

Abstract
Various factors are thought to cause the development and progression of disease in macular degeneration, diabetic retinopathy, and retinitis pigmentosa. Some of the deleterious processes include oxidative stress, hypoxia, metabolic derangement, genetics, and vasculopathy. In this review, we present a unified theory for the pathophysiology of several retinopathies based on the unique and intense metabolism of rod photoreceptors.
Keywords: diabetic retinopathy, macular degeneration, metabolism, retinitis pigmentosa, SIRT6
1. Introduction
Rod photoreceptors consume more energy in darkness than in light.1 Unlike most other neurons, rods do not fire action potentials. Rather, in darkness they exhibit a continuous depolarized state of the rod membrane potential that allows constant neurotransmitter release to activate second-order neurons in the visual pathway.2 The “dark current” is maintained by cyclic-nucleotide-gated (CNG) channels that allow inward flow of cations, approximately 80% Na+ and 15% Ca2+. The Ca2+ influx is balanced by a Na+/Ca2+–K+ exchanger that exchanges four Na+ inward for one Ca2+ and one K+ outward, and the large Na+ influx is balanced by a Na+/K+ ATPase at the inner segment.3 Rods consume up to four times as much adenosine triphosphate (ATP) in darkness as that in light to support the high energy demand of these transporters; one ATP is consumed per Ca2+ exported and one ATP is consumed per three Na+ exported.3 Photoexcitation closes the CNG channel, preventing influx of Na+ and Ca2+ and causing hyperpolarization across the rod membrane, which reduces neurotransmitter release. For reviews of phototransduction, see the works of Yau and Hardie3 and Fain et al.4 Briefly, in rod phototransduction, light activates rhodopsin, which activates the G protein transducin, which in turn activates phos-phodiesterase, which hydrolyzes cyclic guanosine monophosphate (cGMP) to guanosine monophosphate (GMP).5,6,7,8,9 Removal of cGMP from the CNG channel causes channel closure and prevents the influx of cations.3,4
The high energy demand for maintaining the “dark current” causes rods to consume the highest amount of energy among all cell types in the body.10,11,12 A large amount of ATP and Nicotinamide adenine dinucleotide phosphate (NADPH) are needed for recovery of cGMP from photoexcitation and its resynthesis in darkness.1 In darkness, to meet the ATP demands of ion transporters, rods use large amounts of O2 and glucose that are metabolized by both glycolysis and oxidative phosphorylation (Fig. 1).13,14 In light, however, consumption of O2 by rods was shown to decrease by approximately 30% in macaques and approximately 50–70% in cats,10,11,12 indicating a marked reduction in oxidative phosphory-lation in light compared with that in darkness (Fig. 1). In light, there is increased anabolic activity14; mRNA levels are increased four to 10 times,15,16 and outer segments appear to follow a circadian pattern, in which discs are shed more at the onset of light in a 12–hour light–dark cycle irrespective of whether the lights are turned on or not, which is accompanied by an increase in outer segment renewal,17,18 presumably fueled by increased lipid and protein production.
Fig. 1.
