

Abstract
An enzymatic strategy was developed to generate asymmetrically branched N-glycans from natural sources by using a panel of glycosidases and glycosyltransferases. Briefly, LacZ β-galactosidase was employed to selectively trim symmetrically branched N-glycans isolated from bovine fetuin. The yielding stuctures were then converted to asymetrically branched core structures by robust glycosyltransferase for further extension.
Graphical Abstract
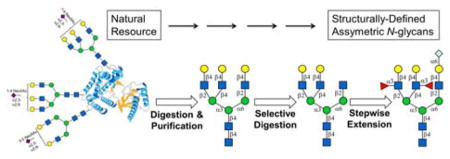
Using a set of glycosidases and glycosyltransferases, an enzymatic strategy was developed to prepare asymmetric N-glycans excluding any chemical procedures.
Asparagine-linked glycosylation (N-glycosylation) is one of the most important post-translational modifications of eukaryotic proteins, and the N-glycan structures have been shown to modulate protein’s structure and function in many ways. For example, N-glycans were found to play key roles in a variety of biological processes, including cell adhesion, immune responses, tumor metastasis, etc.1 On the other hand, N-glycans can influence proteins folding, stability, and immunogenicity, among others.1c, 2 Thus, elucidating the structures and functions of N-glycans is essential for understanding related biological and pathological processes, and access of structurally defined N-glycans is prerequisite for this purpose.
The natural complexity in the biosynthesis of (N-)glycans has led to the generation of very diverse and complex glycan structures.3 Take mouse zona pellucida glycoprotein 3 as an example, 58 distinct N-glycan structures were identified associated with a single glycosylation site (named N-glycan microheterogeneity).4 Another representative example is that 13 high-mannose N-glycans structures were identified within 3 glycomers (Man5GlcNAc2, Man7GlcNAc2 and Man8GlcNAc2) released from bovine ribonuclease B.5 Modern glycobiology studies have indicated that N-glycans with minor structural differences may exhibit distinct protein binding profiles,6 and certain structures may play essential roles in various biological processes,1b, 7 However, in-depth study of such roles have been largely limited by the lack of structurally defined N-glycans in adequate amount.
Current methods for the separation of N-glycan structures have been focusing on small quantities for analytical purposes.8 And due to the diversity and complexity, as well as low abundance of most structures, it is extremely challenging to isolate adequate amounts of N-glycans with specific structures from natural sources. On the other hand, despite numerous efforts on chemical synthesis of N-glycans,9 only a few groups had recently developed strategies for synthesizing libraries of structurally defined N-glycans, including Boons’s general strategy for chemoenzymatic synthesis of asymmetric N-glycans,6, 10 Wong’s modular synthesis of HIV N-glycans,11 and our core synthesis/enzymatic extension (CSEE) strategy to prepare N-glycan isomers.12 Nevertheless, all strategies require expertise and proficient skills for chemical synthesis of key asymmetric N-glycan core or modular structures for further enzymatic diversification.
Exclusively, starting with sialylglycopeptide (SGP) isolated from egg yolk, we were able to generate 36 symmetric bi-antennary N-glycans.10b Herein, we introduce a novel enzymatic strategy to generate asymmetric N-glycans excluding any chemical synthesis. Briefly, two symmetric N-glycan structures, N001 and N301 (Fig. 1A) were firstly separated and purified from bovine fetuin in milligram scale, followed by selective degalactosylation and glycosylation to generate a panel of asymmetric N-glycan core structures (Fig. 1B), which can be further enzymatically extended to asymmetric bi-, tri- and tetra-antennary structures as described previously.6, 12a
Figure 1.

Symmetric (A) and asymmetric (B) N-glycan core structures for the generation of N-glycan libraries. N111 was not prepared in this study.
