

Abstract
Pathological proliferation of retinal blood vessels commonly causes vision impairment in proliferative retinopathies, including retinopathy of prematurity. Dysregulated crosstalk between the vasculature and retinal neurons is increasingly recognized as a major factor contributing to the pathogenesis of vascular diseases. Class 3 semaphorins (SEMA3s), a group of neuron-secreted axonal and vascular guidance factors, suppress pathological vascular growth in retinopathy. However, the upstream transcriptional regulators that mediate the function of SEMA3s in vascular growth are poorly understood. Here we showed that retinoic acid receptor–related orphan receptor α (RORα), a nuclear receptor and transcription factor, is a novel transcriptional regulator of SEMA3E-mediated neurovascular coupling in a mouse model of oxygen-induced proliferative retinopathy. We found that genetic deficiency of RORα substantially induced Sema3e expression in retinopathy. Both RORα and SEMA3E were expressed in retinal ganglion cells. RORα directly bound to a specific ROR response element on the promoter of Sema3e and negatively regulated Sema3e promoter–driven luciferase expression. Suppression of Sema3e using adeno-associated virus 2 carrying short hairpin RNA targeting Sema3e promoted disoriented pathological neovascularization and partially abolished the inhibitory vascular effects of RORα deficiency in retinopathy. Our findings suggest that RORα is a novel transcriptional regulator of SEMA3E-mediated neurovascular coupling in pathological retinal angiogenesis.—Sun, Y., Liu, C.-H., Wang, Z., Meng, S. S., Burnim, S. B., SanGiovanni, J. P., Kamenecka, T. M., Solt, L. A., Chen, J. RORα modulates semaphorin 3E transcription and neurovascular interaction in pathological retinal angiogenesis.
Keywords: retinopathy, SEMA3E, nuclear receptor, neovascularization
Pathological proliferation of retinal blood vessels commonly causes vision impairment in proliferative retinopathies, including retinopathy of prematurity and diabetic retinopathy (1–3). Dysregulated crosstalk between the vasculature and retinal neurons is increasingly recognized as a major factor contributing to the pathogenesis of ocular vascular diseases (4–6). Among many neuron-derived growth factors and guidance cues, semaphorins (SEMAs) are critical regulators of angiogenesis during development and in diseases such as cancer and retinopathy. Among 20 identified SEMAs, class 3 SEMAs (SEMA3s) are one of the most versatile and studied SEMA classes. Originally identified as axonal growth repulsive cues, SEMA3s are a family of cell-surface and secreted proteins capable of regulating cell–cell interactions as well as cell differentiation, morphology, and function (7). They are crucial determinants of tumor growth and metastasis through mediating tumor angiogenesis (8, 9). SEMA3s are all secreted proteins and include 7 members (SEMA3A–G). In the retina, Sema3A, E and F were reported as vaso-repulsive cues in developmental retinal angiogenesis (10–12). In addition, SEMA-3A, C, and E are all involved in pathological retinal neovascularization in eye disease models (13–15). Both SEMA3A and -E are expressed in the retinal ganglion cells (RGCs) (14). However, the upstream factors that mediate the expression of SEMA3s in the retinas to influence neurovascular interaction and pathological retinal angiogenesis are poorly understood.
Retinoic acid receptor–related orphan receptor α (RORα) is a member of the nuclear receptor superfamily of ligand-regulated transcription factors (16). The 3 members of the ROR subfamily—RORα, RORβ, and RORγ—have sequence similarities to the retinoic acid receptor (17, 18), and each receptor is thought to constitutively activate transcription through ligand-independent recruitment of transcriptional coactivators (19). RORα is widely expressed in many tissues, including cerebellar Purkinje cells, liver, thymus, skeletal muscle, skin, lung, adipose tissue, and kidney (20, 21), and thereby controls diverse biological processes, including cerebellum development, immunity, and circadian rhythm (22). Previous studies reported transcriptional regulation of SEMA3A and -F by RORα in epidermal keratinocytes and breast cancer cells, respectively (23, 24). Our previous study (25) showed that RORα is expressed in retinal macrophages and microglia and regulates pathological retinal angiogenesis by modulating SOCS3-dependent inflammation in a mouse model of oxygen-induced retinopathy (OIR), modeling retinopathy of prematurity and the proliferative aspects of diabetic retinopathy in humans. Other studies also identified RORα expression in retinal neurons, including RGCs and photoreceptors (21, 26, 27), yet whether RORα regulates expression of SEMA3s in the retinal neurons to affect retinal angiogenesis is not known.
In this study we investigated whether RORα modulates transcription of SEMA3s and neurovascular coupling in pathological retinal angiogenesis. Here we found that genetic deficiency of RORα significantly induced SEMA3E in the retinas and that RORα transcriptionally suppressed Sema3e expression. RORα induction in OIR is inversely associated with Sema3e suppression in OIR. RORα and SEMA3E are colocalized in RGCs. Suppression of Sema3e using adeno-associated virus (AAV)2 carrying short hairpin RNA (shRNA) targeting Sema3e promotes pathological neovascularization and partially abolished the inhibitory effects of RORα deficiency in retinopathy in mice. Together, these data suggest that RORα transcriptionally regulates SEMA3E to influence neurovascular interaction in pathological retinal angiogenesis.
MATERIALS AND METHODS
Animals
All animal studies were performed according to protocols reviewed and approved by the Institutional Animal Care and Use Committee at the Boston Children’s Hospital. Rora heterozygous Staggerer (Sg/+) mice were obtained from The Jackson Laboratory (002651; Bar Harbor, ME, USA) and bred together to generate homozygous RORα–deficient Staggerer (Sg/Sg) and wild-type (WT) littermates. C57BL/6J mice (000664) were also obtained from The Jackson Laboratory.
OIR
OIR was carried out in neonatal Sg/Sg or WT littermate mice as previously described (28–30), with mouse pups exposed to 75% oxygen at postnatal day (P)7–12 followed by room air (Fig. 1A). At P17, mice were anesthetized, and retinas were dissected followed by either RNA isolation or fluoresceinated isolectin IB4 (Griffonia simplicifolia) (Invitrogen, Carlsbad, CA, USA) staining to visualize vessels on whole-mount retinas. Areas of retinal vaso-obliteration and pathological neovascularization were quantified as a percentage of total retinal areas using Adobe Photoshop (Adobe Systems, San Jose, CA, USA) and ImageJ (National Institutes of Health, Bethesda, MD, USA) according to previous protocols (31). Sg/Sg mice and WT mice were maintained within normal body weight range for OIR (5–7.5 g at P17) (32) through adjusting litter size. Mice with very low body weight (<5 g at P17) were excluded from the study because very low body weight affects OIR outcomes with delayed neovascularization and exaggerated vaso-obliteration (32).
Figure 1.

