

Abstract
Limited understanding of sweet basil (Ocimum basilicum L.) genetics and genome structure has reduced efficiency of breeding strategies. This is evidenced by the rapid, worldwide dissemination of basil downy mildew (Peronospora belbahrii) in the absence of resistant cultivars. In an effort to improve available genetic resources, expressed sequence tag simple sequence repeat (EST-SSR) and single nucleotide polymorphism (SNP) markers were developed and used to genotype the MRI x SB22 F2 mapping population, which segregates for response to downy mildew. SNP markers were generated from genomic sequences derived from double digestion restriction site associated DNA sequencing (ddRADseq). Disomic segregation was observed in both SNP and EST-SSR markers providing evidence of an O. basilicum allotetraploid genome structure and allowing for subsequent analysis of the mapping population as a diploid intercross. A dense linkage map was constructed using 42 EST-SSR and 1,847 SNP markers spanning 3,030.9 cM. Multiple quantitative trait loci (QTL) model (MQM) analysis identified three QTL that explained 37–55% of phenotypic variance associated with downy mildew response across three environments. A single major QTL, dm11.1 explained 21–28% of phenotypic variance and demonstrated dominant gene action. Two minor QTL dm9.1 and dm14.1 explained 5–16% and 4–18% of phenotypic variance, respectively. Evidence is provided for an additive effect between the two minor QTL and the major QTL dm11.1 increasing downy mildew susceptibility. Results indicate that ddRADseq-facilitated SNP and SSR marker genotyping is an effective approach for mapping the sweet basil genome.
Introduction
Sweet basil (Ocimum basilicum L.) is the most widely cultivated and economically salient Ocimum species in the United States and Europe [1]. Annual US revenue generated from sweet basil and other culinary herbs sold as bedding plants are estimated to be 96.8 million dollars [2]. Introduction of basil downy mildew (Peronospora belbahrii) to Europe in 2001 [3] and the United States in 2007 [4] has resulted in wide spread crop destruction and an estimated tens of millions of dollars in economic losses in the US alone [5]. Absence of effective seed treatment or chemical control measures has aided rapid dissemination of this pathogen to major production areas worldwide [6–13]. Lack of economically sustainable conventional, organic or cultural control, potential for fungicide resistant pathogen evolution [7,14] and consistent disease presence in major growing regions create a compelling rationale for the development of genetic resistance to downy mildew in sweet basil.
Multiple publications have identified downy mildew resistance within the Ocimum genus [15,16] and Ben-Naim et al. [17] demonstrated introgression into O. x basilicum F1 hybrids. Although possible, hybridization of commercial O. basilicum varieties with resistant genotypes is largely met with F1 sterility or sexual incompatibility [17,18]. Despite these challenges, an initial characterization of downy mildew resistance was provided by a multi-site field trial that evaluated F2 and backcross generations derived from a cross between downy mildew resistant inbred cultivar Mrihani (MRI) and susceptible inbred Rutgers University breeding line SB22 [19]. These efforts generated important results that have helped to inform an effective breeding program targeting genetic resistance. Nevertheless, a resistant commercial sweet basil variety remains to be seen 15 years after the first report of P. belbahrii, reflecting the difficulties currently associated with genetic improvement of this poorly characterized plant species.
Linkage map construction and subsequent association of DNA markers linked to important traits, or QTL, are becoming essential components of modern plant breeding programs. Although increasingly common in other horticultural species, neither a linkage map nor QTL have been developed for sweet basil. Lack of insight into the rather large [20] and potentially complex O. basilicum genome render sweet basil an unattractive species for genetic studies. Fortunately the rapid rise and plummeting costs of next-generation sequencing (NGS) have made high-throughput single nucleotide polymorphism (SNP) discovery accessible to non-model species [21,22]. Moreover, the introduction of reduced representation NGS strategies through restriction site associated DNA sequencing (RADseq) has revolutionized population-level genetic studies by providing a uniformly distributed subset of the genome across pooled individuals [23,24]. The combination of RADseq techniques with increasingly robust data analysis software [22,25,26] has provided unprecedented access to the genomes of complex species for linkage mapping. This includes polyploid species, which can generally be divided into autopolyploid (whole genome duplication following intraspecific hybridization) or allopolyploid (whole genome duplication following interspecific hybridization) classes. Allopolyploids are more easily targeted for linkage mapping because the segregation of loci usually occurs within divergent sub-genomes, which allows for separation of homologous from homeologous loci [27,28]. By dividing segregating loci among sub-genomes one can obtain a dataset compatible with traditional diploid map construction software packages (Joinmap, RQTL, etc.). Genotyping allopolyploids is typically performed with co-dominant markers such as simple sequence repeats (SSRs) or single nucleotide polymorphisms (SNPs) by isolation of single-locus (single sub-genome) markers [29] or development of a model for allele sub-genome assignment [30,31].
Ocimum basilicum is an outcrossing [32] tetraploid [18,33] that has demonstrated disomic inheritance for multiple traits [34,35], suggesting a diploidized polyploid genome. This allopolyploid hypothesis is supported by cytological evidence that demonstrated preferential pairing of O. basilicum, O. americanum (syn. O. canum) and their F1 hybrid [18]. Furthermore, an initial investigation indicated that basil downy mildew resistance conferred genotype MRI segregates in disomic fashion [19]. Major gene control is a common form of host resistance among plant species and has been demonstrated through QTL discovery in lettuce [36–38], spinach [39], melon [40], and grape [41].
In this study, a set of genic SSR markers were developed from the currently available National Center for Biotechnology Information (NCBI) EST database and used to genotype a sweet basil F2 mapping population from a cross between MRI and downy mildew susceptible genotype SB22 following an allotetraploid segregation model. A double digestion RADseq (ddRADseq) approach was then employed for SNP discovery and de novo genotyping. Genotype data was subjected to a filtration process to retain bi-allelic, homologous polymorphic loci to generate an intercross diploid dataset. Resulting genotype data were used to construct the first linkage map for sweet basil, which is anchored by SSRs and saturated by SNPs. Multiple QTL analyses were performed to identify genomic regions with association to downy mildew (P. belbahrii) resistance.
Materials and methods
Plant material
An F2 mapping population was developed in 2014 from a cross between inbred genotypes MRI (♀) and SB22 (♂) as previously described [19]. SB22 is an inbred line selected for tolerance to Fusarium wilt (Fusarium oxysporum f. sp. basilica) and is highly susceptible to P. belbahrii, while MRI is an inbred line resistant to P. belbahrii. The F1 hybrid and 104 F2 individuals were randomly selected and maintained as vegetative cuttings in Rutgers University research greenhouses (New Brunswick, NJ, U.S.A). This allowed for clones of each individual to be field transplanted for phenotyping across multiple years and locations.
Genotyping
Genomic DNA (gDNA) was extracted from the grandparents, F1 and 104 F2 individuals using ~80 mg of young ground leaf tissue using the E.N.Z.A. SP Plant DNA Kit (Omega BioTek, Norcross, GA). DNA was quantified and assessed for quality by measurement of 260/280 and 260/230 absorbance ratios using a Nanodrop (Thermo Scientific, Waltham, MA).
EST-SSR analysis
The National Center for Biotechnology Information (NCBI) O. basilicum expressed sequence tag (EST) database of 23,845 cDNA sequences was downloaded and assembled using CAP3 software [42] with default settings. The resulting contig and remaining singlet sequences were mined with SciRoKo software [43] for di-, tri- and tetranucleotide repeat sequences with a minimum of 10 nucleotides. SSRs meeting this criteria were selected for the presence of ≤300 bp flanking sequences that were subsequently used for primer pair design with Primer3 software [44]. This pipeline produced 811 putative SSR markers from which a subset of 89 di-, 115 tri-, and 36 tetranucleotide were used in this study. Primer pairs were synthesized (Integrated DNA Technologies, Coralville, IA) with the 5’ end of forward primers appended with the M13 (-21) sequence (5’-TGTAAAACGACGGCCAGT-3’)[45] to facilitate fluorescent labeling of PCR products. The 5’ end of reverse primers were “pig-tailed” with the 5’-GTTTCTT-3’ sequence [46] to ensure consistent polyadenylation across reactions.
PCR amplification for all reactions included 5 ng of gDNA, 10x Ramp-Taq PCR buffer (Denville Scientific, Metuchen, NJ), 2.0 mM MgCl2, 0.25 mM each dNTP (Denville Scientific), 0.5 U Ramp-Taq DNA polymerase (Denville Scientific), 0.5 pmol forward primer, 1.0 pmol reverse primer, and 1.0 pmol fluorescently labeled (FAM, NED, PET, or VIC) M13(-21) primer. Template gDNA was amplified using the following conditions: initial denaturation of 94°C for 5 min, followed by 30 cycles of 94°C for 30 sec, 55°C for 45 sec, 72°C for 45 sec, followed by 20 cycles of 94°C for 30 sec, 53°C for 45 sec, 72°C for 45 sec, followed by a final extension of 72°C for 10 min. GeneScan 600 LIZ (Applied Biosystems) size standard was added to resulting PCR products and separated by capillary electrophoresis on an ABI 3500xL Genetic Analyzer (Life Technologies Corporation, Carlsbad, CA). PCR product fragment length measurement and allele binning and was performed using Genemapper 4.1 software (Applied Biosystems).
PCR was performed in duplicate for 240 SSR markers initially using only MRI and SB22 grandparent gDNA to select for markers resulting in unambiguous PCR products (e.g., absent of any non-specific binding) and polymorphism. Markers fulfilling these criteria were subsequently used to evaluate the F2 mapping population. SSR markers having ≥3 amplicons for either grandparent were discarded and the remaining SSRs (containing either one or two amplicons per grandparent) were tested for chi-square goodness of fit to 1:2:1 or 3:1 diploid segregation models.