Isolation of homogenous symmetric N-glycans N001 and N301 from bovine fetuin
Bovine fetuin is a serum glycoprotein that contributes to the attachment and spreading of cells, which thus has been widely used in cell culture. Bovine fetuin is also considered as a golden standard in glycomics and glycoanalysis, as glycan moieties (6 glycosylation sites) accounts for over one fourth of its molecular weight.13 The 3 N-glycans represent 80% of total carbohydrates, and the 3 O-glycans represent the remaining 20%.13 Among the N-glycans, totally 23 structures were previously identified, including around 80% tri-antennary ones (mass ratio) with one to four Neu5Ac attached (either α2,3-linked or α2,6-linked) to core structure N301, and 16% with zero to two Neu5Ac attached to N001.13 No more monosaccharide residues were found in fetuin other than Neu5Ac, Gal, GlcNAc and Man. While it is challenging to isolate homogenous forms of each glycans in preparative scale, it could be much practical to get mg to grams of core structures N301 and N001 in pure form after glycan trimming.
To test this, 400 mg of bovine fetuin was treated with PNGaseF (release N-glycans, monitored by SDS-PAGE) and neuraminidase from Clostridium perfringens (remove Neu5Ac, monitored by HPLC analysis of N-glycans) in one pot for two days (Supplementary Information III, Fig. S2). Two major N-glycan peaks (Fig. 2A) were found on the HPLC profile (running condition: Waters amide column: 130 Å, 5 μm, 4.6 mm × 250 mm; Solvent A : Water; Solvent B: Acetonitrile; Gradient elution B%: 65% to 50% within 25 min; Flow rate: 1 mL/min; monitor by UV210nm) of the product, and MALDI-MS analysis showed that Peak 1(retention time = 15.0 min) and Peak 2 (retention time = 17.5 min) have m/z of 1663.5902 and 2028.7246 (Fig. 2B&C), corresponding to molecular weight of N001 [M + Na]+ and N301 [M + Na]+, respectively. Please note that, due to existence of interconverting α- and β-anomers of glycans with free reducing end in solution,14 two adjacent bumps or a large shoulder can usually be observed on the glycan peak in HPLC analysis. Such a phenomenon was found and reported repeatedly in HPLC analysis of N-glycans.10a, 12, 15 To purify the two glycans, fetuin protein were firstly precipitated by the addition of ice-cold acetone (3 volumes). After brief centrifugation, the supernatant was then concentrated and subjected to G-25 gel filtration (2.5 cm×100 cm) for rough separation. The N-glycan containing fractions (analyzed by MALDI-MS, Fig. S3) were then subject to HPLC purification as previously reported.12a Totally, 4.1 mg (63% yield, Supplemental Information III) of compound corresponding to peak 1 and 26 mg (65% yield) of compound corresponding to peak 2 were collected. Extensive NMR characterization (Supplemental Information IX) confirmed peak 1 as N001 and peak 2 as N301. It is worth to note that, possible β1,3-linked Gal on β1,4-GlcNAc branch and/or core-fucose were observed on N301,13, 16 however, we did not find any signals corresponding to them on NMR spectra of purified N-glycans, nor did we find impurity peaks on MS and HPLC profiles. One possible explanation is that only minor amount of such glycans exist in bovine fetuin, and HPLC purification steps eliminated these structures.
Figure 2.

HPLC analysis of PNGaseF and neuraminidase treated bovine fetuin (A), and MALDI-MS analysis of HPLC peak 1 (B) and peak 2 (C). The two adjacent bumps of peak 1 and peak 2 represent α- and β-anomer of the free reducing end glycans.
Selective degalactosylation of N001
Branch specificity of several β-galactosidases towards Galβ1,4-GlcNAcβ1,2-Manα1,6-(Galβ1,4-GlcNAcβ1,2-Manα1,3-)Manβ1,4-GlcNAc (N001 without reducing end GlcNAc) were tested, with only Escherichia coli β-galactosidase (LacZ) exhibiting branch preferences (prefers α1,3-Man branch).17 Interestingly, LacZ also had a branch preference towards Galβ1,4-GlcNAcβ1,6-(Galβ1,4-GlcNAcβ1,3-)Gal (prefers β1,6-GlcNAc branch) but not to Galβ1,4-GlcNAcβ1,6-(Galβ1,4-GlcNAcβ1,3-)Galβ1,4-GlcNAc.17–18 For this reason, we first performed a substrate specificity assay of LacZ towards a pair of N-glycan isomers N111 and N211 (Fig. 1).12a The results (Supplemental Information VI) showed that LacZ digested N111 much faster than N211, confirming LacZ has a preference toward the α1,3-Man branch.