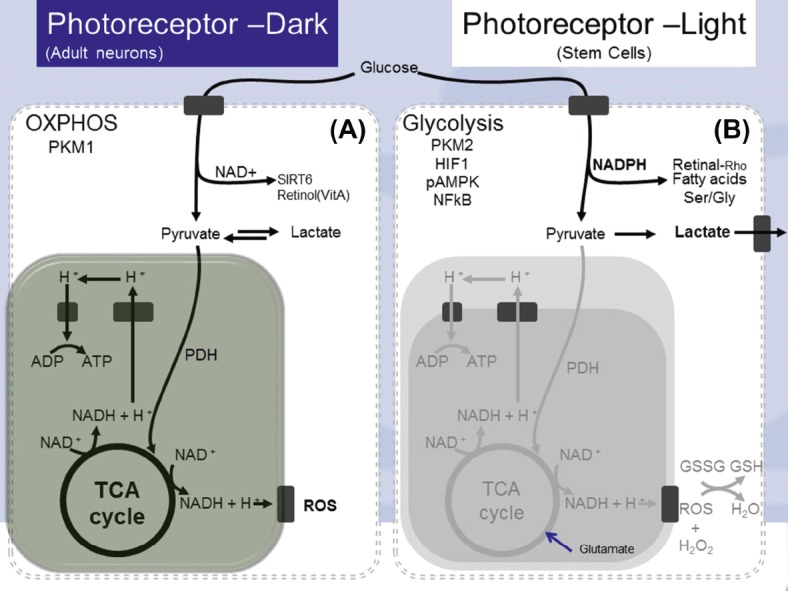
(A) In darkness, photoreceptor metabolism is similar to adult neurons. Glucose is catabolized through glycolysis, the TCA cycle, and oxidative phosphorylation, generating NAD+ and a large amount of ATP along with ROS. (B) In light, photoreceptor metabolism is similar to that of stem cells. Glucose undergoes glycolysis to form pyruvate, which is not further catabolized by the TCA cycle but is metabolized by the pentose phosphate pathway, generating several molecules of NADPH. NADPH is used for anabolic production of fatty acids, proteins, nucleic acids, and 11-cis retinal. ADP = adenosine diphosphate; ATP = adenosine triphosphate; HIF1 = hypoxia inducible factor 1; NFκB = nuclear factor kappa beta; PKM = pyruvate kinase muscle isozyme; ROS = reactive oxygen species; TCA = tricarboxylic acid; NADH = nicotinamide adenine dinucleotide (reduced); NAD+ = nicotinamide adenine dinucleotide (oxidized); PDH = pyruvate dehydrogenase; pAMPK = phosphorylated adenosine monophosphate activated protein kinase; NADPH = nicotinamide adenine dinucleotide phosphate; Ser = serine; Gly = glycine; GSSG = glutathione disulfide; GSH = glutathione.
2. Dark-adapted rod metabolism and the Warburg effect
The anabolic state of light-adapted rods is similar to the Warburg effect seen in cancer cells and stem cells, in which aerobic glycolysis is the exclusive catabolic process used to produce massive amounts of biomolecules for cell growth and division.19,20 Conversely, dark-adapted rods exhibit glycolysis as well as oxidative phosphorylation, which, similar to conventional neuronal metabolism, consumes large amounts of glucose and O2.21 Much of the understanding of the Warburg effect can potentially be applied to rod photoreceptors in light adaptation. The shift from oxidative phosphorylation to glycolysis allows for the production of two NADPH molecules per glucose molecule via the pentose phosphate pathway, which fuels the anabolic processes of the cell, and synthesis of amino acids, nucleic acids, lipids, and carbohydrates.22
3. Cellular control of metabolism via hypoxia inducible factor 1 and SIRT6
Recently, new details of the molecular basis for the switch from aerobic respiration to aerobic glycolysis have been uncovered. It has long been known that a state of hypoxia or low nutrients induces cells to use glycolysis alone, rather than continuing catabolism through the tricarboxylic acid (TCA) cycle and oxidative phosphorylation. This switch to glycolysis is eponymously referred to as the “Pasteur effect.” Recently, some molecular details have been uncovered. The low-oxygen sensing transcription factor, hypoxia inducible factor 1 (HIF1), was shown to activate transcription of genes, the products of which inhibit the TCA cycle and promote glycolysis.23,24 HIF1 drives anaerobic glycolysis and regulates cancer metabolism, and it has been a target for cancer therapy.25,26 The histone deacetylase SIRT6, a member of the sirtuin family, has been shown to have the opposite effect of HIF1 on metabolism and to function as a tumor suppressor.27,28 While SIRT6 and HIF1 have both been touted as possible cancer therapy targets, their role in retinal diseases has yet to be explored. We postulate that activation of HIF1 or inhibition of SIRT6 can cause rods to switch from oxidative phosphorylation to aerobic glycolysis, reducing cellular consumption of ATP and O2 to promote survival by preventing ischemia and oxidative damage.
