiolabeled, Selective Antagonist [3H]PSB-11 to A3 Adenosine Receptors
Membranes (80 μg of protein) were incubated with 8 nM [3H]PSB-11 (23) at 25 °C in a total assay volume of 400 μl for 60 min). Nonspecific binding was measured in the presence of 10 μM Cl-IB-MECA. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandel, Gaithersburg, MD).
Inositol Phosphate Determination
The method used was similar to that of Harden et al. (28). About 24 h after transfection, the cells were harvested by trypsinization and grown in 6-well plates (~106 cells/well; Costar, Cambridge, MA) in Dulbecco’s modified Eagle’s culture medium supplemented with 2 μCi/ml myo-[3H]inositol. After a 24-h labeling period, cells were preincubated in the presence of 3 units/ml ADA for 30 min at 37 °C with 10 mM LiCl and for 20 min at room temperature. The mixtures were swirled to ensure uniformity. Following the addition of the agonist Cl-IB-MECA, the cells were incubated for 30 min at 37 °C and 5% CO2. The supernatants were removed by aspiration, and 750 μl of cold 20 mM formic acid was added to each well. Cell extracts were collected after a 30-min incubation at 4 °C and neutralized with 250 μl of 60 mM NH4OH. The inositol monophosphate fraction was then isolated by anion exchange chromatography (29). The content of each well was applied to a small anion exchange column (AG-1-X8; Bio-Rad) that had been pretreated with 15 ml of 0.1 M formic acid, 3 M ammonium formate, followed by 15 ml of water. The columns were then washed with 15 ml of a solution containing 5 mM sodium borate and 60 mM sodium formate. [3H]Inositol phosphates were eluted twice with 5 ml of 0.1 M formic acid, 0.2 M ammonium formate, and radioactivity was quantified by liquid scintillation counting (LKB Wallace 1215 Rackbeta scintillation counter).
Statistical Analysis
Binding and functional parameters were calculated using the Prism software (GraphPAD, San Diego). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation (30). Data were expressed as means ± S.E.
RESULTS
The A3 receptor cDNAs were initially isolated from rat testis and rat brain cDNA libraries and later from other species (31, 32). Sequence alignments for selected transmembrane domains of four human adenosine receptor subtypes, sheep and rat A3 receptors, and other GPCRs are shown in Fig. 2. The A3 receptor exhibits the lowest degree of identity between species compared with other adenosine receptor subtypes (31, 32). The residues of the human A3 adenosine receptor selected for mutation in this study are shown in boldface type. His95 (3.37) is conserved in A3 receptors from various species including human, sheep and rat, and the corresponding residue in A1 and A2A adenosine receptor (Gln) has been studied (33, 34). Lys152 occurs in the second extracellular loop (EL2). Three residues in TM6 were mutated as follows: Trp243 (6.48), Ser247 (6.52; corresponding to His in human A1 and A2A receptors), and Asn250 (6.55). All of these residues were predicted in molecular modeling (21) to be involved in the ligand recognition in the A3 adenosine receptor. By comparison, we also tested the role of Leu244 (6.49), which was not predicted to be involved in ligand recognition but was located one helical turn below Ser247. Each of these residues was individually replaced with Ala or both Ala and Phe.
Fig. 2. Sequence alignments of selected regions of A3 adenosine receptors and other G protein-coupled receptors.

Residues mutated in this study are shown in boldface type. The standardized numbering system of van Rhee and Jacobson (25) was used to identify residues in the transmembrane domains (TMs) of various receptors. Each residue is identified by two numbers; the first corresponds to the TM in which it is located; the second indicates its position relative to the most conserved residue in that helix, arbitrarily assigned to 50. For example, H3.37 is the histidine in TM3, located 13 residues before the most conserved arginine R3.50; W6.48 corresponds to Trp243.
Ligand Binding Properties of the WT and Mutant Receptors
The affinity of a number of agonists and antagonists belonging to different chemical classes (Fig. 3) were tested in both WT and mutant receptors. As shown in Fig. 4, following the mutation of His95 to Ala, the affinity of the high affinity selective A3 agonist, Cl-IB-MECA, decreased 26-fold. Similar to Cl-IB-MECA, the affinity of other agonists and antagonists with different structures was decreased by the H95A mutation (Table I). The affinity of most test ligands for the H95A mutant receptor was ~10–100-fold lower than that for the WT receptor, suggesting that this residue is important for ligand recognition in the A3 adenosine receptor.
Fig. 3.

Chemical structures of ligands tested in this study.
Fig. 4. Effects of mutations in TM3, TM6, and EL2 on the binding of A3 agonist Cl-IB-MECA.

The agonist radioligand [125I]I-AB-MECA (1.0 nM) was used in the experiment. The data shown are from a representative example out of at least three independent experiments performed in duplicate.
TABLE I.