Next, we monitored the digestion reaction of N001 by LacZ to determine the best time to collect mono-agalactosylated N001 (N111/N211) (Supplementary Information VII). Result showed that with appropriate amounts of LacZ, after 6 hours, all N001 was digested and converted into mono-agalactosylated N001 (85%) and di-agalactosylated N001 (N000, 15%) (Fig. S6). Milligram scale LacZ digestion of N001 were then performed and mono-agalactosylated N001 (Fig. 3B, MALDI-MS m/z peak of 1501.5210) were purified by HPLC as previously reported.12a As our HPLC method cannot distinguish N111 and N211, we applied an enzymatic strategy, using two human glycosyltransferases, α1,6-fucosyltransferase (FUT8) and mannosyl-α1,6-glycoprotein β1,6-N-acetyl-glucosaminyltransferase (MGAT5, also known as GnT-V). FUT8 strictly requires a free GlcNAc on the α1,3-Man branch of N-glycans for activity,19 and MGAT5 requires a free GlcNAc on the α1,6-Man branch.20 We also did detailed substrate specificity study of FUT812b and MGAT5 (Supplementary Information V, Fig. S4) using a library of synthetic N-glycans, which double confirmed previous reports.19–20 Thus, as illustrated in Fig. 3A, FUT8 can fucosylate N211 but not N111, whereas MGAT5 can glycosylate N111 but not N211. MALDI-MS analysis showed that all N-glycans were fucosylated (Fig. 3C, m/z peak corresponding to starting material disappeared, new m/z peak of 1647.5821 corresponding to N6211 [M + Na]+ appeared) in FUT8-catalyzed reaction, whereas no new peak was found corresponding to N4111 in MGAT5-catalyzed reaction (Fig. 3D). These results confirmed that the purified LacZ digestion product of N001 is N211 in pure form.
Figure 3.
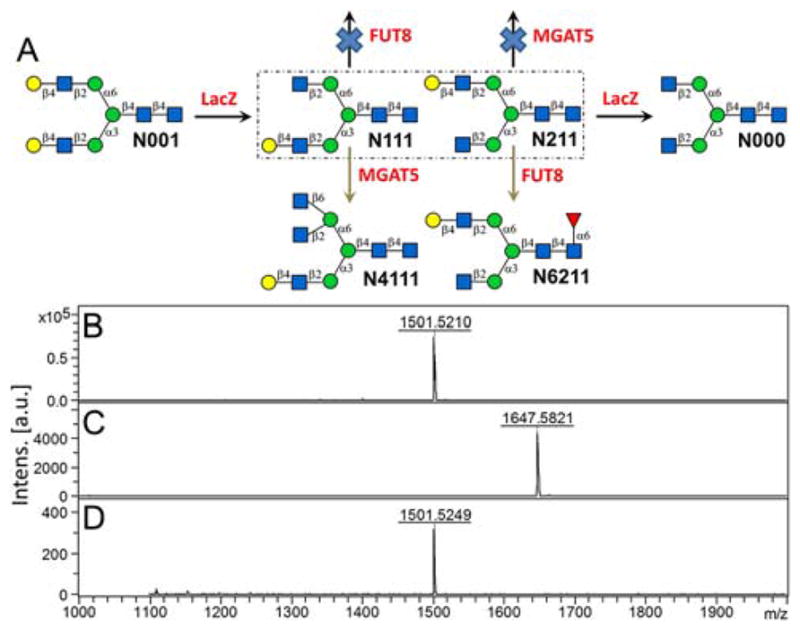
Rationale of enzymatic N-glycan structure identification of monoagalactosylated N001 (A); and MALDI-MS analysis of mono-agalactosylated N001 (B), mono-agalactosylated N001 + FUT8 (C), mono-agalactosylated N001 + MGAT5 (D). LacZ, E. coli β-galactosidase; FUT8, human α1,6-fucosyltransferase; MGAT5, human mannosyl-α1,6-glycoprotein β1,6-N-acetyl-gucosaminyltransferase.