4. Rod energy demand drives retinal degenerative diseases
The high energy cost and O2 demand of dark-adapted rod cells are the bases for a recently developed theory for the mechanism of age-related macular degeneration (AMD), diabetic retinopathy (DR), and retinopathy of prematurity.29 During dark adaptation, partial pressure of oxygen nears zero at the ellipsoid zone where the rod mitochondria reside, which was shown in cats, monkeys, and rats.10,12,30 In low-oxygen settings, such as at a high elevation,31 and in diseases such as polycythemia vera32,33 and partial carotid occlusion,34 loss of dark adaptation is one of the first symptoms to manifest, indicating the high sensitivity of rods to hypoxic insult. Because rods operate in a near hypoxic state, any reduction in the oxygenation of the retina causes release of hypoxic factors, notably vascular endothelial growth factor (VEGF), which leads to neovascularization of either the choroidal (in AMD) or the retinal (in retinopathy of prematurity and DR) vasculature.29
5. Rod metabolism in AMD
Early pathological changes observed in AMD are thickening of Bruch's membrane and deposition of subretinal drusen.35 These lead to a decrease in diffusion capacity of O2 from the chorioca-pillaris to the retinal pigment epithelium (RPE), causing ischemia in the retina. In early disease stages, a manifestation of the ischemic injury is that dark adaptation is reduced compared with healthy age-controlled adults.36 This finding suggests that in AMD, rod function is actually impaired before macular degeneration occurs and before cone function is lost. The histological evidence that rods are lost in the parafoveal region of the retina where they are in the highest density and O2 consumption is greatest37 also supports the theory that hypoxic/ischemic injury causes degeneration of the macula. Subsequent cone photoreceptor death is generally accepted to be caused by rod death, with a rod-derived cone viability factor thought to play a prominent role in this process.38 The RPE releases VEGF normally to sustain the choriocapillaris, and without it, the choriocapillaris atrophies in normal aging.39 In “wet” AMD, usually the late-stage form of the disease, there is substantial neovascularization and subsequent capillary leakage, as opposed to in “dry” AMD, in which RPE atrophy predominates. Anti-VEGF therapies were successful taking advantage of this mechanism to suppress neovascularization in wet AMD.40 In addition, in patients treated for DR with panretinal photocoagulation, the development of wet and dry AMD was shown to have reduced compared with patients with DR without panretinal photocoagulation treatment.41 This may be due to the dramatic decrease in the number of rods, lowering the burden of oxygenating the retina.29 A novel approach to treating AMD, which combats the high energy consumption by rods in darkness, is light therapy, in which patients are exposed to constant low levels of light. This has produced promising results in an early-stage AMD clinical trial.42
6. Rod metabolism in DR
Similarly to AMD, it has been well established that the clinical development of DR begins with a decade-long pre-nonproliferative phase, followed by a nonproliferative phase that continues or becomes proliferative in the end-stage disease.43 The molecular pathophysiology is under constant debate; however, the importance of elevated glucose levels to initiate the disease and VEGF release to cause neovascularization is largely accepted in the field.44 Hyperglycemia has been shown to induce vascular changes in early and late stages of DR through retinal capillary damage from peri-cyte loss and possible alterations in retinal blood vessel diameter, retinal oxygenation, and retinal blood flow.45 Prolonged elevation of glucose level and advanced glycation products have been shown separately to increase VEGF release in RPE.46,47 Arden and others29,42,48,49 have advocated that VEGF release induced by elevated glucose levels and hypoxia is caused by the high energy demand of rod photoreceptors in dark adaptation. In support of their hypothesis, Arden and colleagues50 showed that patients with concurrent diabetes and retinitis pigmentosa (RP) do not develop DR, which, according to them, is due to a decrease in energy demand as a result of loss of rod photoreceptors. Arden and col-leagues42 have also proposed light therapy for treatment of DR, which showed benefit in early disease stages. Other methods to lower energy demand from rod photoreceptors need to be explored to validate the hypothesis that rod energy consumption drives DR.