SNP analysis
An initial experiment was performed to compare two and three enzyme library preparation approaches using gDNA from 22 F2 individuals. The double digestion RADseq libraries were prepared according to Poland et al. [47] using rare-cutting PstI (NEB, USA) and the more common-cutting MspI (NEB, USA). In the three-enzyme digestion, the Poland et al. [47] protocol was modified to include ApeKI, which serves as a cutter to further reduce the complexity of the genome. Due to the addition of this enzyme, a 2 hour, 75°C incubation followed the initial digestion of gDNA with PstI and MspI. For both the two- and three-enzyme approaches, the PstI-complementary forward adapter and MspI-complementary reverse Y-adapters were added to the ligation reaction to ensure that only PstI-MspI fragments would be amplified during PCR, while all other digested fragments (MspI-MspI, MspI-ApeKI, PstI-ApeKI and PstI- PstI) should fail to amplify. Both prior to PCR and following library pooling, samples were mixed with 0.5 v/v Agencourt AMPure XP Beads (Beckman Coulter, USA) and washed with 70% ethanol to remove fragments sized less than 300 bp. All PstI and MspI adaptors included previously published barcodes [47] to uniquely identify individual samples.
All final libraries were prepared using the double digestion method rather than the triple digestion method, which generated library concentrations too low for sequencing. Four separate libraries were prepared for each grandparent to generate a 4x sequencing depth relative to the F1 and F2 progeny. Libraries for MRI (4x), SB22 (4x), F1 and 100 F2 individuals were quantified using Qubit (Life technologies, Grand island, NY). All samples were normalized to 5 ng/uL before pooling. This pool (109-plex) was paired-end sequenced on two Hi-Seq 2000 (Illumina, USA) lanes, once in Rapid-Run mode (2x150bp) and once in High-Output mode (2x100).
The Stacks (v1.3) software [48] pipeline was used to convert raw reads into genotype data. Concatenated single and paired end read fastq files were trimmed to 75 bp, quality filtered with the ‘-q’ flag and de-multiplexed using the default settings of the Stacks process_radtags.pl program. The ustacks program was then used to assemble matching reads across all samples and call SNPs within each group of reads (Stack) to generate individual haplotype alleles. A minimum of 3 matching reads (-m) was required to create a Stack with a maximum nucleotide mismatch allowance (-M) of 3 for F2 and grandparent stacks. The cstacks program generated a Catalog from Stacks that were polymorphic among the grandparents (MRI and SB22) to which F2 progeny haplotypes were matched using the sstacks program to identify putative loci. Loci missing >20 genotyped progeny were excluded from subsequent analyses.
A potential pitfall in polyploid SNP genotyping is the vulnerability of paralogous sequence variants being called as false positive polymorphic loci [27]. In an approach similar to Hohenlohe et al. [28], the Stacks web user interface was used to exclude loci with >1 SNP and significant (p≤0.10) deviation from an 1:2:1 ratio expected for an F2 intercross between dual-homozygous grandparents. This approach was therefore employed to filter paralogs and obtain a bi-allelic SNP dataset for genetic mapping.
Linkage map construction
Linkage map construction was performed using Joinmap 4.1® (Kyazma, NL) [49] with genotype data coded as an F2 intercross population type. Grouping was performed using independence logarithm of odds (LOD) scores from 2 to 15 with a step of 1, after which a minimum LOD score of 10 was used to determine autonomous linkage groups (LGs). Placement of loci was determined by comparison of map orders derived from the multipoint maximum likelihood and regression algorithms in Joinmap 4.1® (Kyazma, NL). An initial map was constructed using the maximum likelihood mapping function with parameters adjusted from the default settings when necessary to allow the algorithm to converge [50]. A second map was generated by regression mapping using a minimum LOD score of 4.0, recombination frequency of 0.35 and ripple jump threshold of 5.0. Maximum likelihood and regression maps were compared to identify suspect loci that might be misplaced. A locus or group of loci demonstrating major differences in map order location were removed to provide robust support for loci placement in the final map, which was estimated by maximum likelihood method.
Phenotyping
Downy mildew response for all individuals in this study was measured over two years and at two field locations by assessing the severity of abaxial leaf sporulation as described by Pyne et al. [19]. Field phenotyping experiment locations were selected based consistent annual disease pressure [19,51] and susceptible check plants. Susceptible control cultivar ‘DiGenova’ was included in the experimental design and overhead irrigation was applied as needed to provide uniform disease severity. Six leaves per individual were randomly sampled and scored using an ordered categorical scale of 0–4 (0 = no sporulation, 1 = 1–10%, 2 = 11–25%, 3 = 26–50%, 4 = 51–100%). Individuals were assigned a value between 0 (lowest possible severity) and 1 (highest possible severity) by dividing by a maximum score of 24. Data were collected in 2014 at Northern (NJSN14) and Southern (NJRA14) New Jersey locations [19] and the Southern New Jersey location in 2015 (NJRA15). All experiments were performed in randomized complete block design with three replications. Each of the 94 genotyped F2 individuals were phenotyped in NJRA14 and NJSN14, while 80 were phenotyped in NJRA15 due to plant death during the one year period between experiments. Phenotypic data from a single date corresponding to the highest F2 population mean disease severity were selected for each unique year x location combination (environment) for subsequent QTL analyses. Phenotype data was square root transformed and individuals not included in 2015 phenotyping were scored as missing for QTL analysis.
QTL analysis
A ‘forward selection’ [52] approach for identification of appropriate QTL models was implemented using the R/qtl package [53]. Single-QTL analysis using standard interval mapping and the Kruskal-Wallis rank-sum test was initially performed using the scanone function to detect genomic regions associated with downy mildew resistance across three environments. Unlike all other QTL analyses, data was not transformed prior to the Kruskal-Wallis test. LOD thresholds for significance of QTL level were determined by separate permutation tests with 1,000 iterations at α = 0.05. 1.5-LOD support intervals were calculated for all significant QTL detected.
A subsequent two-dimensional (2D) analysis was performed using the scantwo function to detect QTL pairs on separate LGs. Permutation tests were again performed with 1,000 iterations to determine significant LOD thresholds for the joint (full), conditional-interactive, interaction, additive, and conditional-additive two QTL models. An additional genome-wide scan for locus pairs within LGs was performed to account for potentially linked QTL.
Finally, a multiple-QTL model (MQM) was implemented using the fitqtl function to fit the appropriate linear model with the QTL detected from single and 2D analyses, represented as main effects. Analysis of variance (ANOVA) was performed to determine significant QTL effects and percentage of phenotypic variance explained (PVE) by each QTL. Genotype means and standard error for significant QTL were calculated and plotted using R/qtl.
Results
EST-SSR markers
Among 240 EST-SSR markers used to genotype the grandparents, 142 primer pairs demonstrated clean (unambiguous) PCR amplification in which 1–4 unique amplicons (alleles) were classified as ‘functional’. Forty primer pairs (28.2%) (Table 1) were polymorphic and could be grouped into three bi-parental dual-homozygous genotypes: (i) one polymorphic, bi-allelic locus in one sub-genome and no locus present in the second sub-genome (Fig 1A); (ii) one monomorphic locus in one sub-genome and one polymorphic, bi-allelic locus in a second sub-genome (Fig 1B); and (iii) one monomorphic locus in one sub-genome and one locus with one allele present or absent (null) in a second sub-genome (Fig 1C). In scenarios (i) and (ii) the polymorphic locus was fit to an F2 segregation ratio of 1:2:1 (a2a2:a2b2:b2b2) while in scenario (iii) the polymorphic locus was fit to an F2 segregation ratio of 3:1 (b2b2:a2a2+a2b2).
Table 1. Description of 42 mapped EST-SSR markers in the MRIxSB22 linkage map indicating nucleotide sequence source, repeat motif, linkage group, centimorgan position and chi-square goodness-of-fit test results.