Selective degalactosylation of N301
Degalactosylation of N301 was slower than that of N001. After 3 days of LacZ incubation, 4 peaks corresponding to N-glycans were observed on HPLC chromatograph (Fig. S7). We then performed degalactosylation of N301 in milligram scale (20 mg), with each peak purified by semi-preparative HPLC. MALDI-MS analysis indicated that peak I (10%), II (73%), III (10%), and IV (7%) are corresponding to N301, mono-agalactosylated N301 (N391 or N392 or N393 or mixture, m/z peak of 1866.6707 as shown in Fig. 4B), di-agalactosylated N301 (N381 or N382 or N383 or mixture, m/z peak of 1704.6011 as shown in Fig. 4E) and N300, respectively. FUT8- and MGAT5-catalyzed reactions were performed to identify structures of agalactosylated N301 as illustrated in Fig. 4A. In MGAT5-catalyzed reaction m/z peaks of 2069.7421 and 2086.5037 corresponding to N4391 [M + Na]+ and [M + K]+ were observed while the peak corresponding to mono-agalactosylated N301 disappeared (Fig. 4C), but in FUT8-catalyzed reaction, a product m/z peak corresponding to N6393 was not observed (Fig. 4D). Such results indicated that purified mono-agalactosylated N301 is N391 in pure form. The structure was further confirmed by NMR characterization (Supplementary Information IX). Similarly, for di-agalactosylated N301, both MGAT5- and FUT8-catalyzed reactions yielded new m/z peaks on MALDI-MS profiles while the m/z peak of the starting glycan disappeared (Fig. 4F&G), suggesting that purified di-agalactosylated N301 is pure N382.
Figure 4.

Rationale of enzymatic N-glycan structure identification of mono- and di-agalactosylated N301 (A); and MALDI-MS analysis of mono-agalactosylated N301 (B), mono-agalactosylated N001 + MGAT5 (C), mono-agalactosylated N001 + FUT8 (D), di-agalactosylated N301 (E), di-agalactosylated N001 + MGAT5 (F), di-agalactosylated N001 + FUT8 (G). LacZ, E. coli β-galactosidase; FUT8, human α1,6-fucosyltransferase; MGAT5, human mannosyl-α1,6-glycoprotein β1,6-N-acetyl-gucosaminyltransferase.
These results indicated that LacZ prefers to digest the Gal residue on the α1,6-Man branch of tri-antennary N-glycan N301, and then the Gal residue on the β1,2-GlcNAcα1,3-Man branch, different from that towards bi-antennary N-glycan N001, which prefers to digest Gal residue on the α1,3-Man branch. Nevertheless, LacZ was successfully used to generate asymmetric bi- and tri-antennary N-glycan core structures (N211, N391 and N382) in mg scales.