7. Rod metabolism in RP
The pathophysiological basis of photoreceptor loss in RP can also be due to excessive energy demand from being in a state that mimics dark adaptation or “equivalent darkness.” Rhodopsin is the most commonly mutated gene responsible for the disease, and mutation of rhodopsin and other phototransduction genes such as the phosphodiesterase (PDE) subunits causes impairment of photoexcitation.51 Mutations of PDE6 that reduce enzyme activity have been shown to cause a disease state in mice that mimics RP and similar diseases,8,9 which may be rescued by the introduction of a wild-type copy of the gene via gene therapy.52 This leads to increased opening of the CNG channel and increased oxidative phosphorylation by rod photoreceptors in a state that mimics dark adaptation. Oxidative phosphorylation creates reactive oxygen species that may cause autophagy or apoptosis.53,54 In addition, one of the earliest histopathological signs of disease is shortening of the outer segments.55 As described above, light-induced outer segment turnover is one of the most important anabolic functions of rod cells, and if the cells are primarily using oxidative phosphorylation, they may not have adequate energy directed for rebuilding the outer segments. Many structural genes, ciliopathy genes, and protein trafficking genes known to be genetically responsible for RP,51 likely contribute to the disease by also preventing adequate outer segment renewal. RP exhibits a characteristic pattern of rod loss beginning at the midperiphery of the retina where rod density is high, and occasionally a bull's-eye pattern of rod loss is formed in the parafoveal region of highest rod density.51,56 This predilection for atrophy in areas of high rod density further supports the notion that a rod process gone awry causes the disease, and we believe that rod cells succumb to the mutations that cause increased energy demand or other insults to the ability to renew outer segments. Treating RP with an agent that maintains rods in a state that mimics light adaptation, in which only glycolysis is used without oxidative phosphorylation, may allow for increased preservation of rod cells’ ability to maintain outer segments and survive. In pre-clinical studies using mice as models of RP, increased mammalian target of rapamycin (mTOR) signaling has been shown to prolong photoreceptor survival.57,58 The mTOR signaling is also known to induce aerobic glycolysis,59 which may explain why increased mTOR signaling is protective for RP.
8. Conclusion
The above evidence gives credence to the theory that overburdening rod metabolism causes several retinopathies. More studies must be performed to further understand the mechanism by which oxidative phosphorylation leads to rod death, and to determine the treatment options based on this knowledge. Whether this theory can be applied to other retinal degenerative diseases is currently being explored.
Footnotes
Conflicts of interest: The authors report no conflicts of interest in this work.