Affinities of various agonists and antagonists in binding experiments at WT and mutant human A3 adenosine receptors
Binding parameters were measured on COS-7-transfected cells as described under “Experimental Procedures.” The binding affinity of various ligands for different receptors was determined by using the agonist radioligand [125I]I-AB-MECA (1.0 nM). Values represent the mean ± S.E. of at least three independent determinations. NA, no activity. A Ki value could not be determined (ND) for the N250A (6.55) mutant receptor, as this mutant receptor did bind to the agonist radioligand [125I]I-AB-MECA or the antagonist radioligand [3H]PSB-11.
|
Ki (nM)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| WT | H95A (3.37) | K152A (EL2) | W243A (6.48) | W243F (6.48) | L244A (6.49) | S247A (6.52) | N250A (6.55) | H272Ea (7.43) | |
| Agonists | |||||||||
| [125I]I-AB-MECA (Kd) | 1.7 ± 0.4 | 5.7 ± 1.2 | 3.7 ± 1.4 | 1.1 ± 0.3 | 1.6 ± 0.2 | 0.85 ± 0.11 | 1.7 ± 0.3 | NA | ND |
| CADO | 523 ± 116 | 58,200 ± 6500 | 323 ± 107 | 334 ± 67 | 321 ± 14 | ND | 927 ± 353 | ND | 11,600 ± 4200 |
| NECA | 249 ± 72 | 3430 ± 900 | 278 ± 44 | 166 ± 77 | 132 ± 45 | 68 ± 14 | 247 ± 60 | ND | 3200 ± 770 |
| Cl-IB-MECA | 2.3 ± 0.6 | 60 ± 15 | 1.7 ± 0.6 | 2.9 ± 0.5 | 1.9 ± 0.6 | 2.5 ± 0.7 | 2.2 ± 0.2 | ND | 20 ± 4 |
| MRS1898 | 1.4 ± 0.4 | 143 ± 36 | 2.3 ± 0.6 | 2.1 ± 0.6 | 1.2 ± 0.3 | 0.73 ± 0.31 | 1.5 ± 0.5 | ND | 9.9 ± 2.6 |
| DBXRM | 2830 ± 570 | 36,300 ± 11,500 | 6030 ± 2320 | 4390 ± 950 | 2640 ± 530 | 7550 ± 2340 | 3040 ± 570 | ND | ND |
| Antagonists | |||||||||
| CGS15943 | 182 ± 71 | 5130 ± 1140 | 571 ± 124 | 1160 ± 340 | 547 ± 87 | 209 ± 48 | 333 ± 144 | ND | 3260 ± 990 |
| MRS1220 | 1.6 ± 0.3 | 976 ± 119 | 5.4 ± 0.7 | 45 ± 13 | 10.2 ± 3.3 | 0.63 ± 0.14 | 3.1 ± 1.4 | ND | ND |
| VUF8504 | 68 ± 19 | 4000 ± 590 | 575 ± 134 | 2570 ± 690 | 1340 ± 370 | 42 ± 15 | 81 ± 26 | ND | 1690 ± 340 |
| XAC | 142 ± 35 | 9440 ± 3260 | 726 ± 214 | 552 ± 79 | 263 ± 46 | 61 - 18 | 165 - 47 | ND | 20,100 – 4500 |
Data are from Jacobson et al. (21).
The Trp residue (6.48) is conserved among all four subtypes of adenosine receptors and a variety of other GPCRs (Fig. 3) and was proposed to be involved in ligand recognition in human A3 adenosine receptors as predicted by molecular modeling. However, the replacement of this Trp by Ala or Phe did not influence the agonist binding affinity, as determined by saturation experiment using [125I]I-AB-MECA. The displacement of [125I]I-AB-MECA binding by five agonists, adenosine derivatives CADO, Cl-IB-MECA, and NECA; the rigid methanocarba derivative MRS1898 (24); and the xanthine riboside N-methyl-1,3-dibutylxanthine 7-β-D-ribofuronamide, was also not affected by mutation of Trp243. The Kd value for [125I]I-AB-MECA and Ki values for other agonists were summarized in Table I.
In contrast to the agonist binding, the binding affinity of the A3 antagonist MRS1220 was diminished ~30-fold for the W243A mutant receptor (Fig. 5). Similar to MRS1220, other test antagonists also showed a marked decrease of binding affinity following the mutation of the Trp residue (Table I), suggesting the involvement, either directly or indirectly, of this residue in antagonist recognition.
Fig. 5. Effects of the Trp mutation on the binding of A3 antagonist MRS1220.

The agonist radioligand [125I]I-AB-MECA (1.0 nM) was used in the experiment. The data shown are from a representative example of at least three independent experiments performed in duplicate.
Following the substitution of Leu244 or Ser247 with Ala, saturation experiments were carried out using [125I]I-AB-MECA as a radioligand. Competitive binding of various ligands to human WT and mutant A3 adenosine receptors demonstrated that the binding parameters for all the agonists and antagonists were only slightly affected (Table I). In contrast to L244A and S247A mutant receptors, the specific binding of [125I]I-AB-MECA (1.0 nM) to the N250A mutant receptor was not detectable. In order to examine the effects of this mutation on antagonist binding, a radiolabeled A3 antagonist [3H]PSB-11 (8 nM, final concentration) was used in the experiment. Similar to the [125I]I-AB-MECA binding, the mutation also resulted in a complete loss of high affinity [3H]PSB-11 binding, suggesting that Asn250 is essential, either directly or indirectly, for ligand recognition in the human A3 adenosine receptor. The proximity of this residue to the putative ligand binding site was predicted using the molecular modeling.