Generation of other core structures and asymmetric N-glycans
Several other asymmetric N-glycan core structures were generated (Fig. S8), including N4391 that derived from N391 via MGAT5-catalyzed reaction, as well as N4382 and N6382 that derived from N382 via MGAT5 and FUT8-catalyzed reactions (Supplementary Information VIII and IV). These core structures are readily available for the synthesis of libraries of asymmetric complex N-glycans. For example, N211 had been used as a core to generate a number of bi-antennary N-glycan isomers with or without core-fucosylation.12 Taking the tri-antennary core N391 as another example, stepwise enzymatic synthesis was performed to generate complex asymmetric N-glycans such as N396 (a similar asymmetric tri-antennary N-glycan was prepared previously via chemoenzymatic approach). As shown in Fig. 5, firstly, in a 0.5 mL reaction system, 4 mg (4 mM) of N391was incubated with GDP-Fuc (10 mM), MnCl2 (5 mM), and 0.1 mg of Helicobacter pylori α1,3-fucosyltransferase (Hp3FT).12a One microliter of the reaction mixture was sampled every 3 hours for analysis. MALDI-MS analysis showed a peak at m/z = 2158.7823, corresponding to N394 [M + Na]+ (Fig. 5B). After 20 hours, the reaction was freeze quenched at −80 °C for 30 min, and condensed for HPLC purification using a water/acetonitrile gradient elution, yielding 4.2 mg of N394 (90% yield). The purified N394 (3 mg) was then utilized for enzymatic syntheses of N396 in two steps, catalyzed by bovine β1,4-galactosyltransferase (bGalT)12a and Photobacterium damselae α2, 6-sialyltransferase (Pd26ST),21 respectively (Supplementary Information VIII for details). NMR analysis was performed to confirm the structure of the final product (2.5 mg, 93% yield for step 1, 85% yield for step 2) (Supplementary Information IX).
Figure 5.
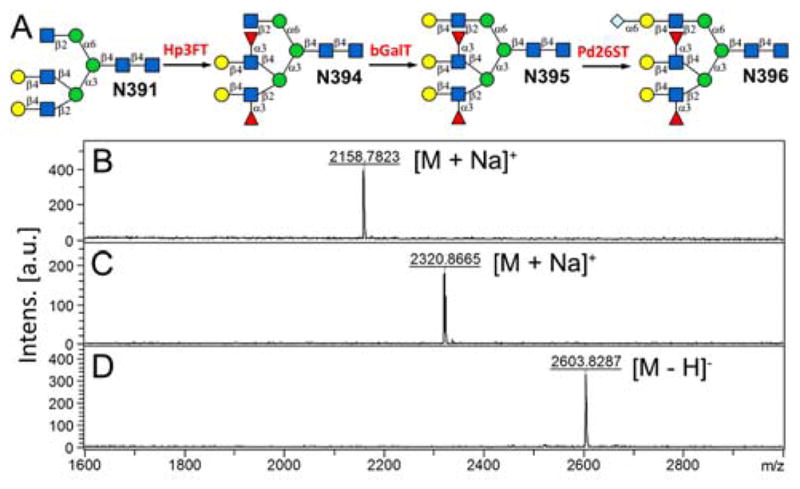
Enzymatic extension of N391 to generate asymmetric tri-antennary glycans (A) and MALDI-MS analysis of N394 (B), N395 (C) and N396 (D). Hp3FT, Helicobacter pylori α1,3-fucosyltransferase; bGalT, bovine β1,3-galactosyltransferase; Pd26ST, Photobacterium damslae a2,6-sialyltransferase.
In summary, we have developed a pure enzymatic strategy to generate asymmetric complex N-glycans excluding chemical procedures. The strategy includes 1) glycosidase (PNGaseF and neuraminidase) assisted separation of bi- and tri-antennary N-glycans in mg scale; 2) β-galactosidase mediated selective degalactosylation and human FUT8- and MGAT5-catalyzed glycosylation to generate asymmetric N-glycan core structures. The yielded core structures can be readily utilized in the synthesis of asymmetrically branched bi-, tri- and tetra-antennary N-glycans. Large-scale preparation of asymmetrically branched N-glycans employing this strategy is undergoing.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health (U01GM0116263 to P.G. Wang and L. Li), National Science Foundation of China (31200605 to W. Guan) and the Key Grant Project of Chinese Ministry of Education (313033). A. D. Calderon is grateful to Graduate Assistance in Areas of National Need (GAANN P200A150308). We are grateful to Dr. Donald Jarvis from the University of Wyoming for providing FUT8 and MGAT5 baculovirus stock (Glycorepository project supported by NIH-NIGMS P41GM103390). We thank Dr. Xi Chen from UC Davis for providing α2,6-sialyltransferase.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental details for expression, purification and substrate specificity study of MGAT5, isolation and digestion of symmetrically branched N-glycans, as well as synthesis, purification and characterization of N-glycans. See DOI: 10.1039/x0xx00000x