RORα negatively regulated SEMA3E in OIR. A, B) In OIR model, Rora mRNA was expressed in WT retina but was barely detectable in Sg/Sg OIR retinas, confirming RORα deficiency in Sg/Sg eyes (A). Both mRNA (n = 6/group; A) and protein (n = 4/group; B) levels of Sema3e were significantly up-regulated in Sg/Sg OIR retinas compared with littermate WT controls at P17. C) There was no significant change in the mRNA expression levels of Sema3a, -c, and -f in Sg/Sg OIR retinas compared with WT retinas except for Sema3d mRNA level, which was induced as well in Sg/Sg eyes with OIR. Data are presented as means ± sem (n = 3/group). N.s., no significance; NV, neovascularization; VO, vaso-obliteration. **P < 0.01, ***P < 0.001.
Quantification of retinal vascular development
Quantitative analysis of retinal vasculature was performed with P5 retinal whole mounts stained with isolectin IB4 (Invitrogen) and obtained using an AxioCam MRm, an AxioObserver.Z1 microscope, and AxioVision 4.6.3.0 software (Zeiss, Oberkochen, Germany). The numbers of endothelial filopodia and vascular branching points were determined in sprouting vascular fronts of all 4 quadrants and then averaged per retina. The vascular area was quantified as a percentage of vascularized area compared with total retinal area. Whole-mount retinal images were obtained at ×10 magnification using the mosaic function; merge images were imported into Adobe Photoshop for further analysis followed by the established protocol (33). For each group, six retinas were analyzed.
RNA isolation and quantitative RT-PCR
Total RNA was extracted from mouse retinas using the RNeasy kit (Qiagen, Germantown, MD, USA) and reverse-transcribed with M-MLV reverse transcriptase (Invitrogen) to generate cDNA. Quantitative PCR (qPCR) was performed using a 7300 Real-time PCR System (Applied Biosystems, Beverly, MA, USA) with reagents from KAPA SYBR Fast qPCR Kits (Kapa Biosystems, Wilmington, MA, USA). Primer sequences of mouse genes were: Rora, 5′-TCCCACCTGGAAACCTGCCAGT-3′ (forward), 5′-CCACGAGCGATCCGCTGACA-3′ (reverse); Sema3a, 5′-TGACGCCCAGCCAGAAGGTCT-3′ (forward), 5′-AGAGTGCCCCGGCCTTTGTC-3′ (reverse); Sema3c, 5′-CAAAATGGCTGGCAAAGATCC-3′ (forward), 5′-TCCCCGGTTCAGGTAGGTG-3′ (reverse); Sema3d, 5′-GATGTCATGTTTCTCGGAACAGA-3′ (forward), 5′-GGTGCTTGAATACCTGAAGCT C-3′ (reverse); Sema3e, 5′-ATCCGGGTTTTGCATCACTAC-3′ (forward), 5′-CTCCAGGTGAAACAGGGGTT-3′ (reverse); Sema3f, 5′-TCGCGCACAGGATTACATCTT-3′ (forward), 5′-ACCGGGAGTTGTACTGATCTG-3′ (reverse); cyclophilin A, 5′-CAGACGCCACTGTCGCTTT-3′ (forward), 5′-TGTCTTTGGAACTTTGTCTGCAA-3′ (reverse). Cyclophilin A was used as internal control, and relative gene expression levels were calculated using ΔΔCt method and normalized to cyclophilin A as endogenous reference.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assay was performed using a ChIP Assay Kit (EMD Millipore, Billerica, MA, USA) according to the manufacturer’s protocol, with minor modifications (25). The ChIP assay was repeated 3 times. Briefly, for each ChIP assay, 30 retinas from P17 C57BL/6J mice with OIR were dissected and pooled in 10 ml ice-cold PBS with protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Chilled retinas were dissociated and crosslinked with 1% formaldehyde for 20 min at room temperature with agitation. Retinas were incubated for an additional 5 min in 125 mM glycine and rinsed twice with ice-cold PBS. Retinas were lysed and sonicated on ice using a Misonix Sonicator 3000. Retina lysates were precleaned using a salmon sperm DNA/protein A agarose-50% slurry, and immunoprecipitation was performed overnight with 4 µg of RORα antibody (ab60134; Abcam, Cambridge, MA, USA) at 4°C with agitation. DNA fragments bound to RORα antibody were eluted from agarose beads with SDS elution buffer (1% SDS, 50 mM Tris-HCl, and 10 mM EDTA buffer) and incubated with 10 mg RNase A at 37°C for 1 h. Proteins digested by incubation with proteinase K (20 mg/ml) at 55°C for 1 h. Precipitated DNA fragments were purified using the quick PCR purification kit (Qiagen). Input (control) samples were isolated similarly and incubated with preclean solution instead of RORα antibody or with rabbit IgG antibody (negative control). Real-time PCR reactions were performed using diluted input, IgG control, or experimental (RORα antibody–precipitated) DNA fragments as template and with primers designed flanking a putative ROR response element (RORE) site in the potential target genes to amplify ∼200-bp products. Enrichment levels significantly higher than nonspecific negative control Hemoglobin β (Hbb) and comparable with or higher than positive control (Opsin 1, medium-wave-sensitive, Opn1mw) were considered to be the positive enrichment threshold.
Luciferase reporter assay
DNA fragment containing putative RORE site in Sema3e promoter regions (covering residues −634 to −638 bp region) was amplified from mouse genomic DNA and then cloned into luciferase report vector pGL2. Mutant construct of Sema3e with deletion in RORE site (−634 to −638 bp) was also constructed with luciferase reporter. The primers for cloning the WT Sema3e promoter were: 5′-AAAAGCTAGCGTCTCAAAGACTATTCCGAAGGATAG-3′ (forward), 5′-AAAACTCGAGCCCTTTCTATGGCCTTTTACCTT T-3′ (reverse). The primers for cloning mutant Sema3e promoter were: 5′-GGATAATCTAAAAACGCGCTGAGGAATCTGAGAGCT T-3′ (forward), 5′-AAGCTCTCAGATTCCTCAGCGCGTTTTTAGATTATCC-3′ (reverse). HEK293T cells were transiently cotransfected with pCDH1-RORα-Flag, WT or mutant pGL2-Sema3e promoter reporter, or Renilla luciferase vector alone as control. SR1001 was synthesized at the Scripps Research Institute as previously described (34–36). SR1001 was dissolved in DMSO, and the pretransfected HEK293T cells were treated overnight. At 36 h after transfection, cells were lysed in passive lysis buffer (Dual-Luciferase assay system; Promega, Fitchburg, WI, USA), and luciferase reporter activities were measured using a Wallac 1420 Multilabel Counter (Perkin Elmer, Boston, MA, USA).
Western blot
A standard immunoblotting protocol was used with minor modifications (25, 37, 38). Briefly, RIPA buffer (Cell Signaling Technology, Danvers, MA, USA) containing protease inhibitor cocktail (78430; Thermo Fisher Scientific, Waltham, MA, USA) was used to lyse the retinal samples or cells. Proteins were separated by electrophoresis using 4–12% NuPage Novex Bis-Tris gels (Invitrogen). The primary antibodies used were β-Actin (A1978; Sigma-Aldrich, Natick, MA, USA) and SEMA3E (SAB2501467; Sigma-Aldrich).