| Markera | Sourceb | Motifc | LG | Position (cM) | Ratio | χ2 | Pd |
|---|---|---|---|---|---|---|---|
| OBNJR3cn391 | Contig3280 | (TCA)5 | 1 | 16.4 | 3:1 | 3.33 | <0.10 |
| OBNJR2cn104 | Contig3401 | (CT)14 | 1 | 19.0 | 3:1 | 0.24 | - |
| OBNJR3sg98 | DY337354 | (CCT)8 | 1 | 19.2 | 3:1 | 2.97 | <0.10 |
| OBNJR3cn201 | Contig1783 | (GAA)6 | 1 | 59.8 | 1:2:1 | 3.34 | - |
| OBNJR2sg34 | DY331634 | (CT)11 | 2 | 32.4 | 1:2:1 | 0.77 | - |
| OBNJR2cn79 | Contig2573 | (AT)9 | 2 | 32.7 | 3:1 | 0.52 | - |
| OBNJR3cn362 | Contig2969 | (TGA)6 | 3 | 78.0 | 1:2:1 | 1.47 | - |
| OBNJR3cn56 | Contig582 | (AGG)7 | 3 | 84.7 | 1:2:1 | 5.16 | <0.10 |
| OBNJR2cn80 | Contig2575 | (TA)12 | 4 | 90.3 | 1:2:1 | 1.68 | - |
| OBNJR2sg21.1 | DY339566 | (TC)13 | 5 | 33.9 | 1:2:1 | 2.67 | - |
| OBNJR3sg19 | DY343509 | (TCA)6 | 6 | 18.9 | 3:1 | 3.49 | <0.10 |
| OBNJR2sg15 | DY340778 | (GA)25 | 6 | 50.8 | 1:2:1 | 2.25 | - |
| OBNJR4sg06 | DY338242 | (ACAA)5 | 7 | 139.4 | 3:1 | 1.65 | - |
| OBNJR2cn78 | Contig2475 | (AT)10 | 8 | 56.4 | 1:2:1 | 1.88 | - |
| OBNJR2sg21.2 | DY339566 | (TC)13 | 10 | 65.7 | 1:2:1 | 1.62 | - |
| OBNJR3cn389 | Contig3254 | (GCA)8 | 11 | 132.0 | 3:1 | 0.86 | - |
| OBNJR4cn11 | Contig1679 | (TCAC)4 | 12 | 144.0 | 3:1 | 0.06 | - |
| OBNJR2cn83 | Contig2631 | (GA)18 | 12 | 144.4 | 1:2:1 | 2.65 | - |
| OBNJR2cn17 | Contig606 | (AT)10 | 12 | 146.6 | 3:1 | 0.23 | - |
| OBNJR2sg04 | DY343638 | (GA)17 | 13 | 40.2 | 1:2:1 | 5.37 | <0.10 |
| OBNJR4cn16 | Contig2294 | (CAAA)4 | 13 | 42.9 | 1:2:1 | 3.61 | - |
| OBNJR2sg119 | DY333250 | (GTA)7 | 13 | 49.7 | 1:2:1 | 10.14 | <0.01 |
| OBNJR2cn29 | Contig1138 | (AC)16 | 13 | 54.6 | 1:2:1 | 2.25 | - |
| OBNJR2sg33 | DY333933 | (AC)16 | 14 | 82.7 | 1:2:1 | 0.03 | - |
| OBNJR4cn15 | Contig2242 | (GCCT)5 | 15 | 0.0 | 1:2:1 | 7.13 | <0.05 |
| OBNJR3cn358 | Contig2910 | (TCC)7 | 15 | 24.0 | 1:2:1 | 2.31 | - |
| OBNJR3cn356 | Contig2907 | (AAG)9 | 15 | 26.8 | 1:2:1 | 0.48 | - |
| OBNJR2sg31 | DY336298 | (AG)9 | 16 | 106.9 | 1:2:1 | 0.07 | - |
| OBNJR3cn328 | Contig2750 | (AAG)6 | 16 | 107.5 | 1:2:1 | 0.76 | - |
| OBNJR3cn192 | Contig1724 | (ACA)8 | 16 | 161.5 | 3:1 | 0.13 | - |
| OBNJR3cn243 | Contig2153 | (GAA)6 | 17 | 0.0 | 3:1 | 0.06 | - |
| OBNJR3sg177 | DY322989 | (TGC)5 | 17 | 2.5 | 3:1 | 4.28 | <0.01 |
| OBNJR3cn401 | Contig3437 | (CAG)7 | 17 | 6.4 | 3:1 | 0.97 | - |
| OBNJR3cn377 | Contig3126 | (TAT)6 | 18 | 30.9 | 1:2:1 | 1.8 | - |
| OBNJR2cn92.2 | Contig3041 | (CT)13 | 18 | 78.1 | 3:1 | 0.84 | - |
| OBNJR3cn54 | Contig573 | (TTA)18 | 19 | 88.7 | 1:2:1 | 0.4 | - |
| OBNJR2cn92.1 | Contig3041 | (CT)13 | 19 | 89.3 | 1:2:1 | 0.64 | - |
| OBNJR3cn217 | Contig1936 | (ATT)6 | 20 | 32.1 | 3:1 | 0.37 | - |
| OBNJR4sg01 | DY343743 | (TCCC)5 | 21 | 91.9 | 1:2:1 | 1.27 | - |
| OBNJR3cn239 | Contig2134 | (TTC)8 | 24 | 58.0 | 1:2:1 | 1.2 | - |
| OBNJR2cn38 | Contig1352 | (CA)13 | 25 | 56.0 | 1:2:1 | 1.2 | - |
| OBNJR2cn73 | Contig2250 | (CT)24 | 26 | 56.5 | 1:2:1 | 7.13 | <0.05 |
aSSR markers failing to map or found to be located at identical positions to other SSR markers are excluded. A single primer set resulting in two independently segregating loci are indicated by marker name followed by either ‘.1’ or ‘.2’
bSSRs are sourced from CAP3-assembled NCBI O. basilicum EST sequence database. NCBI genbank nucleotide accession is provided for SSRs located in EST sequences that could not be assembled into contigs.
cRepeat motif sequence and number reported refer to the original Genbank (parent) sequences
dChi-square goodness-of-fit tests resulting in p < 0.10 indicate evidence for segregation distortion.
Fig 1. Polymorphic EST-SSR genotypes observed among subgenomes of homozygous grandparent genotypes MRI and SB22.

Electropherogram plots for alleles represented as peaks with size (nucleotides) represented along the x-axis. (A) Marker OBNJR2sg34 genotype: One polymorphic locus within a single subgenome (arbitrarily designated with subscript 1) corresponding to MRI genotype a2a2 and SB22 genotype b2b2 with expected F2 segregation ratio 1:2:1 (a2a2:a2b2:b2b2). (B) Marker OBNJR3cn328 genotype: One monomorphic locus corresponding to a single sub-genome genotype a1a1 and one polymorphic locus in another sub-genome (arbitrarily designated with subscript 2) with expected F2 segregation ratio 1:2:1 (a2a2:a2b2:b2b2). (C) Marker OBNJR3cn80 genotype: Monomorphic locus a1a1 and polymorphic locus in another sub-genome corresponding to MRI null genotype -2−2 and SB22 genotype b2b2 with expected F2 segregation ratio 3:1 (-2−2: b2b2).
The majority of mapped EST-SSR markers grouped in scenarios (ii) or (iii) (Fig 1B and 1C) in which two loci (one monomorphic and one polymorphic) are amplified in individual sub-genomes (multi-locus markers). Six EST-SSR markers fit scenario (i) (Fig 1A) in which a single homozygous, polymorphic locus is amplified within a single sub-genome and segregated in 1:2:1 (single locus markers). Two additional scenarios were observed for markers OBNJR2sg21 and OBNJR2cn92, in which two independently segregating loci represented putative homeologs. The former generated two allele pairs (OBNJR2sg21.1 and OBNJR2sg21.2), both segregating independently with each pair fitting a 1:2:1 ratio. The latter generated one pair (OBNJR2cn92.1) fitting 1:2:1 segregation and a second pair (OBNJR2cn92.2) exhibiting amplification in a presence (MRI), absence (SB22) fashion fitting a 3:1 segregation ratio. Thus, 42 EST-SSR markers were generated from 40 primer sets (Table 1; S1 Table). Tests for goodness of fit provided evidence of segregation distortion for 10 (p<0.10) and 5 (p<0.05) EST-SSR markers depending on statistical stringency.
SNP discovery and polymorphic loci development
In an effort to maximize O. basilicum genome complexity reduction double (PstI-MspI) and triple (PstI-MspI + ApeKI) digestion library preparations were compared on a subset of the F2 population (22 individuals). Triple digestion resulted in a mean DNA concentration of 3.4 ng/uL±1.6 considered too low for sequencing. Library preparation without ApeKI (double digestion) resulted in an approximate 3-fold library concentration increase (mean = 15.5ng/uL±3.63) adequate for sequencing. Thus, all libraries sequenced in this study were prepared by the double digestion method.
Illumina sequencing generated a total of 478,850,390 paired end reads with a guanine-cytosine (GC) content of 39–42%. 420,611,900 reads were retained following trimming (75bp), de-multiplexing and quality filtering. Six F2 individual samples had a ~10x lower retained read count relative to other samples and were excluded from subsequent analysis. The average number of reads for the remaining 94 F2 individuals was 4,021,000. The retained read count for the parents was ~4x the F2 read count mean with 16,940,960 and 16,973,580 for MRI and SB22, respectively (S1 Fig).
Alignment of retained reads from both parents resulted in a Catalog containing 195,123 Stacks, 47,842 SNPs and 25,363 polymorphic loci. 3,492 polymorphic loci having less than 20 missing individuals could be mapped back to the parent Catalog. 2,532 polymorphic loci contained exactly 1 SNP (S2 Table). Chi-square test of retained single-SNP loci revealed 565 with evidence for segregation distortion (p<0.10), or 22.3%, which were removed leaving 1,918 loci. Strict Chi-square (p <0.10) filtration of single SNP loci was employed to retain a bi-allelic set of loci [28]. A final set of 64 SNP markers produced identical genotypes and was removed, leaving a total of 1,954 polymorphic SNP and EST-SSR loci prior to linkage grouping.
Linkage map construction
Following removal of markers with poor support for grouping (LOD<10.0; rf<0.35) or placement, the final genetic map contained 1,847 SNP and 42 EST-SSR markers. The overall frequency of aa, ab and bb genotypes was 22.6%, 52.0% and 25.4%, respectively, reflecting an F2 dataset with adherence to Mendelian segregation. Grouping initially yielded 24 LGs as expected for the haploid chromosome set (n = 24). However, two LGs exhibited large and centrally located gaps of 53.5 and 50.4 cM, resulting in unusually high total map distances of 315.2 and 310.5 cM, respectively. Multiple marker placement diagnostics did not provide evidence for any poorly supported markers causing distance inflation, thus these gaps were determined real representations and warranted the division of each LG into two: LG8 (87.1 cM), LG9 (95.6 cM) and LG14 (142.0 cM), LG15 (71.4 cM). Division of these groups had no effect on marker order. The final map yielded 26 LGs (Fig 2) with an average distance of 166.6 cM and a total map distance of 3030.9 cM. SNP and EST-SSR markers were densely populated throughout the map and uniformly distributed among LGs, averaging a 1.6 cM distance between markers (Table 2).
Fig 2. Sweet basil linkage map constructed for MRI x SB22 F2 intercross family.

The map includes 1,847 SNP (black font) and 42 EST-SSR markers (red font) across 26 LGs for a total length of 3030.9 cM. Two pairs of multi-locus EST-SSR (bold/italic/red font) markers represent putative homeologous pairs of loci (LGs 5,10 and 18,19). Blue lines represent 1.5 LOD score confidence intervals located to the left of linkage group locations associated with downy mildew resistance.