References
- 1.(a) Varki A, Lowe JB. In: Essentials of Glycobiology. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]; (b) Zhao YY, Takahashi M, Gu JG, Miyoshi E, Matsumoto A, Kitazume S, Taniguchi N. Cancer Sci. 2008;99:1304. doi: 10.1111/j.1349-7006.2008.00839.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Dwek RA. Chem Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 2.(a) Imperiali B, O’Connor SE. Curr Opin Chem Biol. 1999;3:643. doi: 10.1016/s1367-5931(99)00021-6. [DOI] [PubMed] [Google Scholar]; (b) Helenius A, Aebi M. Science. 2001;291:2364. doi: 10.1126/science.291.5512.2364. [DOI] [PubMed] [Google Scholar]; (c) Helenius A, Aebi M. Annu Rev Biochem. 2004;73:1019. doi: 10.1146/annurev.biochem.73.011303.073752. [DOI] [PubMed] [Google Scholar]
- 3.Krasnova L, Wong CH. Annu Rev Biochem. 2016;85:599. doi: 10.1146/annurev-biochem-060614-034420. [DOI] [PubMed] [Google Scholar]
- 4.Stanley P, Schachter H, Taniguchi N. In: Essentials of Glycobiology. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- 5.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. J Am Soc Mass Spectrom. 2009;20:539. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Chinoy ZS, Ambre SG, Peng W, McBride R, de Vries RP, Glushka J, Paulson JC, Boons GJ. Science. 2013;341:379. doi: 10.1126/science.1236231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Takamatsu S, Shimomura M, Kamada Y, Maeda H, Sobajima T, Hikita H, Iijima M, Okamoto Y, Misaki R, Fujiyama K, Nagamori S, Kanai Y, Takehara T, Ueda K, Kuroda S, Miyoshi E. Glycobiology. 2016;26:1180. doi: 10.1093/glycob/cww067. [DOI] [PubMed] [Google Scholar]; (b) Pang PC, Chiu PC, Lee CL, Chang LY, Panico M, Morris HR, Haslam SM, Khoo KH, Clark GF, Yeung WS, Dell A. Science. 2011;333:1761. doi: 10.1126/science.1207438. [DOI] [PubMed] [Google Scholar]
- 8.(a) Anumula KR. Anal Biochem. 2000;283:17. doi: 10.1006/abio.2000.4645. [DOI] [PubMed] [Google Scholar]; (b) Harvey DJ. Proteomics. 2001;1:311. doi: 10.1002/1615-9861(200102)1:2<311::AID-PROT311>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]; (c) Hua S, An HJ, Ozcan S, Ro GS, Soares S, DeVere-White R, Lebrilla CB. Analyst. 2011;136:3663. doi: 10.1039/c1an15093f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mechref Y. Electrophoresis. 2011;32:3467. doi: 10.1002/elps.201100342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huang Y, Nie Y, Boyes B, Orlando R. J Biomol Tech. 2016;27:98. doi: 10.7171/jbt.16-2703-003. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pu Y, Ridgeway ME, Glaskin RS, Park MA, Costello CE, Lin C. Ana Chem. 2016;88:3440. doi: 10.1021/acs.analchem.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Boltje TJ, Buskas T, Boons GJ. Nat Chem. 2009;1:611. doi: 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Unverzagt C, Kajihara Y. Chem Soc Rev. 2013;42:4408. doi: 10.1039/c3cs35485g. [DOI] [PubMed] [Google Scholar]; (c) Maki Y, Okamoto R, Izumi M, Murase T, Kajihara Y. J Am Chem Soc. 2016;138:3461. doi: 10.1021/jacs.5b13098. [DOI] [PubMed] [Google Scholar]