The Lys residue of EL2 was predicted to be involved in ligand recognition using a molecular model of the A3 adenosine receptor (see below). Following the K152A mutation, the affinity of all the agonists tested was essentially unchanged; however, the antagonist affinity decreased 3–9-fold, suggestive of the possible involvement of this residue in antagonist recognition. The Kd and Ki values were summarized in Table I.
Agonist-induced Phosphoinositide Turnover in COS-7 Cells Expressing WT and Mutant Receptors
The A3 adenosine receptor is coupled to Gi protein, whereas coupling to Gq protein is controversial, because some studies have shown that PLC activation by the A3 adenosine receptor is prevented by pertussis toxin (3, 35). To test the activation of the WT and mutant receptors, we initially attempted to perform a cyclic AMP production assay in transfected COS-7 cells. However, we observed that both NECA and Cl-IB-MECA, at certain concentrations, induced an increase of cyclic AMP production in non-transfected COS-7 cells, suggesting the presence of an endogenous Gs-coupled adenosine receptor, possibly the A2B adenosine receptor. In the same cell line, Cl-IB-MECA had no effect on basal phosphoinositide turnover. Hence, to avoid interference by endogenous receptors, together with the fact that forskolin-stimulated adenylyl cyclase activity was only partly inhibited by A3 receptor agonists as demonstrated elsewhere (36), the PLC assay was used in the determination of functional coupling of the WT and mutant receptors.
Fig. 6 demonstrated that Cl-IB-MECA induced accumulation of inositol phosphates in COS-7 cells expressing WT receptors in a concentration-dependent manner, with an EC50 of 260 ± 49 nM (n = 3). The H95A mutation did not influence the production of inositol phosphates significantly (EC50 = 224 ± 57 nM; n = 3) (p > 0.05 compared with WT). In contrast, no substantial stimulation of phosphoinositide turnover by Cl-IB-MECA was observed in COS-7 cells expressing W243A, W243F, and N250A mutant receptors. In the case of the L244A mutant receptor, Cl-IB-MECA could still stimulate PLC activity but with 36-fold decreased potency (EC50 = 9430 ± 2470 nM; n = 3). No enhancement in basal PLC activity was observed for these mutant receptors.
Fig. 6. Cl-IB-MECA induced phosphoinositide turnover in COS-7 cells expressing WT and mutant human A3 adenosine receptors.
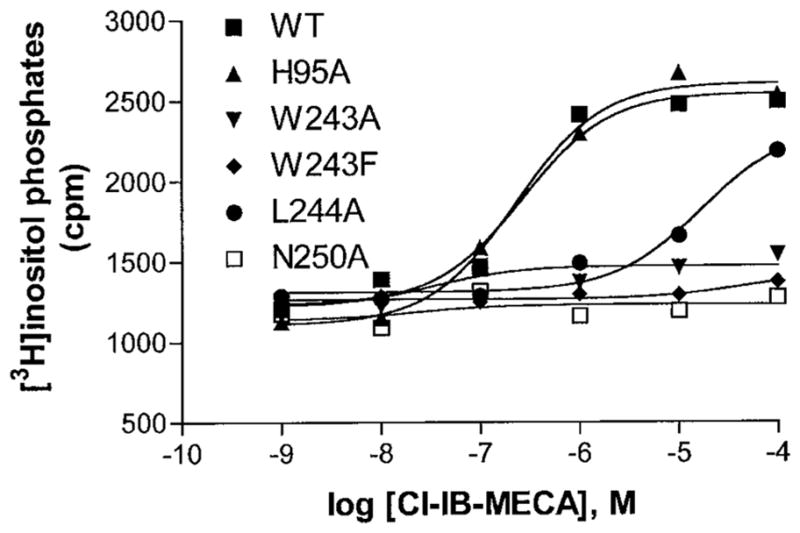
Receptors were transiently expressed in COS-7 cells and used 48 h after transfection. The data shown are from a representative example of three independent experiments.
Effects of a Guanine Nucleotide, GTPγS, on Agonist Binding
To characterize further the interactions between WT and mutant receptors and G proteins, we initially tried to do a membrane binding assay using [35S]GTPγS; however, the signal-to-noise ratio was quite low. Hence, the ability of WT and W243A and W243F mutant receptors to interact with G proteins was further evaluated by measuring the effects of GTPγS on binding of the agonist [125I]I-AB-MECA and on agonist Cl-IB-MECA competition for [3H]PSB-11 binding in membranes of COS-7 cells expressing WT and mutant receptors. As shown in Fig. 7, treatment with GTPγS reduced agonist binding. The inhibitory effect of this GTP analogue on agonist binding to W243A and W243F mutant receptors was less pronounced compared with that of WT receptors, consistent with the impaired ability of these mutant receptors to mediate inositol phosphate production in response to an A3 agonist.
Fig. 7. Effects of GTP analogue on A3 receptor binding.

Shown are the effects of increasing concentrations of GTPγS on binding of the agonist radioligand [125I]I-AB-MECA (1.0 nM) to membranes from COS-7 cells expressing WT and mutant receptors. Results are expressed as percent of binding determined in the absence of GTPγS and are shown as means of values obtained from three experiments performed in duplicate.