Immunohistochemistry
Immunostaining in retinas was performed as described previously (39–41). Briefly, eyes were isolated from Sg/Sg and WT littermate mice with OIR, fixed, and permeabilized. Retinal cross sections were stained with RORα antibody (ab60134; Abcam) and SEMA3E antibody (SAB2501467; Sigma-Aldrich) and counterstained with DAPI followed by imaging with a Fluoview 1000 confocal microscope (Olympus, Waltham, MA, USA).
Generation of AAV2-shSema3e vector and AAV2 virus
Three independent shRNAs against mouse Sema3e were designed using a published algorithm (42). The sequences of the mouse Sema3e shRNAs were 5′-GCAGTAAAGGTAGAAGAATGC-3′, 5′-GCAAGTATGGAACCACCAAAG-3′, and 5′-GGAGGTGGTCGAAGAGCATAA-3′. The Sema3E shRNAs were cloned into a CAGmiR30-GFP plasmid to confirm the Sema3e knock down efficiency in mouse retinas. Recombinant AAV2 vectors were produced as previously described (43, 44). Briefly, AAV vector, rep/cap packaging plasmid, and adenoviral helper plasmid were mixed with polyethylenimine (Sigma-Aldrich) and transfected into HEK293T cells (HCL4517; Thermo Fisher Scientific). Seventy-two hours after transfection, cells were harvested, and the cell pellet was resuspended in virus buffer followed by 3 cycles of freeze–thaw and homogenization. Cell debris was pelleted by centrifuging at 5000 g for 20 min, and the supernatant containing AAV was separated on an iodixanol gradient. Recovered AAV vectors were washed 3 times with PBS using Amicon 100K columns (EMD Millipore). Real-time PCR was used to determine genome titers of the recombinant AAV2. A similar protocol was carried out to generate a shRNA control AAV (AAV2-shControl). Viruses were diluted to various concentrations to evaluate transfection efficiency, and a concentration of ∼2 × 1012 gc/ml was used for the experiments.
Intravitreal injection
Intravitreal injections were performed by inserting an Exmire microsyringe (MS-NE05; ITO Corp., Medellín, Colombia) with 35-gauge needle into the vitreous 1 mm posterior to the corneal limbus after anesthesia (25, 39). Intravitreal injection of 0.5 µl AAV2-shSema3e was performed at P5 in Sg/Sg and WT mice with identical volume of AAV2-shControl injected in the contralateral eye of the same animal. After exposure to OIR, mice were euthanized at P17 for retinal isolation for RT-qPCR of Sema3e expression and for phenotypic analysis of retinopathy development, including quantification of vaso-obliteration and neovascularization.
SR1001 treatment
SR1001 (25 mg/kg body weight) or vehicle control (10% DMSO/10% Tween80/80% H2O) was injected intraperitoneally in C57BL/6J OIR littermate mice from P12 to P17 twice per day. SR1001 was synthesized at the Scripps Research Institute (25, 35). Mice treated with SR1001 had comparable body weight in normal range as controls. For the SR1001 group, intravitreal injection of 0.5 µl AAV2-shSema3e was performed at P5 in mice with an identical volume of AAV2-shControl injected in the contralateral eye of the same animal, whereas for the vehicle control group, AAV2-shControl was intravietreally injected into both eyes at P5. After exposure to OIR, mice were euthanized at P17, and retinas were isolated for either real-time qPCR measurement of Sema3e expression or quantitative analysis of neovascularization.
Statistics
Results were presented as means ± sem for animal studies (n = the number of retinas quantified). For all statistical analysis, an F test (for 2 samples) or Levene’s test (for more than 2 samples) was performed first to assess whether the variance are homogenous. If the variances were homogenous, 2-tailed Student’s t tests (2 groups) or ANOVA (more than 2 groups) were performed assuming equal variances. For nonhomogenous variances, 2-tailed Student’s t test (or ANOVA) assuming unequal variances was performed. Differences were considered significant if P ≤ 0.05. Data shown are representative of at least 3 independent experiments.
RESULTS
Sema3e was induced in RORα–deficient retinas with OIR
To investigate whether RORα regulates the expression of SEMA3 family members, we used Sg/Sg mice, which have a spontaneous mutation in the Rora gene resulting in a premature stop codon. The resulting protein is truncated, leading to the loss of RORα function (20). Sg/Sg mice and littermate WT mice were exposed to 75% oxygen from P7 to P12 to induce proliferative retinopathy in OIR, and the retinas were collected at P17 for analysis. The mRNA levels of Rora were barely detectable in Sg/Sg retinas (Fig. 1A), confirming RORα deficiency. We found that Sema3e mRNA levels were significantly up-regulated in Sg/Sg vs. WT OIR retinas (Fig. 1A). Protein levels of SEMA3E also showed significant induction (∼2.5-fold) in Sg/Sg OIR retinas compared with WT (Fig. 1B). Screening of other Sema3 family members did not show significant change in mRNA levels of Sema3a, -c, and -f, except for Sema3d (Fig. 1C). These results suggest a potential role of RORα in regulating the expression of Sema3e and -d.
Sema3e was a direct transcriptional target of RORα
RORα controls its target gene expression through binding as a monomer to a core DNA consensus sequence termed the RORE, PuGGTCA (16, 45, 46). When coupled with coactivators or corepressors, binding of RORα to a RORE controls transcription of RORα target genes. The promoter sequences on both Sema3d and -e were analyzed, and multiple potential ROREs were identified (Fig. 2A). Next, chromatin immunoprecipitation (ChIP) assays with a RORα antibody were performed in P17 OIR retinas followed by qPCR to quantitate RORα binding to candidate DNA regions. Among the 6 RORE sites tested, binding to RORE site 1 of Sema3e showed the most significant enrichment compared with IgG control (Fig. 2B). Direct binding of RORα to this Sema3e site was the strongest in the ChIP–qPCR assay (P < 0.001) (Fig. 2B), with almost 2-fold enrichment compared with the positive control Opn1mw, a known RORα target (47). There was no significant enrichment of the other 2 Sema3e RORE sites, supporting the specificity of RORE site 1 for RORα binding to Sema3e. In contrast, no enrichment of Sema3d RORE sites was found. Together these data indicate that RORα directly recognizes and binds to the promoter region of Sema3e but not Sema3d.
Figure 2.