- 10.(a) Gagarinov IA, Li T, Torano JS, Caval T, Srivastava AD, Kruijtzer JA, Heck AJ, Boons GJ. J Am Chem Soc. 2017;139:1011. doi: 10.1021/jacs.6b12080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wu Z, Liu Y, Ma C, Li L, Bai J, Byrd-Leotis L, Lasanajak Y, Guo Y, Wen L, Zhu H, Song J, Li Y, Steinhauer DA, Smith DF, Zhao B, Chen X, Guan W, Wang PG. Org Biomol Chem. 2016;14:11106. doi: 10.1039/c6ob01982j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Shivatare SS, Chang SH, Tsai TI, Tseng SY, Shivatare VS, Lin YS, Cheng YY, Ren CT, Lee CC, Pawar S, Tsai CS, Shih HW, Zeng YF, Liang CH, Kwong PD, Burton DR, Wu CY, Wong CH. Nat Chem. 2016;8:338. doi: 10.1038/nchem.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shivatare SS, Chang SH, Tsai TI, Ren CT, Chuang HY, Hsu L, Lin CW, Li ST, Wu CY, Wong CH. J Am Chem Soc. 2013;135:15382. doi: 10.1021/ja409097c. [DOI] [PubMed] [Google Scholar]
- 12.(a) Li L, Liu Y, Ma C, Qu J, Calderon AD, Wu B, Wei N, Wang X, Guo Y, Xiao Z, Song J, Sugiarto G, Li Y, Yu H, Chen X, Wang PG. Chem Sci. 2015;6:5652. doi: 10.1039/c5sc02025e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Calderon AD, Liu Y, Li X, Wang X, Chen X, Li L, Wang PG. Org Biomol Chem. 2016;14:4027. doi: 10.1039/c6ob00586a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Green ED, Adelt G, Baenziger JU, Wilson S, Van Halbeek H. J Biol Chem. 1988;263:18253. [PubMed] [Google Scholar]
- 14.Bertozzi CR, Rabuka D. In: Essentials of Glycobiology. Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME, editors. Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- 15.Li T, Huang M, Liu L, Wang S, Moremen KW, Boons GJ. Chem Eur J. 2016;22:18742. doi: 10.1002/chem.201604999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun S, Shah P, Eshghi ST, Yang W, Trikannad N, Yang S, Chen L, Aiyetan P, Hoti N, Zhang Z, Chan DW, Zhang H. Nat Biotechnol. 2016;34:84. doi: 10.1038/nbt.3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Eijnden DH, Blanken WM, van Vliet A. Carbohydr Res. 1986;151:329. doi: 10.1016/s0008-6215(00)90352-5. [DOI] [PubMed] [Google Scholar]
- 18.Renkonen O, Helin J, Vainio A, Niemela R, Penttila L, Hilden P. Biochem Cell Biol. 1990;68:1032. doi: 10.1139/o90-152. [DOI] [PubMed] [Google Scholar]
- 19.(a) Longmore GD, Schachter H. Carbohydr Res. 1982;100:365. doi: 10.1016/s0008-6215(00)81049-6. [DOI] [PubMed] [Google Scholar]; (b) Voynow JA, Kaiser RS, Scanlin TF, Glick MC. J Biol Chem. 1991;266:21572. [PubMed] [Google Scholar]; (c) Kaminska J, Glick MC, Koscielak J. Glycoconj J. 1998;15:783. doi: 10.1023/a:1006959915435. [DOI] [PubMed] [Google Scholar]; (d) Paschinger K, Staudacher E, Stemmer U, Fabini G, Wilson IB. Glycobiology. 2005;15:463. doi: 10.1093/glycob/cwi028. [DOI] [PubMed] [Google Scholar]
- 20.(a) Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Carbohydr Res. 1993;243:139. doi: 10.1016/0008-6215(93)84087-m. [DOI] [PubMed] [Google Scholar]; (b) Kanie O, Crawley SC, Palcic MM, Hindsgaul O. Bioorg Med Chem. 1994;2:1231. doi: 10.1016/s0968-0896(00)82074-x. [DOI] [PubMed] [Google Scholar]
- 21.Yu H, Chokhawala HA, Huang S, Chen X. Nat Protoc. 2006;1:2485. doi: 10.1038/nprot.2006.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.