A GTP-induced shift of agonist competition for radiolabeled antagonist is another indicator of functional coupling between a receptor and G protein. In this study, we further observed the effect of GTPγS on the competition by agonist Cl-IB-MECA for binding of the antagonist [3H]PSB-11 to WT and mutant receptors. The Ki values for Cl-IB-MECA in the absence and presence 100 μM GTPγS were 2.5 ± 0.4 and 7.4 ± 1.1 nM, respectively, in WT receptors. In W243F mutant receptors, the respective Ki values were 2.3 ± 0.2 and 3.2 ± 0.4 nM. Hence, the GTP analogue induced a 3-fold affinity decrease of Cl-IB-MECA in WT receptors, whereas in the W243F mutant receptor only a 1.4-fold decrease of affinity was observed, consistent with the impaired ability of agonist to activate PLC through this mutant receptor. In the case of the W243A mutant receptor, the agonist affinity shift could not be observed due to the extremely low affinity of this mutant receptor for the antagonist radioligand [3H]PSB-11.
Molecular Modeling
Recently a human A3 receptor model, including the seven transmembrane helical domains (TMs) and EL2, has been constructed (21) in homology to the x-ray structure of bovine rhodopsin (15). The model was used primarily to rationalize the observed effects of the replacement of residue His272 (7.43) by glutamate on the receptor affinity toward A3 agonists and antagonists. Through docking of the A3 agonists and antagonists, we have initially defined the ligand binding environment to include side chains of residues Thr94 (3.36), His95 (3.37), Ser247 (6.52), Gln167 and Lys152 (EL2), and Ser271 (7.42) and Asn274 (7.45), and we suggested differences in the accommodation of agonists bearing different N6 substituents. Thus, although the N6 moiety of CADO was located within H-bonding distance from the amide oxygen of Trp243 and Oγ of Ser247 (2.76 Å and 2.51 Å, respectively), the N6-benzyl substituent of IB-MECA appeared to interact with residues Phe182, Ile186, and Phe187 in TM5. The effect of this bulky N6 substitution on the orientation of the bound agonist was to displace the respective N6 substituent away from the amide oxygen of Trp243. Consequently the 3′-hydroxy substituent of IB-MECA did not seem to interact with His272, whereas such interactions could be observed with the corresponding substituents of CADO or NECA. In addition, the modeled interaction of the agonist N6 moiety with Oγ–Ser247 seemed to orient the adenine ring for aromatic–aromatic interaction with the indole moiety of Trp243. Although this model could be used successfully to formalize the concept of neoceptor with respect to the A3 receptor–agonist interaction (21), further experimental evidence was needed especially for corroborating the putative interactions of the adenine moiety of the agonists.
In the present study ligand accommodation in the A3-binding site was further examined by modeling adducts of the various agonists and antagonists (Table I) with mutant receptors carrying residue replacements at the putative binding site. The results indicated that although the overall positioning of the ligands within the A3 helical bundle was consistent with pharmacological findings, the orientation of the adenine moiety with respect to TM3 and TM6 had to be adjusted. In the modified models of A3–agonist complexes the N6 moiety interacted with Asn250 instead of Ser247, tilting the adenine ring away from the indole moiety of Trp243. This adjustment did not seem to affect the relative positions of the ribose (or the methanocarba (24)) moieties of the different agonists. Fig. 8 shows a molecular model of the human A3 adenosine receptor complex with the nonselective antagonist CGS15943, which is located in proximity to both Trp243 and His95, of which mutation reduced antagonist affinity.
Fig. 8. Molecular model of the nonselective antagonist CGS15943 binding to the human A3 adenosine receptor.
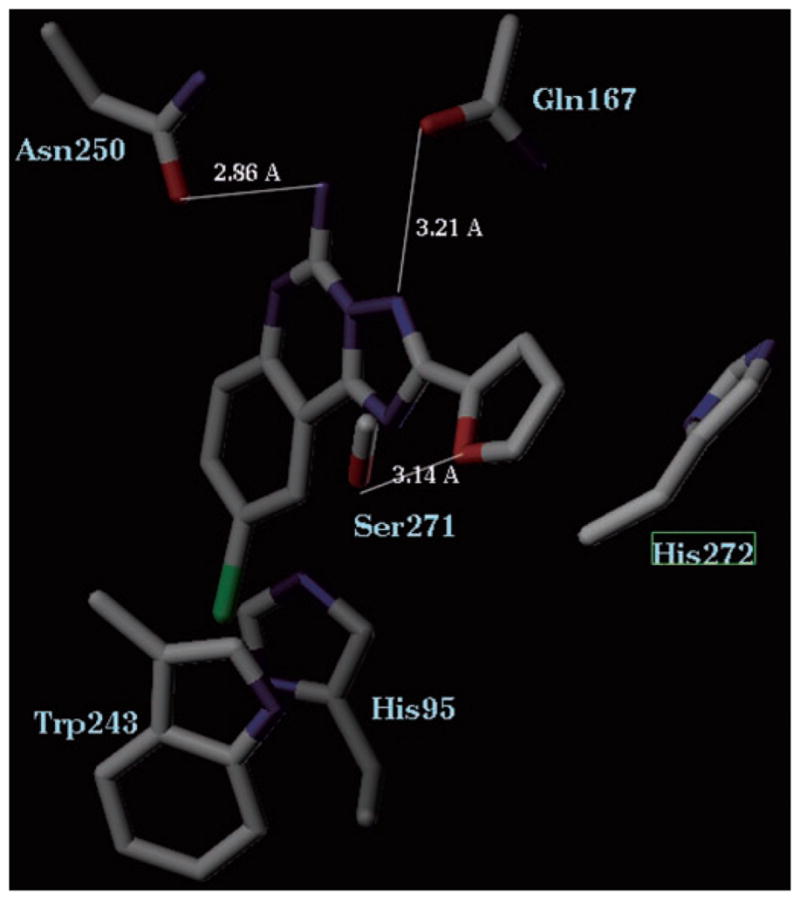
Residues in proximity to this triazoloquinazoline are shown. Blue, nitrogen, red, oxygen, green, chlorine.