Table 2. Summary of the MRIxSB22 F2 linkage map including number of SNPs and EST-SSRs, centimorgan length and average centimorgan distance between markers for each linkage group.
| LG | SNPs | SSRs | Distance (cM) | Average distance between markers (cM) |
|---|---|---|---|---|
| 1 | 158 | 4 | 199.3 | 1.2 |
| 2 | 68 | 2 | 69.1 | 1 |
| 3 | 48 | 2 | 93.9 | 1.9 |
| 4 | 90 | 1 | 112.3 | 1.2 |
| 5 | 71 | 1 | 121.3 | 1.7 |
| 6 | 37 | 2 | 149 | 3.9 |
| 7 | 141 | 1 | 198.7 | 1.4 |
| 8 | 52 | 1 | 87.1 | 1.7 |
| 9 | 45 | 0 | 95.6 | 2.2 |
| 10 | 87 | 1 | 123.8 | 1.4 |
| 11 | 85 | 1 | 160 | 1.9 |
| 12 | 143 | 3 | 156.9 | 1.1 |
| 13 | 57 | 4 | 89.5 | 1.5 |
| 14 | 97 | 1 | 142.9 | 1.5 |
| 15 | 37 | 3 | 71.4 | 1.8 |
| 16 | 105 | 3 | 161.5 | 1.5 |
| 17 | 91 | 3 | 126.8 | 1.4 |
| 18 | 91 | 2 | 110.3 | 1.2 |
| 19 | 65 | 2 | 122.6 | 1.9 |
| 20 | 60 | 1 | 113.7 | 1.9 |
| 21 | 57 | 1 | 110.6 | 1.9 |
| 22 | 40 | 0 | 88.8 | 2.3 |
| 23 | 34 | 0 | 86.3 | 2.6 |
| 24 | 31 | 1 | 79.2 | 2.6 |
| 25 | 37 | 1 | 103.8 | 2.8 |
| 26 | 20 | 1 | 56.5 | 2.8 |
| Overall | 1847a | 42a | 3030.9a | 1.6b |
aTotal
bAverage centimorgan distance
Forty-two SSR markers mapped across 23 of the 26 LGs (Table 2), providing critical PCR-based “anchor” markers for potential comparison to future sweet basil linkage maps. Two primer sets resulted in two pairs of markers (OBNJR2sg21.1, OBNJR2sg21.2 and OBNJR2cn92.1, OBNJR2cn92.2) mapping to 4 LGs and may represent two homeologous chromosome sets (LGs 5,10 and 18,19) (Fig 2).
Downy mildew response
Downy mildew response among individuals evaluated in 2015 was similar to previously reported results in 2014 [19]. Both grandparents were consistent in their differential response to downy mildew with SB22 >0.96 disease severity and MRI < 0.04 disease severity (Fig 3). The F2 population mean for NJRA15 was 0.43 as compared to 0.44 and 0.39 for NJRA14 and NJSN14, respectively. The distribution across three environments demonstrates some skewness towards a disease resistance response as previously described [19] (Fig 3). Square root transformation was applied to F2 phenotypic data, resulting in normally distributed residual variance across the three environments.
Fig 3. Frequency distribution of disease severity in the MRI x SB22 F2 mapping population across three environments.
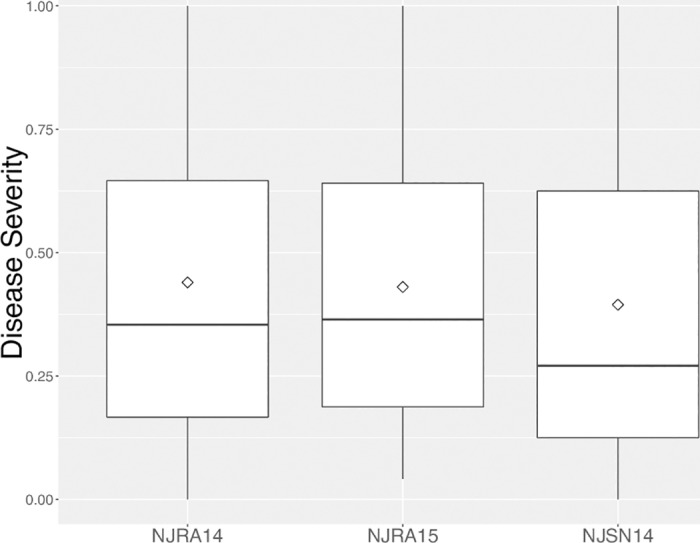
Codes for each environment are shown on the x-axis and correspond to data recorded in 2014 in southern New Jersey (NJRA14), 2015 in southern New Jersey (NJRA15) and northern New Jersey in 2014 (NJSN14). Disease severity measured on a scale in which 0 = lowest possible severity score and 1 = highest possible severity score.
Detection of QTL conferring downy mildew resistance
Initial interval mapping QTL analysis detected one LG region surpassing calculated LOD thresholds (α = 0.05) 3.99, 4.01, and 4.10 corresponding to environments NJSN14, NJRA14 and NJRA15, respectively (Fig 4). Results of the Kruskal-Wallis test confirmed the significance of this region with LOD scores for these respective environments of 3.74, 5.08 and 5.69. A maximum LOD score was associated with the most distal end of LG 11 closest to SNP marker ‘11636’, which was renamed dm11.1. A 1.5 LOD confidence interval spanned approximately 45 cM from the location of dm11.1 (Fig 2).
Fig 4. Detection of major downy mildew resistance QTL dm11.1 across three environments.

LOD scores for genome-wide scan using square-root transformed phenotype data from three environments: NJSN14 (northern New Jersey; 2014), NJRA14 (southern New Jersey; 2014) and NJRA15 (southern New Jersey; 2015). Significant LOD thresholds (α = 0.05) were calculated by permutation tests with 1,000 iterations and are shown with red, dashed horizontal lines.
A 2D genome scan identified two additional QTL, located on LG 9 at 74.9 cM and LG 14 at 73.7 cM, renamed dm9.1 and dm14.1, respectively. Interestingly, these QTL were detected (p<0.05) in environment NJSN14, but not in NJRA14 or NJRA15. The two-QTL model identified in NJSN14 provided evidence that dm9.1 and dm14.1 each act independently with dm11.1, thus forming two pairs: dm9.1, dm11.1 and dm11.1, dm14.1. In both pairs the following two-QTL models were found to be significant: additive (LOD≥8.17; p≤0.003) and additive-conditional (LOD≥4.53; p≤0.016). These results indicate a single-QTL model is inadequate and that downy mildew resistance in at least one environment (NJSN14) should be modeled with multiple QTLs. Strong evidence (p≤0.003) was provided for the pairwise additive model suggesting that the detected locus pairs act additively to affect response to downy mildew in the MRI x SB22 F2 population.
Significance of two QTL pairs provided evidence for 3-QTL in at least one environment (NJSN14), necessitating the inclusion of dm9.1, dm11.1 and dm14.1 in a MQM. Results of the MQM analysis across all three environments indicated that dm11.1 represents a ‘major QTL’ that consistently explained the greatest percentage of phenotypic variance (20.6–28.2%). The dm14.1 QTL was well-supported (LOD = 3.3; p<0.001) in the NJRA14 environment with 10.5% PVE, but was not detected (p = 0.11) in single 2015 environment (NJRA15). Less support (LOD = 2.1; p<0.012) was provided for dm9.1 in NJRA14 and explained 6.5% of phenotypic variance (Table 3). Similarly, in NJRA15 dm9.1 was weakly detected (p = 0.047) with 5.5% PVE. LOD scores and phenotypic effect of these two QTL were more pronounced in environment NJSN14 where dm9.1 (LOD = 5.8; p<0.001) and dm14.1 (LOD = 6.5; p<0.001) explained 16.1% and 18.4%, respectively. Given their variable and generally lower contribution across environments dm9.1 and dm14.1 were considered ‘minor QTL’.
Table 3. Summary of three downy mildew resistance QTL detected using a multiple QTL model (MQM) across three environments.
| QTL | LG | Position (cM) | SNPa | Confidence Interval (cM)b | Environment | LOD | Pc | PVE (%)d |
|---|---|---|---|---|---|---|---|---|
| dm9.1 | 9 | 74.9 | 95799 | 51.9–95.6 | NJSN14 | 5.8 | <0.001 | 16.1 |
| - | - | - | NJRA14 | 2.1 | 0.012 | 6.5 | ||
| - | - | - | NJRA15 | 1.5 | 0.047 | 5.5 | ||
| dm11.1 | 11 | 160.0 | 11636 | 115.3–160.0 | NJSN14 | 7.2 | <0.001 | 20.6 |
| 160.0 | 11636 | 115.3–160.0 | NJRA14 | 6.7 | <0.001 | 23.3 | ||
| 160.0 | 11636 | 114.0–160.0 | NJRA15 | 6.5 | <0.001 | 28.2 | ||
| dm14.1 | 14 | 73.7 | 120555 | 65.3–92.1 | NJSN14 | 6.6 | <0.001 | 18.4 |
| 73.7 | 120555 | 65.3–131.0 | NJRA14 | 3.3 | <0.001 | 10.5 | ||
| - | - | - | NJRA15 | 1.1 | 0.109 | 3.9 |
aSingle nucleotide polymorphism (SNP) marker located in closest proximity to the QTL location
b1.5 LOD score intervals shown or significant (P<0.01) QTL only
cP-values represent the significance of LOD scores determined by permutation tests with 1,000 iterations at α = 0.05
dPercent phenotypic variance explained
Genotypes for QTL detected in the 2D genome scan were examined for their effect on downy mildew response in environment NJSN14 (Fig 5). In each QTL, ‘a’ alleles inherited from resistant parent MRI were associated with a lower F2 mean downy mildew (disease) severity, while the ‘b’ alleles from susceptible parent SB22 were associated with a higher mean disease severity. Individuals with dm11.1 genotypes ‘aa’ or ‘ab’ had similarly low means of 0.25±0.06 and 0.34±0.05, as compared to 0.63±0.07 for the ‘bb’ genotype (Table 4). Proximity of the ‘ab’ mean to the MRI genotype, ‘aa’ (Fig 5B), demonstrated dominant gene effects influence dm11.1-conferred downy mildew resistance. In contrast, the homozygote (‘aa’)—heterozygote (‘ab’)—homozygote (‘bb’) trend for dm9.1 (Fig 5A) and dm14.1 (Fig 5C) appears relatively linear suggesting additive gene effects have greater influence over the response to downy mildew.