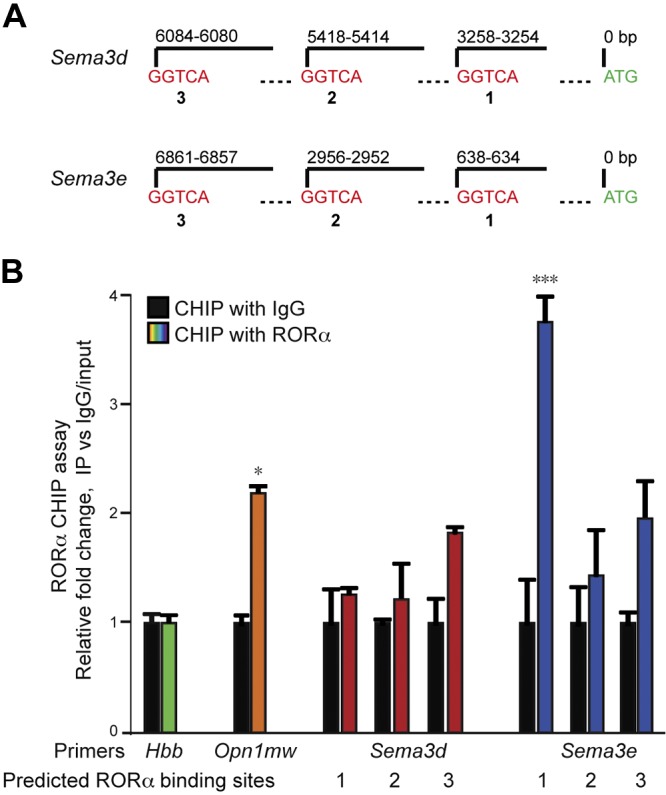
RORα binds to the promoter region of Sema3e. A) Positions of Rora DNA binding elements (i.e., ROREs), with the consensus sequence GGTCA, on the promoters of mouse Sema3d and Sema3e genes. B) ChIP assays were performed with P17 WT OIR retinas, and DNA fragments bound to immunoprecipitated RORα were quantified with qPCR with primers flanking potential RORE sites. RORE site 1 of Sema3e showed the strongest enrichment. None of the other RORE sites in Sema3e and Sema3d analyzed showed significant enrichment compared with IgG control. Data were normalized to IgG control. Primers of Opn1mw was used a positive control, and Hbb was as a negative control (n = 3/group). Data are presented as means ± sem. *P < 0.05, ***P < 0.001.
RORα negatively regulated Sema3e promoter–driven transcription
We next performed luciferase reporter assays to assess the direct effects of RORα on Sema3e promoter activity. Both WT and mutant Sema3e:Luc luciferase reporter constructs were constructed containing the native or mutated RORE site (from −634 to −638 bp) (Fig. 3A) and cotransfected with RORα–expressing vector into HEK293T cells separately. In the WT Sema3e:Luc reporter, expression of RORα significantly reduced the levels of Sema3e promoter activity (>40%) in a dose-dependent manner, as measured by firefly/Renilla luciferase activity (Fig. 3B), but not in mutant Sema3e:Luc reporter, suggesting that binding of RORα to this specific RORE region of Sema3e is critical for its transcriptional suppression.
Figure 3.

Sema3e was a direct transcriptional target of RORα. A) Luciferase reporters with native (WT/Sema3e) or mutated RORα (MUT/Sema3e) binding sites in Sema3e promoter were cloned and cotransfected with varying amounts of RORα–expressing vector into HEK293T cells. RE, responsive element. B) Cotransfection of RORα–expressing vector dose-dependently suppressed WT/Sema3e luciferase expression in pGL2 vector, reflecting the transcriptional activity of WT/Sema3e (covering residues −634 to −638 bp). Luciferase activity from mutant constructs of MUT/Sema3e did not respond to RORα cotransfection. C) SR1001 treatment dose-dependently promoted WT/Sema3e promoter–driven luciferase reporter activity but did not promote MUT/Sema3e RORE reporter constructs (n = 6/group). Data are presented as means ± sem. N.s., no significance. *P < 0.05, **P < 0.01.
RORα regulation of Sema3e transcription was also validated by treatment with a synthetic inverse agonist of RORα/γ, SR1001, which can bind directly to the ligand-binding domain of RORα (35, 36). Binding of SR1001 results in a conformational change that decreases the affinity of RORα for coactivators and increases its affinity for corepressors, leading to inhibition of RORα activity (35, 36). Treatment of SR1001 suppressed pathological retinal neovascularization in the OIR model in our previous study (25). The transcriptional activity of WT/Sema3e:Luc reporter was significantly enhanced with increasing concentrations of SR1001, which showed no significant effects on MUT/Sema3e:Luc reporters (Fig. 3C). Together these results indicate that RORα directly binds to this RORE site in Sema3e promoter to influence Sema3e transcription.
RORα and SEMA3E were inversely expressed in OIR retinas and colocalized in RGCs
To further investigate expression and localization of SEMA3E and RORα in retinopathy, we examined the mRNA expression of Sema3e and Rora at different time points during OIR and found that Sema3e mRNA expression was significantly down-regulated at both P14 and 17 in the hypoxic and proliferative phases, compared with age-matched room air controls (Fig. 4A), consistent with induction of RORα at these time points (25), suggesting inverse regulation of Sema3e and Rora in retinopathy. Consistent with a previous report of SEMA3E expression almost exclusively in retinal RGCs (10, 14), we found that SEMA3E was localized in RGCs and colocalized with RORα in the RGC layer of WT retinas with immunohistochemical staining (Fig. 4B). Together these data suggest that RORα may regulate in retinal RGCs expression of Sema3e, which then acts upon its receptors in pathological neovessels to influence neovascularization.
Figure 4.

RORα and SEMA3E were expressed inversely in OIR and colocalized in RGCs. A) Rora and Sema3e mRNA expression in OIR retinas compared with normoxic retinas at P14 and P17 (n = 6/group). B) RORα and SEMA3E were colocalized in retinal ganglion cell layer. Cross-sections from WT were stained with RORα antibody (green), SEMA3E antibody (red), and nuclear marker DAPI (blue) (n = 6/group). Scale bar, 20 µm. Data are presented as means ± sem. INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; RGC, retinal ganglion cell layer. *P < 0.05.
RORα–SEMA3E axis regulated neurovascular interaction to affect pathological neovascularization in retinopathy
In our previous study, we showed that genetic deficiency of RORα in Sg/Sg mice leads to significant reduction of pathological neovascularization in OIR (25). To determine whether transcriptional suppression of Sema3e by RORα contributes to the vascular effects of RORα in retinal neovascularization, we silenced Sema3e expression with an intravitreal injection of AAV2-carried shRNA targeting Seme3e (AAV2-shSema3e) in both WT and Sg/Sg mice at P5 before exposing mice to OIR. The efficacy of AAV2-shSema3e was validated, showing ∼70% reduction of Sema3e expression in WT retinas compared with AAV2-shControl at P16 with one of the constructs (shSema3e-3), which was used for all subsequent experiments (Fig. 5A). At P17, AAV2-shControl–treated Sg/Sg retinas exhibited significantly reduced pathological neovascularization compared with littermate AAV2-shControl–treated WT retinas in OIR (P < 0.05; n = 8 in WT group, n = 6 in Sg/Sg group) (Fig. 5B, C), similar to the reduced pathological neovessels observed in untreated Sg/Sg vs. WT OIR retinas in our previous study (25). AAV2-shSema3e injection in WT OIR retinas significantly increased the level of neovascularization compared with AAV2-shControl–treated WT retinas (Fig. 5B, C), consistent with a previously suggested role of SEMA3E in suppressing pathological neovessels in OIR (14). Importantly, with AAV2-shSema3e injection in Sg/Sg OIR retinas, pathological neovascularization was significantly increased (P < 0.05; n = 6/group, compared with shControl-treated Sg/Sg retinas) to levels comparable to AAV2-shSema3e–treated WT retinas (no significance; n = 6–8/group). There was no change in vaso-obliteration among all the groups (Fig. 5C). In addition, we evaluated whether AAV2-shSema3e may reverse the pathological neovascularization suppressed by a synthetic inverse agonist of RORα/γ, SR1001 (35), in OIR. SR1001 treatment increased Sema3e expression in the retinas, confirming that RORα inhibition induces Sema3e (Fig. 5D). SR1001 treatment from P12 to P17 significantly reduced pathological neovascularization in WT OIR mice compared with the vehicle-treated control group, consistent with our previous study (25). Moreover, AAV2-shSema3e treatment significantly increased pathological neovascularization in SR1001-treated mice at P17 by about 55% (P < 0.01; n = 11 per group) to the level comparable to littermate vehicle controls (Fig. 5D), further confirming that RORα mediates retinal neovascularization in retinopathy through modulation of Sema3e. Together, these data indicate that the SEMA3E-dependent neurovascular interaction mediates in part the vaso-protective effects of RORα deficiency in retinopathy (Fig. 5E).