References
- 1.Okawa H, Sampath AP, Laughlin SB, Fain GL. ATP consumption by mammalian rod photoreceptors in darkness and in light. Curr Biol. 2008;18:1917–1921. doi: 10.1016/j.cub.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakanishi S. Second-order neurones and receptor mechanisms in visual- and olfactory-information processing. Trends Neurosci. 1995;18:359–364. doi: 10.1016/0166-2236(95)93929-r. [DOI] [PubMed] [Google Scholar]
- 3.Yau KW, Hardie RC. Phototransduction motifs and variations. Cell. 2009 Oct 16;139(2):246–264. doi: 10.1016/j.cell.2009.09.029. http://dx.doi.org/10.1016/j.cell.2009.09.029 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fain GL, Hardie R, Laughlin SB. Phototransduction and the evolution of pho-toreceptors. Curr Biol. 2010;20:R114–R124. doi: 10.1016/j.cub.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tsang SH, Woodruff ML, Chen CK, et al. GAP-independent termination of photoreceptor light response by excess gamma subunit of the cGMP-phos-phodiesterase. J Neurosci. 2006;26:4472–4480. doi: 10.1523/JNEUROSCI.4775-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsang SH, Woodruff ML, Janisch KM, Cilluffo MC, Farber DB, Fain GL. Removal of phosphorylation sites of gamma subunit of phosphodiesterase 6 alters rod light response. J Physiol. 2007;579(Pt 2):303–312. doi: 10.1113/jphysiol.2006.121772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woodruff ML, Janisch KM, Peshenko IV, Dizhoor AM, Tsang SH, Fain GL. Modulation of phosphodiesterase6 turnoff during background illumination in mouse rod photoreceptors. J Neurosci. 2008;28:2064–2074. doi: 10.1523/JNEUROSCI.2973-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsang S, Woodruff M, Lin C, et al. Effect of the ILE86TER mutation in the γ subunit of cGMP phosphodiesterase (PDE6) on rod photoreceptor signalling. Cell Signal. 2012;24:181–188. doi: 10.1016/j.cellsig.2011.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tsang SH, Woodruff ML, Jun L, et al. Transgenic mice carrying the H258N mutation in the gene encoding the beta-subunit of phosphodiesterase-6 (PDE6B) provide a model for human congenital stationary night blindness. Hum Mutat. 2007;28:243–254. doi: 10.1002/humu.20425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Linsenmeier RA. Effects of light and darkness on oxygen distribution and consumption in the cat retina. J Gen Physiol. 1986;88:521–542. doi: 10.1085/jgp.88.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahmed J, Braun RD, Dunn R, Jr, Linsenmeier RA. Oxygen distribution in the macaque retina. Invest Ophthalmol Vis Sci. 1993;34:516–521. [PubMed] [Google Scholar]
- 12.Birol G, Wang S, Budzynski E, Wangsa-Wirawan ND, Linsenmeier RA. Oxygen distribution and consumption in the macaque retina. Am J Physiol Heart Circ Physiol. 2007;293:H1696–H1704. doi: 10.1152/ajpheart.00221.2007. [DOI] [PubMed] [Google Scholar]
- 13.Wong-Riley MT. Energy metabolism of the visual system. Eye Brain. 2010;2:99–116. doi: 10.2147/EB.S9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hurley J, Chertov A, Lindsay K, et al. Energy metabolism in the vertebrate retina. In: Furukawa T, Hurley JB, Kawamura S, editors. Vertebrate Photoreceptors. Japan: Springer; 2014. pp. 91–137. [Google Scholar]
- 15.Schmidt SY, Blanks JC, Sandberg MA. Enhancement of (polyA+)RNA synthesis in light in isolated intact photoreceptor cells of the rat. Exp Eye Res. 1985;41:159–170. doi: 10.1016/0014-4835(85)90020-x. [DOI] [PubMed] [Google Scholar]
- 16.Hollyfield JG, Basinger SF. RNA metabolism in the retina in relation to cyclic lighting. Vis Res. 1980;20:1151–1155. doi: 10.1016/0042-6989(80)90053-x. [DOI] [PubMed] [Google Scholar]
- 17.Basinger S, Hoffman R, Matthes M. Photoreceptor shedding is initiated by light in the frog retina. Science (New York, NY) 1976;194:1074–1076. doi: 10.1126/science.1086510. [DOI] [PubMed] [Google Scholar]