Fig 5. Effect and interaction plots for three QTL detected in environment NJSN14.

MRI x SB22 F2 genotype means (circles) ± 1 SE (error bars) for (A) minor QTL dm9.1, (B) major QTL dm11.1 and (C) minor dm14.1. Two-QTL genotype by genotype means ± 1 SE for (D) dm11.1 by dm9.1 and (E) dm11.1 by dm14.1. Allele ‘a’ is inherited from downy mildew resistant grandparent MRI and allele ‘b’ is inherited from susceptible grandparent SB22. Error bars represent ±1 SE.
Table 4. F2 means for downy mildew (disease) response in environment NJSN14 according to QTL genotype.
| QTL | aa | ab | bb |
|---|---|---|---|
| dm9.1 | 0.21±0.08 | 0.37±0.05 | 0.54±0.06 |
| dm11.1 | 0.25±0.06 | 0.34±0.05 | 0.66±0.07 |
| dm14.1 | 0.21±0.07 | 0.35±0.04 | 0.63±0.06 |
The genotypes of dm9.1 (Fig 5A) and dm14.1 (Fig 5C) were evaluated in combination with dm11.1 genotypes and demonstrated a similar effect across interacting genotype classes. In the case of dm9.1 in combination with dm11.1, dual ‘aa’ homozygotes conferred the strongest resistance (0.14±0.08) while the dual ‘bb’ homozygotes result in highest mean disease severity (0.90±0.11). When dm11.1 is considered with dm14.1 dual ‘aa’ and ‘bb’ homozygotes result in a mean response of 0.18±0.11 and 0.97±0.11, respectively. An intermediate phenotype was previously hypothesized for the MRI x SB22 F2 population [19] and is supported in the presence of 3 ‘b’ alleles in both cases of QTL pairs (Fig 5D and 5E). Three ‘b’ alleles can be achieved with one heterozygous locus, one homozygous ‘bb’ locus and the reciprocal (genotypes at each locus switched). In the case of a heterozygous dm11.1 and homozygous dm14.1, mean response is 0.59±0.07 compared to 0.69±0.09 for the reciprocal (Table 5). When considered with their associated standard errors, these two genotype scenarios appear to be associated with an intermediate response (Fig 5E). Results are similar for dm11.1 and dm14.1, where the range is 0.65±0.08–0.66±0.11 (Table 5). Investigation of segregating alleles at multiple loci demonstrates that while dm11.1 is most influential, it is not acting independently and a more complete informative model must include additive minor effect QTL.
Table 5. F2 means for downy mildew (disease) response in environment NJSN14 according to two-QTL genotype by genotype combinations.
| dm9.1 | dm14.1 | |||||
|---|---|---|---|---|---|---|
| dm11.1 | aa | ab | bb | aa | ab | bb |
| aa | 0.14±0.12 | 0.19±0.11 | 0.30±0.07 | 0.18±0.08 | 0.20±0.08 | 0.38±0.10 |
| ab | 0.18±0.10 | 0.27±0.05 | 0.67±0.11 | 0.16±0.11 | 0.22±0.07 | 0.60±0.07 |
| bb | 0.35±0.15 | 0.654±0.08 | 0.90±0.11 | 0.30±0.11 | 0.69±0.09 | 0.98±0.11 |
Both dm9.1 and dm14.1 separately fit a two-QTL additive model with dm11.1. Means for each genotype by genotype effect colored on a scale from green (low disease severity) to yellow (intermediate disease severity) to red (high disease severity).
Discussion
EST-SSR and SNP genotyping
Until recently, linkage mapping of non-model species relied heavily upon PCR-based markers such as SSRs and AFLPs, providing valuable but costly genotype data that were often inadequate for achieving dense genome coverage. Reduced representation sequencing has substantially decreased the economic and bioinformatics hurdles required to genotype and map plant species lacking genomic resources [54–56]. Little to no genomic sequence data have been made available for basil with the exception of a recent Ocimum sanctum draft genome assembly estimated to be 386 Mbp [57]. This massive disparity in O. sanctum genome size relative to O. basilicum suggests massive accumulation of genomic content and genetic divergence likely to exceed a threshold (1–5%) [58] at which mapping short reads would be successful. The O. sanctum assembly is therefore unlikely to provide utility for read alignment or validation of physical marker positions, leaving O. basilicum currently without a reference genome.
Implementation of PstI-MspI ddRADseq [47] facilitated high-throughput de novo SNP discovery and made feasible the construction of a first generation sweet basil linkage map. The Stacks de novo genotyping software pipeline performed effectively with ddRADseq sequence data, generating over 25,000 polymorphic loci between grandparents without a reference genome. Strict filtration for single-SNP markers from dual-homozygous grandparents absent of F2 segregation distortion (p<0.10) resulted in a greater than 10-fold reduction in the final number of mappable loci. However, 1,887 bi-allelic SNP markers were identified, successfully generating a dataset imputable as a diploid intercross.
The 22.3% rate of segregation distortion (p<0.10) observed for the MRIxSB22 F2 population is comparable to that of better characterized allopolyploids such as strawberry (22.4%. p < 0.05) [59], wheat (34%) [60] and peanut (39.1%, P<0.05) [61]. Deviation from expected segregation ratios can be attributed to various reproductive biological factors [62,63] and are typically higher in mapping populations derived from interspecific crosses [64,65]. F1 progeny obtained from hybridization of MRI and SB22 exhibited no sterility suggesting both grandparents belong to the same species, O. basilicum. Furthermore, preferential pairing is demonstrated by the frequent occurrence of predictable disomic inheritance patterns of EST-SSR loci (1:2:1 and 3:1), indicating divergent subgenomes that are less likely to exhibit multivalent chromosome behavior during meiosis. Instead, SNP markers failing to fit expected genotype class segregation patterns may be due to mistaken merging of sequence reads from homeologs subsequently called as homologous polymorphic loci within the same subgenome [22,27]. Availability of known diploid ancestors for the Gossypium sp. AT and DT subgenomes facilitated identification of putative SNP locus homologs [31]. In the absence of such resources, precautionary removal of loci with poor chi square goodness of fit to a 1:2:1 ratio was necessary to avoid inclusion of potential false-positive loci.
Although not critical to saturation of the linkage map, development and mapping of 42 EST-SSRs (28.1% of functional markers) (Table 1) provided needed evidence for disomic inheritance. A similar approach was recently employed to determined disomic inheritance of SSR markers in S1 populations of Cynodon dactylon [66]. Eighty-seven of the 142 (61.3%) functional EST-SSR markers amplified two or more alleles, which is comparable to the 66.2% reported in a comparison inbred Brassica species [29]. In an inbred allotetraploid the maximum number of alleles represented by a single SSR marker in one genotype should not exceed two (Fig 1). Occurrence of 3 or 4 alleles per locus in a single grandparent (7.3% in SB22 and 8.7% in MRI) suggests a small percentage of heterozygous loci in one (3 alleles) or both (4 alleles) subgenomes. Although these SSR markers were not mappable, observation of tri- and tetra-allelic loci provide further supporting evidence for an allotetraploid genome structure. In the absence of knowledge concerning O. basilicum genome structure, initial EST-SSR genotype information provided needed evidence for disomic inheritance that could be fit to a traditional diploid intercross model for further investigation.
A first generation sweet basil linkage map
This study resulted in a sweet basil linkage map with 1,847 SNP and 42 SSR markers covering 3030.9 cM. The 26 LGs reported include two LG sets (8,9 and 14,15) that were originally merged and >300 cM in length. Observation of ~50 cM gaps in these two LGs and evidence of weak linkages among markers on either side of each gap informed the decision to divide these two LGs. This conservative approach avoided the possibility of mistakenly combining separate chromosomes and generated 26 highly supported LGs with evenly distributed markers (Table 2) and no major gaps (Fig 3). A similar result was recently reported for cultivated strawberry from a ddRADseq-based linkage mapping generating 31 LGs, three greater than the expected 28 for the known haploid chromosome set (n = 28) [56].
EST-SSR markers were successfully distributed across 23 of the 26 linkage groups in this study (Fig 1). SSR markers derived from genic sequence databases such as EST libraries are more likely to be transferrable across diverse germplasm and are thus ideal ‘anchor’ markers for comparison of linkage maps across populations [29,67]. Clustering of 2–4 EST-SSR markers was common on the MRI x SB22 linkage map. Among 7 LGs, SSRs (between 2 and 3) mapped within very short intervals (2.3 ± 1.7 cM) (Fig 2). The O. basilicum NCBI EST library is based largely on tissue-specific cDNA sequences from transcripts related to synthesis of secondary metabolites [68]. The occurrence of the EST-SSR groupings suggest they may be derived from transcripts contributing to a given biosynthetic pathway, potentially clustered in a single genomic region. Interestingly, two multi-locus SSRs (OBNJR2sg21 and OBNJR2cn92) mapped to 4 unique LGs with no evidence for segregation distortion (p>0.10) (Table 1). These four LGs potentially represent two homeologous chromosome sets (LGs 5,10 and 18,19), however, further investigation is needed to build support for this hypothesis.