Figure 5.

RORα-Sema3e axis regulated neurovascular interaction in pathological retinal angiogenesis. A) AAV2-shSema3e (shSema3e) effectively knocked down retinal Sema3e expression compared with AAV2-sh control (shCtrl) at P16. The knockdown efficiency was validated by real-time qPCR analysis (left) and immunofluorescence analysis (right). AAV2-shSema3e-3 was selected for experiments in panel B (n = 6/group). B) Representative P17 retinal whole-mounts from Sg/Sg and WT mice with OIR treated with AAV2-shSema3e and AAV2-shCtrl in contralateral eyes, stained with isolectin IB4 (red) for blood vessels. Selected retinal areas (white box) were enlarged to show pathological neovessels (bottom panel). Scale bars, 500 μm (top) and 125 μm (bottom). C) The levels of vaso-obliteration (left panel) were comparable between AAV2-shSema3e and AAV2-shCtrl treated P17 OIR retinas in both Sg/Sg and WT groups. Quantified levels of pathological neovascularization (right panel) were significantly decreased in Sg/Sg OIR retinas compared with WT, both with AAV2-shCtrl treatment. AAV2-shSema3e treatment largely abolished the decrease of pathological neovascularization in Sg/Sg OIR retinas compared with AAV2-shCtrl–treated Sg/Sg retinas. Levels of neovascularization were expressed as percentage of total retinal areas (n = 8 in WT group; n = 6 in Sg/Sg group). D) SR1001 treatment induced retinal expression of Sema3e (n = 5/group), which was suppressed by AAV-shSema3e treatment. AAV-shSema3e treatment significantly increased the levels of pathological neovascularization in SR1001-treated OIR mice compared with AAV-shCtrl (n = 11/group). E) Schematic illustration of the role of neuronal RORα in pathological ocular neovascularization. In normal retinas, RORα levels are low and SEMA3E expression levels are high, which suppresses blood vessel growth and maintains quiescence of normal retinal vessels. In retinopathy, RORα induction in RGC represses transcription of SEMA3E, leading to decreased SEMA3E levels and relieving its inhibition of pathological vessel growth, thereby resulting in increased pathological neovascularization. Targeted suppression of RORα may promote SEMA3E signaling–mediated neurovascular interaction and protect pathological neovascularization in retinopathy. Data are presented as means ± sem. BV, blood vessel; n.s., no significance; NV, neovessel; RGC, retinal ganglion cell. *P < 0.05, **P < 0.01, ***P < 0.001.
RORα deficiency does not affect developmental retinal angiogenesis or Sema3e expression during development
Mouse retinal vasculature begins to develop after birth, with the superficial vascular layer extending radially from optic nerve head and reaching the retinal periphery by P7. The deep layers of retinal vessels are fully developed by ∼20 d after birth (48, 49). SEMA3E was suggested to mediate retinal vascular development through regulating VEGF function (10). We next evaluated whether RORα affects normal retinal vessel development via SEMA3E. In contrast to OIR, there was no significant difference in normal retinal vascular development between WT and Sg/Sg retinas at P5, as analyzed by the vascular coverage rate, tip cell filopodia number, and vascular branch density (Supplemental Fig. 1A–C). Consistent with the lack of vascular difference, Sema3e mRNA expression also showed comparable levels between WT and Sg/Sg retinas at P5 (Supplemental Fig. 1D). Together, these results support the notion that RORα regulates SEMA3E-dependent neurovascular interaction in the pathological context of OIR but not during developmental retinal angiogenesis.
DISCUSSION
Neurovascular interactions are important in the maintenance of the nervous system, and dysfunction in this relationship is detrimental to the microenvironmental homeostasis of neurovascular unit and tropical support of neurons in disease, such as stroke (50), Alzheimer’s disease (51), and epilepsy (52). In the retina, which is part of the CNS, accumulating evidence suggests that dysregulated crosstalk between the vasculature and retinal neuroglia, photoreceptors, and other neural cells in diabetes contributes to the pathogenesis of diabetic retinopathy (4–6). SEMA3 is a major class of neuronal and vascular guidance cues that mediate neurovascular coupling in retinopathy. In the mouse model of oxygen-induced proliferative retinopathy, inhibition of SEMA3E-PlexinD1 signaling resulted in an increase of pathological extraretinal neovessels, whereas activation of SEMA3E-PlexinD1 signaling suppressed the outgrowth of extraretinal vessels via acting through PlexinD1 and downstream small GTPase RhoJ signaling, both of which are highly expressed in growing extraretinal vessels (14). In humans, lower aqueous levels of SEMA3E were observed in patients with diabetic retinopathy compared with control groups, and SEMA3E levels were negatively correlated with VEGF levels (53). In addition, inhibition of SEMA3E-PlexinD1 signaling enhances angiogenesis and blood-flow recovery in a mouse model of hind-limb ischemia (54), all suggesting a suppressive role of SEMA3E-PlexinD1 in VEGF-induced angiogenesis and a protective role of SEMA3E-PlexinD1 in ischemic retinopathy against VEGF-induced extraretinal neovascularization. Therapies targeting SEMA3E are potentially promising approaches for the treatment of microvascular diseases, particularly microvascular retinopathies (7). In this study, we identified RORα as a critical upstream regulator of SEMA3E expression, which thereby mediates SEM3E-dependent neurovascular coupling in retinopathy. RORα directly recognizes and binds to a specific ROR response element on the promoter of Sema3e and negatively regulates Sema3e transcription, which can affect retinopathy development via signaling through its receptor complexes located on pathological neovessels (Fig. 5E).