- 18.LaVail MM. Kinetics of rod outer segment renewal in the developing mouse retina. J Cell Biol. 1973;58:650–661. doi: 10.1083/jcb.58.3.650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Menendez JA, Joven J, Cufi S, et al. The Warburg effect version 2.0: metabolic reprogramming of cancer stem cells. Cell Cycle (Georgetown, TX) 2013;12:1166–1179. doi: 10.4161/cc.24479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science (New York, NY) 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Belanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Nelson DL, Nelson DL, Lehninger AL, Cox MM. Lehninger Principles of Biochemistry. New York: W.H. Freeman; 2008. [Google Scholar]
- 23.Marin-Hernandez A, Gallardo-Perez JC, Ralph SJ, Rodriguez-Enriquez S, Moreno-Sanchez R. HIF-1α modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev Med Chem. 2009;9:1084–1101. doi: 10.2174/138955709788922610. [DOI] [PubMed] [Google Scholar]
- 24.Kim J-W, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong L, D’Urso A, Toiber D, et al. The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1α. Cell. 2010;140:280–293. doi: 10.1016/j.cell.2009.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sebastián C, Zwaans BM, Silberman DM, et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185–1199. doi: 10.1016/j.cell.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arden GB, Sidman RL, Arap W, Schlingemann RO. Spare the rod and spoil the eye. Br J Ophthalmol. 2005;89:764–769. doi: 10.1136/bjo.2004.062547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lau JC, Linsenmeier RA. Oxygen consumption and distribution in the Long-Evans rat retina. Exp Eye Res. 2012;102:50–58. doi: 10.1016/j.exer.2012.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McFarland RA, Evans JN. Alterations in dark adaptation under reduced oxygen tensions. Am J Physiol. 1939;127:37–50. [Google Scholar]
- 32.Havelius U, Bergqvist D, Hindfelt B, Krakau T., II Improved dark adaptation after carotid endarterectomy. Evidence of a long-term ischemic penumbra? Neurology. 1997;49:1360–1364. doi: 10.1212/wnl.49.5.1360. [DOI] [PubMed] [Google Scholar]
- 33.Havelius U, Bergqvist D, Falke P, Hindfelt B, Krakau TI. Impaired dark adaptation in symptomatic carotid artery disease. Neurology. 1997;49:1353–1359. doi: 10.1212/wnl.49.5.1353. [DOI] [PubMed] [Google Scholar]
- 34.Havelius U, Berglund S, Falke P, Hindfelt B, Krakau T. Impaired dark adaptation in polycythemia. Improvement after treatment. Acta Ophthalmol Scand. 2000;78:53–57. doi: 10.1034/j.1600-0420.2000.078001053.x. [DOI] [PubMed] [Google Scholar]
- 35.Feigl B. Age-related maculopathy—linking aetiology and pathophysiological changes to the ischaemia hypothesis. Prog Retin Eye Res. 2009;28:63–86. doi: 10.1016/j.preteyeres.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 36.Owsley C, Jackson GR, Cideciyan AV, et al. Psychophysical evidence for rod vulnerability in age-related macular degeneration. Invest Ophthalmol Vis Sci. 2000;41:267–273. [PubMed] [Google Scholar]
- 37.Curcio CA. Photoreceptor topography in ageing and age-related maculopathy. Eye (London, England) 2001;15(Pt 3):376–383. doi: 10.1038/eye.2001.140. [DOI] [PubMed] [Google Scholar]
- 38.Leveillard T, Sahel JA. Rod-derived cone viability factor for treating blinding diseases: from clinic to redox signaling. Sci Transl Med. 2010;2 doi: 10.1126/scitranslmed.3000866. 26ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Witmer AN, Vrensen GF, Van Noorden CJ, Schlingemann RO. Vascular endo-thelial growth factors and angiogenesis in eye disease. Prog Retin Eye Res. 2003;22:1–29. doi: 10.1016/s1350-9462(02)00043-5. [DOI] [PubMed] [Google Scholar]