Downy mildew resistance QTL detection
One major QTL, dm11.1, and two minor QTL, dm9.1 and dm14.1, were associated with response to downy mildew in the MRI x SB22 F2 mapping population. Major QTL dm11.1 was located on the most distal end of LG 11 (160.0 cM), close to SNP ‘11636’ and explained 20.6–28.2% of phenotypic variance across three environments. The contribution of minor QTL dm9.1 and dm14.1 was lower in NJRA14 and NJRA15 where the combined PVE was 17.0% and 9.4%, respectively. The 2014 F2 mean disease severity in southern NJ (NJRA14) was significantly higher (p<0.05) than northern NJ (NJSN14) [19]. Despite this difference in disease pressure all three QTL were detected (p<0.05) in both 2014 NJ locations. However, the PVE in NJRA14 by minor QTL decreased substantially relative to NJSN14, while PVE by dm11.1 was slightly increased by 3.3% (Table 3). Only dm9.1 and dm11.1 were detected in the 2015 environment NJRA15, while dm14.1 could not be identified (p = 0.109). PVE of 28.2% for dm11.1 was highest in this environment, while that of dm9.1 was comparable to NJRA14 (Table 3). Higher F2 mean disease severities of 0.44 and 0.43 for the southern NJ location in 2014 and 2015, respectively, suggest an interaction of disease pressure and/or location with QTL effect. However, it should be noted that the population size was reduced to 80 individuals in the NJRA15, which may have affected QTL resolution in this environment. PVE of dm11.1 was negatively correlated with dm9.1 and dm14.1 across all environments (Table 3), suggesting that the effect of minor and major QTL may be inversely related when subject to different environmental conditions (eg. disease severity).
When considered in isolation, this dm11.1 would appear to act as a single-dominant gene in which one ‘a’ allele is sufficient to confer a resistant downy mildew response (≤0.34±0.05) (Table 5) (Fig 5B) as has been observed in Brassica spp. [69], spinach [39] grape [41]. However, the single, dominant gene hypothesis (0–0.33 = resistant individual and 0.34 ≤ susceptible individual) was previously rejected by Pyne et al. [19] (Chi-squared test, p <0.01) in F2 and backcross populations using phenotypic data from NJSN14 and NJRA14, concluding that at least one additional locus was affecting downy mildew response. In this study, detection of additional dm9.1 and dm14.1 in additive-two and multiple QTL models support the previous phenotypic-based findings.
Greatest support for QTL dm9.1 and dm14.1 (LOD> 5.8; p<0.001) occurred in environment NJSN14 (northern New Jersey; 2014) where these QTL were associated with 34.5% of PVE (Table 3). Consideration of genotype effects for both QTL in this environment demonstrates a severe consequence (high susceptibility) for individuals with dual homozygous ‘b’ alleles (Fig 5D and 5E) in which susceptibility is additive as F2 mean disease severity surpasses the maximum (0.66±0.07) observed for any individual ‘bb’ QTL genotype (Fig 5D and 5E). This result is supported by previously identified highly significant (p<0.001) positive additive (‘bb’) x additive (‘bb’) effects from a joint scaling test using the MRI x SB22 full-sibling family indicating the presence of homozygous loci with an increase in susceptibility [19]. Successive subtraction of SB22 ‘b’ alleles from either of the dm11.1-dm9.1 or dm11.1-dm14.1 QTL pairs detected in 2D QTL analysis results in an incremental reduction of F2 mean disease severity (i.e., increased resistance). This two-QTL system therefore provides evidence for at least three downy mildew response classes: susceptible (4 ‘b’ alleles), intermediate (3 ‘b’ alleles) and resistant (0–2 ‘b’ alleles) (Fig 5D and 5E).
Dominant gene action conferred by the ‘a’ allele in dm11.1 appears to be capable of countering the susceptibility effect of ‘b’ alleles in either minor QTL when 1–2 ‘b’ alleles are present. Resistance begins to break down, however, with the accumulation 3 or 4 ‘b’ alleles in either QTL (Fig 5D and 5E). Again, these results are supported by a previously described positive additive (‘bb’) x dominant (‘ab’) effect [19] through resistance of the heterozygous dm11.1 locus being reduced by homozygous ‘bb’ loci in either minor QTL. The resistance-reducing effect of a ‘bb’ genotype in dm11.1 with an ‘aa’ genotype in dm9.1 was greater than the reciprocal (‘aa’ genotype in dm11.1 with ‘bb’ dm9.1) (Fig 5D). In contrast, comparison of F2 means for reciprocal, opposing homozygous genotype combination in dm11.1 and dm14.1 resulted in similar mean disease severity (Fig 5E). In both cases, a relatively high SE (0.07–0.10) demonstrates variability in downy mildew response when the recessive ‘bb’ dm11.1 genotype is present with the ‘aa’ genotype of either minor QTL, resulting in some loss of resistance response within the population. Chi-square goodness of fit to complementary and recessive epistatic gene models in F2 and backcross generations from phenotypic data suggested dominant effects were needed to confer resistance. A dominant (‘ab’) x dominant (‘ab’) gene effect was thus expected but unsupported (p = 0.769) [19]. QTL in this study provide evidence for a more complex gene model with a major, single dominant and two minor, additive QTL (Table 3).
It is clear that applied resistance breeding would benefit from ensuring germplasm have the dm11.1 ‘aa’ (MRI) genotype as a heterozygous locus at dm11.1 will result in loss of resistance through segregation during self-pollinated seed propagation. Given the potential increased susceptibility effect of the ‘b’ allele, its removal from each QTL (‘aa’ genotype) would be preferable to ensure a high level of stable resistance. A similar model was identified in the GR x Ice RIL population in which the homozygous Iceberg genotype at two QTL conferred significantly higher resistance to downy mildew [70].
Quantitative and qualitative forms of downy mildew resistance have been reported in multiple plant species such as Cucumis spp. where sources of host resistance have been identified and characterized for decades [71]. Quantitative resistance, while less susceptible to breakdown, is subject to greater variation [71] across environments as was observed for the QTL dm9.1 and dm14.1 detected in this study (9.4–34.5% PVE). A similar range of PVE (15–30%) for downy mildew response was reported for two additive QTL in cucumber F2:3 families across three environments [40]. Qualitative downy mildew resistance, while more prone to breakdown, is less vulnerable to environmental interaction and often associated with gene-for-gene host-pathogen interaction. Major QTL dm11.1 was detected in a distal region of LG 11 with resistance conferred from grandparent MRI by the dominant ‘a’ allele. The downy mildew resistance dominant locus Rpv3 was also detected in a distal chromosomal region of V. vinifera ‘Bianca’ known to contain NBS-LRR gene clusters [41]. Development of a de novo metatranscriptomics pipeline [72] for O. basilicum provides a platform for identification of resistant gene motifs, which could potentially be mapped to genomic regions containing QTL such as dm11.1 for functional characterization.
RADseq approaches have changed the landscape of linkage and QTL mapping for non-model plant species by introducing low-cost, high-throughput, de novo SNP discovery. The inexpensive acquisition of tens or hundreds of thousands of SNP markers allow researchers working with poorly understood genomes to be ‘picky’, applying strict filtration to retain >1,000 high-quality SNPs with predictable segregation patterns. This reduces the burden to generate large numbers of labor-intensive, costly markers such as SSRs, instead utilizing a smaller subset to serve as ‘anchor’ markers for subsequent map comparison. This genotyping approach high-SNP/low-SSR genotyping approach has facilitated map construction in peach [55], strawberry [56], sesame [73] and lentil [74]. In this study, the power of this approach is further demonstrated through the development of a sweet basil linkage map and QTL detection.
Conclusions
Genetic study of non-model, horticultural species such as sweet basil are often neglected due to perceived low economic importance; however, this renders such crops vulnerable to rapid and wide-spread decline upon introduction of new plant pathogens. P. belbahrii now causes worldwide economic losses with no available resistant sweet basil cultivars. In this study, a set of EST-SSR markers were developed and mapped in the MRI x SB22 F2 sweet basil mapping population providing molecular evidence of disomic inheritance. Effective filtration of ddRADseq SNP markers generated 1,847 bi-allelic, polymorphic markers in the absence of a reference genome. This novel genotyping approach facilitated construction of the first linkage map for sweet basil. The utility of this map was demonstrated through identification of one major and two minor QTL associated with downy mildew resistance, largely supporting a previous report using phenotypic data only. Results provide the first steps towards the development of molecular tools for accelerated sweet basil breeding strategies.
Supporting information
(XLSX)
(XLSX)
Distribution (bar graph) of Stacks, SNPs and Polymorphic Loci identified in the MRIxSB22 F2 population.
(PDF)
Acknowledgments
Funds for this research and in support of a graduate assistantship/doctoral degree for RMP were provided by United States United Department of Agriculture (USDA) Special Crops Research Initiative Award No.2011-51181-30646. Additional funds were provided by USDA National Institute of Food and Agriculture Award No. 2016-68004-24931, and the Binational Agricultural Research & Development (BARD) Award No. US-4947-16R. We thank the New Jersey Agricultural Experiment Station, the New Use Agriculture and Natural Plant Products Program and the Rutgers Cooperative Extension Service for also providing funds in support of this work (NIFA HATCH project reports NJAES 12131 and 1005685). We thank Dr. Todd Wehner (North Carolina State University) for his advice throughout the course of this project. We thank Mark Peacos, and Ed Dager for their assistance with greenhouse and field work.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funds for this research and in support of a graduate assistantship/doctoral degree for RMP were provided by United States United Department of Agriculture (USDA) Special Crops Research Initiative Award No. 2011-51181-30646. Additional funds were provided by USDA National Institute of Food and Agriculture Award No. 2016-68004-24931, and the Binational Agricultural Research & Development (BARD) Award No. US-4947-16R. We thank the New Jersey Agricultural Experiment Station, the New Use Agriculture and Natural Plant Products Program and the Rutgers Cooperative Extension Service for also providing funds in support of this work (NIFA HATCH project reports NJAES 12131 and 1005685).
References
- 1.Simon J.E, Quinn J, Murray JG. Basil: A Source of Essential Oils In: Janick J.; Simon JE, editor. Advances in New Crops. Portland, OR: Timber Press; 1990. pp. 484–489. [Google Scholar]
- 2.USDA. United States Department of Agriculture Census of Agriculture. https://www.agcensus.usda.gov. (Accessed October 1, 2016).