Our findings indicate that RORα and SEMA3E are both localized in RGCs, which play a pivotal role in physiological and pathological retinal angiogenesis (5). In mice lacking RGCs, no retinal vascular network is developed (55). The contribution of RGCs to the maintenance of retinal blood vessels was also established through experimental models of retinal regeneration induced by ischemia-reperfusion (56, 57), where RGC apoptosis and/or thinning of the inner retina precedes subsequent capillary degeneration (56, 58). These findings strongly suggest that RGCs play an important functional role in maintaining the normal structure and function of retinal blood vessels. Our work established a novel transcriptional regulatory mechanism of RORα–SEMA3E axis in the retinal ganglion cell layer that play a significant role in mediating neurovascular crosstalk in pathological retinal angiogenesis during retinopathy. However, RORα does not appear to affect SEMA3E expression during retinal development or developmental angiogenesis, which may be explained by the hypoxia- and hypoxia-inducible factor–responsive nature of RORα (59, 60). It is conceivable that only under certain pathological conditions of ischemia or hypoxia, RORα becomes highly induced to exert suppression on SEMA3E to result in exacerbation of pathological neovascularization, whereas under normal developmental conditions RORα is likely expressed only at a low basal levels that are not sufficient to significantly affect Sema3E expression and developmental retinal angiogenesis. In addition to RGCs, expression of RORα was identified in retinal photoreceptors (26, 27) and macrophages/microglia (25). Whereas expression of RORα was reported in human vascular cells (61, 62), previously we observed only low levels of RORα expression in retinal blood vessels.
Sema3e suppression in Sg/Sg OIR retinas and SR1001-treated WT OIR retinas largely reversed pathological neovascularization suppression by RORα deficiency or inhibition (Fig. 5B, C), yet other mechanisms may also be at work at the same time. Our previous findings showed (25) that macrophage-derived RORα controls retinal pathologic neovascularization through SOCS3-mediated retinal inflammation, indicating a diverse role of RORα in a variety of retinal cell types. Together, these findings suggest that neuronal RORα may synergistically work together with macrophage RORα in mediating pathological neovascularization in oxygen-induced retinopathy through regulating SEMA3E-dependent neurovascular coupling in addition to inflammatory control. In addition to RORα, another member of the ROR family, RORγ, is hypoxia sensitive, and SR1001 is a dual RORα/γ inverse agonist, binding to the ligand binding domains of both RORα and RORγ (35). Suppression of RORγ also suppressed pathological retinal neovascularization through the IL-17A/VEGF axis (63), indicating an increasingly recognized role of these nuclear receptor family proteins in pathological retinal angiogenesis.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Drs. Lois Smith, Zhongjie Fu, and Yan Gong (all from Boston Children’s Hospital) for helpful discussions and Peyton Morss, Thomas Fredrick, and Nicholas Saba (all from Boston Children’s Hospital) for excellent technical assistance. This work was supported by U.S. National Institutes of Health (NIH) National Eye Institute Grant R01 EY024963; the BrightFocus Foundation; a Boston Children’s Hospital Career Development Award; the Boston Children’s Hospital Ophthalmology Foundation and Massachusetts Lions Eye Research Fund (to J.C.), NIH National Institute of Allergy and Infectious Diseases Grant 1R01 AI116885-01A1 (to L.A.S.); and funds from the Margaret Q. Landenberger Research Foundation (to L.A.S.). The authors declare no conflicts of interest.
Glossary
- AAV
adeno-associated virus
- AAV2-shControl
shRNA control AAV
- ChIP
Chromatin immunoprecipitation
- OIR
oxygen-induced retinopathy
- qPCR
quantitative PCR
- RGC
retinal ganglion cell
- RORα
retinoic acid receptor–related orphan receptor α
- RORE
ROR response element
- SEMA
semaphorin
- SEMA3
class 3 semaphorin
- Sg/+
Rora heterozygous Staggerer
- Sg/Sg
RORα–deficient Staggerer
- shRNA
short hairpin RNA
- WT
wild type
Footnotes
This article includes supplemental data. Please visit http://www.fasebj.org to obtain this information.
AUTHOR CONTRIBUTIONS
Y. Sun and J. Chen conceived and designed research, collected and analyzed data, and wrote the manuscript; C.-H. Liu, Z. Wang, S. S. Meng, and S. B. Burnim collected and analyzed data; J. P. SanGiovanni, T. M. Kamenecka, and L. A. Solt provided expert advice and reagents, analyzed data, and wrote the manuscript; and all authors edited and approved the manuscript.
REFERENCES
- 1.Antonetti D. A., Klein R., Gardner T. W. (2012) Diabetic retinopathy. N. Engl. J. Med. 366, 1227–1239 [DOI] [PubMed] [Google Scholar]
- 2.Hartnett M. E., Penn J. S. (2013) Mechanisms and management of retinopathy of prematurity. N. Engl. J. Med. 368, 1162–1163 [DOI] [PubMed] [Google Scholar]
- 3.Chen J., Smith L. E. (2007) Retinopathy of prematurity. Angiogenesis 10, 133–140 [DOI] [PubMed] [Google Scholar]
- 4.Kern T. S. (2014) Interrelationships between the retinal neuroglia and vasculature in diabetes. Diabetes Metab. J. 38, 163–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakahara T., Mori A., Kurauchi Y., Sakamoto K., Ishii K. (2013) Neurovascular interactions in the retina: physiological and pathological roles. J. Pharmacol. Sci. 123, 79–84 [DOI] [PubMed] [Google Scholar]
- 6.Qian H., Ripps H. (2011) Neurovascular interaction and the pathophysiology of diabetic retinopathy. Exp. Diabetes Res. 2011, 693426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Worzfeld T., Offermanns S. (2014) Semaphorins and plexins as therapeutic targets. Nat. Rev. Drug Discov. 13, 603–621 [DOI] [PubMed] [Google Scholar]
- 8.Nasarre P., Gemmill R. M., Drabkin H. A. (2014) The emerging role of class-3 semaphorins and their neuropilin receptors in oncology. Onco Targets Ther. 7, 1663–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Serini G., Bussolino F., Maione F., Giraudo E. (2013) Class 3 semaphorins: physiological vascular normalizing agents for anti-cancer therapy. J. Intern. Med. 273, 138–155 [DOI] [PubMed] [Google Scholar]
- 10.Kim J., Oh W. J., Gaiano N., Yoshida Y., Gu C. (2011) Semaphorin 3E-Plexin-D1 signaling regulates VEGF function in developmental angiogenesis via a feedback mechanism. Genes Dev. 25, 1399–1411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buehler A., Sitaras N., Favret S., Bucher F., Berger S., Pielen A., Joyal J. S., Juan A. M., Martin G., Schlunck G., Agostini H. T., Klagsbrun M., Smith L. E., Sapieha P., Stahl A. (2013) Semaphorin 3F forms an anti-angiogenic barrier in outer retina. FEBS Lett. 587, 1650–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ochsenbein A. M., Karaman S., Proulx S. T., Berchtold M., Jurisic G., Stoeckli E. T., Detmar M. (2016) Endothelial cell-derived semaphorin 3A inhibits filopodia formation by blood vascular tip cells. Development 143, 589–594 [DOI] [PubMed] [Google Scholar]