- 40.Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA) Br J Ophthalmol. 2014;98:1144–1167. doi: 10.1136/bjophthalmol-2014-305702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zylbermann R, Landau D, Rozenman Y, Abrahami S, Pollack A. Exudative age-related macular degeneration in patients with diabetic retinopathy and its relation to retinal laser photocoagulation. Eye (London, England) 1997;11(Pt 6):872–875. doi: 10.1038/eye.1997.224. [DOI] [PubMed] [Google Scholar]
- 42.Arden GB, Gunduz MK, Kurtenbach A, et al. A preliminary trial to determine whether prevention of dark adaptation affects the course of early diabetic retinopathy. Eye (London, England) 2010;24:1149–1155. doi: 10.1038/eye.2009.328. [DOI] [PubMed] [Google Scholar]
- 43.Ciulla TA, Amador AG, Zinman B. Diabetic retinopathy and diabetic macular edema: pathophysiology, screening, and novel therapies. Diabetes Care. 2003;26:2653–2664. doi: 10.2337/diacare.26.9.2653. [DOI] [PubMed] [Google Scholar]
- 44.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 45.Pournaras CJ, Rungger-Brandle E, Riva CE, Hardarson SH, Stefansson E. Regulation of retinal blood flow in health and disease. Prog Retin Eye Res. 2008;27:284–330. doi: 10.1016/j.preteyeres.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 46.Sone H, Kawakami Y, Okuda Y, et al. Vascular endothelial growth factor is induced by long-term high glucose concentration and up-regulated by acute glucose deprivation in cultured bovine retinal pigmented epithelial cells. Bio-chem Biophys Res Comm. 1996;221:193–198. doi: 10.1006/bbrc.1996.0568. [DOI] [PubMed] [Google Scholar]
- 47.Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–1224. doi: 10.1172/JCI1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arden GB, Wolf JE, Tsang Y. Does dark adaptation exacerbate diabetic reti-nopathy? Evidence and a linking hypothesis. Vision Res. 1998;38:1723–1729. doi: 10.1016/s0042-6989(98)00004-2. [DOI] [PubMed] [Google Scholar]
- 49.Arden GB, Sivaprasad S. Hypoxia and oxidative stress in the causation of diabetic retinopathy. Curr Diabetes Rev. 2011;7:291–304. doi: 10.2174/157339911797415620. [DOI] [PubMed] [Google Scholar]
- 50.Arden GB. The absence of diabetic retinopathy in patients with retinitis pig-mentosa: implications for pathophysiology and possible treatment. Br J Ophthalmol. 2001;85:366–370. doi: 10.1136/bjo.85.3.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 52.Davis RJ, Tosi J, Janisch KM, et al. Functional rescue of degenerating photore-ceptors in mice homozygous for a hypomorphic cGMP phosphodiesterase 6 b allele (Pde6bH620Q) Invest Ophthalmol Vis Sci. 2008;49:5067–5076. doi: 10.1167/iovs.07-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Rad Biol Med. 2010;48:749–762. doi: 10.1016/j.freeradbiomed.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fariss RN, Li Z-Y, Milam AH. Abnormalities in rod photoreceptors, amacrine cells, and horizontal cells in human retinas with retinitis pigmentosa. Am J Ophthalmol. 2000;129:215–223. doi: 10.1016/s0002-9394(99)00401-8. [DOI] [PubMed] [Google Scholar]
- 56.Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol. 1990;292:497–523. doi: 10.1002/cne.902920402. [DOI] [PubMed] [Google Scholar]
- 57.Tsang SH, Chan L, Tsai YT, et al. Silencing of tuberin enhances photoreceptor survival and function in a preclinical model of retinitis pigmentosa (an American Ophthalmological Society thesis) Trans Am Ophthalmol Soc. 2014;112:103–115. [PMC free article] [PubMed] [Google Scholar]
- 58.Punzo C, Kornacker K, Cepko CL. Stimulation of the insulin/mTOR pathway delays cone death in a mouse model of retinitis pigmentosa. Nat Neurosci. 2009;12:44–52. doi: 10.1038/nn.2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun Q, Chen X, Ma J, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]