The effects of replacement of His95 were consistent with the model with the imidazole moiety adjacent to the 2-position of the agonist adenine ring. Yet, the interaction seemed to depend upon the precise juxtaposition of the ligand, because affinity of the H95A receptor toward I-AB-MECA was only 3-fold lower compared with that of the WT receptor, whereas for NECA the ratio was 14-fold. Although both ligands lacked substitution at the 2-position, I-AB-MECA was shifted toward TM5 (relative to NECA) and therefore its adenine ring was further removed from the residue at position 95. Whereas the 100-fold affinity decrease of the H95A receptor toward CADO could be attributed to interaction with the 2-chloro substituent, comparison of the relative affinities of MRS1898 and MRS1939 did not support such a conclusion (Table II). In addition, the 2-chloro substituent did not seem to contribute to the affinity of either MRS1898 or of Cl-IB-MECA toward the A3 receptor (Table II). Thus, although the model could accommodate small substituents at the 2-position of the adenine ring, the specific role of the 2-chloro substituent, which appeared in many of the A3 agonists, in binding to the receptor remains unclear.
Table II.
Examination of the influence of 2-chloro substitution on the effects of mutangenesis on agonist affinity
| Agonista |
Ki, nM
|
KI mutant/KI WT for 2-H | 2-Cl analogue | |
|---|---|---|---|---|
| WT | H95A | |||
| Adenosine | 111 | |||
| IB-MECA | 4.5 ± 1.6 | 44 ± 12 | 9.8 | 26 |
| MRS1939 | 1.6 ± 0.3 | 147 ± 45 | 92 | 100 |
The 2-H analogue is listed in this column. The corresponding 2-chloro analogues are CADO, Cl-IB-MECA, and MRS 1898, respectively. The binding affinity of adenosine is not measurable directly, due to the need to add ADA.
DISCUSSION
In this study we used a combination of mutagenesis, radio-ligand binding, functional activity (PLC), and molecular modeling approaches in order to identify residues important for ligand recognition in the human A3 adenosine receptor. A change in the binding affinity due to a mutation can be due to an indirect effect on receptor structure and does not necessarily prove that the residue in question directly interacts with the ligand.
The most deleterious of the mutations with respect to binding affinity and functional potency was N250A (6.55), indicating that Asn250 is crucially involved in either human A3 adenosine receptor-ligand recognition or in maintaining receptor structure. This asparagine residue is conserved among all four subtypes of adenosine receptors and a variety of other G protein-coupled receptors. Consistent with the present result, the mutation of the corresponding residue in the human A2A adenosine receptor (N253A) also caused a drastic decrease of the affinity for both agonists and antagonists (18).
Although less critically required for ligand binding, His95 (3, 37) contributed significantly to the binding of most ligands tested in the present study. In A1 adenosine receptors (the residue at the homologous position is Gln), the mutation (Q92A) also demonstrated its critical role in ligand recognition (34). However, in the human A2A adenosine receptor, the mutation of the corresponding residue into Ala (Q89A) increased both agonist and antagonist affinity (33). Similar to the results obtained here, Perlman et al. (37) have demonstrated in a site-directed mutagenesis study that the mutation of the corresponding residue in murine thyrotropin-releasing hormone receptor (N110A) also decreased agonist binding. Similar results have also been reported in a study of human D2 dopamine receptors, in which the antagonist binding affinity was significantly diminished after the mutation of the corresponding residue (T119C) (38).
Interestingly, we have identified a mutant receptor W243A (6.48) that bound agonist strongly but was functionally inactive. The function of this conserved residue has been extensively studied in a variety of G protein-coupled receptors, and it is suggested that this residue plays a subtle role in receptor binding and activation. The importance of this Trp residue in GPCR function was first emphasized by the study of its effects in rhodopsin, in which this residue forms part of the retinal-binding pocket (39). In contrast to the results obtained here, the mutation of the equivalent Trp residue in the mouse δ-opioid receptor (40) impaired the binding of most ligands. In the rat AT1A receptor, the mutation of the homologous residue (W253A) decreased agonist but not antagonist binding (41). In rat m3 muscarinic receptor, the corresponding W503F mutation decreased both agonist and antagonist binding; however, receptor activation was not affected. Consistent with the present study, the mutation of the corresponding residue in the murine thyrotropin-releasing hormone receptor (W279A) did not affect agonist binding (42).