- 13.Yang W. J., Hu J., Uemura A., Tetzlaff F., Augustin H. G., Fischer A. (2015) Semaphorin-3C signals through Neuropilin-1 and PlexinD1 receptors to inhibit pathological angiogenesis. EMBO Mol. Med. 7, 1267–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukushima Y., Okada M., Kataoka H., Hirashima M., Yoshida Y., Mann F., Gomi F., Nishida K., Nishikawa S., Uemura A. (2011) Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Invest. 121, 1974–1985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyal J. S., Sitaras N., Binet F., Rivera J. C., Stahl A., Zaniolo K., Shao Z., Polosa A., Zhu T., Hamel D., Djavari M., Kunik D., Honoré J. C., Picard E., Zabeida A., Varma D. R., Hickson G., Mancini J., Klagsbrun M., Costantino S., Beauséjour C., Lachapelle P., Smith L. E., Chemtob S., Sapieha P. (2011) Ischemic neurons prevent vascular regeneration of neural tissue by secreting semaphorin 3A. Blood 117, 6024–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jetten A. M. (2009) Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal. 7, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giguère V., Tini M., Flock G., Ong E., Evans R. M., Otulakowski G. (1994) Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 8, 538–553 [DOI] [PubMed] [Google Scholar]
- 18.Hirose T., Smith R. J., Jetten A. M. (1994) ROR gamma: the third member of ROR/RZR orphan receptor subfamily that is highly expressed in skeletal muscle. Biochem. Biophys. Res. Commun. 205, 1976–1983 [DOI] [PubMed] [Google Scholar]
- 19.Kojetin D. J., Burris T. P. (2014) REV-ERB and ROR nuclear receptors as drug targets. Nat. Rev. Drug Discov. 13, 197–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamilton B. A., Frankel W. N., Kerrebrock A. W., Hawkins T. L., FitzHugh W., Kusumi K., Russell L. B., Mueller K. L., van Berkel V., Birren B. W., Kruglyak L., Lander E. S. (1996) Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 379, 736–739 [DOI] [PubMed] [Google Scholar]
- 21.Steinmayr M., André E., Conquet F., Rondi-Reig L., Delhaye-Bouchaud N., Auclair N., Daniel H., Crépel F., Mariani J., Sotelo C., Becker-André M. (1998) Staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc. Natl. Acad. Sci. USA 95, 3960–3965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kallen J., Schlaeppi J. M., Bitsch F., Delhon I., Fournier B. (2004) Crystal structure of the human RORalpha Ligand binding domain in complex with cholesterol sulfate at 2.2 A. J. Biol. Chem. 279, 14033–14038 [DOI] [PubMed] [Google Scholar]
- 23.Kamata Y., Tominaga M., Sakaguchi A., Umehara Y., Negi O., Ogawa H., Takamori K. (2015) Retinoid-related orphan receptor α is involved in induction of semaphorin 3A expression in normal human epidermal keratinocytes. J. Dermatol. Sci. 79, 84–86 [DOI] [PubMed] [Google Scholar]
- 24.Xiong G., Wang C., Evers B. M., Zhou B. P., Xu R. (2012) RORα suppresses breast tumor invasion by inducing SEMA3F expression. Cancer Res. 72, 1728–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sun Y., Liu C. H., SanGiovanni J. P., Evans L. P., Tian K. T., Zhang B., Stahl A., Pu W. T., Kamenecka T. M., Solt L. A., Chen J. (2015) Nuclear receptor RORα regulates pathologic retinal angiogenesis by modulating SOCS3-dependent inflammation. Proc. Natl. Acad. Sci. USA 112, 10401–10406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ino H. (2004) Immunohistochemical characterization of the orphan nuclear receptor ROR alpha in the mouse nervous system. J. Histochem. Cytochem. 52, 311–323 [DOI] [PubMed] [Google Scholar]
- 27.Tosini G., Davidson A. J., Fukuhara C., Kasamatsu M., Castanon-Cervantes O. (2007) Localization of a circadian clock in mammalian photoreceptors. FASEB J. 21, 3866–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith L. E., Wesolowski E., McLellan A., Kostyk S. K., D’Amato R., Sullivan R., D’Amore P. A. (1994) Oxygen-induced retinopathy in the mouse. Invest. Ophthalmol. Vis. Sci. 35, 101–111 [PubMed] [Google Scholar]
- 29.Michan S., Juan A. M., Hurst C. G., Cui Z., Evans L. P., Hatton C. J., Pei D. T., Ju M., Sinclair D. A., Smith L. E., Chen J. (2014) Sirtuin1 over-expression does not impact retinal vascular and neuronal degeneration in a mouse model of oxygen-induced retinopathy. PLoS One 9, e85031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu C. H., Wang Z., Sun Y., SanGiovanni J. P., Chen J. (2016) Retinal expression of small non-coding RNAs in a murine model of proliferative retinopathy. Sci. Rep. 6, 33947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stahl A., Connor K. M., Sapieha P., Willett K. L., Krah N. M., Dennison R. J., Chen J., Guerin K. I., Smith L. E. (2009) Computer-aided quantification of retinal neovascularization. Angiogenesis 12, 297–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stahl A., Chen J., Sapieha P., Seaward M. R., Krah N. M., Dennison R. J., Favazza T., Bucher F., Löfqvist C., Ong H., Hellström A., Chemtob S., Akula J. D., Smith L. E. (2010) Postnatal weight gain modifies severity and functional outcome of oxygen-induced proliferative retinopathy. Am. J. Pathol. 177, 2715–2723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Connor K. M., Krah N. M., Dennison R. J., Aderman C. M., Chen J., Guerin K. I., Sapieha P., Stahl A., Willett K. L., Smith L. E. (2009) Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 4, 1565–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y., Solt L. A., Kojetin D. J., Burris T. P. (2012) Regulation of p53 stability and apoptosis by a ROR agonist. PLoS One 7, e34921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Solt L. A., Kumar N., Nuhant P., Wang Y., Lauer J. L., Liu J., Istrate M. A., Kamenecka T. M., Roush W. R., Vidović D., Schürer S. C., Xu J., Wagoner G., Drew P. D., Griffin P. R., Burris T. P. (2011) Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 472, 491–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Solt L. A., Banerjee S., Campbell S., Kamenecka T. M., Burris T. P. (2015) ROR inverse agonist suppresses insulitis and prevents hyperglycemia in a mouse model of type 1 diabetes. Endocrinology 156, 869–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z., Liu C. H., Sun Y., Gong Y., Favazza T. L., Morss P. C., Saba N. J., Fredrick T. W., He X., Akula J. D., Chen J. (2016) Pharmacologic activation of wnt signaling by lithium normalizes retinal vasculature in a murine model of familial exudative vitreoretinopathy. Am. J. Pathol. 