- 3.Belbahri L, Calmin G, Pawlowski J, Lefort F. Phylogenetic analysis and real time PCR detection of a presumbably undescribed Peronospora species on sweet basil and sage. Mycol Res. 2005;109: 1276–1287. doi: 10.1017/S0953756205003928 [DOI] [PubMed] [Google Scholar]
- 4.Roberts PD, Raid RN, Harmon PF, Jordan SA, Palmateer AJ. First Report of Downy Mildew Caused by a Peronospora sp. on Basil in Florida and the United States. Plant Dis. Scientific Societies; 2009;93: 199 doi: 10.1094/PDIS-93-2-0199B [DOI] [PubMed] [Google Scholar]
- 5.Wyenandt C a, Simon JE, Pyne RM, Homa K, McGrath MT, Zhang S, et al. Basil Downy Mildew (Peronospora belbahrii): Discoveries and Challenges Relative to Its Control. Phytopathology. 2015;105: 885–94. doi: 10.1094/PHYTO-02-15-0032-FI [DOI] [PubMed] [Google Scholar]
- 6.Blomquist CL, Rooney-Latham S, Nolan PA. First Report of Downy Mildew on Field-Grown Sweet Basil Caused by a Peronospora sp. in San Diego County, California. Plant Dis. Scientific Societies; 2009;93: 968 doi: 10.1094/PDIS-93-9-0968A [DOI] [PubMed] [Google Scholar]
- 7.Cohen Y, Vaknin M, Ben-Naim Y, Rubin AE, Galperin M. First Report of the Occurrence and Resistance to Mefenoxam of Peronospora belbahrii, Causal Agent of Downy Mildew of Basil (Ocimum basilicum) in Israel . Plant Dis. 2013; 97: 692 doi: 10.1094/PDIS-12-12-1126-PDN [DOI] [PubMed] [Google Scholar]
- 8.Garibaldi A, Minuto A, Minuto G, Gullino ML. First Report of Downy Mildew on Basil (Ocimum basilicum) in Italy. Plant Dis. Scientific Societies; 2004;88: 312 doi: 10.1094/PDIS.2004.88.3.312A [DOI] [PubMed] [Google Scholar]
- 9.Kanetis L, Vasiliou A, Neophytou G, Samouel S, Tsaltas D. First Report of Downy Mildew Caused by Peronospora belbahrii on Sweet Basil (Ocimum basilicum) in Cyprus. Plant Dis. Scientific Societies; 2013;98: 283 doi: 10.1094/PDIS-07-13-0759-PDN [DOI] [PubMed] [Google Scholar]
- 10.McLeod A, Coertze S, Mostert L. First Report of a Peronospora Species on Sweet Basil in South Africa. Plant Dis. Scientific Societies; 2006;90: 1115 doi: 10.1094/PD-90-1115A [DOI] [PubMed] [Google Scholar]
- 11.Nagy G, Horváth A. Occurrence of Downy Mildew Caused by Peronospora belbahrii on Sweet Basil in Hungary. Plant Dis. Scientific Societies; 2011;95: 1034 doi: 10.1094/PDIS-04-11-0329 [DOI] [PubMed] [Google Scholar]
- 12.Saude C, Westerveld S, Filotas M, McDonald MR. First Report of Downy Mildew Caused by Peronospora belbahrii on Basil (Ocimum spp.) in Ontario. Plant Dis. Scientific Societies; 2013;97: 1248 doi: 10.1094/PDIS-01-13-0026-PDN [DOI] [PubMed] [Google Scholar]
- 13.Kong XY, Wang S, Wan SL, Xiao CL, Luo F, Liu Y. First Report of Downy Mildew on Basil (Ocimum basilicum) in China. Plant Dis. Scientific Societies; 2015;99: 1642 doi: 10.1094/PDIS-01-15-0077-PDN [Google Scholar]
- 14.Pintore I, Gilardi G, Gullino ML, Garibaldi A. Detection of mefenoxam-resistant strains of Peronospora belbahrii, the causal agent of basil downy mildew, transmitted through infected seeds. Phytoparasitica; 2016; doi: 10.1007/s12600-016-0538-x [Google Scholar]
- 15.Pyne RM, Koroch AR, Wyenandt CA, Simon JE. A Rapid Screening Approach to Identify Resistance to Basil Downy Mildew (Peronospora belbahrii). HortScience. 2014;49: 1041–1045. [Google Scholar]
- 16.Djalali Farahani-Kofoet R, Römer P, Grosch R. Systemic spread of downy mildew in basil plants and detection of the pathogen in seed and plant samples. Mycol Prog. 2012;11: 961–966. doi: 10.1007/s11557-012-0816-z [Google Scholar]
- 17.Ben-Naim Y, Lidan F, Cohen Y. Resistance against basil downy mildew in Ocimum species. Phytopathology. 2015; 2710 doi: 10.1094/PHYTO-11-14-0295-R [DOI] [PubMed] [Google Scholar]
- 18.Pushpangadan P. and Sobti S.N. Cytogenetical studies in the genus Ocimum. I. Origin of O. americanum, cytotaxonomical and experimental proof. Cytologia. 1982. 47: 575–583. [Google Scholar]
- 19.Pyne RM, Koroch AR, Wyenandt CA, Simon JE. Inheritance of Resistance to Downy Mildew in Sweet Basil. J Am Soc Hortic Sci. 2015;140: 396–403. [Google Scholar]
- 20.Koroch AR, Wang W, Michael TP, Dudai N, Simon JE, Belanger FC. Estimation of nuclear DNA content of cultivated Ocimum species by using flow cytometry. Isr J Plant Sci. 2010;58: 183–189. doi: 10.1560/IJPS.59.3–4.183 [Google Scholar]
- 21.Davey JW., Hohenlohe PA, Etter PD, Boone JQ, Catchen JM, Blaxter ML. Genome-wide genetic marker discovery and genotyping using next-generation sequencing. Nat Rev Genet. 2011;12: 499–510. doi: 10.1038/nrg3012 [DOI] [PubMed] [Google Scholar]
- 22.Clevenger J, Chavarro C, Pearl SA, Ozias-Akins P, Jackson SA. Single nucleotide polymorphism identification in polyploids: A review, example, and recommendations. Mol Plant; 2015;8: 831–846. doi: 10.1016/j.molp.2015.02.002 [DOI] [PubMed] [Google Scholar]
- 23.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, et al. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. Public Library of Science; 2008;3: e3376 doi: 10.1371/journal.pone.0003376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One. 2011;6: 1–10. doi: 10.1371/journal.pone.0019379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA. Stacks: An analysis tool set for population genomics. Mol Ecol. 2013;22: 3124–3140. doi: 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS One. 2014;9 doi: 10.1371/journal.pone.0090346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Christensen K a, Brunelli JP, Lambert MJ, DeKoning J, Phillips RB, Thorgaard GH. Identification of single nucleotide polymorphisms from the transcriptome of an organism with a whole genome duplication. BMC Bioinformatics. 2013;14: 325 doi: 10.1186/1471-2105-14-325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hohenlohe PA, Amish SJ, Catchen JM, Allendorf FW, Luikart G. Next-generation RAD sequencing identifies thousands of SNPs for assessing hybridization between rainbow and westslope cutthroat trout. Mol Ecol Resour. 2011;11: 117–122. doi: 10.1111/j.1755-0998.2010.02967.x [DOI] [PubMed] [Google Scholar]
- 29.Li H, Younas M, Wang X, Li X, Chen L, Zhao B, et al. Development of a core set of single-locus SSR markers for allotetraploid rapeseed (Brassica napus L.). Theor Appl Genet. 2013;126: 937–947. doi: 10.1007/s00122-012-2027-z [DOI] [PubMed] [Google Scholar]
- 30.van Dijk T, Noordijk Y, Dubos T, Bink MC, Meulenbroek BJ, Visser RG, et al. Microsatellite allele dose and configuration establishment (MADCE): an integrated approach for genetic studies in allopolyploids. BMC Plant Biol. BioMed Central Ltd; 2012;12: 25 doi: 10.1186/1471-2229-12-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logan-Young CJ, Yu JZ, Verma SK, Percy RG, Pepper AE. SNP Discovery in Complex Allotetraploid Genomes (Gossypium spp., Malvaceae) Using Genotyping by Sequencing. Appl Plant Sci. 2015;3: 1400077 doi: 10.3732/apps.1400077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nation RG, Janick J, Simon JE. Estimation of outcrossing in basil. Hortic Sci. 1992;27: 1221–1222. [Google Scholar]
- 33.Khosla MK, Tawi J, Group S. Karyomorphological Studies in Genus Ocimum. Cytologia (Tokyo). 1985;50: 253–263. [Google Scholar]
- 34.Phippen WB, Simon JE. Anthocyanin inheritance and instability in purple basil (Ocimum basilicum L.). J Hered. 2000;91: 289–296. doi: 10.1093/jhered/91.4.289 [DOI] [PubMed] [Google Scholar]
- 35.Chaimovitsh D.;Dudai N., Putievesky E.; Ashri A. Inheritance of Resistance to Fusarium Wilt in Sweet Basil. Phytopathology. 1983;90: 58–60. doi: 10.2135/cropsci1983.0011183X002300010010x [DOI] [PubMed] [Google Scholar]
- 36.Jeuken M, Lindhout P. Lactuca saligna, a non-host for lettuce downy mildew (Bremia lactucae), harbors a new race-specific Dm gene and three QTLs for resistance. Theor Appl Genet. 2002;105: 384–391. doi: 10.1007/s00122-002-0943-z [DOI] [PubMed] [Google Scholar]
- 37.Zhang NW, Lindhout P, Niks RE, Jeuken MJW. Genetic dissection of Lactuca saligna nonhost resistance to downy mildew at various lettuce developmental stages. Plant Pathol. 2009;58: 923–932. doi: 10.1111/j.1365-3059.2009.02066.x [Google Scholar]
- 38.den Boer E, Pelgrom KTB, Zhang NW, Visser RGF, Niks RE, Jeuken MJW. Effects of stacked quantitative resistances to downy mildew in lettuce do not simply add up. Theor Appl Genet. 2014;127: 1805–1816. doi: 10.1007/s00122-014-2342-7 [DOI] [PubMed] [Google Scholar]
- 39.Irish BM, Correll JC, Feng C, Bentley T, de Los Reyes BG. Characterization of a resistance locus (Pfs-1) to the spinach downy mildew pathogen (Peronospora farinosa f. sp. spinaciae) and development of a molecular marker linked to Pfs-1. Phytopathology. 2008;98: 894–900. doi: 10.1094/PHYTO-98-8-0894 [DOI] [PubMed] [Google Scholar]