The major consequence of the W243A and W243F mutations was functional inactivation of the A3 receptor, indicating that Trp243 may play a pivotal role in receptor transduction of the signal. The diminished effects of a guanine nucleotide on agonist binding in the Trp243 mutant receptors indicated impairment of the coupling of the receptor to G protein. Activation of GPCRs involves positive heterotropic, long range interactions between agonist- and G protein-binding sites, and several lines of evidence suggested that the movements of TM6 were critical in this process (43–45). Molecular modeling suggested that the Trp243 was in the binding pocket, where it might occupy a strategic position and might act as a switch in TM6-mediated structural transition from the resting to active state. It is possible that the replacement of this residue with Ala or Phe left the switch in the “off ” position, resulting in functional inactivation and uncoupling. The result also suggested that this conserved Trp residue did not play the same role in agonist binding and receptor activation.
A recent study (46) demonstrated that the W256A mutation of the homologous residue in the human B2 bradykinin receptor resulted in a marked decrease of the affinity of nonpeptide antagonists but not agonists, which is in fair agreement with the present study. These results suggested that this conserved residue might be specifically involved in antagonist recognition in some GPCRs. By comparison, mutation of residue Leu244 (at a neighboring position to Trp243) to Ala did not influence the antagonist binding. These insights may prove useful in future efforts to design A3 receptor antagonists by a rational approach.
In the present study, the mutation of the serine residue (S247A) in the TM6 of the human A3 adenosine receptor did not influence agonist binding and only slightly affected antagonist binding, which is consistent with the results from the mutagenesis study of the bovine A1 adenosine receptor. The mutation of the corresponding residue (H251L) diminished antagonist binding ~4-fold, but agonist binding was essentially not affected (47). This result was also consonant with the results of mutation of the corresponding residue (H265A) in the human NK1 receptor, which decreased antagonist binding but did not affect the agonist binding (48). In contrast to these results, the corresponding H250A mutation in the human A2A receptor diminished agonist and antagonist binding as well as receptor activation (18). In rat m3 muscarinic receptors, the binding of agonist and antagonist was also impaired by the homologous N507A mutation (49). Similarly, in the m3 receptor, the agonist binding was decreased after the homologous R283A mutation in the murine thyrotropin-releasing hormone receptor (42). These results suggested that the residue at this position contributed unequally in different receptors. It was also further suggested that different receptor subtypes or different receptors might have quite specific structural and functional characteristics.
It is interesting that the H95A mutation impaired agonist binding but not agonist-induced receptor activation, whereas the W243A mutation impaired functional coupling but did not diminish agonist affinity. The mutation of residue Leu244 (adjacent to Trp243) to Ala resulted in nearly the same agonist binding affinity as that of the WT receptor, but the activation was only partly impaired, also suggesting the involvement in the receptor activation process. Clearly these residues contribute differently to ligand binding and receptor activation processes.
In the modified models of A3–agonist complexes the ligands did not interact directly with residue Trp243. Yet this residue is essential for receptor activation, suggesting that binding of agonist may induce reorientation of its side chain accompanying the receptor transition from an inactive to an active state. Unlike the case of agonists, residue Trp243 seemed to participate in accommodation of antagonists, apparently through aromatic–aromatic interactions. This conclusion was based upon the differential effect on affinity toward antagonists upon replacement of Trp243 with Phe and Ala (Table I). According to the data from mutagenesis and from molecular modeling, the binding sites of A3 agonists and antagonists mostly overlap. Thus, the lack of participation of Trp243 in agonist binding probably resulted from a different size and nature of the adenine ring as compared with the aromatic systems of the antagonists. According to the models, these differences seemed to account also for a better interaction of antagonists with residue His95 and with Lys152.
The models of A3-ligand complexes suggested that motion of the Trp243 side chain may be one of the primary molecular events taking place upon ligand binding. In the inactive (dark) conformation of rhodopsin the side chain of the analogous Trp256 was oriented along the TM6 helical axis and interacted with the ring moiety of retinal. If the Trp243 side chain were to adopt a somewhat similar orientation in the free A3 receptor, its Cη2 would be within interaction distance from His95. Upon binding, the Trp243 side chain rotated out of the way of the incoming ligand and assumed a new conformation to position the indole moiety adjacent to Phe239. In this new conformation Trp243 did not interact directly with agonists, yet the aromatic–aromatic interaction with Phe239 may stabilize the active conformation of the receptor.
In conclusion, the binding modes of nucleoside agonists and non-nucleoside antagonists at human A3 receptors are different. Residues needed for antagonist but not agonist binding include Tyr243 (6.48) and Lys152 (EL2). Residues needed for both antagonist and agonist binding include His95 (3.37), His272 (7.43), and Asn250 (6.55), which is critical. Furthermore, it is possible to separate the structural bases for binding and activation processes. Trp243 is required for activation of the receptor but not for binding of agonists.