186, 2588–2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Liu C. H., Sun Y., Gong Y., Fu Z., Evans L. P., Tian K. T., Juan A. M., Hurst C. G., Mammoto A., Chen J. (2014) Endothelial TWIST1 promotes pathological ocular angiogenesis. Invest. Ophthalmol. Vis. Sci. 55, 8267–8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen J., Connor K. M., Aderman C. M., Willett K. L., Aspegren O. P., Smith L. E. (2009) Suppression of retinal neovascularization by erythropoietin siRNA in a mouse model of proliferative retinopathy. Invest. Ophthalmol. Vis. Sci. 50, 1329–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen J., Michan S., Juan A. M., Hurst C. G., Hatton C. J., Pei D. T., Joyal J. S., Evans L. P., Cui Z., Stahl A., Sapieha P., Sinclair D. A., Smith L. E. (2013) Neuronal sirtuin1 mediates retinal vascular regeneration in oxygen-induced ischemic retinopathy. Angiogenesis 16, 985–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun Y., Ju M., Lin Z., Fredrick T. W., Evans L. P., Tian K. T., Saba N. J., Morss P. C., Pu W. T., Chen J., Stahl A., Joyal J. S., Smith L. E. (2015) SOCS3 in retinal neurons and glial cells suppresses VEGF signaling to prevent pathological neovascular growth. Sci. Signal. 8, ra94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park Y. K., Park S. M., Choi Y. C., Lee D., Won M., Kim Y. J. (2008) AsiDesigner: exon-based siRNA design server considering alternative splicing. Nucleic Acids Res. 36, W97–W103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grieger J. C., Choi V. W., Samulski R. J. (2006) Production and characterization of adeno-associated viral vectors. Nat. Protoc. 1, 1412–1428 [DOI] [PubMed] [Google Scholar]
- 44.Vandenberghe L. H., Xiao R., Lock M., Lin J., Korn M., Wilson J. M. (2010) Efficient serotype-dependent release of functional vector into the culture medium during adeno-associated virus manufacturing. Hum. Gene Ther. 21, 1251–1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jetten A. M., Kurebayashi S., Ueda E. (2001) The ROR nuclear orphan receptor subfamily: critical regulators of multiple biological processes. Prog. Nucleic Acid Res. Mol. Biol. 69, 205–247 [DOI] [PubMed] [Google Scholar]
- 46.Lechtken A., Zündorf I., Dingermann T., Firla B., Steinhilber D. (2006) Overexpression, refolding, and purification of polyhistidine-tagged human retinoic acid related orphan receptor RORalpha4. Protein Expr. Purif. 49, 114–120 [DOI] [PubMed] [Google Scholar]
- 47.Fujieda H., Bremner R., Mears A. J., Sasaki H. (2009) Retinoic acid receptor-related orphan receptor alpha regulates a subset of cone genes during mouse retinal development. J. Neurochem. 108, 91–101 [DOI] [PubMed] [Google Scholar]
- 48.Stahl A., Connor K. M., Sapieha P., Chen J., Dennison R. J., Krah N. M., Seaward M. R., Willett K. L., Aderman C. M., Guerin K. I., Hua J., Löfqvist C., Hellström A., Smith L. E. (2010) The mouse retina as an angiogenesis model. Invest. Ophthalmol. Vis. Sci. 51, 2813–2826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J., Liu C.-H., Sapieha P. (2016) Retinal vascular development. In Anti-Angiogenic Therapy in Ophthalmology (Stahl A., ed.), pp. 1–19, Springer International Publishing, New York: [Google Scholar]
- 50.Guo S., Kim W. J., Lok J., Lee S. R., Besancon E., Luo B. H., Stins M. F., Wang X., Dedhar S., Lo E. H. (2008) Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc. Natl. Acad. Sci. USA 105, 7582–7587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Iadecola C. (2004) Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat. Rev. Neurosci. 5, 347–360 [DOI] [PubMed] [Google Scholar]
- 52.Nishijima T., Piriz J., Duflot S., Fernandez A. M., Gaitan G., Gomez-Pinedo U., Verdugo J. M., Leroy F., Soya H., Nuñez A., Torres-Aleman I. (2010) Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 67, 834–846 [DOI] [PubMed] [Google Scholar]
- 53.Kwon S. H., Shin J. P., Kim I. T., Park D. H. (2015) Aqueous levels of angiopoietin-like 4 and semaphorin 3E correlate with nonperfusion area and macular volume in diabetic retinopathy. Ophthalmology 122, 968–975 [DOI] [PubMed] [Google Scholar]
- 54.Moriya J., Minamino T., Tateno K., Okada S., Uemura A., Shimizu I., Yokoyama M., Nojima A., Okada M., Koga H., Komuro I. (2010) Inhibition of semaphorin as a novel strategy for therapeutic angiogenesis. Circ. Res. 106, 391–398 [DOI] [PubMed] [Google Scholar]
- 55.Sapieha P., Sirinyan M., Hamel D., Zaniolo K., Joyal J. S., Cho J. H., Honoré J. C., Kermorvant-Duchemin E., Varma D. R., Tremblay S., Leduc M., Rihakova L., Hardy P., Klein W. H., Mu X., Mamer O., Lachapelle P., Di Polo A., Beauséjour C., Andelfinger G., Mitchell G., Sennlaub F., Chemtob S. (2008) The succinate receptor GPR91 in neurons has a major role in retinal angiogenesis. Nat. Med. 14, 1067–1076 [DOI] [PubMed] [Google Scholar]
- 56.Zheng L., Gong B., Hatala D. A., Kern T. S. (2007) Retinal ischemia and reperfusion causes capillary degeneration: similarities to diabetes. Invest. Ophthalmol. Vis. Sci. 48, 361–367 [DOI] [PubMed] [Google Scholar]
- 57.Ueda K., Nakahara T., Hoshino M., Mori A., Sakamoto K., Ishii K. (2010) Retinal blood vessels are damaged in a rat model of NMDA-induced retinal degeneration. Neurosci. Lett. 485, 55–59 [DOI] [PubMed] [Google Scholar]
- 58.Ueda K., Nakahara T., Mori A., Sakamoto K., Ishii K. (2013) Protective effects of TGF-β inhibitors in a rat model of NMDA-induced retinal degeneration. Eur. J. Pharmacol. 699, 188–193 [DOI] [PubMed] [Google Scholar]
- 59.Miki N., Ikuta M., Matsui T. (2004) Hypoxia-induced activation of the retinoic acid receptor-related orphan receptor alpha4 gene by an interaction between hypoxia-inducible factor-1 and Sp1. J. Biol. Chem. 279, 15025–15031 [DOI] [PubMed] [Google Scholar]
- 60.Kim E. J., Yoo Y. G., Yang W. K., Lim Y. S., Na T. Y., Lee I. K., Lee M. O. (2008) Transcriptional activation of HIF-1 by RORalpha and its role in hypoxia signaling. Arterioscler. Thromb. Vasc. Biol. 28, 1796–1802 [DOI] [PubMed] [Google Scholar]
- 61.Besnard S., Heymes C., Merval R., Rodriguez M., Galizzi J. P., Boutin J. A., Mariani J., Tedgui A. (2002) Expression and regulation of the nuclear receptor RORalpha in human vascular cells. FEBS Lett. 511, 36–40 [DOI] [PubMed] [Google Scholar]
- 62.Migita H., Satozawa N., Lin J. H., Morser J., Kawai K. (2004) RORalpha1 and RORalpha4 suppress TNF-alpha-induced VCAM-1 and ICAM-1 expression in human endothelial cells. FEBS Lett. 557, 269–274 [DOI] [PubMed] [Google Scholar]
- 63.Talia D. M., Deliyanti D., Agrotis A., Wilkinson-Berka J. L. (2016) Inhibition of the nuclear receptor RORγ and interleukin-17A suppresses neovascular retinopathy: involvement of immunocompetent microglia. Arterioscler. Thromb. Vasc. Biol. 36, 1186–1196 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.