- 40.Wang Y, VandenLangenberg K, Wehner TC, Kraan PAG, Suelmann J, Zheng X, et al. QTL mapping for downy mildew resistance in cucumber inbred line WI7120 (PI 330628). Theor Appl Genet. 2016;7120 doi: 10.1007/s00122-016-2719-x [DOI] [PubMed] [Google Scholar]
- 41.Bellin D, Peressotti E, Merdinoglu D, Wiedemann-Merdinoglu S, Adam-Blondon AF, Cipriani G, et al. Resistance to Plasmopara viticola in grapevine “Bianca” is controlled by a major dominant gene causing localised necrosis at the infection site. Theor Appl Genet. 2009;120: 163–176. doi: 10.1007/s00122-009-1167-2 [DOI] [PubMed] [Google Scholar]
- 42.Huang X, Madan a. CAP 3: A DNA sequence assembly program. Genome Res. 1999;9: 868–877. doi: 10.1101/gr.9.9.868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kofler R, Schlötterer C, Lelley T. SciRoKo: A new tool for whole genome microsatellite search and investigation. Bioinformatics. 2007;23: 1683–1685. doi: 10.1093/bioinformatics/btm157 [DOI] [PubMed] [Google Scholar]
- 44.Rozen S, Skaletksy H. Primer 3 on the WWW for general uses and for biologist programmers. Methods Mol Biol. 2000; 365–386. [DOI] [PubMed] [Google Scholar]
- 45.Schuelke M. An economic method for the fluorescent labeling of PCR fragments. Nat Biotechnol. 2000;18: 233–234. doi: 10.1038/72708 [DOI] [PubMed] [Google Scholar]
- 46.Brownstein MJ, Carpten JD, Smith JR. Modulation of non-templated nucleotide addition by Taq DNA polymerase: Primer modifications that facilitate genotyping. Biotechniques. 1996;20: 1004–1010. doi: 10.2144/000113156 [DOI] [PubMed] [Google Scholar]
- 47.Poland JA, Brown PJ, Sorrells ME, Jannink JL. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Catchen JM, Amores A., Hohenlohe P, Cresko W, Postlethwait JH, De Koning D-J. Stacks: Building and Genotyping Loci De Novo From Short-Read Sequences. Genes|Genomes|Genetics. 2011;1: 171–182. doi: 10.1534/g3.111.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van Ooijen JW. JoinMap ® 4. JoinMap.; Software for the calculation of genetic linkage maps in experimental populations. Version 4.1. Wageningen, Netherlands. Kyazma; 2006.
- 50.Jansen J, De Jong AG, Van Ooijen JW. Constructing dense genetic linkage maps. Theor Appl Genet. 2001;102: 1113–1122. doi: 10.1007/s001220000489 [Google Scholar]
- 51.Wyenandt CA, Simon JE, McGrath MT, Ward DL. Susceptibility of basil cultivars and breeding lines to downy mildew (Peronospora belbahrii). HortScience. 2010;45: 1416–1419. [Google Scholar]
- 52.Broman KW. Review of statistical methods for QTL mapping in experimental crosses. Lab Anim (NY). 2001;30: 44–52. [PubMed] [Google Scholar]
- 53.Broman KW, Wu H, Sen Ś, Churchill GA. R/qtl: QTL mapping in experimental crosses. Bioinformatics. 2003;19: 889–890. doi: 10.1093/bioinformatics/btg112 [DOI] [PubMed] [Google Scholar]
- 54.Henning JA, Gent DH, Twomey MC, Townsend MS, Pitra NJ, Matthews PD. Genotyping-by-sequencing of a bi-parental mapping population segregating for downy mildew resistance in hop (Humulus lupulus L.). Euphytica. Springer Netherlands; 2016;208: 545–559. doi: 10.1007/s10681-015-1600-3 [Google Scholar]
- 55.Bielenberg DG, Rauh B, Fan S, Gasic K, Abbott AG, Reighard GL, et al. Genotyping by sequencing for SNP-based linkage map construction and QTL analysis of chilling requirement and bloom date in peach [Prunus persica (L.) Batsch]. PLoS One. 2015;10: 1–14. doi: 10.1371/journal.pone.0139406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davik J, Sargent DJ, Brurberg MB, Lien S, Kent M, Alsheikh M. A ddRAD based linkage map of the cultivated Strawberry, Fragaria xananassa. PLoS One. 2015;10: 1–10. doi: 10.1371/journal.pone.0137746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rastogi S, Kalra A, Gupta V, Khan F, Lal RK, Tripathi AK, et al. Unravelling the genome of Holy basil: an “incomparable” “elixir of life” of traditional Indian medicine. BMC Genomics. 2015;16: 413 doi: 10.1186/s12864-015-1640-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7 doi: 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Isobe SN, Hirakawa H, Sato S, Maeda F, Ishikawa M, Mori T, et al. Construction of an integrated high density simple sequence repeat linkage map in cultivated strawberry (Fragariax ananassa) and its applicability. DNA Res. 2013;20: 79–92. doi: 10.1093/dnares/dss035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alheit KV, Reif JC, Maurer HP, Hahn V, Weissmann EA, Miedaner T, et al. Detection of segregation distortion loci in triticale (x Triticosecale Wittmack) based on a high-density DArT marker consensus genetic linkage map. BMC Genomics. 2011;12: 380 doi: 10.1186/1471-2164-12-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou X, Xia Y, Ren X, Chen Y, Huang L, Huang S, et al. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq). BMC Genomics. 2014;15: 351 doi: 10.1186/1471-2164-15-351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fishman L, Willis JH. A novel meiotic drive locus almost completely distorts segregation in Mimulus (monkeyflower) hybrids. Genetics. 2005;169: 347–353. doi: 10.1534/genetics.104.032789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Taylor DR, Ingvarsson PK. Common features of segregation distortion in plants and animals. Genetica. 2003;117: 27–35. doi: 10.1023/A:1022308414864 [DOI] [PubMed] [Google Scholar]
- 64.Manrique-Carpintero NC, Coombs JJ, Veilleux RE, Buell CR, Douches DS. Comparative Analysis of Regions with Distorted Segregation in Three Diploid Populations of Potato. G3 Genes|Genomes|Genetics. 2016;6: 2617–2628. doi: 10.1534/g3.116.030031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reflinur, Kim B, Jang SM, Chu S-H, Bordiya Y, Akter MB, et al. Analysis of segregation distortion and its relationship to hybrid barriers in rice. Rice (N Y). 2014;7: 3 doi: 10.1186/s12284-014-0003-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Guo Y, Wu Y, Anderson JA, Moss JQ, Zhu L. Disomic inheritance and segregation distortion of SSR markers in two populations of Cynodon dactylon (L.) Pers. var. dactylon. PLoS One. 2015;10: 1–10. doi: 10.1371/journal.pone.0136332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gonzalo MJ, Oliver M, Garcia-Mas J, Monfort A, Dolcet-Sanjuan R, Katzir N, et al. Simple-sequence repeat markers used in merging linkage maps of melon (Cucumis melo L.). Theor Appl Genet. 2005;110: 802–811. doi: 10.1007/s00122-004-1814-6 [DOI] [PubMed] [Google Scholar]
- 68.Gang DR, Wang J, Dudareva N, Nam KH, Simon JE, Lewinsohn E, et al. An Investigation of the Storage and Biosynthesis of Phenylpropenes in Sweet Basil. Plant Physiol. 2001;125: 539–555. doi: 10.1104/pp.125.2.539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vicente JG, Gunn ND, Bailey L, Pink DAC, Holub EB. Genetics of resistance to downy mildew in Brassica oleracea and breeding towards durable disease control for UK vegetable production. Plant Pathol. 2012;61: 600–609. doi: 10.1111/j.1365-3059.2011.02539.x [Google Scholar]
- 70.Simko I, Atallah AJ, Ochoa OE, Antonise R, Galeano CH, Truco MJ, et al. Identification of QTLs conferring resistance to downy mildew in legacy cultivars of lettuce. Sci Rep. 2013;3: 2875 doi: 10.1038/srep02875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olczak-Woltman H, Marcinkowska J, Niemirowicz-Szczytt K. The genetic basis of resistance to downy mildew in Cucumis spp.-latest developments and prospects. J Appl Genet. 2011;52: 249–255. doi: 10.1007/s13353-011-0030-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guo L, Allen KS, Deiulio GA, Zhang Y, Madeiras AM, Wick RL, et al. A de-novo-assembly-based Data Analysis Pipeline for Plant Obligate Parasite Metatranscriptomic Studies. Front Plant Sci. 2016;7: 925 doi: 10.3389/fpls.2016.00925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu K, Liu H, Yang M, Tao Y, Ma H, Wu W, et al. High-density genetic map construction and QTLs analysis of grain yield-related traits in Sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014;14: 274 doi: 10.1186/s12870-014-0274-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ates D, Sever T, Aldemir S, Yagmur B, Temel HY, Kaya HB, et al. Identification QTLs controlling genes for se uptake in lentil seeds. PLoS One. 2016;11 doi: 10.1371/journal.pone.0149210 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
Distribution (bar graph) of Stacks, SNPs and Polymorphic Loci identified in the MRIxSB22 F2 population.
(PDF)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.