Acknowledgments
We thank Dr. Neli Melman (NIDDK) for assistance in radioligand binding assays. We thank Dr. Ad IJzerman (Leiden/Amsterdam Center for Drug Research, Leiden, The Netherlands) for the gift of 4-methoxy-N-[3-(2-pyridinyl)-1-isoquinolinyl]benzamide, Dr. Kyeong Lee (NIDDK) for preparing MRS1898, and Dr. Bruce Liang (University of Pennsylvania, Philadelphia, PA) for helpful discussions. Proofreading and graphics by Kelly Soltysiak (NIDDK) is appreciated.
Footnotes
The abbreviations used are: GPCR, G protein-coupled receptor; AB-MECA, N6-(4-aminobenzyl)-5′-N-methylcarboxamidoadenosine; ADA, adenosine deaminase; CADO, 2-chloroadenosine; CGS15943, 5-amino-9-chloro-2-(2-furyl)-1,2,4-triazolo[1,5-c]quinazoline; Cl-IB-MECA, 2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarbamoyladenosine; GTPγS, guanosine 5′-γ-thiotriophosphate; IB-MECA, N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine; MRS1939, (1′R,2′R,3′S,4′R,5′S)-4-{6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0] hexane-2,3-diol; MRS1898, (1′R,2′R,3′S,4′R,5′S)-4-{2-chloro-6-[(3-iodophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo [3.1.0]hexane-2,3-diol; MRS1220, N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo [1,5-c]quinazolin-5-yl]benzene-acetamide; NECA, 5′-N-ethylcarboxamido adenosine; PSB-11, 8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2.1-i]purin-5-one; PLC, phospholipase C; WT, wild type; TM, transmembrane.
References
- 1.Zhou QY, Li C, Olah ME, Johnson RA, Stiles GL, Civelli O. Proc Natl Acad Sci U S A. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Salvatore SA, Tilley SL, Latour AM, Fletcher DS, Koller BH, Jacobson MA. J Biol Chem. 2000;275:4429–4434. doi: 10.1074/jbc.275.6.4429. [DOI] [PubMed] [Google Scholar]
- 3.Olah ME, Stiles GL. Pharmacol Ther. 2000;85:55–75. doi: 10.1016/s0163-7258(99)00051-0. [DOI] [PubMed] [Google Scholar]
- 4.Fredholm BB, Arslan G, Halldner L, Kull B, Schulte G, Wasserman W. Naunyn-Schmiedeberg’s Arch Pharmacol. 2000;362:364–374. doi: 10.1007/s002100000313. [DOI] [PubMed] [Google Scholar]
- 5.Linden J. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 6.Ramkumar V, Stiles GL, Beaven MA, Ali H. J Biol Chem. 1993;268:16887–16890. [PubMed] [Google Scholar]
- 7.Sajjadi FG, Takabayashi K, Foster AC, Domingo RC, Firestein GS. J Immunol. 1996;156:3435–3442. [PubMed] [Google Scholar]
- 8.Kaiser SM, Quinn RJ. Drug Disc Today. 1999;4:542–551. doi: 10.1016/s1359-6446(99)01421-x. [DOI] [PubMed] [Google Scholar]
- 9.von Lubitz DK, Lin RC, Popik P, Carter MF, Jacobson KA. Eur J Pharmacol. 1994;263:59–67. doi: 10.1016/0014-2999(94)90523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.von Lubitz DK, Ye W, McClellan J, Lin RC. Ann N Y Acad Sci. 1999;890:93–106. doi: 10.1111/j.1749-6632.1999.tb07984.x. [DOI] [PubMed] [Google Scholar]
- 11.Liang BT, Jacobson KA. Proc Natl Acad Sci U S A. 1998;95:6995–6999. doi: 10.1073/pnas.95.12.6995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardo L, Ballesteros JA, Osman R, Weinstein H. Proc Natl Acad Sci U S A. 1992;89:4009–4012. doi: 10.1073/pnas.89.9.4009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hibert MF, Trumpp-Kallmeyer S, Hoflack J, Bruinvels A. Trends Pharmacol Sci. 1993;14:7–12. doi: 10.1016/0165-6147(93)90106-t. [DOI] [PubMed] [Google Scholar]
- 14.Unger VM, Hargrave PA, Baldwin JM, Schertler GF. Nature. 1997;389:203–206. doi: 10.1038/38316. [DOI] [PubMed] [Google Scholar]
- 15.Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp TE, Yamamoto M, Miyano M. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- 16.Barbhaiya H, McClain R, IJzerman AP, Rivkees SA. Mol Pharmacol. 1996;50:1635–1642. [PubMed] [Google Scholar]
- 17.Tucker AL, Robeva AS, Taylor HE, Holeton D, Bockner M, Lynch KR, Linden J. J Biol Chem. 1994;269:27900–27906. [PubMed] [Google Scholar]
- 18.Kim J, Wess J, van Rhee AM, Schoneberg T, Jacobson KA. J Biol Chem. 1995;270:13987–13997. doi: 10.1074/jbc.270.23.13987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao ZG, Jiang Q, Jacobson KA, IJzerman AP. Biochem Pharmacol. 2000;60:661–668. doi: 10.1016/s0006-2952(00)00357-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beukers MW, den Dulk H, van Tilburg EW, Brouwer J, IJzerman AP. Mol Pharmacol. 2000;58:1349–1356. doi: 10.1124/mol.58.6.1349. [DOI] [PubMed] [Google Scholar]
- 21.Jacobson KA, Gao ZG, Chen A, Barak D, Kim SA, Lee K, Link A, van Rompaey P, van Calenbergh S, Liang BT. J Med Chem. 2001;44:4125–4136. doi: 10.1021/jm010232o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen A, Gao ZG, Barak D, Liang BT, Jacobson KA. Biochem Biophys Res Commun. 2001;284:596–601. doi: 10.1006/bbrc.2001.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Müller CE, Diekmann M, Thorand M, Ozola V. Bioorg Med Chem Lett. 2001;12:501–503. doi: 10.1016/s0960-894x(01)00785-5. [DOI] [PubMed] [Google Scholar]
- 24.Lee K, Ravi RG, Ji XD, Marquez VE, Jacobson KA. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Rhee AM, Jacobson KA. Drug Dev Res. 1996;37:1–38. doi: 10.1002/(SICI)1098-2299(199601)37:1<1::AID-DDR1>3.0.CO;2-S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cullen BR. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 28.Harden TK, Hawkins PT, Stephens L, Boyer JL, Downes P. Biochem J. 1988;252:583–593. doi: 10.1042/bj2520583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berridge MJ, Dawson RM, Downes CP, Heslop JP, Irvine RF. Biochem J. 1983;212:473–482. doi: 10.1042/bj2120473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng Y, Prusoff WH. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 31.Olah ME, Stiles GL. Annu Rev Pharmacol Toxicol. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- 32.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Proc Natl Acad Sci U S A. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Q, van Rhee AM, Kim J, Yehle S, Wess J, Jacobson KA. Mol Pharmacol. 1996;50:512–521. [PMC free article] [PubMed] [Google Scholar]
- 34.Rivkees SA, Barbhaiya H, IJzerman AP. J Biol Chem. 1999;274:3617–3621. doi: 10.1074/jbc.274.6.3617. [DOI] [PubMed] [Google Scholar]
- 35.Abbracchio MP, Brambilla R, Ceruti S, Kim HO, von Lubitz DK, Jacobson KA, Cattabeni F. Mol Pharmacol. 1995;48:1038–1045. [PubMed] [Google Scholar]
- 36.Jacobson KA, Park KS, Jiang JL, Kim YC, Olah ME, Stiles GL, Ji XD. Neuropharmacology. 1997;36:1157–1165. doi: 10.1016/s0028-3908(97)00104-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perlman JH, Laakkonen L, Osman R, Gershengorn MC. J Biol Chem. 1994;269:23383–23386. [PubMed] [Google Scholar]
- 38.Javitch JA, Fu D, Chen J, Karlin A. Neuron. 1995;14:825–831. doi: 10.1016/0896-6273(95)90226-0. [DOI] [PubMed] [Google Scholar]
- 39.Nakayama TA, Khorana HG. J Biol Chem. 1990;265:15762–15769. [PubMed] [Google Scholar]
- 40.Befort K, Tabbara L, Kling D, Maigret B, Kieffer BL. J Biol Chem. 1996;271:10161–10168. doi: 10.1074/jbc.271.17.10161. [DOI] [PubMed] [Google Scholar]
- 41.Yamano Y, Ohyama K, Kikyo M, Sano T, Nakagomi Y, Inoue Y, Nakamura N, Morishima I, Guo DF, Hamakubo T, Inagami T. J Biol Chem. 1995;270:14024–14030. doi: 10.1074/jbc.270.23.14024. [DOI] [PubMed] [Google Scholar]
- 42.Perlman JH, Laakkonen L, Osman R, Gershengorn MC. Mol Pharmacol. 1995;47:480–484. [PubMed] [Google Scholar]
- 43.Dunham TD, Farrens DL. J Biol Chem. 1999;274:1683–16890. doi: 10.1074/jbc.274.3.1683. [DOI] [PubMed] [Google Scholar]
- 44.Mouledous L, Topham CM, Moisand C, Mollereau C, Meunier JC. Mol Pharmacol. 2000;57:495–502. doi: 10.1124/mol.57.3.495. [DOI] [PubMed] [Google Scholar]
- 45.Gether U. Endocr Rev. 2000;21:90–113. doi: 10.1210/edrv.21.1.0390. [DOI] [PubMed] [Google Scholar]
- 46.Marie J, Richard E, Pruneau D, Paquet JL, Siatka C, Larguier R, Poncé C, Vassault P, Groblewski T, Maigret B, Bonnafous JC. J Biol Chem. 2001;276:41100–41111. doi: 10.1074/jbc.M104875200. [DOI] [PubMed] [Google Scholar]
- 47.Olah ME, Ren H, Ostrowski J, Jacobson KA, Stiles GL. J Biol Chem. 1992;267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- 48.Zoffmann S, Gether U, Schwartz TW. FEBS Lett. 1993;336:506–510. doi: 10.1016/0014-5793(93)80865-r. [DOI] [PubMed] [Google Scholar]
- 49.Bluml K, Mutschler E, Wess J. J Biol Chem. 1994;269:18870–18876. [PubMed] [Google Scholar]